(Carregando Índice)... (Carregando Índice)... |
Você está em:
Inicial  acp-medicine Hematologia
acp-medicine Hematologia
Última revisão: 28/06/2012
Comentários de assinantes: 0
Robert T. Means Jr., MD, FACP
Professor of Internal Medicine and Executive Vice Dean, University of Kentucky College of Medicine, Lexington, KY
Artigo original: Means Jr., RT. Red blood cell function and disorders of iron metabolism. ACP Medicine. 2008;1-18.
[The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2011 Decker Intellectual Properties Inc. All Rights Reserved.]
Agradecimentos: Figura 1 – Seward Hung. Figura 2 – Marcia Kammerer. Figura 3 – Seward Hung. Figura 4 – Dimitry Schidlovsky.
Tradução: Soraya Imon de Oliveira.
Revisão técnica: Dr. Euclides Furtado de Albuquerque Cavalcanti.
A hemácia, ou eritrócito, consiste em um disco bicôncavo anucleado, cujo diâmetro mede aproximadamente 7 mcm. A principal função da hemácia é trocar dióxido de carbono por oxigênio na circulação pulmonar, trocar oxigênio por dióxido de carbono nos tecidos periféricos e transportar o dióxido de carbono de volta para os pulmões. A composição físico-química das hemácias otimiza esta função. O estado de oxigenação tecidual, por sua vez, regula a produção de hemácias.
A estrutura das hemácias é projetada para otimizar o trânsito através da microvasculatura, com a finalidade de trocar oxigênio. A membrana celular do eritrócito é uma estrutura flexível, constituída por uma bicamada lipídica que contém proteínas integrais. Estas proteínas ancoram a membrana a um esqueleto proteico subjacente, que mantém a forma discoidal bicôncava da célula.1 Esta forma otimiza a passagem da célula pela circulação e facilita as trocas gasosas através do endotélio capilar. As vias metabólicas ativas nas hemácias também sustentam a função de troca gasosa. A geração de 2,3-difosfoglicerato (2,3-DFG) regula a afinidade entre a hemoglobina e o oxigênio,2 enquanto outros mecanismos metabólicos dão suporte à sobrevida da hemácia mantendo condições ideais de ambiente redox intracelular e de estabilidade osmótica.3,4 Anormalidades envolvendo estas enzimas acarretam hemólise via produção de danos oxidativos ou osmóticos.
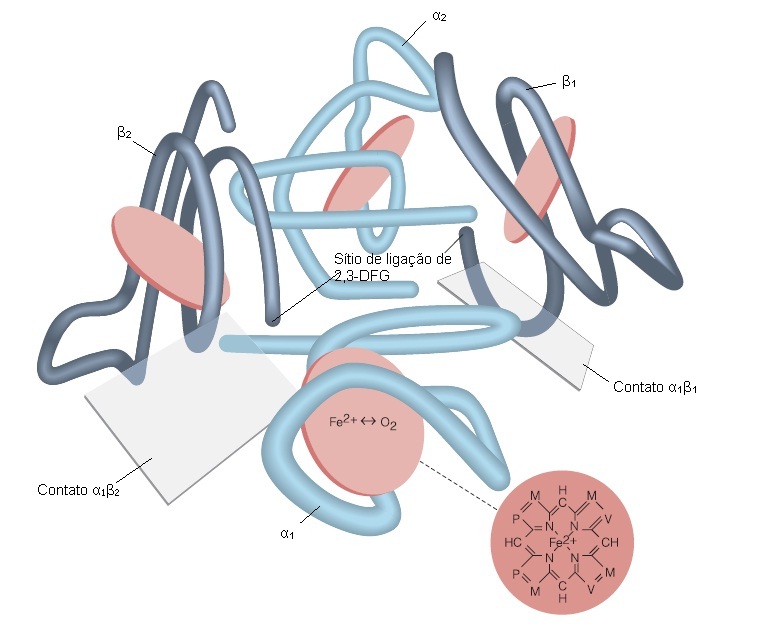
A hemoglobina é a principal proteína intracelular da hemácia e a molécula responsável pela verdadeira função de troca gasosa. A hemoglobina é uma molécula esférica, composta por 2 pares de cadeias de globina diferentes, com um grupo heme, a ferroprotoporfirina IX, ligado de forma covalente a um sítio específico em cada cadeia.5 A principal hemoglobina do adulto, a hemoglobina A, é formada a partir de um par de cadeias alfa (cada uma contendo 141 aminoácidos) e um par de cadeias beta (contendo 146 aminoácidos cada), sendo representada como alfa-2-beta-2 [Figura 1].

Figura 1. Modelo da molécula de hemoglobina, mostrando o relativo alinhamento das cadeias alfa (azul claro) e das cadeias beta (azul escuro). O 2,3-difosfoglicerato (2,3-DFG), um intermediário glicolítico, liga-se na cavidade central da hemoglobina e estabiliza a forma desoxigenada estabelecendo ligações cruzadas com as cadeias beta e, assim, diminuindo a afinidade pelo oxigênio da hemoglobina. Note-se que as cadeias alfa e beta estão em contato em 2 pontos. Na oxigenação, o movimento do átomo de ferro internamente no plano do grupo heme (discos vermelhos) parece desencadear outras alterações estruturais nas subunidades alfa e beta, conforme a molécula assume a conformação oxigenada. Há um deslizamento ao nível da interface alfa-2-beta-2, e o espaço existente entre as 2 cadeias beta é estreitado na oxiemoglobina. O detalhe mostra a estrutura do heme.
M = metil. P = ácido propiônico. V = vinil.
A configuração da hemoglobina muda com a oxigenação e desoxigenação.2,6 A configuração desoxi da hemoglobina é estabilizada por meio da ligação de prótons e 2,3-DFG, um ânion altamente carregado. Com a oxigenação de uma subunidade, estas ligações são sequencialmente quebradas, e a alteração resultante na estrutura terciária aumenta a afinidade pelo oxigênio das subunidades não oxigenadas remanescentes (cooperatividade).5 Conforme o oxigênio é liberado nos tecidos, uma reversão deste processo diminui a afinidade pelo oxigênio, facilitando sua liberação.
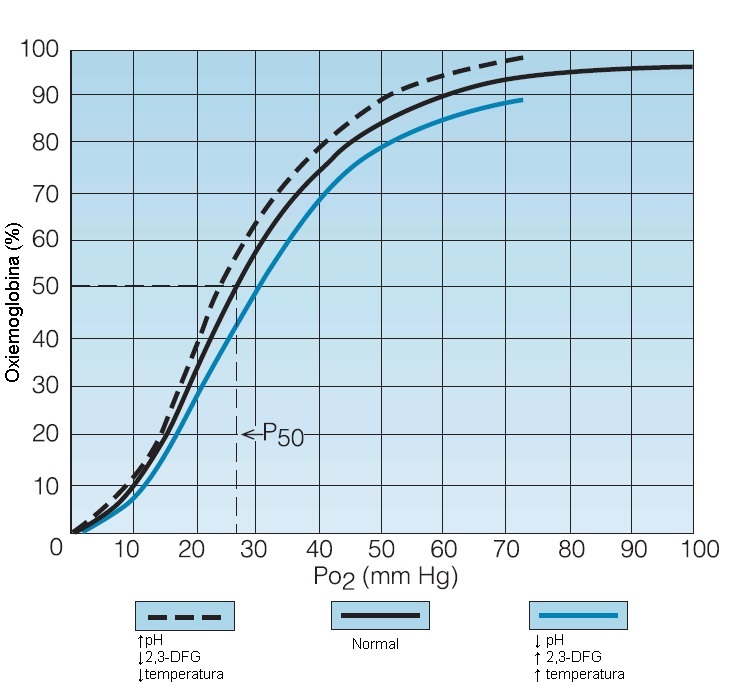
A relação existente entre a tensão de oxigênio (Po2) a que as hemácias (ou uma solução de hemoglobina) são expostas e o grau de saturação da hemoglobina com oxigênio descreve uma curva sigmoide referida como curva de dissociação oxigênio-hemoglobina5 [Figura 2]. Ao nível do mar e com uma Po2 da ordem de 90 mmHg, o grau de saturação da hemoglobina é de aproximadamente 97%. A uma Po2 de 60 mmHg, a hemoglobina encontra-se saturada em cerca de 90%, enquanto uma Po2 de 30 mmHg representa uma saturação de 60%. Estas relações constituem a base da utilização do monitoramento não invasivo da saturação do oxigênio em substituição à determinação da Po2 dos gases no sangue arterial.
Figura 2. Curva de dissociação oxigênio-hemoglobina normal (linha preta sólida) é deslocada por alterações de temperatura, pH e concentração intracelular de 2,3-difosfoglicerato (2,3-DFG).
P50 = saturação de oxigênio de 50%. Po2 = tensão de oxigênio.
As curvas de dissociação oxigênio-hemoglobina são tipicamente caracterizadas pela Po2 em que a saturação de oxigênio é 50% (P50). A P50 é um indicador da afinidade pelo oxigênio e, sob condições fisiológicas normais, vale aproximadamente 26 mmHg para a hemoglobina normal. Quando a afinidade pelo oxigênio aumenta, a P50 diminui (ou seja, a hemoglobina retém mais oxigênio a qualquer Po2). Uma forma de expressar isto é dizer que a curva de dissociação oxigênio-hemoglobina está “deslocada para a esquerda”.
As alterações de pH modificam a afinidade da hemoglobina pelo oxigênio (efeito de Bohr).2 Nos pulmões, a eliminação do dióxido de carbono eleva o pH, aumentando a afinidade e a captação do oxigênio. Já nos tecidos, a captação de dióxido de carbono diminui o pH, diminuindo a afinidade pelo oxigênio e, assim, facilitando sua liberação.
O intermediário glicolítico 2,3-DFG, que está presente em eritrócitos maduros mais ou menos na mesma concentração intracelular da hemoglobina, constitui o principal regulador alostérico da afinidade pelo oxigênio.5 As concentrações de 2,3-DFG estão inversamente relacionadas à afinidade pelo oxigênio. Na hipóxia aguda, as concentrações de 2,3-DFG aumentam em questão de horas e isto acarreta a diminuição da afinidade da hemoglobina pelo oxigênio (desloca a curva de dissociação oxigênio-hemoglobina para a direita), permitindo a intensificação da liberação de oxigênio. O aumento da concentração de 2,3-DFG promove a distribuição de oxigênio aos tecidos, mas também impede a aquisição de oxigênio nos pulmões. Portanto, esta é uma resposta efetiva de curta duração. Contudo, seus benefícios dependem da distribuição adequada de oxigênio aos pulmões e de uma função cardiopulmonar efetiva.
A temperatura também altera a afinidade da hemoglobina pelo oxigênio. Uma elevação da temperatura aumenta a P50, deslocando a curva de dissociação para a direita e intensificando a liberação de oxigênio. Uma diminuição da temperatura exerce o efeito contrário. Outros fatores capazes de afetar a curva de dissociação oxigênio-hemoglobina são a concentração intracelular de carboxiemoglobina (a afinidade pelo oxigênio aumenta com o aumento da proporção de carboxiemoglobina) e de dióxido de carbono (de maneira independente e, todavia, paralela ao seu efeito sobre o pH).
Para que o oxigênio seja efetivamente distribuído aos tecidos, é preciso haver adequada perfusão sanguínea tecidual (que requer a existência de uma infraestrutura cardiovascular satisfatória) e concentração apropriada de hemácias no sangue (expressa como hematócrito ou concentração sanguínea de hemoglobina). A produção de hemácias está sujeita à regulação por feedback (retroalimentação) pela oxigenação tecidual. Um hematócrito diminuído é percebido como hipóxia pelas células renais peritubulares produtoras de eritropoetina. As subunidades alfa e beta do fator hipóxia-induzível-1 (HIF-1 – em inglês, hypoxia-inducible factor-1), então, sofrem dimerização e translocam-se para o núcleo, onde promovem transcrição da eritropoetina. Quando o hematócrito aumenta, este processo é desligado.7 Pacientes que possuem hemoglobinas de alta afinidade, sejam congênitas ou adquiridas, desenvolvem policitemia porque a diminuída liberação de oxigênio ao nível renal é percebida como anemia pelas células produtoras de eritropoetina.8
Os eritrócitos também contribuem para a regulação da perfusão tecidual. A hemoglobina varre e libera óxido nítrico sob as mesmas condições que estão associadas à liberação de oxigênio. Os efeitos vasodilatadores do óxido nítrico, podem, então, intensificar a perfusão tecidual e a distribuição do oxigênio.5,9
Após distribuir o oxigênio, a hemoglobina liga-se ao dióxido de carbono.10 A desoxiemoglobina apresenta uma afinidade maior pelo dióxido de carbono do que a oxiemoglobina, o que facilita o descarregamento do dióxido de carbono dos tecidos e sua excreção pulmonar.10 A maior parte do dióxido de carbono oriundo dos capilares teciduais é transportada para os pulmões na forma de bicarbonato, sendo que aproximadamente 10% é transportado como complexo carbamino ligado de maneira reversível às cadeias de globina.
Desde a última edição deste texto, avanços consideráveis foram alcançados em termos de compreensão da biologia do metabolismo do ferro e do modo como este é alterado nas doenças. No corpo, o ferro transporta e armazena oxigênio, transporta elétrons, catalisa reações no metabolismo oxidativo e sustenta o crescimento e a proliferação celulares. Apesar de o aspecto predominante no distúrbio clínico da deficiência de ferro ser a subprodução de hemoglobina, o corpo também é incapaz de produzir quantidade suficiente de outros complexos de ferro-porfirina, metaloenzimas e outros compostos contendo ferro, para sustentar as funções normais. Isto é particularmente relevante no caso das crianças e de indivíduos desnutridos.11 No outro extremo do espectro, o excesso de ferro nos tecidos pode catalisar reações de produção de radicais livres que podem danificar membranas celulares, proteínas e ácidos nucleicos, levando a uma progressiva disfunção celular e orgânica.
