(Carregando Índice)... (Carregando Índice)... |
Última revisão: 08/08/2012
Comentários de assinantes: 0
Michael D. Lockshin, MD
Professor of Medicine and Obstretrics-Gynecology, Weill-Cornell Medical Center, New York, NY; Director, Barbara Volcker Center, Hospital for Special Surgery, New York, NY
Artigo original: Lockshin MD. Systemic lupus erythematosus. ACP Medicine. 2008;1-18.
[The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2011 Decker Intellectual Properties Inc. All Rights Reserved.]
Agradecimentos: Figura 2b – ©2002 Hospital for Special Surgery. www.Rheumatology.HSS.edu. Todos os direitos reservados. Usada com permissão.
Tradução: Soraya Imon de Oliveira
Revisão técnica: Dr. Euclides Furtado de Albuquerque Cavalcanti
O lúpus é uma doença autoimmune crônica caracterizada pela formação de autoanticorpos dirigidos contra os antígenos nucleares, que causam uma variedade de anormalidades clínicas e laboratoriais, como erupções, artrite, leucopenia e trombocitopenia, alopecia, febre, nefrite e doença neurológica. A maioria ou todos os sintomas do lúpus agudo são atribuíveis ao ataque imunológico dos órgãos afetados. Muitas complicações da doença a longo prazo são atribuíveis tanto à doença como ao seu tratamento.1
O termo lúpus aplica-se a diversas variantes da doença [Tabela 1], das quais o lúpus eritematoso sistêmico (LES) é a mais grave e a mais comum. O LES representa o protótipo de uma doença autoimune sistêmica, envolvendo múltiplos sistemas orgânicos de modos patologicamente similares. Caracteristicamente, os pacientes com LES progridem ao longo de vários períodos de inflamação ativa (exacerbações) e períodos de quiescência (remissão), sendo que ambos podem ocorrer de maneira espontânea. A exacerbação e a quiescência também podem ser induzidas. Os motivos que levam a este curso variável são desconhecidos. A exposição solar intensa, reações medicamentosas e infecções são circunstâncias que induzem as exacerbações. O tratamento tem como objetivo induzir a remissão.
Tabela 1. Aspectos característicos que diferenciam as formas de lúpus das doenças lúpus-símile
|
Sistema orgânico |
Lúpus |
AR |
Síndrome de Sjögren | |||
|
LES |
Discoide |
Fármaco-induzido |
Neonatal | |||
|
Pele |
Erupções específicas, alopecia, úlceras mucosas, telangiectasia periungueal |
Erupções específicas, alopecia, úlceras mucosas |
Erupção |
Erupção |
Nódulos subcutâneos |
Ressecamento ocular e bucal |
|
Articulações |
Artrite não destrutiva simétrica |
– |
Artrite não destrutiva simétrica |
– |
Artrite destrutiva simétrica |
Artrite destrutiva simétrica |
|
Renal |
Glomerulonefrite, insuficiência renal |
– |
– |
– |
Amiloidose (tardia) |
Disfunção tubular |
|
SNC |
Convulsões, psicose, disfunção cognitiva, AVC, mielopatia, neuropatia |
– |
– |
– |
Neuropatia periférica |
Neuropatia craniana e periférica |
|
Cardíaco |
– |
– |
– |
Bloqueio cardíaco |
– |
– |
|
Sangue |
|
|
|
|
|
– |
|
FAN |
Positivo forte, qualquer padrão |
Pode ser positivo |
Positivo forte |
Positivo |
Pode ser positivo |
Positivo |
|
Complemento |
Baixa intensidade com doença renal ou anemia hemolítica |
Normal |
Normal |
Normal ou baixo |
Comumente elevado |
Normal |
|
Autoanticorpos diagnósticos |
Anti-dsDNA, anti-Sm |
– |
Anti-histona |
Anti-SS-A, anti-SS-B |
Anti-IgG (fator reumatoide), anti-CCP |
Anti-SS-A, anti-SS-B |
|
Outros autoanticorpos |
Anti-SS-A (Ro), anti-SS-B (La), anti-RNP, anti-IgG, anti-ssDNA |
Anti-ssDNA |
Anti-histona |
Anti-RNP |
– |
Anti-IgG |
|
Outras anormalidades |
Leucopenia, trombocitopenia, hemólise |
– |
Leucopenia |
Trombocitopenia, hemólise |
Leucocitose, trombólise |
Hiperglobulinemia |
Tabela 1 (cont.). Aspectos característicos que diferenciam as formas de lúpus das doenças lúpus-símile
|
Sistema orgânico |
DMTC |
DITC |
SAAFP |
Esclerodermia |
DLS |
Dermatomiosite |
|
Pele |
Esclerodactilia |
– |
Livedo reticular |
Esclerodermia, telangiectasia periungueal |
– |
Erupção específica, telangiectasia periungueal |
|
Articulações |
Artrite não destrutiva simétrica |
Artrite não destrutiva simétrica |
– |
Artrite precoce, simétrica, transiente |
Artrite não destrutiva simétrica |
– |
|
Renal |
– |
– |
Microangiopatia trombótica |
Crise renal esclerodérmica |
– |
– |
|
SNC |
– |
– |
AVC, mielopatia |
Crise hipertensiva |
– |
Miopatia |
|
Cardíaco |
– |
– |
– |
– |
– |
– |
|
Sangue |
– |
– |
– |
– |
– |
– |
|
FAN |
Positivo, pontilhado |
Pode ser positivo |
Pode ser positivo |
Positivo, pontilhado, nucleolar, centromérico |
Pode ser positivo |
Positivo |
|
Complemento |
Normal |
Normal |
Normal |
Normal |
Normal |
Normal |
|
Autoanticorpos diagnósticos |
Anti-RNP |
– |
Anticardiolipina, anticoagulante lúpico |
Anti-Scl-70, anticentrômero (topoisomerase-1) |
– |
Anti-Jo-1 |
|
Outros autoanticorpos |
– |
– |
Anti-beta2-glicoproteína |
– |
– |
– |
|
Outras anormalidades |
– |
– |
Trombocitopenia |
Renina elevada durante a crise |
– |
CPK e aldolase elevadas |
FAN = fator antinuclear; anti-CCP= anti-peptídeo citrulinado cítrico; AVC = acidente vascular cerebral; SNC = sistema nervoso central; CPK = creatinina fosfoquinase; dsDNA = DNA de fita dupla (double-stranded DNA); DLS = doença lúpus-símile; DMTC = doença mista do tecido conectivo; SAFP = síndrome antifosfolipíde primária; AR = artrite reumatoide; RNP = ribonucleoproteína; ssDNA = DNA de fita única (single-stranded DNA); LES = lúpus eritematoso sistêmico; DITC = doença indiferenciada do tecido conjuntivo.
O LES pode ocorrer como uma síndrome por sobreposição, que compartilha certos aspectos com outras doenças autoimunes, como a doença do tecido conectivo indiferenciado ou mista, dermatomiosite, síndrome de Sjögren, artrite reumatoide e esclerodermia. As doenças autoimunes órgão-específicas (p. ex., tireoidite, anemia hemolítica autoimune e trombocitopenia idiopática) frequentemente acompanham e podem fazer parte do LES.
O lúpus também pode se manifestar apenas como uma doença cutânea. O lúpus discoide manifesta-se como uma erupção destrutiva que deixa cicatriz, sem sintomas sistêmicos nem autoanticorpos.2 O lúpus cutâneo subagudo abrange uma característica erupção policíclica persistente, sintomas viscerais relativamente mínimos e exames de sangue fortemente positivos.
Fármacos como a procainamida e alguns anticonvulsivos induzem uma síndrome lúpus-símile, denominada lúpus fármaco-induzido.3 Em casos pouco comuns, alguns indivíduos (que muitas vezes são parentes de pacientes com lúpus) apresentam testes sanguíneos positivos para lúpus, no entanto seu estado clínico é normal. Na ausência de sintomas, estes indivíduos não são considerados como portadores do lúpus.