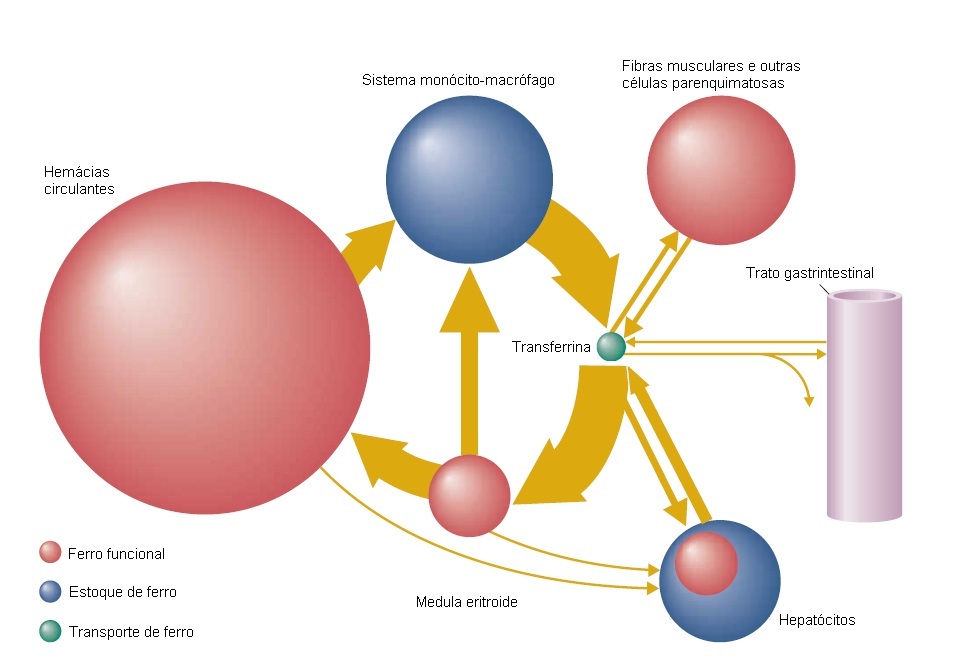
A concentração de ferro no corpo humano é cuidadosamente regulada e mantida em níveis normais de cerca de 40 mg de Fe/kg nas mulheres e 50 mg de Fe/kg nos homens. O equilíbrio do ferro resulta da diferença entre a quantidade de ferro captada pelo corpo e a quantidade perdida [Figura 3]. Os sítios fisiológicos de perda de ferro são as células mortas esfoliadas da pele ou mucosas, bem como a menstruação nas mulheres em idade fértil. Assim, como os seres humanos são incapazes de excretar ferro em excesso de modo regulado, o equilíbrio de ferro é fisiologicamente regulado por meio do controle de sua absorção. Os dois fatores principais a influenciar a absorção de ferro são os níveis de estoque corporal e a extensão da eritropoese.12 Quando os estoques de ferro aumentam, a absorção diminui. Do mesmo modo, se as reservas diminuem, a absorção aumenta. A absorção do ferro também aumenta com o aumento da atividade eritropoética, sobretudo quando a eritropoese é inefetiva. A maior parte do ferro existente no corpo encontra-se na forma de hemoglobina e está contida no eritron, que é um pool funcional constituído pela massa de hemácias circulantes e precursores eritroides da medula óssea. As hemácias circulantes contêm mais de 95% do total de ferro em forma de eritron. A via predominante de fluxo de ferro interno é um fluxo de sentido único, a partir da proteína de transporte de ferro plasmática transferrina para o eritron e, então, através do sistema monócito-macrófago e de volta à transferrina [Figura 3]. O eritron usa cerca de 80% do ferro que passa pelo compartimento da transferrina diariamente. O ferro restante que passa por este compartimento segue em grande parte para os estoques, com proporções menores sendo utilizadas para síntese da mioglobulina ou na síntese de outras proteínas que contêm ferro. Normalmente, a maioria deste ferro é utilizada na síntese de hemoglobina e retorna à circulação junto às hemácias. Quantidades significativas de ferro são armazenadas na ferritina, entram na constituição das enzimas que contêm ferro presentes nas células eritroides imaturas, ou são perdidas nos produtos de uma eritropoese inefetiva. Ao fim de seu tempo de sobrevida, as hemácias senescentes são fagocitadas por macrófagos especializados junto ao baço, medula óssea e fígado que, por sua vez, devolvem a maior parte do ferro ao compartimento da transferrina, onde o ciclo é reiniciado. A fagocitose de eritrócitos não funcionais ou velhos contribui para quase todo o estoque de ferro normalmente encontrado nos macrófagos do fígado, medula óssea e baço. Em contraste, as células parenquimatosas do fígado podem captar o ferro oriundo da transferrina ou deixá-lo seguir para a transferrina plasmática. Sob condições fisiológicas normais, a reciclagem de ferro é bastante eficiente. Menos de 0,05% do ferro corporal total é adquirido ou perdido diariamente.
Figura 3. Suprimento e estoques de ferro do corpo. A figura consiste em uma representação esquemática das vias de movimentação do ferro em um indivíduo adulto. A área de cada círculo é proporcional à quantidade de ferro contida no compartimento correspondente, e a largura de cada seta é proporcional ao fluxo diário de ferro de um compartimento para outro. A principal porção do conteúdo de ferro é encontrada junto ao eritron, na forma de ferro da hemoglobina (28 mg/kg nas mulheres; 32 mg/kg nos homens) destinado ao transporte e distribuição de oxigênio. Pequenas quantidades de ferro de eritron (< 1 mg/kg) também estão presentes em enzimas heme e não heme localizadas nas hemácias em desenvolvimento. O ferro funcional restante é encontrado sob a forma de ferro da mioglobulina (4 mg/kg nas mulheres; 5 mg/kg nos homens) na musculatura e como enzimas contendo ferro e enzimas ferro-dependentes (1 a 2 mg/kg) presentes em todas as células do corpo. Pequenas quantidades de ferro são depositadas junto à ferritina, nas células eritroides, porém a maior parte das reservas de ferro (5 a 6 mg/kg nas mulheres; 10 a 12 mg/kg nos homens) permanece no pool de armazenamento pela ação de hepatócitos e macrófagos residentes no fígado, medula óssea, baço e músculos. A pequena fração de ferro transportado (cerca de 0,2 mg/kg) no plasma e no líquido extracelular está ligada à proteína transferrina, que carrega o ferro por todo o corpo para atender às necessidades teciduais.
O ferro da dieta é absorvido em sua forma ferrosa (FeII) no duodeno, e transportado dentro dos enterócitos pelo transportador metálico divalente 1 (DMT1 – em inglês, divalent metal transporter 1). O ferro férrico (FeIII) da dieta precisa ser convertido na forma ferrosa por enzimas existentes na superfície luminal. O ferro dietético na forma de heme é captado para dentro da célula pela proteína transportadora de heme-1 (HCP-1, heme carrier protein-1).13 Uma vez no interior do enterócito, o ferro é exportado pela ferroportina para a circulação, onde se liga à transferrina.
A transferrina é o transportador fisiológico do ferro através do plasma e do líquido extracelular.14 A apotransferrina, que consiste na transferrina sem ferro ligado, é uma glicoproteína de cadeia única que possui dois lobos estruturalmente similares. A ligação de um íon férrico a um destes lobos origina a transferrina monoférrica. A ligação de íons em ambos os lobos resulta na transferrina diférrica. A saturação da transferrina é dada pela proporção de sítios de ligação ao ferro disponíveis na transferrina que estão ocupados por átomos de ferro, sendo expressa em percentual. Após distribuir o ferro às células, a apotransferrina é reciclada de volta para o plasma para atuar novamente como transportador de ferro.
Os receptores específicos de transferrina, que são encontrados na membrana de superfície de todas as células nucleadas, fornecem a via de entrada para o ferro ligado à transferrina.14 A afinidade dos receptores de transferrina pela transferrina diférrica é maior, sendo intermediária para a transferrina monoférrica e quase desprezível para a apotransferrina. Estas diferenças de afinidade contribuem para a eficiência da distribuição do ferro. Até agora, foram identificadas 2 formas de receptor de transferrina, que foram denominadas receptores de transferrina 1 e 2.15,16 Ambas consistem em subunidades pareadas, cada qual capaz de ligar uma molécula de transferrina. Embora suas estruturas extracelulares sejam bastante semelhantes, as 2 formas do receptor de transferrina apresentam diferenças funcionais importantes. A afinidade da ligação entre o receptor de transferrina 1 e a transferrina diférrica é aproximadamente 25 a 30 vezes maior do que a afinidade da ligação desta com o receptor de transferrina 2. A expressão do receptor de transferrina 1, mas não a do receptor de transferrina 2, é regulada pelos níveis intracelulares de ferro (ver adiante). O receptor de transferrina 1 é expresso em todas as células que necessitam de ferro, enquanto o receptor de transferrina 2 é mais intensamente expresso nos hepatócitos e células eritroides em desenvolvimento. O receptor de transferrina 1 é necessário à vida nos mamíferos;17 O receptor de transferrina 2 não pode compensar a ausência do receptor de transferrina 1. Aparentemente, o receptor de transferrina 2 exerce outras funções na manutenção da homeostasia do ferro (ver adiante).
A distribuição do ferro ligado à transferrina começa com a ligação de 2 moléculas de transferrina mono ou diférricas a um receptor de transferrina localizado na superfície celular.14 O complexo ferro-transferrina/receptor de transferrina, então, move-se para dentro da célula no interior de um endossomo, onde libera o ferro. O ferro dissociado é captado pelo DMT1 e transportado através da membrana endossômica para ser utilizado ou armazenado junto à célula.17
O armazenamento celular de ferro requer a participação da ferritina, que é uma proteína encontrada no citoplasma de quase todas as células. A ferritina é uma espécie de concha esférica que consegue estocar até 4.500 átomos de ferro em seu interior.18 A ferritina atua como uma reserva de ferro prontamente acessível, adquirida pela célula a partir do excesso diante de suas necessidades imediatas, e mantém o ferro em um estado que não contribui para a geração de radicais livres.19 A ferritina é encontrada de maneira predominante nas células dedicadas ao armazenamento de ferro (p. ex., macrófagos e hepatócitos) e nas células que necessitam de maior quantidade de ferro para sintetizar compostos contendo ferro (p. ex., células eritroides em desenvolvimento). A apoferritina, ou ferritina sem ferro ligado, é composta por 24 subunidades oblongas, que são designadas como H (do inglês heavy, pesada) e L (do inglês light, leve). As moléculas de ferritina contendo uma proporção maior de subunidades H parecem ser mais ativas no metabolismo do ferro. As moléculas de ferritina mais abundantes em subunidades L aparentemente são utilizadas para armazenar ferro por tempo prolongado.18
Os macrófagos localizados junto à medula óssea, ao fígado e ao baço fazem o reconhecimento seletivo e a fagocitose dos eritrócitos senescentes ou danificados.20 A hemoglobina contida nestes eritrócitos sofre precipitação oxidativa e catabolismo rápido em heme.
Qualquer hemoglobina liberada no plasma em decorrência de hemólise intravascular é rapidamente ligada pela haptoglobina. Em seguida, o complexo haptoglobina/hemoglobina liga-se ao receptor scavenger (do inglês scavenger = removedor) de hemoglobina (CD163) localizado na superfície do macrófago.21 O complexo haptoglobina/hemoglobina, então, é endocitado e sofre degradação lisossômica, com consequente liberação do heme. Em contraste, o heme liberado no plasma em decorrência de hemólise intramedular ou extravascular é ligado pela hemopexina e levado até o hepatócito. A captação, que é mediada por um receptor heme-hemopexina-específico (CD91),22 é seguida de endocitose e digestão lisossômica.
O heme oriundo de ambas as fontes é degradado por uma enzima microssomal denominada heme oxigenasse, com consequente formação de biliverdina IXa, monóxido de carbono e ferro.23 Este ferro, por sua vez, é disponibilizado para ser estocado como ferritina ou exportado para o plasma.
Diante de uma saturação menor de transferrina plasmática, o ferro é mobilizado a partir do macrófago ou hepatócito. A exportação de ferro a partir dos macrófagos ou hepatócitos é mediada pela ferroportina, em grande parte como descrito anteriormente, para o enterócito duodeonal. O ferro, então, liga-se à apotransferrina e é disponibilizado para uso na eritropoese, entre outras utilidades. A maior parte da mobilização ocorre a partir dos macrófagos, com o fígado fornecendo uma proporção bem menor.
A hepcidina é um peptídeo de 27 kDa, produzido no fígado, que pode ser detectado no sangue e na urina. A hepcidina apresenta propriedades antimicrobianas e é um componente do sistema imunológico inato.24 Nos últimos anos, ficou claro que a hepcidina é um regulador importante da homeostasia do ferro. A hepcidina liga-se à ferroportina e promove sua internalização e degradação, prevenindo, desse modo, a exportação do ferro a partir dos enterócitos, hepatócitos ou macrófagos.25 A injeção de hepcidina está associada a uma redução da concentração sérica ou plasmática de ferro.26 O papel central da hepcidina na mobilização do ferro é confirmado pelas proteínas que regulam sua expressão, entre as quais o produto do gene da hemocromatose (HFE), hemojuvelina e receptor de transferrina 2.13 Mutações envolvendo estes genes, assim como as mutações no gene da própria hepcidina, produzem hemocromatose.27 O ferro desregula a expressão de hepcidina, embora os mecanismos pelos quais isto ocorra sejam obscuros.28
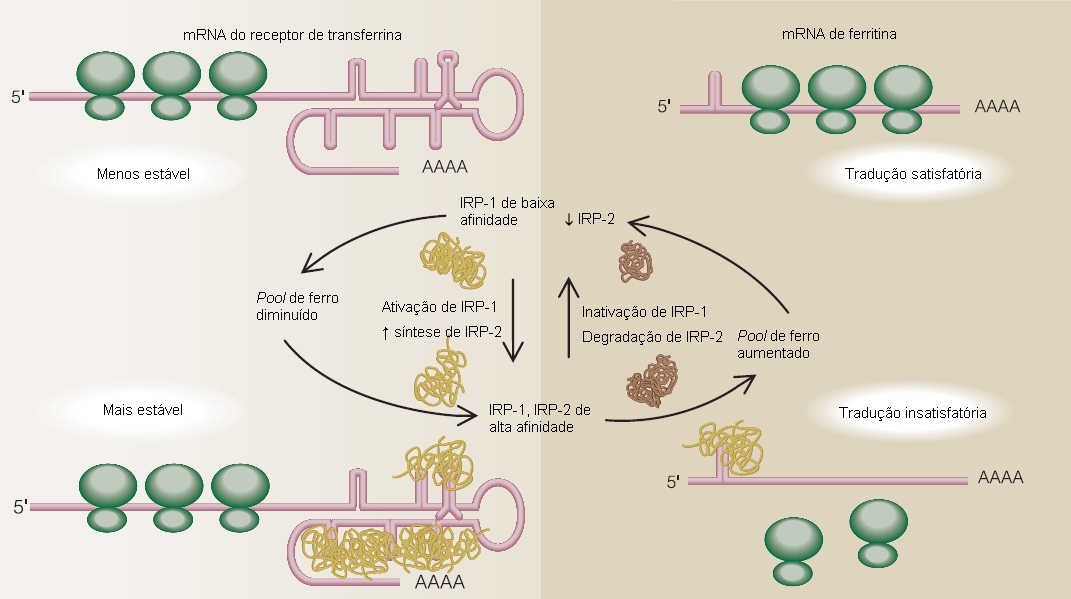
Apesar de a saída do ferro (dos eritrócitos, hepatócitos ou macrófagos) ser principalmente regulada pela hepcidina e seus efeitos sobre a ferroportina, a captação e o armazenamento celular de ferro são regulados via síntese de receptores de transferrina e ferritina. Este processo é coordenado pelas proteínas reguladoras de ferro (IRP-1 e IRP-2) e suas interações com o RNA mensageiro (mRNA) das proteínas que regulam [Figura 4]. Quando os níveis intracelulares de ferro estão baixos, a IRP-1 e a IRP-2 se ligam aos elementos da haste-alça do mRNA, conhecidos como elementos ferro-responsivos presentes nos transcritos.29 A síntese do receptor da transferrina é regulada pelo controle da estabilidade do mRNA do receptor citoplasmático de transferrina, enquanto a síntese de ferritina é regulada por meio do controle da tradução do mRNA da ferritina sem modificação da quantidade de mRNA de ferritina presente no citoplasma. Como resultado, os efeitos das IRP sobre os receptores da ferritina e da transferrina tornam-se recíprocos, permitindo que o ferro autorregule o equilíbrio entre ferro estocado e ferro circulante. As IRP também regulam os mRNAs de outras proteínas envolvidas na captação, disponibilização, liberação e uso do ferro.30-32
Figura 4. Regulação da expressão do receptor de transferrina e da ferritina pelas proteínas reguladoras de ferro, IRP-1 e IRP-2.
mRNA = RNA mensageiro.
A deficiência de ferro deve ser entendida como um estágio no espectro do equilíbrio negativo do ferro. O equilíbrio negativo do ferro tem lugar quando as necessidades corporais de ferro excedem seu suprimento. O ferro é necessário para se obter a reposição da perda fisiológica basal que ocorre via células de mucosa ou de pele esfoliadas, que é de aproximadamente 14 mcg/kg/dia.33 Em mulheres que menstruam, é preciso compensar uma perda adicional de ferro da ordem de 20 mg que ocorre a cada ciclo menstrual. Esta perda basal atinge menos de 1 mg/dia em homens de estatura mediana, discretamente menor do que a perda observada em mulheres pós-menopáusicas, e é igual a cerca de 1,5 mg/dia nas mulheres que menstruam. O ferro também é necessário ao crescimento em crianças e durante a gestação. A perda de ferro patológica (geralmente atribuível a sangramentos) também pode criar demandas para uma maior ingesta de ferro. O equilíbrio negativo do ferro apresenta estágios sequenciais, que são destacados na Tabela 1. Se um estado de equilíbrio positivo de ferro não é restaurado, a progressão entre estes estágios se torna inevitável. Uma diminuição das reservas de ferro sem nenhuma alteração das quantidades de compostos de ferro funcionais é referida como estoques de ferro diminuídos. Quando as reservas de ferro são esgotadas, o indivíduo apresenta deficiência de ferro. A eritropoese ferro-deficiente – que consiste na produção de eritrócitos com utilização do ferro imediatamente absorvido ou reciclado na ausência de reservas de ferro, porém sem queda da concentração de hemoglobina nem do hematócrito aos níveis de anemia – por vezes é considerada um estágio à parte ou um estágio parcial de deficiência de ferro. Quando o equilíbrio negativo de ferro persiste, o indivíduo apresenta anemia ferropriva. É somente neste estágio final que as hemácias hipocrômicas microcíticas estão presentes.
Tabela 1. Alterações nas reservas e na distribuição do ferro com aumento ou diminuição do conteúdo corporal de ferro
|
Condição |
Ferro Medular (0 a 6+) |
Ferro hepático (mcmol/g, peso seco) |
Ferritina plasmática (mcg/L) |
Receptor de transferrina plasmático (mg/L) |
Ferro plasmático (mcg/dL) |
Saturação da transferrina (%) |
Hemácias |
|
Anemia ferropriva |
0 |
< 3 |
< 12 |
Elevado |
Diminuído |
< 10 |
Microcíticas, hipocrômicas |
|
Eritropoiese ferro-deficiente |
0 a traços |
< 3 |
< 20 |
Elevado |
Diminuído |
< 16 |
Normal |
|
Deficiência de ferro |
0 a traços |
3 |
< 20 |
Normal |
Diminuído |
< 30 |
Normal |
|
Estoques de ferro diminuídos |
1+ |
< 10 |
< 25 |
Normal |
Normal a levemente diminuído |
30 |
Normal |
|
Normal |
2 a 3+ |
15 ± 5 |
Normal |
Normal |
Normal |
35 ± 15 |
Normal |
|
Anemia por doença crônica |
2 a 4+ |
? |
Normal ou elevada |
Normal ou elevado |
Diminuído |
16 a 30 |
Normal ou levemente microcíticas |
|
Estoques de ferro aumentados, hemocromatose |
2 a 3+ |
100 |
Elevada* |
Normal |
Elevado |
> 60 |
Normal |
|
Estoques de ferro aumentados; sobrecarga de ferro transfusional |
4+ |
20 |
Elevada |
Normal |
Elevado |
> 50 |
Normal |
|
Sobrecarga maciça de ferro; hemocromatose associada ao HLA |
3 a 4+ |
500 |
Acentuadamente elevada* |
Normal |
Elevado |
> 60 |
Normal |
|
Sobrecarga maciça de ferro; sobrecarga de ferro transfusional |
6+ |
300 |
Acentuadamente elevada |
Normal |
Elevado |
> 90 |
Normal |
*Pode estar normal em alguns pacientes.
Onde existem duas possibilidades descritas, a mais comum foi sublinhada.
A deficiência de ferro é a deficiência nutricional mais comum em todo o mundo. Sua prevalência é mais alta nos países em desenvolvimento, onde 30 a 70% da população pode estar afetada. Em comparação, a prevalência geral da deficiência de ferro é inferior a 20% nos países industrializados da Europa e da América do Norte.34 Este panorama reflete aspectos nutricionais e econômicos, além de problemas médicos endêmicos, entre os quais as infestações por ancilostomídeos.35 Em todas as populações, a prevalência é maior entre mulheres em idade fértil e entre crianças. Nos Estados Unidos, o Centers for Disease Control and Prevention (CDC) estimam que a prevalência da deficiência de ferro é maior entre crianças pequenas com 1 a 2 anos de idade (7%) e entre meninas adolescentes e mulheres adultas na faixa etária de 12 a 49 anos (9 a 16%). Neste último grupo, a prevalência entre mulheres hispânicas ou de etnia africana é, a grosso modo, 2 vezes maior do que a prevalência entre mulheres brancas não hispânicas. A prevalência da deficiência de ferro entre garotas na pré-menarca e meninos na mesma faixa etária é grosseiramente a mesma (4 a 5%). Entre os homens com mais de 16 anos de idade, a prevalência é de 2 a 3%. Entre as mulheres pós-menopáusicas, a prevalência é 2 a 3 vezes maior, provavelmente refletindo uma deficiência de ferro adquirida ao longo dos anos de menstruação e idade fértil.36
A deficiência de ferro pode resultar de aumento das necessidades, suprimento inadequado ou ambas as condições [Tabela 2]. A perda de sangue é a causa mais comum de aumento das necessidades de ferro que leva à deficiência de ferro. Nos homens, a deficiência de ferro quase sempre resulta da perda de sangue gastrintestinal. Em mulheres na pós-menopausa, a deficiência de ferro pode refletir uma perda de ferro descompensada anterior associada às menstruações ou pode refletir um sangramento gastrintestinal. Nestas duas populações, a possibilidade de perda de sangue gastrintestinal deve ser investigada.37 Em mulheres que não menstruam, é comum a perda de sangue geniturinária ser responsável pelo aumento das necessidades de ferro. O uso de anticoncepcionais orais tendem a diminuir a perda de sangue menstrual, enquanto os dispositivos intrauterinos tendem a aumentar o sangramento menstrual. Outras causas de sangramento geniturinário e sangramento no trato respiratório também podem aumentar os requerimentos de ferro do corpo [Tabela 2]. Para os doadores de sangue, cada doação resulta na perda de 200 a 250 mg de ferro.38 Durante os períodos de crescimento na lactação, infância e adolescência, a necessidade de ferro pode ultrapassar o suprimento de ferro disponibilizado a partir da dieta e das reservas corporais.39 Além disso, crianças alimentadas com leite de vaca integral podem desenvolver um sangramento gastrintestinal que conduz à deficiência de ferro.40 A quantidade de ferro consumida pelo crescimento tecidual fetal durante a gestação, bem como pelo sangramento que ocorre durante o parto e pós-parto, é da ordem de 750 a 900 mg. No 3º trimestre, os requerimentos de ferro maternos diários são cerca de 5 vezes maiores àqueles da fase pré-gestação.41 A amamentação aumenta a necessidade de ferro do corpo em cerca de 0,5 a 1 mg/dia.
Um suprimento de ferro insuficiente pode contribuir para o desenvolvimento de deficiência de ferro. Em bebês e mulheres com altos requerimentos de ferro, as dietas pobres em ferro biodisponível aumentam o risco de desenvolvimento de deficiência de ferro.42 Em crianças maiores, homens e mulheres em pós-menopausa, um suprimento dietético de ferro pobre quase nunca é o único fator responsável pela deficiência de ferro. Sendo assim, outras etiologias devem ser investigadas. O comprometimento da absorção de ferro é causa incomum da deficiência deste nutriente. Em alguns pacientes, a má absorção intestinal de ferro constitui o único aspecto significativo de uma má absorção mais generalizada43 [Tabela 2]. Cirurgias gástricas, sobretudo a ressecção gástrica parcial ou total e a gastrenterostomia de desvio duodenal, podem resultar em deficiência de ferro.44 Nestes pacientes, embora a absorção do ferro dietético possa ser precária, os sais de ferro terapêuticos geralmente são bem absorvidos, de modo que a deficiência de ferro pode ser prontamente corrigida. Mulheres com altos requerimentos de ferro em decorrência da menstruação não raramente consomem dietas que contêm pouco ferro biodisponível, além de inibidores da absorção do ferro (p. ex., cálcio). Uma mutação comum no gene da transferrina (denominado G277S) foi associada à redução da concentração de transferrina circulante. Ainda não está claro se isto tem algum significado clínico.45,46
Tabela 2. Causas da deficiência de ferro
|
|
|
Perda de sangue |
|
Trato gastrintestinal |
|
Lesões hemorrágicas (p. ex., hérnia de hiato, úlceras esofágicas, gastrite, duodenite, úlcera péptica, colelitíase, sangramento intra-hepático, enteropatia inflamatória, diverticulose, hemorroidas ou pólipos adenomatosos, infecção por Helicobacter pylori) |
|
Malignidade gastrintestinal oculta |
|
Ingesta crônica de fármacos (p. ex., álcool, salicilatos, esteroides e fármacos anti-inflamatórios não hormonais) |
|
Infecções helmínticas (p. ex., ancilostomídeos, Schistosoma mansoni, Schistosoma japonicum ou infecção severa com Trichuris trichiura) |
|
Outras (p. ex., púrpura vascular com escorbuto, pâncreas aberrante, divertículo de Meckel, telangiectasia hemorrágica hereditária e outras ectasias vasculares intestinais ou polipose colônica) |
|
Trato geniturinário |
|
Perda de sangue menstrual |
|
Outras (p. ex., malignidades uterinas de fibroides, cálculos, infarto, infecção por Schistosoma haematobium, doença inflamatória, malignidade do trato urinário ou hemoglobinúria crônica ou hemossidenúria resultante de hemoglobinúria paroxística noturna ou hemólise intravascular crônica) |
|
Trato respiratório |
|
Hemoptise recorrente crônica |
|
Siderose pulmonar idiopática |
|
Síndrome de Goodpasture |
|
Doação de sangue |
|
Crescimento |
|
Bebês, bebês prematuros |
|
Crianças |
|
Adolescentes |
|
Gestação e lactação |
|
Suprimento de ferro inadequado |
|
Dietas pobres em ferro biodisponível |
|
Comprometimento da absorção do ferro |
|
Má absorção intestinal (p. ex., esteatorreia, espru, doença celíaca, enterite difusa, gastrite atrófica com acloridria, ou pica) |
|
Cirurgia gástrica |
Pacientes com deficiência de ferro podem ser assintomáticos e ter seu distúrbio reconhecido somente em função dos resultados anormais de exames laboratoriais realizados por outro motivo. Alguns pacientes procuram atendimento médico por causa de manifestações de um distúrbio subjacente que produziu deficiência de ferro, mas podem não apresentar achados resultantes dessa deficiência. Outros pacientes, ainda, podem apresentar sintomas inespecíficos que são comuns a todos os tipos de anemia, como fraqueza, tontura, fadiga fácil, palidez ou irritabilidade. A deficiência de ferro também pode estar relacionada a achados não associados à anemia, tais como estomatite angular, glossite, membrana ou estreitamento esofágico pós-cricoide, além de atrofia gástrica. É provável que estes achados reflitam os processos subjacentes que estão levando à deficiência de ferro, em vez da própria deficiência de ferro em si. Uma esclera azulada e coiloníquia (unhas em colher) são achados bastante incomuns ao exame físico, considerados específicos para a deficiência de ferro, podendo ser observados também em outras síndromes clínicas.