Cerca de 1/3 dos pacientes lúpicos possuem anticorpos antifosfolípides, que induzem formação de coágulos sanguíneos e morte fetal. Este anticorpo e os sintomas compatíveis, na ausência de lúpus clínico, são referidos como síndrome antifosfolípide (SAF).4
O lúpus neonatal consiste numa síndrome em que se observa erupção, trombocitopenia e bloqueio cardíaco congênito em bebês nascidos de mães portadoras de anticorpos contra os antígenos SS-A (Ro) e SS-B (La).5 Esta síndrome não evolui para LES.
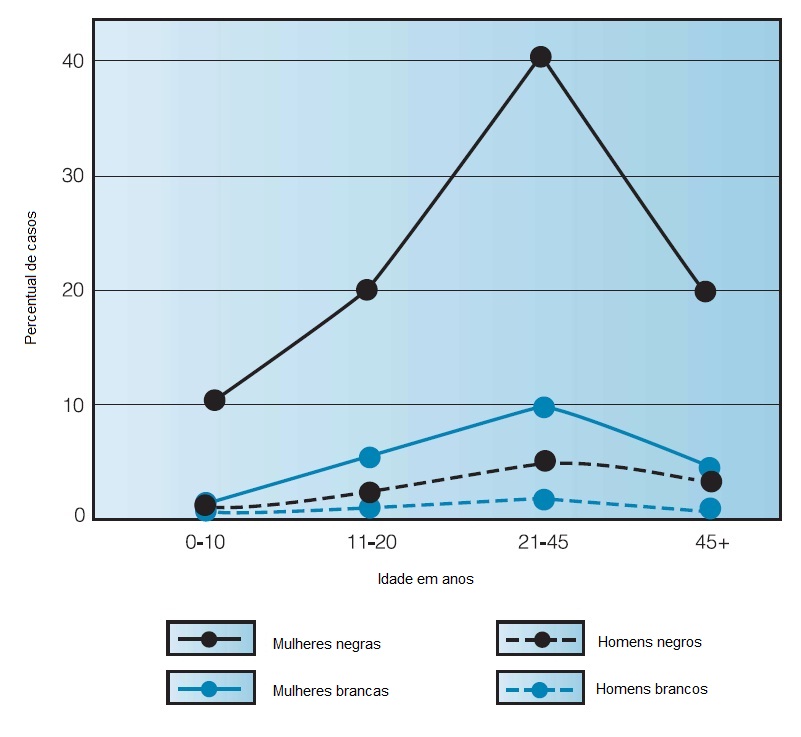
O LES é primariamente uma doença que afeta mulheres jovens. A predominância feminina ainda não foi explicada. Mulheres com idade entre 15 e 45 anos são mais comumente afetadas. Nesta faixa etária, a proporção mulheres:homens está entre 6:1 e 9:1. Indivíduos afro-americanos apresentam uma propensão quatro vezes maior de desenvolver lúpus em comparação aos brancos.6 A incidência da doença nas populações asiáticas, hispânicas e de americanos nativos situa-se entre aquelas observadas nas populações de negros e de brancos [Figura 1]. Até o momento, nenhuma explicação convincente indicou o motivo pelo qual os afro-americanos são afetados com maior frequência. As diferenças raciais relacionadas à incidência do LES persistem quando as diferenças socioeconômicas são controladas. O grau de severidade do LES é semelhante em homens e mulheres. A sobrevida geral é menor entre os afro-americanos.7

Figura 1. Distribuição da incidência do lúpus eritematoso sistêmico (LES) por idade, sexo e raça.6
Os aspectos familiares do lúpus são marcantes: aproximadamente 10% dos indivíduos afetados possuem familiares com lúpus ou outra doença autoimune. A suscetibilidade ao lúpus é maior entre indivíduos com deficiências genéticas específicas [ver Outras Suscetibilidades Genéticas, adiante].
Anticorpos circulantes contra uma ampla lista de autoantígenos caracterizam o LES. Os anticorpos antinucleares, usualmente definidos por imunofluorescência, são encontrados em altos níveis em quase todos os pacientes lúpicos. Os testes de detecção de fatores antinúcleo (FAN) são utilizados como teste de triagem (sensível, porém inespecífico) da doença. Os autoanticorpos contra constituintes nucleares – dirigidos primariamente contra o DNA de fita dupla (nativo), mas também contra o DNA de fita única (desnaturado), histonas, proteínas ribonucleares e outros antígenos nucleares (p. ex., antígeno de Smith [Sm]) – confirmam o diagnóstico e é provável que sejam patogênicos. Estes autoanticorpos, por exemplo, causam glomerulonefrite via indução de inflamação, ao se depositarem como imunocomplexos fixadores de complemento sobre as membranas basais glomerulares (MBGs) ou ligando-se diretamente à MBG.8 Tanto modelos de experimentação animal como observações clínicas sugerem que os autoanticorpos ativadores de complemento mediam a glomerulonefrite, hemólise e trombocitopenia associadas ao lúpus. Os anticorpos dirigidos contra as proteínas ligadoras de fosfolipídio (beta-2-glicoproteína I, protrombina e outras) mediam a trombose e a perda fetal. A erupção lúpica e a artrite estão menos nitidamente ligadas aos autoanticorpos, porém os imunorreagentes e a inflamação aparecem em biópsias de amostras relevantes, primariamente junto à membrana basal.9 Entre as manifestações do lúpus, o lúpus neurológico é menos nitidamente causado por anticorpos anti-DNA ou por outros autoanticorpos. Entretanto, o anticorpo antirribossômico P pode definir os distúrbios do humor no lúpus neurológico, enquanto os anticorpos contra o receptor do glutamato podem mediar algumas formas de disfunção cognitiva.10,11 Todavia, foi descartada a hipótese de que o anticorpo contra o receptor de glutamato teria utilidade clínica no diagnóstico da disfunção cognitiva associada ao LES.12
Pacientes com lúpus frequentemente apresentam defeitos genéticos envolvendo o processamento de imunocomplexos. Isto sugere que o LES surge em decorrência da eliminação incompleta ou imprópria de material exógeno.13 Estes defeitos incluem as anormalidades do complemento (deficiências de C1q, C2 ou C4), receptor de Fc, vias apoptóticas e células fagocíticas.14 A depuração defeituosa dos imunocomplexos pode resultar na sua persistência em grandes quantidades,15 sendo que os autoanticorpos podem atuar como mecanismo protetor e neutralizá-los. Outras teorias de patogênese argumentam que as predisposições genéticas promotoras de respostas de células T auxiliares de tipo 2 ou de desregulação de citocinas constituem os defeitos subjacentes que levam ao desenvolvimento do LES.
Em modelos experimentais animais, foram identificados vários defeitos imunes que podem causar LES. Estes defeitos incluem anormalidades genéticas que afetam toda a imunorreatividade (apoptose [Fas, Fas-ligante, bcl-2]), ativação da célula B (FcyRIIB, SHP-1, CD22, CD19, PD-1, Lyn, Blys-1), ativação da célula T (TGF-beta, TGF-beta-R, PD-1), proliferação celular (p21, Fli-1) e citocinas (IFN-gama, IL-4, IL-10, TNF-alfa), bem como anormalidades genéticas que afetam a depuração de autoantígenos (C1q, C4, SAP, DNAse-1).16,17 Dados fornecidos por estudos de arranjos de genes indicam fortemente que os genes codificadores do interferon sofrem uma regulação positiva marcante em pacientes com LES ativo.18 Esta área está se desenvolvendo com muita rapidez. Atualmente, sabe-se que o transdutor de sinal e ativador da transcrição 4 (STAT4, signal transducer and activator of transcription 4), fator regulador do interferon 5 (IRF5, interferon regulatory factor 5) e integrina alfa M (componente do complemento 3 e subunidade de receptor 3) (ITGAM, integrin alpha M), talvez combinados, estão estreitamente associados à suscetibilidade ao lúpus.19-25
Estudos sobre o LES envolvendo gêmeos e famílias evidenciaram que esta doença possui um forte componente hereditário. O HLA de tipos DR3 e DR4 predomina em pacientes com LES.26 Foram identificados loci de suscetibilidade específicos localizados nos cromossomos 1, 4 e 7, entre outros.27 Indivíduos com deficiências genéticas de complemento parecem ser mais suscetíveis ao desenvolvimento de lúpus.28 Os alelos específicos para o receptor FcIII gama aumentam a gravidade da nefrite lúpica, particularmente em brancos.29
Apesar de haver suspeitas antigas acerca da existência de um agente deflagrador infeccioso de LES, nenhuma infecção isolada foi encontrada até agora. Observou-se a exposição universal (numa idade em que uma exposição de 50% é a regra) de crianças com LES ao vírus Epstein-Barr, sugerindo uma possível ligação entre este vírus e a doença.30 É possível identificar autoanticorpos em amostras de soro com uma antecedência de até 10 anos em relação à manifestação dos primeiros sintomas do LES. Os autoanticorpos aparecem primeiro como um anticorpo específico e, depois, tornam-se generalizados pouco antes da manifestação clínica inicial. Ainda não se sabe se esta progressão reflete a resposta a uma infecção ou à autoimunidade.31
Alguns pesquisadores atribuem a predominância feminina do LES e sua ocorrência durante a idade fértil ao efeito de regulação positiva exercido pelo estrogênio sobre o sistema imune. Este fenômeno é amplamente demonstrável in vitro. Entretanto, este argumento se aplica à autoimunidade em geral, não especificamente ao lúpus, e falha em explicar o motivo pelo qual outras doenças autoimunes apresentam proporções mulher:homem significativamente menos acentuadas. Além disso, a reposição de estrogênio durante a pós-menopausa e o uso de anticoncepcionais não alteram de modo significativo a incidência ou a severidade do LES. Isto também ocorre durante a gravidez. Diferenças mínimas de incidência ou suscetibilidade foram relatadas, ocasionalmente.32-34 Explicações alternativas para a elevada proporção mulher:homem incluem um mecanismo de limiar estrogênio-sensível27 ou a existência de diferenças sexuais relacionadas à exposição a agentes exógenos (embora nenhuma tenha sido sugerida de maneira convincente).35 Relatos de pacientes com síndrome de Klinefelter e LES levaram os pesquisadores a considerar o possível envolvimento do hipogonadismo masculino na patogênese do lúpus.
A maior parte das informações atualmente disponíveis sobre a patogênese do LES enfoca a regulação positiva ou negativa de componentes da resposta imune, controle genéticos da imunidade e potenciais agentes etiológicos. Entretanto, o dano gerado a longo prazo na doença crônica, a partir da lesão tecidual ou do tratamento, tem a mesma importância de uma doença inflamatória aguda. Alguns elementos do dano são nitidamente atribuíveis à terapia: osteoporose, osteonecrose, cataratas e rupturas de tendão, condições associadas à terapia prolongada com corticosteroides. Outros elementos do dano resultam diretamente dos aspectos inflamatórios e imunológicos da doença, que acarretam necrose e formação de cicatriz nos tecidos: insuficiência renal progressiva, doença articular destrutiva e infartos cerebrais. Muitas destas complicações são mais sérias em pacientes cuja etnia é minoritária ou pertencentes às classes socioeconômicas mais baixas.36,37 Existem ainda outros elementos de cronicidade que apresentam uma relação incerta com a doença e o tratamento: aterosclerose acelerada,38 doença cardíaca valvar, disfunção cognitiva e disfunção psicossocial.
O American College of Rheumatology (ACR) definiu critérios para a classificação do LES [Tabela 2].39 Embora os critérios do ACR sejam úteis para garantir a uniformidade dos pacientes descritos nos periódicos médicos, estes critérios frequentemente são utilizados erroneamente como critérios diagnósticos. Para pacientes individuais, estes critérios estão associados a altas taxas de resultados falso-negativos e falso-positivos.40 Exemplificando, um paciente que apresente como únicas manifestações da doença uma nefrite lúpica comprovada por biópsia, positividade para FAN e anticorpos anti-Sm não atende aos critérios do ACR. Por outro lado, um paciente com artrite reumatoide e positividade para FAN, baixa positividade para anticorpos anti-DNA e leucopenia (decorrente da síndrome de Felty) corresponde aos critérios estabelecidos pelo ACR. Como regra geral a existência de uma doença característica afetando um determinado sistema orgânico (rins, articulações, pele) e acompanhada de altos títulos de anticorpo anti-dsDNA ou anti-Sm é suficiente para estabelecer o diagnóstico clínico.
Tabela 2. Critérios do American College of Rheumatology para classificação do lúpus eritematoso sistêmico (LES).*
|
Erupção malar |
|
Erupção discoide |
|
Fotossensibilidade |
|
Úlceras orais |
|
Artrite |
|
Serosite (pleurite ou pericardite) |
|
Distúrbio renal (proteinúria > 0,5 g/dia ou cilindros celulares) |
|
Distúrbio neurológico (convulsões ou psicose) |
|
Distúrbio hematológico (anemia hemolítica, leucopenia, linfopenia ou trombocitopenia) |
|
Distúrbio imunológico (anticorpos anti-DNA, anti-Sm ou anti-fosfolipídio [anticardiolipina, anticoagulante lúpico ou ensaio biológico com resultado falso-positivo para sífilis]) |
|
Anticorpo antinuclear (FAN) |
Estes critérios não são diagnósticos.
*É necessário que quatro critérios sejam atendidos para incluir um paciente na coorte de LES de um estudo científico.
Na prática clínica, o diagnóstico de LES baseia-se em uma combinação de ensaios de detecção de autoanticorpos, manifestações clínicas e exames laboratoriais avaliando os sistemas orgânicos afetados. As manifestações clínicas do lúpus são muitas. Os pacientes que apresentam atividade ou danos associados ao lúpus podem ser assintomáticos ou exibir achados que refletem os sistemas orgânicos específicos envolvidos [Tabela 3].
Tabela 3. Anormalidades detectadas durante o exame físico nas formas aguda e crônica de LES.*
|
Órgão |
Doença aguda |
Doença crônica | ||
|
Comum |
Incomum |
Comum |
Incomum | |
|
Geral |
Febre, perda de peso |
Astenia |
Caquexia |
|
|
Pele |
Erupção malar, erupções em outros locais, alopecia |
Telangiectasia periungueal, vasculite |
Erupção malar, erupções em outros locais, alopecia, estrias, atrofia, alteração pigmentar |
Telangiectasia periungueal, úlceras cutâneas |
|
Nodos |
Linfadenopatia |
|
|
|
|
Mamas |
|
|
|
|
|
Olhos |
|
Exsudatos, hemorragias retinianas |
Alterações hipertensivas |
Exsudatos, hemorragias retinianas |
|
Orelhas |
|
Erupção no canal auditivo, diminuição da audição |
|
Formação de cicatriz dentro do canal auditivo, diminuição da audição |
|
Nariz |
|
Ulceração septal |
|
Perfuração septal |
|
Garganta |
|
Úlceras mucosas (palato duro) |
|
Cicatrização mucosa (palato duro) |
|
Tórax |
|
Estertores, atrito pleural, efusão |
|
Estertores, efusão |
|
Coração e vasos |
Fenômeno de Raynaud |
Atrito pericárdico, aumento cardíaco |
Fenômeno de Raynaud, doença valvar |
aumento cardíaco, insuficiência valvar, arritmia |
|
Abdome |
|
Hepatomegalia, esplenomegalia |
|
Hepatomegalia, esplenomegalia, ascite |
|
Músculos |
Fraqueza, sensibilidade |
|
Fraqueza, atrofia, ruptura de tendão |
|
|
Ossos |
|
|
Fraturas (vértebras, quadril), osteonecrose |
|
|
Articulações |
Sinovite, movimentação restrita |
|
Sinovite, deformidade, movimentação restrita |
Deformidades de Jaccoud |
|
Neuromotor |
|
AVC, mononeurite múltipla, convulsão |
|
AVC, mononeurite múltipla, convulsões |
|
Neurossensorial |
|
Neuropatia periférica, mononeurite múltipla |
|
Neuropatia periférica, mononeurite múltipla |
|
Cognitivo |
Depressão |
Psicose, demência |
Depressão |
Psicose, demência |
*Esta tabela não é abrangente nem inclui anormalidades raras.