Em indivíduos adultos, a observação de sintomas não hematológicos atribuíveis à deficiência de ferro é controversa. Há relatos de alta prevalência de deficiência de ferro entre pacientes com síndrome das pernas inquietas, um distúrbio neurológico caracterizado pela urgência de movimentar as pernas (acatisia).47 O sintoma não hematológico mais bem estabelecido da deficiência de ferro é a pica, ou ingesta de substâncias não nutricionais.48,49 A fagofagia, ou pica com gelo, é o tipo mais fortemente associado à deficiência de ferro e desaparece logo depois que a terapia com ferro é iniciada.48 Outros sintomas não hematológicos da deficiência de ferro em adultos, como diminuição da tolerância ao exercício e do desempenho no trabalho independente dos efeitos da anemia, foram hipotetizados e estão sendo discutidos.50-53 Em crianças, o quadro é mais nítido: a deficiência de ferro parece afetar adversamente o crescimento, desenvolvimento motor, comportamento e função cognitiva. Estas anormalidades podem não ser revertidas pelo tratamento tardio.54
A anemia ferropriva (por deficiência de ferro) é a única anemia hipocrômica microcítica associada à falta de reservas de ferro. Em outros distúrbios hipocrômicos microcíticos, os estoques de ferro medulares permanecem normais ou aumentam. Medidas indiretas dos níveis corporais de ferro podem ser utilizadas para identificar uma sequência característica de alterações que ocorrem conforme o ferro corporal em níveis normais de repleção cai e atinge os níveis observados na anemia ferropriva [Tabela 2]. Uma concentração sérica ou plasmática de ferro baixa é consistente com a deficiência de ferro, mas também pode ser observada em diversas circunstâncias clínicas, como na anemia por doença crônica. Concentração sérica ou plasmática de transferrina elevada é um achado razoavelmente específico para deficiência de ferro, porém é bastante insensível, em particular no indivíduo doente.55,56 Se a concentração sérica de transferrina estiver alta, uma saturação de transferrina inferior a 15% é fortemente preditiva de deficiência de ferro. Contudo, a saturação da transferrina tem menos utilidade quando a concentração sérica de transferrina está normal ou diminuída.56 As concentrações séricas de ferro e de transferrina são diminuídas pela inflamação, independentemente dos estoques de ferro.57
A determinação da concentração sérica de ferritina constitui o teste isolado mais útil para a detecção da deficiência de ferro, uma vez que as concentrações séricas de ferritina diminuem conforme as reservas corporais de ferro declinam.58 Uma concentração sérica de ferritina abaixo de 20 mcg/L é diagnóstica da completa ou quase inexistência de estoques de ferro no corpo, independentemente da metodologia laboratorial adotada. Em contraste, uma concentração sérica de ferritina normal não confirma a existência de reservas de ferro, pois a concentração sérica de ferritina pode aumentar de modo independente em relação ao conteúdo corporal de ferro, como consequência de infecção, inflamação, doença hepática, malignidade e outras condições.57 Pode haver confusão na interpretação das concentrações séricas de ferritina em laboratórios que relatam faixas de valores normais diferentes para homens e mulheres. O menor valor da faixa normal para mulheres é tipicamente bem menor do que o valor correspondente para os homens, porque a população “normal” em que este valor é determinado inclui um número pequeno, porém significativo, de mulheres com deficiência de ferro que ainda não estão anêmicas. A condição clínica de deficiência de ferro deve ser diagnosticada em homens e mulheres com base no limite inferior da faixa normal estabelecida para os homens.
A concentração sérica de receptor de transferrina solúvel reflete a massa de receptor de transferrina celular corporal total, que, por sua vez, é amplamente um reflexo da massa de precursores eritroides.59 A expressão do receptor de transferrina em precursores eritroides aumenta na deficiência de ferro.60 Como consequência, essa expressão é elevada em indivíduos com deficiência de ferro e em geral não é alta naqueles com síndromes que frequentemente são confundidas com a deficiência de ferro, entre as quais a anemia por doença crônica.61,62 Entretanto, a concentração sérica de receptor de transferrina geralmente também constitui um indicador da atividade eritropoética63 e isto limita sua especificidade como teste isolado único para a deficiência de ferro. Este teste parece ser altamente sensível e específico para a deficiência de ferro quando seus resultados são expressos de forma combinada com a concentração sérica de ferritina, em geral em proporção com o logaritmo da concentração sérica de ferritina.56,64
Os índices de hemácias tradicionais, como o volume corpuscular médio (VCM), a concentração média de hemoglobina corpuscular (CHCM) ou a variação no tamanho das hemácias (RDW - Red Cell Distribution Width), possuem alguma utilidade para o diagnóstico da deficiência de ferro, mas podem estar anormais em outros distúrbios e, portanto, são inespecíficos.65 Um volume corpuscular médio baixo com uma concentração média de hemoglobina corpuscular normal e uma variação no tamanho das hemácias também normal favorecem o diagnóstico de talassemia, em vez de anemia ferropriva. Outros índices de hemácias que podem ser derivados por gerações mais modernas de analisadores hematológicos automatizados, como a proporção de hemácias hipocrômicas e hemoglobina corpuscular de reticulócitos, parecem ter alguma utilidade na identificação da eritropoese ferro-deficiente, até mesmo antes do desenvolvimento de anemia ferropriva.66,67 Seu papel na prática ainda está evoluindo. A protoporfirina eritrocitária livre, também chamada de zinco protoporfirina, encontra-se elevada na deficiência de ferro, porém é um parâmetro raramente utilizado na prática.68
Embora o exame de medula óssea seja atualmente realizado em raras ocasiões e somente para avaliar a condição do ferro, o diagnóstico da deficiência de ferro quase sempre pode ser verificado por meio da avaliação direta dos estoques intracelulares de ferro em esfregaços de aspirado de medula óssea corados com azul da Prússia, ou por um método equivalente. O diagnóstico de deficiência de ferro é estabelecido quando não há reservas de ferro na amostra. Ao contrário, este diagnóstico é excluído quando a hemossiderina (um grânulo intracelular que armazena moléculas contendo ferro) é encontrada.
É importante reconhecer que o tratamento da deficiência de ferro envolve a investigação da etiologia da perda de ferro. Em jovens que menstruam ou em mulheres que passaram por um parto recente e não apresentam evidências clínicas de perda de sangue, é razoável atribuir uma deficiência de ferro, respectivamente, às menstruações ou ao nascimento do bebê. No caso de outras pacientes, geralmente é necessário realizar uma investigação gastrintestinal (com endoscopia e colonoscopia).69,70 A terapia para anemia ferropriva não só corrige o déficit de hemoglobina como repõe as reservas de ferro [Tabela 3]. As terapias de reposição oral e parenteral são ambas efetivas, mas a administração de ferro por via oral constitui o tratamento de escolha para quase todos os pacientes. A terapia com administração de ferro por via oral é efetiva, segura e econômica.71 Por isso, o uso da reposição de ferro por via endovenosa, que está associado a certo grau de risco de desenvolvimento de reações adversas locais e sistêmicas (além de ser inconveniente e caro), deve ser reservado para os pacientes que realmente não são capazes de absorver ou tolerar o ferro por via oral, para aqueles cujos requerimentos de ferro não podem ser atendidos pela terapia oral devido a uma perda de sangue crônica, sangramento descontrolado ou outro tipo de perda de sangue ou, ainda, nos casos em que a reposição oral tenha se mostrado significativamente menos efetiva do que a reposição endovenosa (como nos pacientes em hemodiálise72,73). A reposição de ferro por via intramuscular deve ser evitada, por causa da quantidade limitada que pode ser administrada e da absorção pouco confiável. Na anemia ferropriva severa, em alguns casos, é necessário realizar transfusões de hemácias para prevenir a isquemia cardíaca ou cerebral. Em alguns casos, as transfusões de hemácias também podem ser necessárias quando o paciente apresenta uma taxa crônica de perda de ferro superior à taxa de reposição possibilitada pela terapia parenteral.
Tabela 3. Terapia de reposição de ferro
|
Terapia com ferro oral |
|
Indicação |
|
Tratamento de escolha para casos de anemia ferropriva |
|
Terapia inicial para corrigir a anemia ferropriva |
|
Sal de ferro ferroso (p. ex., sulfato ferroso) administrado à parte das refeições, em 3 doses individuais (p. ex., 3 comprimidos de sulfato ferroso, 325 mg 3 x/dia; comprimidos de gliconato ferroso, 300 mg 3 x/dia) até obter a normalização do hematócrito e do volume corpuscular médio |
|
Terapia continuada para reposição dos estoques de ferro |
|
Sal de ferro ferroso administrado 1 a 3 x/dia, até a concentração plasmática de ferritina se tornar > 50 mcg/L (muitas vezes, são necessários 6 meses ou mais de tratamento) |
|
Tratamento dos efeitos colaterais |
|
Os efeitos colaterais gastrintestinais são mais comuns (10 a 20% dos pacientes ) e geralmente podem ser tratados de maneira sintomática, por meio da administração de ferro junto ou imediatamente após as refeições, ou via redução da frequência da dose |
|
Terapia com ferro parenteral |
|
Indicações |
|
Quando a perda de sangue descontrolada e crônica gera uma necessidade de ferro que não pode ser suprida pela terapia com ferro oral |
|
Má absorção do ferro |
|
Intolerância ao ferro oral, mesmo após o regime de dosagem ter sofrido repetidas modificações |
|
Riscos |
|
Reações anafiláticas imediatadas, prejudiciais à vida |
|
Reações do tipo doença do soro tardias, porém severas, acompanhadas de febre, urticária, adenopatia, mialgias e artralgias |
|
Exacerbação da artrite reumatoide e condições correlatas |
|
Precauções |
|
Ferro dextran ou gliconato férrico: recomenda-se administrar uma dose de teste de 0,5 mL com antecedência mínima de 1 hora em relação a cada injeção de ferro dextran. Mesmo assim, esta medida preventiva tem valor limitado |
|
Ferro sucrose: não requer dose de teste |
|
A pré-medicação com difenidramina e acetaminofeno pode ser utilizada, mas não é obrigatoriamente necessária |
|
Administração e dosagem |
|
Cálculo do déficit de ferro |
|
Déficit de ferro estimado (mg) = [(45 – hematócrito do paciente)/100 × peso corporal (kg) × 70 mL de sangue/kg] (déficit de hemácias circulantes) + [5 × peso corporal (kg)] (déficit de reservas de ferro) |
|
Ferro dextran: administrado por via endovenosa (100 a 200 mg/injeção) ou como infusão de uma dose total |
|
Gliconato férrico: administrado por via endovenosa (125 mg/injeção) |
|
Ferro sucrose: administrada por via endovenosa (100 a 200 mg/injeção) |
|
As recomendações dos fabricantes e as recomendações de estudos recentes para o tratamento devem ser atentamente revisadas, antes do fornecimento de ferro parenteral |
Embora a maioria dos pacientes não apresente dificuldade para tomar ferro por via oral, 10 a 20% desenvolvem efeitos colaterais relacionados ao ferro – apresentando, principalmente, queixas gastrintestinais. Na maioria das vezes estas podem ser solucionadas com a mudança da preparação de ferro utilizada, de sulfato ferroso para gliconato ferroso ou fumarato ferroso. Preparações com revestimento entérico são bem toleradas, mas podem ser fracamente absorvidas.74 A administração de ácido ascórbico pode intensificar a absorção de ferro oral.75
As preparações de ferro endovenosas são classificadas em duas categorias amplas: preparações de ferro dextran e sacaratos de ferro não dextran. A vantagem do ferro dextran reside na possibilidade de administrar grandes quantidades de ferro elementar de uma vez só,76 enquanto a desvantagem consiste no risco associado de reações sistêmicas, entre as quais a anafilaxia. As preparações parenterais de ferro não dextran estão associadas a um risco significativamente menor de desenvolvimento de reações sistêmicas graves, mas somente podem ser fornecidas a uma dosagem de reposição de 125 a 200 mg de ferro elementar a cada administração.77 Recomenda-se aplicar doses de teste antes da 1ª administração de ferro dextran ou gliconato férrico, mas não antes da administração de ferro sucrose. Não está bem estabelecido se as doses de teste predizem ou previnem acuradamente o desenvolvimento de anafilaxia, sobretudo quando o paciente é pré-medicado com difenidramina e acetaminofeno antes de se iniciar a terapia.78-80
A deficiência de ferro quase sempre pode ser tratada de maneira efetiva. O alívio dos sintomas costuma ser alcançado nos primeiros dias de tratamento. Nos casos de deficiência de ferro sem complicação, a resposta hematológica inicial – uma reticulocitose leve – geralmente tem início em 3 a 5 dias após a instituição da terapia, atinge o máximo em 8 a 10 dias e declina subsequentemente. Após a 1ª semana, a concentração de hemoglobina começa a aumentar e em geral normaliza dentro de 6 semanas. É possível que a microcitose não seja totalmente resolvida por até 4 meses. Se a deficiência de ferro for tratada à base de ferro oral a uma dosagem de 200 mg/dia ou menos, a concentração sérica de ferritina permanecerá abaixo de 12 mcg/L até a anemia ser corrigida. A partir daí, os níveis séricos de ferritina aumentam gradualmente, à medida que as reservas de ferro vão sendo repostas. Se a resposta à terapia com ferro não for completa e característica, é obrigatório rever e reavaliar o paciente. Um dos problemas mais comuns é confundir a anemia por doença crônica com a anemia ferropriva. A primeira (ver Anemia: defeitos de produção) está associada à diminuição da concentração sérica de ferro mesmo diante de reservas adequadas. A anemia por doença crônica está associada à ativação de citocinas mediadoras da inflamação, sendo que a hipoferremia associada parece ser induzida pela hepcidina.91 Este tipo de anemia não responde à terapia de reposição de ferro.82 Outros fatores coexistentes que podem impedir a resposta à reposição de ferro incluem as deficiências nutricionais, a doença hepática ou renal, os distúrbios infecciosos, inflamatórios ou malignos, ou a perda de sangue oculto contínua.