Os sinais e sintomas acumulam-se ao longo do tempo nos pacientes com LES [Tabela 4].41 Num dado momento, especialmente no início da doença, somente algumas manifestações estão presentes. Artrite, mal-estar, citopenias e erupções constituem os achados iniciais mais proeminentes. A nefrite (com insuficiência renal), artrite, osteoporose e osteonecrose (complicações associadas aos corticosteroides), doença neurológica, aterosclerose acelerada e doença valvar cardíaca passam a ser dominantes nas fases tardias do curso da doença. Com a atividade da doença e seu tratamento, há também um elevado risco de desenvolvimento de infecções oportunísticas. Para fins conceituais, é mais fácil considerar a atividade e as manifestações da doença separadamente para cada sistema orgânico afetado.
Tabela 4. Frequências de várias manifestações do LES conforme o estágio da doença.33
|
Manifestação |
Doença em estágio inicial (%) |
Doença em estágio tardio (%) |
|
Artrite |
46-53 |
83-95 |
|
Erupções |
9-11 |
81-88 |
|
Febre |
3-5 |
77 |
|
Úlceras mucosas |
— |
7-23 |
|
Alopecia |
— |
37-45 |
|
Serosite |
5 |
63 |
|
Inflamação pulmonar |
— |
9 |
|
Anormalidades no teste de função pulmonar |
1 |
— |
|
Vasculite |
— |
21-27 |
|
Miosite |
— |
5 |
|
Osteoporose |
— |
Alto |
|
Osteonecrose |
— |
7-24 |
|
Leucopenia |
41-66 |
41-66 |
|
Trombocitopenia |
2 |
19-45 |
|
Anemia |
2 |
57-73 |
|
Anormalidades do SNC |
3 |
55-59 |
|
Nefrite |
6 |
31-53 |
|
Insuficiência renal |
< 1 |
20 |
SNC = sistema nervoso central.
Mal-estar, artralgia, mialgia e febre (usualmente baixa), além de perda de peso são manifestações comuns do LES ativo. Alguns pacientes apresentam febre alta (> 40 ºC). No entanto, mesmo com febre alta, os calafrios são incomuns e, quando ocorrem, sugerem a ocorrência de infecção. Assim como as manifestações órgão-específicas do lúpus, os sintomas variam consideravelmente durante o dia e no decorrer das semanas. Em cerca de 1/3 dos pacientes com lúpus, a exposição solar (geralmente intensa) induz exacerbação sistêmica. A exposição solar leve ou de curta duração não é prejudicial à maioria dos pacientes.
Até metade dos pacientes com lúpus manifestam certo grau de alopecia. Tipicamente, esta condição assume a forma de cabelos frontais quebradiços e adelgaçamento, que melhoram quando o paciente recupera a saúde. A alopecia pode ser severa. As erupções discoides produzem placas focais de perda de cabelo.
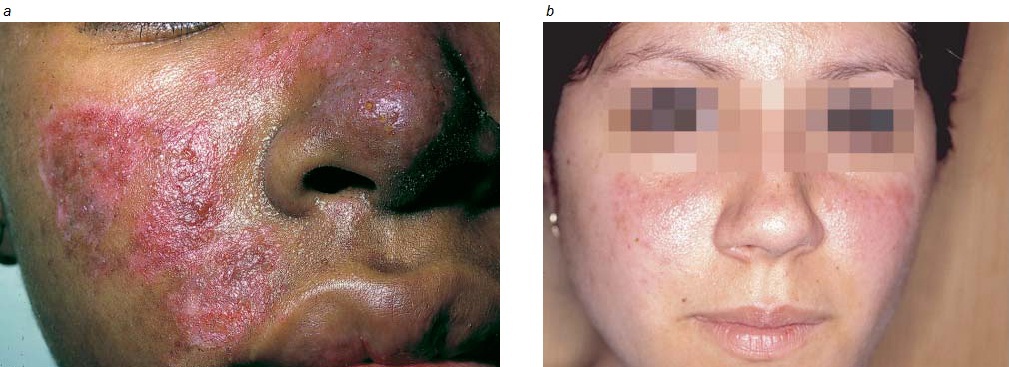
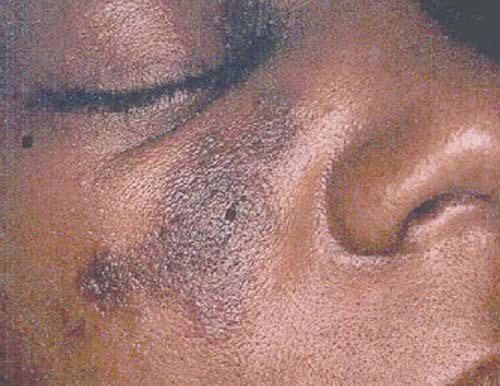
A maioria dos pacientes desenvolve uma erupção em algum momento, ao longo do curso da doença. A conhecida erupção cutânea em forma de asa de borboleta, que ocorre nas bochechas e através da ponte do nariz [Figura 2], é observada apenas em uma minoria dos pacientes. Contudo, a maioria das erupções envolve a face de algum modo. É comum também haver envolvimento da ponta do queixo, lábio superior, sobrancelhas e raiz do cabelo. A erupção pode consistir apenas de um eritema, pode ser papular e escamosa, ou pode ainda ser profundamente pigmentada (erupção discoide). As erupções discoides, que podem formar cicatriz, são hiperpigmentadas ao longo da circunferência e frequentemente despigmentadas no centro [Figura 3]. Os pacientes com erupções discoides podem ter lúpus discoide ou LES. Outros tipos de erupção ocorrem apenas no LES.
Figura 2. A maioria das erupções lúpicas envolve a face (a). A erupção lúpica em forma de asa de borboleta, em ambas as bochechas e atravessando a ponte do nariz (b), é observada apenas em uma minoria dos pacientes.
Figura 3. As erupções do lúpus discoide são hiperpigmentadas ao longo da circunferência e frequentemente despigmentadas no centro.
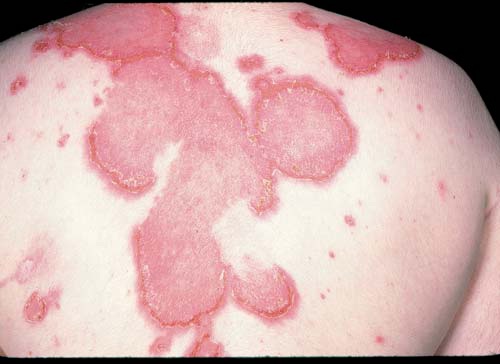
As erupções inflamatórias diagnósticas são caracterizadas por lesões que exibem um aspecto saliente, escamoso e relativamente uniforme. Estas erupções podem ulcerar e apresentam bordas bem definidas. As erupções menos diagnósticas surgem nas superfícies extensoras da porção superior dos braços, na porção mais exposta do pescoço e ombros, bem como nas superfícies extensoras dos cotovelos e dedos da mão. Erupções deste tipo usualmente são eritematosas maculares. As erupções eritematosas evoluem ao longo de dias ou semanas e muitas vezes deixam uma área de hiperpigmentação ao regredirem. Podem parecer mais proeminentes quando há febre ou na gravidez. As erupções vasculíticas (geralmente pequenas pápulas ulcerativas) ocorrem nas extremidades extensoras dos cotovelos. As lesões vasculares eritematosas, que são dolorosas, ocorrem na porção distal dos dedos e palmas das mãos (lúpus pérnio). Uma erupção persistente e policíclica, afetando primariamente o tronco e associada de modo específico aos anticorpos anti-SS-A, é conhecida como lúpus cutâneo subagudo [Figura 4]. Uma lesão subcutânea dolorosa, profundamente endurecida e apresentando sensibilidade é denominada lúpus profundo.
Figura 4. A erupção do lúpus cutâneo subagudo é policíclica e persistente.
Alguns pacientes desenvolvem apenas um eritema com as distribuições típicas das erupções lúpicas. Diferentemente das outras, as erupções eritematosas em si não são diagnósticas de lúpus, mas têm caráter aditivo em termos de informação diagnóstica geral. As erupções lúpicas são frequentemente confundidas com rosáceas (embora estas sejam mais oleosas e papulares), erupções polimórficas leves e reações alérgicas.
As úlceras nasais crônicas e as úlceras bucais indolores recorrentes [Figura 5] (em particular junto ao palato duro, mas também nas gengivas e mucosa bucal) são características da doença mais severa. As úlceras mucosas causam mais irritação do que dor intensa. A telangiectasia periungueal e as pequenas úlceras vasculíticas localizadas nos cotovelos também são observadas na doença mais grave. O lúpus pérnio é uma forma de vasculite.
Figura 5. Úlceras bucais indolores, encontradas mais frequentemente no palato duro, mas também nas gengivas e mucosa bucal, são características do lúpus mais severo.
A linfadenopatia é comum na doença ativa. Os linfonodos são de pequeno tamanho e a linfonodomegalia é generalizada (observada em varreduras de tomografia computadorizada [TC] do abdome ou do tórax). Sua resolução ocorre rapidamente nos pacientes que iniciam a terapia com corticosteroide para tratamento de outras manifestações do LES.
O LES está associado a uma gama de distúrbios cardiopulmonares [Tabela 5] que usualmente produzem sintomas ou achados físicos anormais. É desnecessário submeter pacientes assintomáticos a uma avaliação para doença cardiovascular.
Tabela 5. Manifestações cardiopulmonares do LES.
|
Pleuropericardite |
|
Endocardite de Libman-Sacks |
|
Insuficiência valvar |
|
Estenose valvar |
|
Miocardiopatia isquêmica |
|
Aterosclerose acelerada |
|
Síndrome antifosfolípide |
|
Doença cardíaca hipertensiva |
|
Vasculite |
|
Doença cardíaca hipertensiva |
|
Hipertensão pulmonar |
|
Insuficiência arterial periférica |
|
Vasculite |
|
Aterosclerose |
|
Síndrome antifosfolípide |
|
Trombose venosa periférica |
|
Fenômeno de Raynaud |
|
Bloqueio cardíaco congênito completo (em recém-nascidos com lúpus eritematoso neonatal) |
A pleurocardite costuma ser sintomática, porém geralmente não é prejudicial à vida. Faz com que o paciente sinta dor ao respirar e também causa uma elevação do diafragma que é evidente durante o exame físico ou na radiografia. A atelectasia nas bases dos pulmões é audível como crepitações finas ou visível ao raio X como linhas horizontais (atelectasia em placa). Os testes de função pulmonar podem refletir uma capacidade de difusão reduzida, volumes pulmonares diminuídos e menor elasticidade pulmonar.
Também ocorrem efusões pleurais que, todavia, não costumam ser extensas. Na toracocentese, as células LE (leucócitos polimorfonucleares contendo debris nucleares ingeridos, que são vistos como uma inclusão redonda e homogênea) podem ser detectadas por meio da coloração do sobrenadante pelo método de Wright.
Entre os pacientes, uma minoria desenvolve insuficiência respiratória a partir da fibrose pulmonar. Uma manifestação rara é conhecida como pulmão lúpico (também conhecido por pneumonite lúpica). Esta manifestação consiste numa doença inflamatória dos pulmões ou hemorragia pulmonar, ambas prejudiciais à vida.42 Embora seja incomum, a hipertensão pulmonar é uma condição séria que ocorre com maior frequência em pacientes com fenômeno de Raynaud intenso, embolismo pulmonar recorrente ou fibrose pulmonar.
A ecocardiografia transtorácica revela a doença cardíaca valvar em até 30% dos paciente com LES de longa duração, sendo que a ecocardiografia transesofágica demonstra uma frequência significativamente maior.43 As lesões de Libman-Sacks ocorrem primariamente na valva mitral, mas também afetam a valva aórtica e, em raros casos, as valvas pulmonar e tricúspide. A doença valvar sintomática pode ser mais comum em pacientes com anticorpos antifosfolípides.44 As efusões ou espessamentos pericárdicos ocorrem durante a doença ativa, contudo, são incomuns. Pequenas efusões pericárdicas, que podem ser sintomáticas ou assintomáticas, ocorrem com frequência em pacientes com LES ativo. Efusões extensas com risco de vida são incomuns.
Aterosclerose significativa constitui um risco no lúpus de longa duração,45,46 levando ao infarto do miocárdio e desenvolvimento de outras manifestações obstrutivas vasculares antes dos 40 anos de idade. O infarto do miocárdio também pode ser causado por uma vasculite coronariana, porém isto é menos comum. É importante considerar a possibilidade de aterosclerose, ao lado da vasculite e da trombose (causada por anticorpos antifosfolípides), em pacientes com lúpus de longa duração que se queixam de insuficiência vascular.
A miocardite não isquêmica difusa também pode ocorrer. Recém-nascidos com síndrome do LES neonatal podem apresentar bloqueio cardíaco congênito completo, podendo morrer de insuficiência cardíaca congestiva.
A artrite do LES é tipicamente dolorosa, transiente e simétrica, envolvendo os punhos, articulações pequenas das mãos, cotovelos, joelhos e tornozelos. O inchaço e a vermelhidão são modestos. Menos frequentemente, a artrite do LES apresenta-se como uma oligoartrite assimétrica ou sob a forma de uma poliartrite sustentada e intensamente inflamatória semelhante àquela observada na artrite reumatoide. Embora possa haver deformação em consequência do afrouxamento ligamentoso (subluxações reversíveis, artropatia de Jaccoud),47 não é comum haver uma destruição articular semelhante à artrite reumatoide.
A miosite inflamatória ocorre primariamente em pacientes que apresentam aspectos sobrepostos com esclerodermia ou dermatomiosite. Esta condição se manifesta como uma miopatia proximal, com elevação moderada dos níveis séricos de enzimas musculares, em que os exames de eletromiografia, imagem de ressonância magnética (IRM) e biópsia de músculo (quando realizados) apresentam resultados similares àqueles obtidos na dermatomiosite. Entretanto, níveis enzimáticos anormais associados à sensibilidade ou fraqueza muscular proximal são suficientes o diagnóstico em um paciente com LES estabelecido.
O lúpus não envolve o osso diretamente. Entretanto, pode haver envolvimento ósseo secundário à insuficiência de sistemas orgânicos (insuficiência renal), doença grave (osteoporose por inatividade ou estado catabólico) ou tratamento (necrose avascular ou osteoporose induzida por corticosteroide).
A osteoporose manifesta-se em forma de fraturas atraumáticas das vértebras ou ossos longos e constitui uma ameaça grave aos pacientes com LES, mesmo no caso de mulheres pré-menopáusicas, devido ao uso frequente de corticosteroides no tratamento e por causa da inatividade consequente à poliartrite e à doença sistêmica.