No espectro do equilíbrio do ferro, a sobrecarga de ferro ocorre diante de um fornecimento de ferro excessivo sustentado, que ultrapassa os requerimentos de ferro, provocando as características alterações funcionais, de transporte e de armazenamento do ferro. Conforme discutido anteriormente, a única regulação fisiológica das reservas de ferro do corpo ocorre em termos de absorção dietética de ferro. O corpo não conta com formas de eliminação do excesso de ferro. Um indivíduo desenvolve sobrecarga de ferro quando a regulação da absorção intestinal de ferro não funciona normalmente (como ocorre na hemocromatose hereditária) ou sob condições que desviam totalmente a absorção intestinal de ferro (como na transfusão de sangue crônica). A sobrecarga de ferro pode ser classificada como um distúrbio primário ou secundário [Tabela 4].
Tabela 4. Causas da sobrecarga de ferro
|
Primária |
|
Hemocromatose hereditária |
|
Associada ao HFE (tipo 1) |
|
Hemocromatose juvenil (tipo 2) |
|
Associada à hemojuvelina (tipo 2A) |
|
Associada à hepcidina (tipo 2B) |
|
Associada ao receptor de transferrina 2 (tipo 3) |
|
Associada à ferroportina 1 (tipos 4A e 4B) |
|
Atransferrinemia |
|
Aceruloplasminemia |
|
Mutação em DMT1 |
|
Secundária |
|
Anemias por sobrecarga de ferro (anemias refratárias com medula eritroide hipercelular) |
|
Doença hepática crônica |
|
Porfiria cutânea tarda |
|
Sobrecarga dietética de ferro africana* |
|
Ingesta medicinal de ferro* |
|
Sobrecarga parenteral de ferro |
|
Sobrecarga transfusional de ferro |
|
Sobrecarga inadvertida de ferro por injeções terapêuticas |
|
Perinatal |
|
Hemocromatose neonatal |
|
Tirosemia hereditária (hipermetionemia) |
|
Síndrome cerebral-hepato-renal |
|
Síndrome GRACILE (Fellman) |
|
Sequestro focal de ferro |
|
Hemossiderose pulmonar idiopática |
|
Hemossiderose renal |
|
Associada a anormalidades neurológicas |
|
Neurodegeneração associada à pantotenato quinase (anteriormente conhecida como síndrome de Hallervorden-Spatz) |
|
Neuroferritinopatia |
|
Ataxia de Friedreich |
GRACILE = growth retardation (retardo do crescimento), aminoaciduria (aminoacidúria), cholestasis (colestase), iron overload (sobrecarga de ferro), lactacidosis (lactacidose), early death (morte precoce).
*Pode ter um componente genético.
Quando a extensão do acúmulo de ferro excede a capacidade do corpo de sequestrar com segurança o ferro extra, o indivíduo desenvolve padrões de dano tecidual característicos. As manifestações precisas da sobrecarga de ferro dependem da anormalidade subjacente responsável pela condição, mas em geral são governadas pelos seguintes fatores: magnitude da carga de ferro corporal; velocidade com que o conteúdo corporal de ferro aumentou; distribuição do excesso de ferro entre os sítios de armazenamento nos macrófagos e os depósitos potencialmente mais prejudiciais junto às células parenquimatosas; e coexistência de condições que possam melhorar (p. ex., deficiência de ascorbato) ou piorar (p. ex., consumo de álcool, hepatite, porfiria cutânea tarda) o resultado. As consequências mais comuns da sobrecarga de ferro são: doença hepática, doença pancreática (associada ao diabetes melito), disfunção cardíaca, outros distúrbios endócrinos atribuíveis à infiltração da hipófise pelo ferro (em particular, a disfunção gonadal), artropatia e, em algumas formas de sobrecarga de ferro, anormalidades neurológicas específicas.
Na sobrecarga de ferro primária, a anomalia patofisiológica reside na regulação da homeostasia do ferro. Estes distúrbios podem ser classificados como mutações que afetam a hepcidina ou sua função ou regulação (os quatro tipos de hemocromatose hereditária) e defeitos em genes reguladores de outros aspectos do transporte ou armazenamento de ferro.
Epidemiologia. A hemocromatose associada ao HFE hereditária é o distúrbio genético mais comumente observado entre os descendentes de norte-europeus.83 Dados do estudo Hemochromatosis and Iron Overload Screening (HEIRS) indicam que 0,44% da população branca dos Estados Unidos é homozigota para C282Y, que é a mutação em HFE mais fortemente associada à hemocromatose clínica. A prevalência dos heterozigotos para C282Y é de aproximadamente 10% na população branca. A outra mutação que comumente afeta o HFE, a H63D, embora na verdade seja mais prevalente, está associada a um fenótipo mais brando (2%). A heterozigosidade composta por C282Y e H63D é observada em 2,4% da população branca.84
Etiologia e genética. Mutações em HFE (mais comumente, C282Y ou H63D) estão presentes em 85 a 90% dos pacientes com hemocromatose hereditária, nos Estados Unidos. A frequência varia em outras regiões geográficas e se aproxima de 100% em certas áreas (p. ex., Austrália).85 A proteína HFE é um regulador da expressão de hepcidina. Mutações de sentido incorreto (missense) em HFE, como C282Y, diminuem a produção de hepcidina e, assim, levam à intensificação da mobilização de ferro a partir dos enterócitos para a circulação.13,86 A hemocromatose associada ao HFE consiste em uma doença autossômica recessiva de penetrância variável. Uma significativa sobrecarga de ferro é observada predominantemente em indivíduos homozigotos, embora os heterozigotos compostos para C282Y e H63D possam apresentar evidências de sobrecarga de ferro, em especial quando possuem um fator subjacente capaz de exacerbar a deposição de ferro.87 Embora os indivíduos heterozigotos para mutações em HFE apresentem anormalidades envolvendo marcadores clínicos do metabolismo do ferro, que os distinguem dos indivíduos normais, geralmente não apresentam evidências de sobrecarga de ferro.88
Inicialmente, o padrão de deposição de ferro na hemocromatose associada ao HFE é predominantemente parenquimal, com o ferro se acumulando primeiro nos hepatócitos. Subsequentemente, o ferro acumula-se no pâncreas, no coração e em outros órgãos.85,89 Caracteristicamente, os níveis de ferro nos macrófagos da medula óssea podem estar normais ou até diminuídos, apesar da severa deposição parenquimal de ferro.90
A hemocromatose juvenil consiste em um raro distúrbio autossômico recessivo, no qual se observa que o indivíduo afetado desenvolve uma severa sobrecarga de ferro antes de completar 30 anos de idade. Os pacientes frequentemente apresentam uma disfunção orgânica avançada, que é atribuível à sobrecarga de ferro (miocardiopatia, hipogonadismo, tolerância à glicose comprometida), no momento da manifestação.91 Foram descritos dois subtipos: o tipo 2A, causado por mutações que afetam a hemojuvelina reguladora da hepcidina;92 e o tipo 2B, causado por mutações no próprio gene da hepcidina.27
A hemocromatose hereditária de tipo 3 é um raro distúrbio autossômico recessivo, que também pode produzir uma significativa deposição de ferro precocemente ao longo da vida, mas em geral não no mesmo grau observado na hemocromatose juvenil.86 A doença é produzida por mutações que afetam o receptor de transferrina-293 e está associada a níveis diminuídos de hepcidina.94
A hemocromatose hereditária de tipo 4 é autossômica dominante e resulta de mutações no gene da ferroportina, que a tornam resistente à internalização e degradação em resposta à hepcidina.95,96 Existem dois subtipos: no subtipo 4A a saturação de transferrina é relativamente baixa e a sobrecarga de ferro está amplamente confinada ao sistema reticuloendotelial; o subtipo 4B assemelha-se à hemocromatose associada ao HFE.97,98
Manifestações clínicas da hemocromatose hereditária. Indivíduos homozigotos para hemocromatose hereditária podem não apresentar manifestações clínicas distintivas, sobretudo quando mais jovens. Os pacientes procuram atenção médica para serem avaliados quanto a ocorrências diversas: anormalidades bioquímicas envolvendo a função hepática ou existência de hepatomegalia; para avaliação do diabetes ou hipogonadismo; em decorrência de uma artropatia durante a juventude; ou porque uma avaliação de fadiga inespecífica conduziu à determinação dos níveis de ferro (muitas vezes, em meio à investigação de uma possível anemia ferropriva).99 Este tipo de apresentação inespecífica atualmente é mais comum do que a tríade clássica de sinais clínicos em vigência de doença hepática: diabetes melito, pigmentação cutânea e insuficiência gonadal. Embora a combinação de disfunção pancreática e pigmentação cutânea seja referida como “diabetes de bronze”, a cor da pele é mais tipicamente cinza azulada pálida. A artropatia, que radiograficamente se parece com a pseudogota, pode ser uma manifestação inicial da doença e é observada com frequência em pacientes com doença avançada.100 Pode haver desenvolvimento de insuficiência cardíaca, e o paciente pode apresentar até mesmo os sintomas exibidos pelos indivíduos homozigotos, particularmente na hemocromatose juvenil.
É comum os estoques corporais de ferro se encontrarem aumentados (do conteúdo normal de 1 g ou menos para 15 a 20 g ou mais) no momento em que os sintomas de dano parenquimal se manifestam, em geral na metade ou fase tardia da vida. Os aumentos adicionais do conteúdo corporal de ferro podem ser fatais, embora alguns pacientes consigam tolerar um acúmulo de ferro total de 40 a 50 g ou mais.101 Os homens são afetados mais precocemente do que as mulheres, que provavelmente são protegidas pelas perdas de ferro ocorridas durante as menstruações e partos. Fatores ambientais (p. ex., conteúdo de ferro da dieta, doação ou perda de sangue, e consumo de álcool) e distúrbios coexistentes (p. ex., hepatite viral) podem influenciar significativamente a velocidade e severidade do dano orgânico.89 Dada a variedade de fatores ambientais ou demográficos que podem influenciar as manifestações de hemocromatose, não é surpreendente que a penetrância da hemocromatose associada ao HFE hereditária seja obscura. Alguns estudos sugeriram que a sobrecarga de ferro real está presente em apenas 1% dos indivíduos homozigotos para C282Y.102 No entanto, estudos mais recentes sugeriram que 28,4% dos indivíduos homozigotos do sexo masculino (ainda que apenas 1,2% dos indivíduos homozigotos do sexo feminino) apresentam sobrecarga de ferro.103
Rastreamento e diagnóstico. O rastreamento para detecção de hemocromatose hereditária pode usar métodos bioquímicos ou genotípicos e é indicada para pacientes com doença hepática inexplicável ou achados potencialmente associados à sobrecarga de ferro (p. ex., endocrinopatia ou artropatia) e para os parentes em primeiro grau de pacientes com hemocromatose comprovada.104 No momento, os testes genéticos clinicamente disponíveis limitam-se a avaliar as mutações em HFE. A avaliação bioquímica pode fornecer evidências bioquímicas da existência de sobrecarga de ferro, mas também é capaz de identificar indivíduos com defeitos genéticos que ainda não levaram ao desenvolvimento de uma sobrecarga de ferro.99 Entretanto, nenhum teste isolado nem combinação de testes identificará todos os pacientes geneticamente suscetíveis ao desenvolvimento de sobrecarga de ferro.105 A varredura genotípica em busca das mutações em HFE mais comuns (C282Y e H63D) em populações de descendentes de norte-europeus é capaz de identificar a vasta maioria dos pacientes que apresentam risco de desenvolver uma sobrecarga primária de ferro. A genotipagem deve substituir o teste bioquímico na avaliação de irmãos de um indivíduo homozigoto para C282Y. Os pais homozigotos para C282Y podem ter preocupações relacionadas às consequências não médicas do rastreamento de seus filhos: embora desejem identificar uma criança com risco de desenvolver sobrecarga de ferro, também desejam evitar a possibilidade em potencial de a criança deixar de ter a possibilidade de se cadastrar em um seguro de saúde. Nestes casos, testar o outro genitor é uma estratégia que pode conduzir a uma investigação mais seletiva dos filhos. Contudo, o teste para mutação em HFE não constitui uma estratégia efetiva quando se trata de populações em que o HFE é menos predominante,85 pois não detectará outras mutações genéticas e não avalia a extensão da sobrecarga de ferro nem a penetrância clínica. Embora tenha sido proposta o rastreamento da população, por meio de uma avaliação bioquímica de indivíduos assintomáticos como parte das medidas de manutenção da saúde geral, esta ação ainda é controversa devido às incertezas quanto à penetrância da doença, entre outros fatores.106,107 O United States Public Health Service atualmente não recomenda este rastreamento.108
A determinação da saturação da transferrina sérica geralmente é recomendada como ensaio de rastreamento bioquímico inicial.89,109 Embora os laboratórios individuais possam adotar suas próprias faixas de referência, um valor repetido acima de 45% para mulheres ou de 50% para homens costuma ser aceito como valor limiar para realização de investigações adicionais.110 A concentração sérica de ferritina, então, é medida como indicador bioquímico de sobrecarga de ferro. Na ausência de fatores complicadores, concentrações de ferritina acima de 300 mcg/L sugerem que as reservas de ferro estão aumentadas.110 Alguns defendem o uso de uma concentração de ferritina de 1.000 mcg/L, argumentando que seria um indicador mais específico de pacientes com risco de desenvolver doença clínica.111 Nos Estados Unidos, assim como em outras áreas de prevalência moderada ou alta de hemocromatose associada ao HFE, o teste genético deveria ser considerado em casos de pacientes que apresentam elevações anormais da saturação da transferrina, ferritina sérica ou ambas.