A necrose avascular (osteonecrose) é mais frequente em pacientes que apresentaram uma exacerbação severa tratada com com corticosteroides em altas doses. No entanto, esta complicação pode se desenvolver em pacientes que jamais foram tratados à base de corticosteroides. A observação de aspectos cushingoides marcantes durante o tratamento com esteroide, bem como o fenômeno de Raynaud podem ser preditivos da ocorrência de necrose avascular.48 A cabeça femoral é o sítio mais comumente envolvido, porém os ombros, tornozelos, punhos, metacarpos e diáfises de ossos longos também são vulneráveis.49 De forma típica, as áreas afetadas tornam-se doloridas na ocorrência inicial de infarto e, novamente, anos depois, quando o osso necrótico sofre colapso. A manifestação mais típica da osteonecrose é uma dor súbita no quadril que ocorre 2 a 3 vezes/ano, após uma exacerbação significativa do lúpus. Alguns pacientes que recebem infusões de altas doses endovenosas (IV) de metilprednisolona se queixam de uma dor intensa em sítios osteonecróticos preexistentes, durante e logo após a infusão. A diminuição da dose de corticosteroide no momento em que a dor ocorre não afeta o curso da complicação.
A disfunção esofágica raramente é observada no LES. Ocorre primariamente em pacientes com fenômeno de Raynaud severo ou com doença por sobreposição à esclerodermia. Pode haver desenvolvimento de úlcera gastroduodenal em decorrência do tratamento, todavia sem ligação direta com o LES. A isquemia envolvendo o intestino delgado e o intestino grosso pode ser resultante de uma vasculite sistêmica, e se manifesta como angina abdominal, pneumatose intestinal, infarto ou perfuração ou pseudo-obstrução. A isquemia intestinal constitui uma complicação rara, que ocorre apenas nos pacientes com doença mais severa.50 Também há desenvolvimento das síndromes de paresia gástrica e de dismotilidade intestinal.
A diverticulite desenvolve-se com frequência em pacientes com LES de longa duração, especialmente após o tratamento prolongado com corticosteroides. Os sintomas de diverticulite são facilmente mascarados por essa terapia. Em consequência, abscessos ou perfurações diverticulares com frequência são erroneamente diagnosticados, em especial em pacientes jovens com LES.
Pode haver desenvolvimento de hepatite química subsequente ao uso de fármacos anti-inflamatórios não hormonais (AINH) (os pacientes com lúpus parecem ser especialmente suscetíveis a este efeito colateral) ou outros medicamentos, como a azatioprina.51 Ocasionalmente, os pacientes desenvolvem hepatite autoimune concomitante ou cirrose biliar primária. Na ausência de outras causas, é incomum haver anormalidades envolvendo as enzimas hepáticas em decorrência do LES.
A leucopenia é um aspecto tão comum no LES que sua ausência, na doença não tratada, deve levantar suspeita de um diagnóstico incorreto ou da existência de infecção ou necrose tecidual concomitante. Usualmente, as contagens de linfócitos exibem quedas maiores do que as contagens de granulócitos: em geral, observa-se uma contagem de leucócitos em torno de 3.500/mm3, contendo 10% de linfócitos. Este grau de leucopenia raramente expõe os pacientes a um risco sério de infecção. Em geral, não há necessidade de administrar fator estimulador de colônias de granulócitos-macrófagos. Há relatos anedóticos de que a administração deste agente pode induzir exacerbação lúpica.
A trombocitopenia observada no LES geralmente é de baixo grau, com contagens plaquetárias acima de 50.000/mm3. Contudo, é possível que ocorra uma trombocitopenia severa, sendo que a púrpura trombocitopênica idiopática (PTI) pode ser uma manifestação inicial do LES.
O LES pode resultar em anemia por doença crônica e anemia por hemólise autoimune. A anemia por doença crônica em pacientes com LES pode responder à administração de eritropoietina recombinante.
Os sinais e sintomas neurológicos representam um dos aspectos mais sérios e menos compreendidos do LES. As manifestações neurológicas primárias do LES consistem em doença cerebral generalizada e focal (usualmente vascular), mielopatia, neuropatia periférica, mononeurite múltipla e disfunção cognitiva. Também podem ocorrer eventos neurológicos secundários, que incluem convulsões decorrentes de hipertensão ou hemorragia, delirium causado por fármacos ou uremia, abscessos no cérebro ou medula espinal, e acidente vascular cerebral (AVC) consequente a um ateroma ou êmbolo. Atribuir um sintoma neurológico específico ao lúpus ativo (tratável com imunossupressão), em oposição a uma complicação do LES ou ao seu tratamento (tratável com a amenização do problema agressor), requer uma investigação aprofundada e um discernimento clínico acurado. Confusões relacionadas aos critérios diagnósticos para o lúpus neurológico levaram o ACR a publicar uma nomenclatura e critérios de definição de caso para estas síndromes.52
Pacientes com LES queixam-se frequentemente de disfunção cognitiva progressiva, como confusão, esquecimento e o conhecido “pensamento nebuloso”.53,54 Estudos retrospectivos e de seção transversal relatam uma elevada frequência de mau desempenho nos testes de função cognitiva, em particular nas esferas da função executiva, memória de curta duração e processamento verbal.55,56 Não se sabe se este déficit resulta de um ataque imunológico ao cérebro (por anticorpos antineuronais ou de outro tipo) ou de uma doença vascular difusa. O déficit raramente progride para demência em estágio avançado. As anormalidades envolvendo a substância branca são mais comuns do que as anormalidades corticais.11
As dores de cabeça são comuns no LES. Foi descrita uma forma especial de enxaqueca, denominada dor de cabeça lúpica, mas ainda é discutido se esta manifestação constitui uma entidade definível.
Convulsões, AVCs, neuropatias cranianas (incluindo a cegueira) e disfunção cerebelar são condições possíveis no LES. Considera-se que estes eventos resultem de uma obstrução vascular, mas também podem ocorrer em pacientes sem diátese trombótica, embolismo, aterosclerose nem vasculite conhecidos. O AVC constitui uma das manifestações mais comuns da síndrome antifosfolípide. As convulsões ocorrem com mais frequência na doença severamente ativa, febril e multissistêmica. Neste caso, as convulsões geralmente não persistem depois que a doença é controlada.57
A mielite transversal exibe dois padrões: (1) início abrupto, com progressão dentro de algumas horas após a manifestação do primeiro sintoma, muitas vezes introduzida por uma dor disestésica ardente nas pernas; e (2) progressão mais lenta, com interrupções, piorando com o passar dos dias. A menos que sejam imediata e agressivamente tratadas, ambas as formas podem evoluir para uma paraparesia ou paraplegia. Embora existam poucos dados diretos sustentando esta hipótese, é provável que a primeira forma represente uma obstrução vascular acompanhada de isquemia da medula espinal, enquanto a segunda forma se manifesta por meio de uma doença inflamatória. É obrigatório excluir a possibilidade da existência de uma lesão ocupando espaço em todos estes pacientes.
Uma mielopatia lentamente progressiva e intermitente, bastante parecida com a esclerose múltipla (EM) (conhecida como mielopatia do tipo EM ou esclerose lúpica), desenvolve-se em alguns pacientes com lúpus. Não existe uma forma definitiva para excluir a ocorrência concomitante de EM nestes pacientes, a não ser pela associação da mielopatia ao LES e por sua falha em progredir da mesma forma como usualmente ocorre com a EM. Nesta forma de mielopatia lúpica, o exame do líquido cerebrospinal pode revelar a existência de faixas oligoclonais, contudo as análises de ressonância magnética (RM) são atípicas para EM.
A neuropatia em bota-e-luva consiste numa lesão de progressão lenta que tende a ocorrer em pacientes com doença ativa contínua. Sua patogênese é obscura. Pode resultar de um ataque imune direto aos nervos periféricos ou de uma obstrução vasculítica dos vasos sanguíneos que irrigam os nervos. A perda abrupta da função motora e sensorial, como a ocorrência súbita de pé caído ou de punho caído, é diagnosticada como mononeurite múltipla. Trata-se de uma manifestação bastante grave, que indica a existência de vasculite nos vasos dos nervos, além de implicar a vasculite sistêmica.