Uma vez que o teste genético tenha identificado um paciente homozigoto para a mutação C282Y (isto é, C282Y/C282Y), uma saturação de transferrina elevada estabelece o diagnóstico de hemocromatose hereditária clinicamente expressa. Isto em geral também é válido para indivíduos heterozigotos compostos para C282Y e H63D, bem como para indivíduos homozigotos para H63D, apesar da necessidade de considerar a possibilidade de que outro fator ambiental ou comportamental (particularmente o consumo crônico de álcool ou a hepatite C) esteja contribuindo para a sobrecarga de ferro.88,112 Não há teste genético específico disponível para pacientes que não são homozigotos nem heterozigotos compostos para as mutações em HFE. A biópsia hepática com determinação quantitativa de ferro, que no passado era o padrão-ouro do diagnóstico da hemocromatose hereditária, ainda pode exercer algum papel no caso destes pacientes. O cálculo do índice de ferro hepático – a concentração hepática de ferro (expressa em mcmol de Fe/g de peso seco de fígado) dividida pela idade do paciente (em anos) – pode ser útil para distinguir os indivíduos homozigotos para hemocromatose hereditária daqueles heterozigotos ou dos pacientes com conteúdo corporal de ferro aumentado associado à doença hepática crônica (geralmente alcoólica). Na ausência de outras causas de sobrecarga de ferro, um índice de ferro hepático maior que 1,9 é considerado evidência de hemocromatose hereditária.113,114 Abordagens não invasivas para avaliação da deposição de ferro hepática, como a imagem de ressonância magnética, ainda estão sendo desenvolvidas.115
Em indivíduos homozigotos para C282Y que apresentam evidência bioquímica de sobrecarga de ferro, o exame de biópsia hepática tornou-se desnecessário para estabelecer o diagnóstico. Entretanto, este exame continua sendo indicado para a avaliação de cirrose em pacientes que apresentam risco aumentado de desenvolver esta complicação. Entre estes pacientes, estão aqueles com hepatomegalia, níveis séricos de ferritina maiores que 1.000 mcg/L ou concentração sérica elevada de aspartato transaminase.116
Tratamento. A terapia de remoção de ferro é sempre indicada para pacientes com hemocromatose hereditária, mesmo quando uma cirrose ou dano orgânico já se instalaram, porque a progressão adicional da doença pode ser desacelerada ou retardada, e também seja possível amenizar uma parte da disfunção orgânica.104 O tratamento de escolha para a hemocromatose hereditária consiste na flebotomia para reduzir os níveis de ferro corporais a concentrações normais (ou quase normais) e mantê-los dentro da faixa da normalidade.86 Foi descrita a substituição da flebotomia por agentes quelantes de ferro em casos de pacientes com hemocromatose hereditária e insuficiência cardíaca hemodinamicamente instável.117 A terapia à base de flebotomia deve ser iniciada assim que o diagnóstico de homozigose para hemocromatose hereditária for estabelecido. Seu adiamento apenas aumenta o risco de dano orgânico por sobrecarga de ferro. O programa de flebotomia deve remover 500 mL de sangue (contendo 200 a 250 mg de ferro) 1 vez/semana ou, no caso de pacientes com sobrecarga considerável, 2 vezes/semana, até o estado do paciente se aproximar de uma deficiência de ferro. Antes de cada flebotomia, é preciso determinar o hematócrito ou a concentração de hemoglobina. A princípio, o hematócrito e os níveis de hemoglobina sofrem uma queda de aproximadamente 10% em relação aos valores iniciais, contudo podem aumentar em seguida, conforme a taxa de eritropoese aumenta para atender às demandas da flebotomia. Medidas dos níveis séricos de ferritina, ferro e saturação da transferrina devem ser obtidas regularmente, para se acompanhar o progresso da remoção do ferro. À medida que o ferro é removido, a concentração sérica de ferritina diminui progressivamente, contudo a saturação da transferrina sérica continuará elevada até as reservas de ferro estarem quase esgotadas. O objetivo é atingir uma concentração de ferritina inferior a 50 mcg/L. Há quem argumente que uma concentração de ferritina de 10 mcg/L é mais apropriada. O paciente pode necessitar de tratamento prolongado. A remoção de uma carga corporal de ferro moderada, da ordem de 20 g, requer a realização de cerca de 100 flebotomias – 2 anos, quando realizada semanalmente. Depois que a meta inicial é alcançada, é necessário instituir um programa vitalício de flebotomia de manutenção (habitualmente, 3 a 4 vezes/ano), com o intuito de prevenir o reacúmulo da carga de ferro e manter a concentração sérica de ferritina abaixo de 50 mcg/L.
Quando a terapia de flebotomia remove a carga de ferro antes de o paciente desenvolver diabetes melito ou cirrose, a expectativa de vida é normal.118 No entanto, se houver desenvolvimento de cirrose, o risco de carcinoma hepatocelular torna-se mais de 200 vezes maior.89 Na hemocromatose hereditária, os hepatocarcinomas desenvolvem-se quase exclusivamente em pacientes com cirrose hepática e constituem a causa final de morte em 20 a 30% destes indivíduos, mesmo após a remoção bem-sucedida da carga de ferro. Pacientes com hemocromatose e cirrose, ou pacientes que apresentam alto risco de desenvolver cirrose, devem passar por um rastreamento a cada 6 meses para detecção de hepatocarcinomas, utilizando-se a determinação das concentrações séricas de alfafetoproteína e a ultrassonografia como modalidades de avaliação.86
A atransferrinemia e a aceruloplasminemia são distúrbios autossômicos recessivos associados à sobrecarga de ferro. Na atransferrinemia, o ferro da dieta é prontamente absorvido e circula como ferro plasmático não ligado à transferrina, porém não pode ser utilizado na eritropoese devido à falta de meios fisiológicos de transporte para dentro das células eritroides em desenvolvimento. Os indivíduos afetados morrem, a menos que recebam infusão de transferrina ou transfusão de sangue.119 Na aceruloplasminemia, a deficiência de atividade de ceruloplasmina ferroxidase resulta no acúmulo de ferro no fígado, pâncreas e cérebro, com menores quantidades de ferro em excesso no baço, coração, rins, tireoide e retina. Os pacientes apresentam neurodegeneração progressiva da retina e gânglios basais, além de desenvolverem diabetes melito ao atingirem a meia-idade.120 Também existem diversos relatos isolados de famílias com sobrecarga primária de ferro associada a mutações em genes relacionados ao equilíbrio do ferro, entre os quais o gene codificador de DMT1.121
A sobrecarga primária de ferro resulta de doenças que envolvem componentes do sistema de homeostasia do ferro. A sobrecarga secundária de ferro resulta de distúrbios envolvendo outros sistemas ou tecidos, como as hemácias ou o fígado.122
As anemias por sobrecarga de ferro incluem as anormalidades primárias de produção de hemácias (como a anemia diseritropoética congênita, algumas formas de anemia sideroblástica e as síndromes mielodisplásicas) e algumas anemias hemolíticas congênitas (como a deficiência de piruvato quinase) e talasseminas ou hemoglobinopatias. Em pacientes com anemias por sobrecarga de ferro, observa-se o desenvolvimento de uma severa sobrecarga de ferro que resulta parcialmente do aumento da absorção gastrintestinal de ferro, da deposição de ferro oriundo de hemólise intramedular ou intravascular, e de transfusões de hemácias. A extensão da eritropoese inefetiva, em vez do grau de severidade da anemia, parece determinar a taxa de acúmulo de ferro, de modo que pacientes com anemia apenas discreta ou leve podem desenvolver uma sobrecarga de ferro severa.123 Este efeito parece ser mediado pela regulação negativa da hepcidina.124 As manifestações clínicas e a patologia que podem se desenvolver em pacientes com anemias por sobrecarga de ferro em consequência da deposição de ferro são similares àquelas observadas na hemocromatose hereditária, incluindo doença hepática, diabetes melito, distúrbios endócrinos e disfunção cardíaca.
Doença hepática crônica. Alguns pacientes com doença hepática crônica, incluindo aqueles com cirrose alcoólica e com desvio porto-cava, podem desenvolver graus mínimos ou moderados de acúmulo de ferro, como resultado da absorção aumentada do ferro da dieta.89 Os mecanismos envolvidos são obscuros, mas é possível que estejam relacionados ao aumento da expressão de DMT1.125 As reservas corporais de ferro estão apenas um pouco aumentadas, tipicamente em 2 a 4 g.
Porfiria cutânea tarda. Pacientes sintomáticos com porfiria cutânea tarda – um tipo de porfiria hepática – geralmente apresentam uma modesta elevação dos níveis corporais de ferro, que resulta, namaioria dos casos, de um aumento da absorção gastrintestinal.122 Em pacientes descendentes de europeus, as mutações em HFE são comuns e podem contribuir para a patogênese de ambas as formas, familiar e esporádica, do distúrbio. Uma infecção concomitante pelo vírus da hepatite C também pode exacerbar a sobrecarga de ferro.126 A frequência das manifestações cutâneas significativas da porfiria cutânea tarda pode ser diminuída pela flebotomia, tendo como meta alcançar níveis de ferritina similares àqueles adotados em casos de hemocromatose. Como a sobrecarga de ferro é tipicamente menor, um esquema menos agressivo de flebotomia em geral é aceitável.
Esteato-hepatite não alcoólica. A sobrecarga de ferro caracterizada pelo aumento dos níveis séricos de ferritina com nível normal de saturação da transferrina, associada à existência de anomalias no metabolismo da glicose ou de lipídios, ou de ambos, foi descrita em pacientes com esteato-hepatite não alcoólica.127 Relatos subsequentes questionaram esta associação, sugerindo que os relatos anteriores refletiam apenas uma coincidência de outros fatores.128
Sobrecarga de ferro dietético africana. Foi descrita a ocorrência, na África subsaariana, de uma sobrecarga de ferro associada à ingesta significativamente aumentada de ferro dietético, oriundo de uma bebida caseira preparada a partir da fermentação do milho em tambores de ferro. O acúmulo de ferro pode ser tão extenso quanto aquele encontrado em indivíduos com hemocromatose hereditária, sendo que os pacientes podem desenvolver doença hepática (com cirrose e hepatocarcinoma), doença pancreática (com diabetes melito), distúrbios endócrinos e disfunção cardíaca.129 Embora a ingesta aumentada de ferro a partir da dieta tenha sido considerada por muito tempo como a única causa do aumento da absorção de ferro observado neste distúrbio, uma série de estudos de heredogramas sugeriu a existência de um componente genético que possivelmente estaria envolvido e poderia ser comum nas populações de descendentes de africanos.130
Ingesta de ferro medicinal. A ingesta de suplementos de ferro certamente contribui para o acúmulo corporal de ferro em pacientes com distúrbios de sobrecarga de ferro. No entanto, ainda é incerta a extensão em que o ferro administrado por via oral pode aumentar as reservas corporais de ferro em indivíduos normais. A maioria dos relatos de caso que descrevem acúmulos de ferro em pacientes que tomaram ferro medicinal durante longos períodos antecipa a descrição de muitos defeitos na absorção do ferro,131 de modo que não se pode excluir o potencial envolvimento de uma mutação determinante de sobrecarga de ferro ainda não identificada nestes indivíduos.
Etiologia e diagnóstico. A abordagem clínica das hemoglobinopatias, síndromes de talassemia e distúrbios de insuficiência medular, como as síndromes mielodisplásicas e a anemia aplásica, pode requerer transfusões periódicas. Isso pode levar ao acúmulo progressivo do ferro contido nas hemácias transfundidas.132 Se a anemia dependente de transfusão incluir hiperplasia eritroide com eritropoese inefetiva, a aumentada absorção gastrintestinal de ferro pode somar-se ao acúmulo de ferro. Nestes casos, a captação de ferro da dieta pode ser minimizada pela supressão da eritropoese mediante a instituição de um programa adequado de transfusão. O acúmulo de ferro a partir das transfusões ocorre inicialmente com predominância em sítios de macrófagos e, subsequentemente, via redistribuição para os tecidos parenquimatosos. Como os seres humanos não contam com meios fisiológicos de eliminar o excesso de ferro, o ferro contido nas hemácias transfundidas é progressivamente acumulado e, em determinadas ocasiões, causa danos ao fígado, coração, pâncreas e outros órgãos. A morte geralmente é consequente à insuficiência cardíaca. Em pacientes mais jovens, a carga de ferro resulta em falha de crescimento e, na adolescência, em retardo ou ausência de maturação sexual. O ferro administrado por via parenteral pode somar-se à carga de ferro em pacientes com anemias microcíticas refratárias que são erroneamente diagnosticados como tendo deficiência de ferro.
Tratamento. Cerca de 200 a 250 mg de ferro são adicionados à carga corporal de ferro por cada unidade de hemácias transfundidas. A severidade da toxicidade do ferro parece estar relacionada à magnitude da carga corporal de ferro. Uma deposição de ferro severa, que resulta em danos cardíacos, hepáticos e endócrinos, é geralmente observada em pacientes que receberam 100 unidades ou mais de sangue (cerca de 20 a 25 g de ferro).133,134 Obviamente, a flebotomia não representa uma alternativa, e a única forma de prevenir ou tratar a sobrecarga de ferro consiste na terapia com agentes quelantes capazes de se complexar ao ferro e, assim, permitir sua excreção. Em pacientes com anemia falciforme, a transfusão de troca ou eritrocitaférese pode limitar os aumentos da carga de ferro. Entretanto, a quelação ainda se faz necessária para diminuir as reservas de ferro.135
Existem 3 agentes quelantes potencialmente disponíveis, mas apenas 2 (deferoxamina e desferasirox) tiveram o uso aprovado nos Estados Unidos. A deferoxamina é disponibilizada há muito tempo, de modo que seu uso já foi substancialmente experimentado. Quando os pacientes são totalmente complacentes com seu uso, a deferoxamina pode mobilizar de maneira efetiva e segura tanto o ferro hepático como o ferro cardíaco, que são excretados na urina e nas fezes.136 Contudo, este agente deve ser administrado por via parenteral, e isto limita a aderência dos pacientes. Tipicamente, a deferoxamina é administrada como infusão subcutânea prolongada, por um período de 8 a 10 horas, todas as noites. A dose habitual é 30 a 40 mg/kg/dia. Também é possível administrá-la como infusão endovenosa contínua.137 A falta de complacência com o uso de deferoxamina estimulou a procura por agentes quelantes orais. O deferasirox, administrado 1 vez/dia por via oral, a uma dose de 20 a 30 mg/kg, liga-se ao ferro e promove sua excreção nas fezes. O deferasirox diminui os níveis de ferro hepático a concentrações comparáveis às obtidas com doses-padrão deferoxamina.138 A deferiprona (também chamada de L1) foi o primeiro quelante de ferro oral a ser desenvolvido. Embora seja amplamente utilizado em outros países, seu uso não foi aprovado nos Estados Unidos. O ferro ligado à deferiprona é excretado na urina. Sua eficácia na mobilização do ferro é comparável à da deferoxamina,139 podendo ser mais eficaz do que este agente na mobilização do ferro cardíaco.140
Para prevenir a sobrecarga de ferro, a terapia de quelação deve ser considerada em casos de pacientes com propensão a necessitarem de transfusões crônicas e que tenham recebido mais de 20 unidades de hemácias ou para aqueles cuja concentração sérica de ferritina esteja acima de 1.000 mcg/L. A audiometria e o exame ocular devem ser realizados no momento basal. Na terapia com deferoxamina, estes exames devem ser repetidos anualmente. As concentrações séricas de ferritina devem ser seguidas no mínimo a cada 3 meses, embora estas determinações sejam menos acuradas como indicador de deposição hepática do que o exame de biópsia.141 Uma avaliação direta anual das reservas hepáticas de ferro deve ser considerada, seja por exame de biópsia ou por análise de imagem de ressonância magnética. Do mesmo modo, deve ser considerada a realização de uma avaliação anual do ferro cardíaco via análise de imagem de ressonância magnética.142 O uso de quelantes de ferro orais está associado ao risco de toxicidade hepática e requer o acompanhamento mensal da concentração de alanina aminotransferase. O deferasirox também apresenta certo risco de toxicidade renal, e seu uso requer o acompanhamento mensal dos níveis séricos de creatinina. A deferiprona está associada ao desenvolvimento de agranulocitose, e seu uso demanda a realização semanal de contagens sanguíneas com diferencial.
A hemocromatose neonatal consiste em um grupo heterogêneo de distúrbios associados à doença hepática congênita severa e à presença de depósitos de ferro no fígado, bem como no pâncreas, coração e outros sítios extra-hepáticos, com alguns casos apresentando evidências de herança autossômica recessiva. Este grupo inclui anomalias metabólicas raras associadas a uma deposição de ferro anormal, incluindo a tirosinemia hereditária (hipermetioninemia), síndrome cerebral-hepato-renal de Zellweger, síndrome trico-hepato-entérica e síndrome GRACILE (ou Fellman).143
O sequestro focal de ferro é observado em outros distúrbios raros, entre os quais a hemossiderose pulmonar idiopática e a hemossiderose renal. Este tipo de deposição de ferro anormal está associado à ocorrência das anormalidades neurológicas encontradas na ataxia de Friedreich, neurodegeneração associada à pantotenato quinase (antigamente conhecida como síndrome de Hallervorden-Spatz) e neuroferritinopatia.144 Por fim, a hiperferritinemia com catarata congênita autossômica dominante é um distúrbio do metabolismo do ferro em que os membros afetados da família apresentam cataratas nucleares bilaterais de aparecimento precoce e concentrações séricas de ferritina moderadamente elevadas.145 Nestes pacientes, os níveis corporais de ferro permanecem normais, embora as elevadas concentrações séricas de ferritina frequentemente levem à suspeita de sobrecarga.
O autor recebeu honorários por sua participação em um simpósio patrocinado pela Beckman-Coulter, fabricante de analisadores hematológicos e testes diagnósticos, inclusive alguns que foram discutidos neste capítulo.
O autor também deseja agradecer ao Dr. Gary Brittenham, com quem está em dívida, cujo notável capítulo escrito para a edição anterior deste trabalho serviu de base para esta revisão.
1. Discher DE, Carl P. New insights into red cell network structure, elasticity, and spectrin unfolding—a current review. Cell Mol Biol Lett 2001;6:593–606.
2. Yonetani T, Park SI, Tsuneshige A, et al. Global allostery model of hemoglobin. Modulation of O(2) affi nity, cooperativity, and Bohr effect by heterotropic allosteric effectors. J Biol Chem 2002;277:34508–20.
3. Siems WG, Sommerburg O, Grune T. Erythrocyte free radical and energy metabolism. Clin Nephrol 2000;53 (1 Suppl):S9–17.
4. Joiner CH, Lauf PK. Ouabain binding and potassium transport in young and old populations of human red cells. Membr Biochem 1978;1:187–202.
5. Hsia CC. Respiratory function of hemoglobin. N Engl J Med 1998;338:239–47.
6. Henry ER, Bettati S, Hofrichter J, et al. A tertiary two-state allosteric model for hemoglobin. Biophys Chem 2002;98: 149–64.
7. Zhu H, Jackson T, Bunn HF. Detecting and responding to hypoxia. Nephrol Dial Transplant 2002;17 Suppl 1:3–7.
8. Wajcman H, Galacteros F. Hemoglobins with high oxygen affi nity leading to erythrocytosis. New variants and new concepts. Hemoglobin 2005;29:91–106.
9. Gladwin MT. Role of the red blood cell in nitric oxide homeostasis and hypoxic vasodilation. Adv Exp Med Biol 2006;588:189–205.
10. Geers C, Gros G. Carbon dioxide transport and carbonic anhydrase in blood and muscle. Physiol Rev 2000;80: 681–715.
11. Hercberg S, Galan P. Biochemical effects of iron deprivation. Acta Paediatr Scand Suppl 1989;361:63–70.
12. Finch C. Regulators of iron balance in humans. Blood 1994;84:1697–702.
13. Dunn LL, Rahmanto YS, Richardson DR. Iron uptake and metabolism in the new millennium. Trends Cell Biol 2007;17:93–100.
14. Aisen P. The transferrin receptor and the release of iron from transferrin. Adv Exp Med Biol 1994;356:31–40.
15. Aisen P. Transferrin receptor 1. Int J Biochem Cell Biol 2004;36:2137–43.
16. Trinder D, Baker E. Transferrin receptor 2: a new molecule in iron metabolism. Int J Biochem Cell Biol 2003;35:292–6.
17. Andrews NC. A genetic view of iron homeostasis. Semin Hematol 2002;39:227–34.
18. Koorts AM, Viljoen M. Ferritin and ferritin isoforms I: structure-function relationships, synthesis, degradation and secretion. Arch Physiol Biochem 2007;113:30–54.
19. Koorts AM, Viljoen M. Ferritin and ferritin isoforms II: protection against uncontrolled cellular proliferation, oxidative damage and infl ammatory processes. Arch Physiol Biochem 2007;113:55–64.
20. Clark MR, Shohet SB. Red cell senescence. Clin Haematol 1985;14:223–57.
21. Ascenzi P, Bocedi A, Visca P, et al. Hemoglobin and heme scavenging. IUBMB Life 2005;57:749–59.
22. Hvidberg V, Maniecki MB, Jacobsen C, et al. Identifi cation of the receptor scavenging hemopexin-heme complexes. Blood 2005;106:2572–9.
23. Kikuchi G, Yoshida T, Noguchi M. Heme oxygenase and heme degradation. Biochem Biophys Res Commun 2005;338:558–67.
24. Park CH, Valore EV, Waring AJ, et al. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem 2001;276:7806–10.
25. Nemeth E, Tuttle MS, Powelson J, et al. Hepcidin regulates iron effl ux by binding to ferroportin and inducing its internalization. Science 2004;306:2090–3.
26. Rivera S, Nemeth E, Gabayan V, et al. Synthetic hepcidin causes rapid dose-dependent hypoferremia and is concentrated in ferroportin-containing organs. Blood 2005;106: 2196–9.
27. Roetto A, Papanikolaou G, Politou M, et al. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat Genet 2003;33:21–2.
28. Gehrke SG, Herrmann T, Kulaksiz H, et al. Iron stores modulate hepatic hepcidin expression by an HFEindependent pathway. Digestion 2005;72:25–32.
29. Ponka P, Beaumont C, Richardson DR. Function and regulation of transferrin and ferritin. Semin Hematol 1998;35:35–54.
30. Cairo G, Recalcati S, Pietrangelo A, et al. The iron regulatory proteins: targets and modulators of free radical reactions and oxidative damage. Free Radic Biol Med 2002;32:1237–43.
31. Hubert N, Hentze MW. Previously uncharacterized isoforms of divalent metal transporter (DMT)-1: implications for regulation and cellular function. Proc Natl Acad Sci U S A 2002;99:12345–50.
32. Abboud S, Haile DJ. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J Biol Chem 2000;275:19906–12.
33. Brock JH, Halliday JW, Pippard MJ, Powell LW, editors. Iron metabolism in health & disease. Philadelphia: W.B. Saunders Co., 1994;427–448.
34. Ramakrishnan U, Yip R. Experiences and challenges in industrialized countries: control of iron def ciency in industrialized countries. J Nutr 2002;132:820S–4S.
35. Yip R. Iron supplementation: country level experiences and lessons learned. J Nutr 2002;132:859S–61S.
36. Iron def ciency—United States, 1999-2000. MMWR Morb Mortal Wkly Rep 2002;51:897–9.
37. Wang SA, Fadare O, Nagar A, et al. Gastrointestinal endoscopic f ndings in men with unexplained anemia and low normal ferritin values. Am J Hematol 2006;81:324–7.
38. Djalali M, Neyestani TR, Bateni J, et al. The effect of repeated blood donations on the iron status of Iranian blood donors attending the Iranian blood transfusion organization. Int J Vitam Nutr Res 2006;76:132–7.
39. Brotanek JM, Halterman JS, Auinger P, et al. Iron def ciency, prolonged bottle-feeding, and racial/ethnic disparities in young children. Arch Pediatr Adolesc Med 2005;159:1038–42.
40. Fernandes SM, de Morais MB, Amancio OM. Intestinal blood loss as an aggravating factor of iron def ciency in infants aged 9 to 12 months fed whole cow’s milk. J Clin Gastroenterol 2008;42:152–6.
41. Milman N. Iron and pregnancy—a delicate balance. Ann Hematol 2006;85:559–65.
42. Zimmermann MB, Hurrell RF. Nutritional iron def ciency. Lancet 2007;370:511–20.
43. Baccini F, Spiriti MA, Vannella L, et al. Unawareness of gastrointestinal symptomatology in adult coeliac patients with unexplained iron-def ciency anaemia presentation. Aliment Pharmacol Ther 2006;23:915–21.
44. Beyan C, Beyan E, Kaptan K, et al. Post-gastrectomy anemia: evaluation of 72 cases with post-gastrectomy anemia. Hematology 2007;12:81–4.
45. Lee PL, Halloran C, Trevino R, et al. Human transferrin G277S mutation: a risk factor for iron def ciency anaemia. Br J Haematol 2001;115:329–33.
46. Sarria B, Navas-Carretero S, Lopez-Parra AM, et al. The G277S transferrin mutation does not affect iron absorption in iron def cient women. Eur J Nutr 2007;46:57–60.
47. Merlino G, Valente M, Seraf ni A, et al. Restless legs syndrome: diagnosis, epidemiology, classif cation and consequences. Neurol Sci 2007;28 Suppl 1:S37–46.
48. Rector WG Jr. Pica. its frequency and signif cance in patients with iron-def ciency anemia due to chronic gastrointestinal blood loss. J Gen Intern Med 1989;4: 512–3.
49. Moore DF Jr, Sears DA. Pica, iron def ciency, and the medical history. Am J Med 1994;97:390–3.
50. Rector WG Jr, Fortuin NJ, Conley CL. Non-hematologic effects of chronic iron def ciency. A study of patients with polycythemia vera treated solely with venesections. Medicine (Baltimore) 1982;61:382–9.
51. Haas JD, Brownlie T. Iron def ciency and reduced work capacity: a critical review of the research to determine a causal relationship. J Nutr 2001;131:676S–88S.
52. Okonko DO, Grzeslo A, Witkowski T, et al. Effect of intravenous iron sucrose on exercise tolerance in anemic and nonanemic patients with symptomatic chronic heart failure and iron def ciency FERRIC-HF: a randomized, controlled, observer-blinded trial. J Am Coll Cardiol 2008;51:103–12.
53. Risser WL, Lee EJ, Poindexter HB, et al. Iron def ciency in female athletes: its prevalence and impact on performance. Med Sci Sports Exerc 1988;20:116–21.
54. Grantham-McGregor S, Ani C. A review of studies on the effect of iron def ciency on cognitive development in children. J Nutr 2001;131:649S–66S.
55. Hawkins RC. Total iron binding capacity or transferrin concentration alone outperforms iron and saturation indices in predicting iron def ciency. Clin Chim Acta 2007;380:203–7.
56. Means RT, Allen J, Sears DA, et al. Serum soluble transferrin receptor and the prediction of marrow aspirate results in a heterogeneous group of patients. Clin Lab Haematol 1999;21:161–7.
57. Cunietti E, Chiari MM, Monti M, et al. Distortion of iron status indices by acute infl ammation in older hospitalized patients. Arch Gerontol Geriatr 2004;39:35–42.
58. Lipshitz DA, Cook JD, Finch CA. A clinical evaluation of serum ferritin as an index of marrow iron stores. N Engl J Med 1974;290:1213–6.
59. Baynes RD, Shih YJ, Cook JD. Mechanism of production of the serum transferrin receptor. Adv Exp Med Biol 1994;356: 61–8.
60. Abe Y, Muta K, Nishimura J, et al. Regulation of transferrin receptors by iron in human erythroblasts. Am J Hematol 1992;40:270–5.
61. Skikine BS, Flowers CH, Cook JD. Serum transferrin receptor: a quantitative measure of tissue iron def ciency. Blood 1990;75:1870–6.
62. Chua E, Clague JE, Sharma AK, et al. Serum transferrin receptor assay in iron def ciency anaemia and anaemia of chronic disease in the elderly. QJM 1999;92:587–94.
63. Beguin Y, Clemons GK, Pootrakul P, et al. Quantitative assessment of erythropoiesis and functional classif cation of anemia based on measurements of serum transferrin receptor and erythropoietin. Blood 1993;81:1067–76.
64. Punnonen K, Irjala K, Rajamaeki A. Serum transferrin receptor and its ratio to serum ferritin in the diagnosis of iron def ciency. Blood 1997;89:1052–7.