Cerca de metade dos pacientes com lúpus desenvolve nefrite lúpica, sendo que, no geral, aproximadamente 10% dos casos evoluem para diálise ou transplante. A nefrite lúpica manifesta-se como proteinúria (ou uma urinálise anormal), hipertensão ou elevação dos níveis séricos de creatinina, sempre em graus variáveis. Nos estágios iniciais, a nefrite lúpica é assintomática. Nos estágios mais avançados, ocorrem edema, anemia, hipertensão sintomática e uremia sintomática. Os pacientes que apresentam as formas inflamatórias de nefrite usualmente são hipocomplementêmicos. A maioria destes pacientes apresenta níveis elevados de anticorpos anti-DNA ou anti-Sm. Os sinais ou sintomas de doença ativa em outros sistemas orgânicos não necessariamente acompanham a nefrite lúpica.
A classificação patológica da nefrite lúpica estabelecida pela Organização Mundial da Saúde foi revisada. Os critérios revisados diferem dos critérios adotados nas classificações antigas por incluírem as biópsias normais, formação de cicatriz e alterações tubulointersticiais. Adicionalmente, incorporam as informações fornecidas pelos estudos de imunofluorescência e microscopia eletrônica [Tabela 6].58 Esta classificação possui índices de atividade e cronicidade da doença, que delineiam a necrose aguda, infiltração inflamatória, formação de crescente, formação de cicatriz e atrofia tubular, para fornecer mais informações prognósticas.9 A microscopia eletrônica revela a existência de depósitos de imunocomplexo junto aos espaços epiteliais, na nefrite lúpica membranosa, e junto aos espaços subendoteliais, na nefrite lúpica proliferativa, bem como em localizações mesangiais. Também aparecem estruturas tubulorreticulares características, consideradas produtos da degradação do RNA. Os estudos de imunofluorescência revelam a existência de depósitos de IgG, IgM e C3 com as mesmas distribuições. Também observa-se uma inflamação ou proliferação endotelial.
Tabela 6. Classificação da nefrite lúpica estabelecida pela International Society of Nephrology/Renal Pathology Society 2003.
|
Classe |
Denominação |
Descrição |
Apresentação clínica |
|
I |
Mesangial mínima |
Normal à microscopia comum, com imunodeposição mesangial detectada por imunofluorescência. |
Urinálise e função normais. |
|
II |
Mesangial proliferativa |
Células infiltrativas e proliferação do mesângio. |
Proteinúria leve, celúria e função normal. |
|
III A |
Focal ativa |
Células infiltrativas e deposição de imunocomplexos em partes do glomérulo, bem como em < 50% dos glomérulos (ativa) ou cicatrizes (crônica). |
Proteinúria variável, celúria e função normal. |
|
III A/C |
Focal ativa e crônica | ||
|
III C |
Focal crônica | ||
|
IV A |
Difusa e ativa (A), global (G), segmentar (S), crônica (C) |
Células infiltrativas e deposição moderada de imunocomplexos em todo o glomérulo e em = 50% dos glomérulos com lesões segmentares ou globais, com ou sem cicatriz. |
Proteinúria variável, celúria; diminuição de função frequentemente severa. |
|
IV G | |||
|
IV-S A/C | |||
|
IV-G A/C | |||
|
IV-S C | |||
|
IV-G C | |||
|
V |
Membranosa |
Imunodeposição global ou segmentar subendotelial detectada por microscopia comum, imunofluorescência ou microscopia eletrônica, com ou sem lesões mesangiais. |
Proteinúria acentuada e diminuição lenta da função. |
|
VI |
Esclerótica avançada |
= 90% dos glomérulos globalmente esclerosados e sem atividade residual. |
Proteinúria variável, celúria, diminuição de função. |
Embora a nefrite lúpica possa se apresentar como anúria, hipertensão aguda, ou retenção de líquidos, é mais frequentemente notada pela primeira vez através de uma urinálise anormal. Quando não tratada, a nefrite lúpica evolui para insuficiência renal no decorrer de alguns meses ou anos. É necessário realizar o exame de biópsia primeiramente quando o resultado fornecido por este exame puder alterar o tratamento. A urinálise e os exames de bioquímica sanguínea fornecem resultados que apresentam correlação apenas grosseira com os achados da biópsia [Tabela 7].
Tabela 7. Prováveis achados de biópsia renal conforme os resultados de urinálise e creatinina sérica.
|
Urinálise |
Níveis de creatinina |
Patologia mais provável | |
|
Proteína |
Células | ||
|
Ausente |
Ausentes |
Normal |
Normal ou mesangial |
|
Escassa |
Leucócitos |
Normal |
Mesangial, proliferativa focal |
|
Moderada |
Leucócitos, hemácias |
Normal |
Mesangial, proliferativa focal ou difusa |
|
Moderada |
Leucócitos, hemácias, cilindros |
Normal ou elevada |
Proliferativa focal ou difusa |
|
Severa |
Leucócitos, hemácias, cilindros |
Normal ou elevada |
Proliferativa difusa, membranoproliferativa |
|
Severa |
Poucas |
Normal ou elevada |
Membranosa |
|
Moderada |
Leucócitos |
Normal ou elevada |
Doença intersticial (tubular) (em pacientes com acidose ou anormalidades eletrolíticas) |
Aproximadamente 25% dos bebês nascidos de mães com anticorpos anti-SS-A (RO) ou anti-SS-B (LA) desenvolvem uma erupção fotossensível ou trombocitopenia, ambas transientes. Dentre estes bebês, um número bastante pequeno desenvolve bloqueio cardíaco congênito completo in utero. Tanto as manifestações cardíacas como as manifestações cutâneas constituem a síndrome lúpica neonatal. Qualquer uma destas manifestações pode surgir de maneira independente. A síndrome parece resultar da passagem transplacentária de anticorpos maternos, e se extingue com o desaparecimento destes anticorpos. O bloqueio cardíaco, no entanto, persiste e pode ser letal. O provável alvo destes anticorpos são antígenos expressos de forma transiente no sistema condutor fetal. Os sinais deflagradores de apoptose e fibrose estão positivamente regulados nesta síndrome.59
Entre 1/3 e metade dos pacientes lúpicos possuem anticorpos anticardiolipina, anticoagulante lúpico, ou ambos. Quando um destes anticorpos está presente em níveis elevados, os pacientes são suscetíveis à doença tromboembólica recorrente, trombocitopenia, livedo reticular e doença cardíaca valvar. As mulheres são suscetíveis a perdas recorrentes de gestação. Estes sintomas, combinados aos resultados positivos dos exames de sangue, constituem a síndrome antifosfolípide (SAF).60 O lúpus não necessariamente está presente no momento em que a SAF é diagnosticada. Quando o lúpus e a SAF coexistem, ambos os diagnósticos são considerados. Pesquisas atuais sugerem que o verdadeiro antígeno para a síndrome é a proteína ligadora de fosfolipídios beta2-glicoproteína I, em vez dos próprios fosfolipídios de carga negativa. Em alguns pacientes, anticorpos contra uma proteína ligadora de fosfolipídios alternativa (p. ex., protrombina) induzem a mesma síndrome. As evidências de lesão ou ativação endotelial, como detecção de células endoteliais circulantes, parece estar associada a episódios tromboembólicos. Os sítios de trombose não são inflamatórios e podem ser melhor tratados com anticoagulação do que por imunossupressão. Entretanto, estudos sobre perda de gestação em modelos de experimentação animal indicam que a ativação do complemento é um componente essencial da lesão fetal, sugerindo novamente o possível envolvimento do sistema imune inato no desenvolvimento da SAF.61
Os exames de líquidos corporais podem resultar anormais em pacientes com LES [Tabela 8], mas não é regra. Havendo suspeita de lúpus, devem ser realizados o teste de FAN, hemograma completo e urinálise. Se os resultados destes exames forem todos normais, a possibilidade de LES é excluída. Por outro lado, como usualmente também há suspeita de doenças lúpus-símile, com frequência uma medida eficaz consiste em obter, durante a primeira consulta, os seguintes exames: velocidade de hemossedimentação (VHS) ou níveis de proteína C reativa (PCR); ensaios para detecção de anticorpos contra dsDNA, Sm, RNP, SS-A e SS-B; tempo de tromboplastina parcial (ou outro teste de triagem para anticoagulante lúpico) e anticorpos anticardiolipina; além de um perfil bioquímico que inclua testes de função hepática e níveis séricos de creatinina.