65. Piedras J, Alvarez E, Herrera FM, et al. Clinical usefulness of mean corpuscular volume and red cell distribution width in iron def cient blood donors. Rev Invest Clin 1993;45:469–72.
66. Mast AE, Blinder MA, Lu Q, et al. Clinical utility of the reticulocyte hemoglobin content in the diagnosis of iron def ciency. Blood 2002;99:1489–91.
67. Ervasti M, Kotisaari S, Heinonen S, et al. Use of advanced red blood cell and reticulocyte indices improves the accuracy in diagnosing iron def ciency in pregnant women at term. Eur J Haematol 2007;79:539–45.
68. Lowenstein W, Cals MJ, De JC, et al. Free erythrocyte protoporphyrin assay in the diagnosis of iron def ciency in the anemic aged subject. A prospective study of 103 anemic patients. Ann Med Interne (Paris) 1991;142:13–6.
69. Ioannou GN, Spector J, Scott K, et al. Prospective evaluation of a clinical guideline for the diagnosis and management of iron def ciency anemia. Am J Med 2002;113: 281–7.
70. Rockey DC. Gastrointestinal tract evaluation in patients with iron def ciency anemia. Semin Gastrointest Dis 1999; 10:53–64.
71. McDiarmid T, Johnson ED. Clinical inquiries. Are any oral iron formulations better tolerated than ferrous sulfate? J Fam Pract 2002;51:576.
72. Pollak VE, Lorch JA, Means RT Jr. Unanticipated favorable effects of correcting iron defi ciency in chronic hemodialysis patients. J Investig Med 2001;49:173–83.
73. Macdougall IC. Strategies for iron supplementation: oral versus intravenous. Kidney Int 1999;Suppl 69:S61–6.
74. Rudinskas L, Paton TW, Walker SE, et al. Poor clinical response to enteric-coated iron preparations. CMAJ 1989; 141:565–6.
75. Teucher B, Olivares M, Cori H. Enhancers of iron absorption: ascorbic acid and other organic acids. Int J Vitam Nutr Res 2004;74:403–19.
76. Auerbach M, Witt D, Toler W, et al. Clinical use of the total dose intravenous infusion of iron dextran. J Lab Clin Med 1988;111:566–70.
77. Fishbane S, Kowalski EA. The comparative safety of intravenous iron dextran, iron saccharate, and sodium ferric gluconate. Semin Dial 2000;13:381–4.
78. Monaghan MS, Glasco G, St JG, et al. Safe administration of iron dextran to a patient who reacted to the test dose. South Med J 1994;87:1010–2.
79. Lewinter P. Iron-dextran anaphylaxis: occurrence after a negative test dose. J Med Soc N J 1970;67:116–7.
80. Van Wyck DB, Cavallo G, Spinowitz BS, et al. Safety and effi cacy of iron sucrose in patients sensitive to iron dextran: North American clinical trial. Am J Kidney Dis 2000;36:88–97.
81. Ganz T. Hepcidin, a key regulator of iron metabolism and mediator of anemia of infl ammation. Blood 2003;102: 783–8.
82. Means RT. Recent developments in the anemia of chronic disease. Curr Hematol Rep 2003;2:116–21.
83. Milman N, Pedersen P. Evidence that the Cys282Tyr mutation of the HFE gene originated from a population in southern Scandinavia and spread with the Vikings. Clin Genet 2003;64:36–47.
84. Adams PC, Reboussin DM, Barton JC, et al. Hemochromatosis and iron-overload screening in a racially diverse population. N Engl J Med 2005;352:1769–78.
85. Bomford A. Genetics of haemochromatosis. Lancet 2002; 360:1673–81.
86. Adams PC, Barton JC. Haemochromatosis. Lancet 2007; 370:1855–60.
87. Walsh A, Dixon JL, Ramm GA, et al. The clinical relevance of compound heterozygosity for the C282Y and H63D substitutions in hemochromatosis. Clin Gastroenterol Hepatol 2006;4:1403–10.
88. Bulaj ZJ, Griffen LM, Jorde LB, et al. Clinical and biochemical abnormalities in people heterozygous for hemochromatosis. N Engl J Med 1996;335:1799–805.
89. Powell LW. Hereditary hemochromatosis and iron overload diseases. J Gastroenterol Hepatol 2002;17 Suppl: S191–5.
90. Blitzer BL, Weiss GB, Osbaldiston GW, et al. Early idiopathic hemochromatosis with absent stainable bone marrow iron stores. Gastroenterology 75:886–8, 1978.
91. Camaschella C, Roetto A, De GM. Juvenile hemochromatosis. Semin Hematol 2002;39:242–8.
92. Le GG, Scotet V, Ka C, et al. The recently identifi ed type 2A juvenile haemochromatosis gene (HJV), a second candidate modifi er of the C282Y homozygous phenotype. Hum Mol Genet 2004;13:1913–8.
93. Roetto A, Totaro A, Piperno A, et al. New mutations inactivating transferrin receptor 2 in hemochromatosis type 3. Blood 2001;97:2555–60.
94. Nemeth E, Roetto A, Garozzo G, et al. Hepcidin is decreased in TFR2 hemochromatosis. Blood 2005;105:1803–6.
95. De D, I, Ward DM, Nemeth E, et al. The molecular basis of ferroportin-linked hemochromatosis. Proc Natl Acad Sci U S A 2005;102:8955–60.
96. Drakesmith H, Schimanski LM, Ormerod E, et al. Resistance to hepcidin is conferred by hemochromatosisassociated mutations of ferroportin. Blood 2005;106: 1092–7.
97. Cremonesi L, Forni GL, Soriani N, et al. Genetic and clinical heterogeneity of ferroportin disease. Br J Haematol 2005;131:663–70.
98. Koyama C, Wakusawa S, Hayashi H, et al. A Japanese family with ferroportin disease caused by a novel mutation of SLC40A1 gene: hyperferritinemia associated with a relatively low transferrin saturation of iron. Intern Med 2005;44:990–3.
99. Adams PC, Kertesz AE, Valberg LS. Clinical presentation of hemochromatosis: a changing scene. Am J Med 1991; 90:445–9.
100. Askari AD, Muir WA, Rosner IA, et al. Arthritis of hemochromatosis. Clinical spectrum, relation to histocompatibility antigens, and effectiveness of early phlebotomy. Am J Med 1983;75:957–65.
101. Adams PC, Deugnier Y, Moirand R, et al. The relationship between iron overload, clinical symptoms, and age in 410 patients with genetic hemochromatosis. Hepatology 1997;25:162–6.
102. Beutler E, Felitti VJ. The clinical penetrance of hereditary hemochromatosis. Hepatology 2003;37:711.
103. Allen KJ, Gurrin LC, Constantine CC, et al. Iron-overloadrelated disease in HFE hereditary hemochromatosis. N Engl J Med 2008;358:221–30.
104. Powell LW, Dixon JL, Ramm GA, et al. Screening for hemochromatosis in asymptomatic subjects with or without a family history. Arch Intern Med 2006;166:294–301.
105. Waalen J, Beutler E. Hereditary hemochromatosis: screening and management. Curr Hematol Rep 2006;5: 34–40.
106. Anderson RT, Wenzel L, Walker AP, et al. Impact of hemochromatosis screening in patients with indeterminate results: the Hemochromatosis and Iron Overload Screening Study. Genet Med 2006;8:681–7.
107. Gagne G, Reinharz D, Lafl amme N, et al. Hereditary hemochromatosis screening: effect of mutation penetrance and prevalence on cost-effectiveness of testing algorithms. Clin Genet 2007;71:46–58.
108. Screening for hemochromatosis: recommendation statement. Ann Intern Med 2006;145:204–8.
109. McGrath JS, Deugnier Y, Moirand R, et al. A nomogram to predict C282Y hemochromatosis. J Lab Clin Med 2002;140: 6–8.
110. Barton JC, Acton RT, Dawkins FW, et al. Initial screening transferrin saturation values, serum ferritin concentrations, and HFE genotypes in whites and blacks in the Hemochromatosis and Iron Overload Screening Study. Genet Test 2005;9:231–41.
111. Waalen J, Felitti VJ, Gelbart T, et al. Screening for hemochromatosis by measuring ferritin levels: a more effective approach. Blood 2008;111:3373–6.
112. Tung BY, Emond MJ, Bronner MP, et al. Hepatitis C, iron status, and disease severity: relationship with HFE mutations. Gastroenterology 2003;124:318–26.
113. Nash S, Marconi S, Sikorska K, et al. Role of liver biopsy in the diagnosis of hepatic iron overload in the era of genetic testing. Am J Clin Pathol 2002;118:73–81.
114. Shaheen NJ, Bacon BR, Grimm IS. Clinical characteristics of hereditary hemochromatosis patients who lack the C282Y mutation. Hepatology 1998;28:526–9.
115. Alustiza JM, Castiella A, De J, et al. Iron overload in the liver diagnostic and quantifi cation. Eur J Radiol 2007;61: 499–506.
116. Guyader D, Jacquelinet C, Moirand R, et al. Noninvasive prediction of fi brosis in C282Y homozygous hemochromatosis. Gastroenterology 1998;115:929–36.
117. Fabio G, Minonzio F, Delbini P, et al. Reversal of cardiac complications by deferiprone and deferoxamine combination therapy in a patient affected by a severe type of juvenile hemochromatosis (JH). Blood 2007;109:362–4.
118. Niederau C, Fischer R, Sonnenberg A, et al. Survival and causes of death in cirrhotic and in noncirrhotic patients with primary hemochromatosis. N Engl J Med 1985;313: 1256–62.
119. Aslan D, Crain K, Beutler E. A new case of human atransferrinemia with a previously undescribed mutation in the transferrin gene. Acta Haematol 2007;118:244–7.
120. Skidmore FM, Drago V, Foster P, et al. Aceruloplasminemia with progressive atrophy without brain iron overload: treatment with oral chelation. J Neurol Neurosurg Psychiatry 2008;79:467–70.
121. Mims MP, Guan Y, Pospisilova D, et al. Identifi cation of a human mutation of DMT1 in a patient with microcytic anemia and iron overload. Blood 2005;105:1337–42.
122. Bottomley SS. Secondary iron overload disorders. Semin Hematol 1998;35:77–86.
123. Cazzola M, Beguin Y, Bergamaschi G, et al. Soluble transferrin receptor as a potential determinant of iron loading in congenital anaemias due to ineffective erythropoiesis. Br J Haematol 1999;106:752–5.
124. Gardenghi S, Marongiu MF, Ramos P, et al. Ineffective erythropoiesis in beta-thalassemia is characterized by increased iron absorption mediated by down-regulation of hepcidin and up-regulation of ferroportin. Blood 2007;109: 5027–35.
125. Stuart KA, Anderson GJ, Frazer DM, et al. Increased duodenal expression of divalent metal transporter 1 and iron-regulated gene 1 in cirrhosis. Hepatology 2004;39: 492–9.
126. Chiaverini C, Halimi G, Ouzan D, et al. Porphyria cutanea tarda, C282Y, H63D and S65C HFE gene mutations and hepatitis C infection: a study from southern France. Dermatology 2003;206:212–6.
127. George DK, Goldwurm S, MacDonald GA, et al. Increased hepatic iron concentration in nonalcoholic steatohepatitis is associated with increased fi brosis. Gastroenterology 1998;114:311–8.
128. Uraz S, Aygun C, Sonsuz A, et al. Serum iron levels and hepatic iron overload in nonalcoholic steatohepatitis and chronic viral hepatitis. Dig Dis Sci 2005;50:964–9.
129. Friedman BM, Baynes RD, Bothwell TH, et al. Dietary iron overload in southern African rural blacks. S Afr Med J 1990;78:301–5.
130. Walker AR, Segal I. Iron overload in Sub-Saharan Africa: to what extent is it a public health problem? Br J Nutr 1999;81:427–34.
131. Wheby MS. Medicinal iron-induced hepatic cirrhosis: reversal by phlebotomy: studies on pathogenesis. Trans Am Clin Climatol Assoc 1978;89:100–8.
132. Files B, Brambilla D, Kutlar A, et al. Longitudinal changes in ferritin during chronic transfusion: a report from the Stroke Prevention Trial in Sickle Cell Anemia (STOP). J Pediatr Hematol Oncol 2002;24:284–90.
133. Porter JB. Practical management of iron overload. Br J Haematol 2001;115:239–52.
134. Olivieri NF, Brittenham GM. Iron-chelating therapy and the treatment of thalassemia. Blood 1997;89:739–61.
135. Hilliard LM, Williams BF, Lounsbury AE, et al. Erythrocytapheresis limits iron accumulation in chronically transfused sickle cell patients. Am J Hematol 1998;59: 28–35.
136. Gabutti V, Piga A. Results of long-term iron-chelating therapy. Acta Haematol 1996;95:26–36.
137. Anderson LJ, Westwood MA, Holden S, et al. Myocardial iron clearance during reversal of siderotic cardiomyopathy with intravenous desferrioxamine: a prospective study using T2* cardiovascular magnetic resonance. Br J Haematol 2004;127:348–55.
138. Cappellini MD, Cohen A, Piga A, et al. A phase 3 study of deferasirox (ICL670), a once-daily oral iron chelator, in patients with beta-thalassemia. Blood 2006;107:3455–62.
139. Hoffbrand AV, Cohen A, Hershko C. Role of deferiprone in chelation therapy for transfusional iron overload. Blood 2003;102:17–24.
140. Maggio A, D’Amico G, Morabito A, et al. Deferiprone versus deferoxamine in patients with thalassemia major: a randomized clinical trial. Blood Cells Mol Dis 2002;28: 196–208.
141. Olivieri NF. Progression of iron overload in sickle cell disease. Semin Hematol 2001;38:57–62.
142. Pennell DJ. T2* magnetic resonance and myocardial iron in thalassemia. Ann N Y Acad Sci 2005;1054:373–8.
143. Knisely AS, Mieli-Vergani G, Whitington PF. Neonatal hemochromatosis. Gastroenterol Clin North Am 2003;32: 877–vii.
144. Ponka P. Rare causes of hereditary iron overload. Semin Hematol 2002;39:249–62.
145. Girelli D, Olivieri O, Gasparini P, et al. Molecular basis for the hereditary hyperferritinemia-cataract syndrome. Blood 1996;87:4912–3.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.