Tabela 8. Exames de líquidos corporais comumente anormais no LES.
|
Teste |
Anormalidade |
Interpretação |
|
Contagem sanguínea |
Anemia normocrômica, leucopenia (leucócitos ˜ 3.000/mm3; trombocitopenia) |
LES ativo |
|
VHS e PCR |
Elevadas |
LES ativo |
|
Urinálise |
Proteinúria, hematúria, leucocitúria, cilindrúria |
Nefrite lúpica ativa |
|
Coombs e contagem de reticulócitos |
Positivo; alta |
Anemia hemolítica |
|
TTPA, TVVRd |
Altos |
Se confirmado com teste de mistura, anticoagulante lúpico |
|
FAN |
Fortemente positivo |
Positivo em quase todos os pacientes, na doença ativa; inespecífico para o lúpus. |
|
Anticorpo anti-dsDNA |
Fortemente positivo |
Positivo em 2/3 a 3/4 dos pacientes com doença ativa; é diagnóstico de lúpus. |
|
Anticorpo anti-Sm |
Positivo |
Positivo em 1/4 a 1/3 dos pacientes; é diagnóstico de lúpus. |
|
Anticorpos anti -SS-A, anti-SS-B e anti-RNP |
Positivos |
Positivo em 1/3 dos pacientes; inespecífico. |
|
Anticorpo anticardiolipina |
Positivo |
Síndrome antifosfolípide |
|
Complemento C3, C4 e CH50 |
Baixos |
Nefrite lúpica provável; também, anemia hemolítica e crioglobulinemia |
|
Crioglobulina |
Presente |
LES ativo |
|
Ureia e creatinina sérica |
Elevados |
Nefrite lúpica severa, toxicidade farmacológica |
|
Testes de função hepática |
Elevados |
Toxicidade farmacológica (raramente, LES ativo) |
|
Células e proteína em LCR |
Elevados |
Presentes em uma minoria de pacientes com LES-SNC |
|
Líquido sinovial |
Leucócitos = 5.000-10.000/mm3; nível normais de glicose |
Artrite lúpica |
|
Líquido pleural, líquido pericárdico |
Leucócitos = 5.000-10.000/mm3; nível normais de glicose; níveis baixos de complemento; presença de células LE |
Serosite lúpica |
TTPA = tempo de tromboplastina parcial ativado; SNC = sistema nervoso central; PCR = proteína C reativa; LCR = líquido cefalorraquidiano; TVVRd = tempo de veneno de víbora de Russell diluído; VHS = velocidade de hemossedimentação; LES = lúpus eritematoso sistêmico.
O ensaio de detecção de fator anti-núcleo (FAN) consiste num teste de triagem para lúpus que resulta quase sempre positivo em níveis elevados (> 1:80) em pacientes com doença ativa não tratada. No entanto, um resultado positivo desse teste por si só não confirma um diagnóstico de lúpus. Somente o anticorpo anti-dsDNA e os anticorpos anti-Sm, quando presentes em níveis elevados, são diagnósticos de lúpus. Os níveis de anticorpo anti-dsDNA e complemento atuam a grosso modo como guias da atividade da doença, porém muitos pacientes continuam bem por longos períodos, mesmo apresentando resultados de teste severamente anormais. A hipocomplementenemia reflete a existência de nefrite lúpica proliferativa, mas não espelha outros aspectos do LES, como a nefrite lúpica membranosa. O VHS, assim como os níveis de proteína C reativa, permanece elevada em muitos pacientes com LES que apresentam bem controlados.
A avaliação do envolvimento neurológico no LES é complexa. Varreduras de RM, geralmente com contraste, são indicadas diante de qualquer suspeita clínica de doença envolvendo o SNC, como convulsões, disfunção cognitiva, novos episódios de dor de cabeça severa, coreia ou sintomas de AVC. As varreduras de TC são bem menos definitivas, exceto em casos de AVC. A angiografia cerebral ou angiografia por ressonância magnética (ARM) raramente é útil.
As varreduras de TC e RM do cérebro com frequência mostram atrofia e infartos (sendo que estes últimos incluem áreas hiperintensas na substância branca). Estas lesões apresentam correlação fraca com outras doenças neurológicas diferentes das síndromes de AVC.11 A tomografia por emissão de pósitrons com fluorodesoxiglicose, espectrografia por ressonância magnética e tomografia computadorizada por emissão de fóton único frequentemente fornecem resultados anormais, mesmo em casos de pacientes assintomáticos, que apresentam pouca correlação com a maioria das doenças neuropsiquiátricas severas.62 A interpretação de achados anormais de pacientes assintomáticos é incerta.63,64
A avaliação vascular é indicada para os casos em que há suspeita clínica de obstrução de vasos de médio calibre. Os exames de ultrassonografia, Doppler, angiografia e ARM podem revelar a doença tromboembólica a causada por anticorpos antifosfolípides ou de aterosclerose. A vasculite de pequenos vasos observada no LES usualmente está além da resolução destas tecnologias.
Todos os pacientes com lúpus devem se submeter a uma urinálise, de preferência durante a cada consulta clínica, porque a doença renal pode reaparecer a qualquer momento. Todos os pacientes que apresentam qualquer tipo de anormalidade de urinálise, ureia sanguínea ou níveis séricos de creatinina devem monitorar proteinúria e depuração de creatinina de 24 horas, no máximo a cada 6 meses. É importante considerar os resultados do teste renal dentro de um contexto: níveis séricos de creatinina de 1,2 mg/dL podem estar dentro da faixa da normalidade do laboratório, porém numa mulher jovem com peso aproximado de 50 kg, níveis desta ordem são considerados altos e é provável que sejam anormais. Uma depuração de creatinina em queda sempre demanda avaliação, mesmo quando o paciente está clinicamente bem.
Biópsia renal. A indicação primária para realização de um exame de biópsia renal é para ajudar o médico a tomar decisões sobre o tratamento. Embora pacientes assintomáticos com urinálise normal possam apresentar resultados anormais de biópsia, ainda não está claro se o tratamento destes pacientes melhora o resultado. O paciente em bom estado apresentando resultados normais de urinálise e função renal geralmente não precisa ser submetido ao exame de biópsia de rim, mesmo que tenha níveis elevados de anticorpo anti-DNA e baixos níveis de complemento. O paciente bastante debilitado e com doença multissistêmica, incluindo resultados anormais de urinálise e função renal, provavelmente passará por um tratamento agressivo de qualquer modo e, assim, dispensa o exame de biópsia de rim. O paciente com doença sistêmica branda, anormalidades urinárias leves ou ambas as condições geralmente precisa passar pelo exame de biópsia, porque a decisão de adotar um tratamento moderado ou agressivo pode depender do resultado deste exame. Ocasionalmente, uma biópsia é examinada para descrever uma doença incurável em estágio terminal e, dessa forma, permitir a suspensão da terapia. Entretanto, a ultrassonografia renal usualmente pode fornecer a mesma informação.
O monitoramento cardíaco com ecocardiografia ou testes de estresse é desnecessário na rotina. Devido à alta frequência de aterosclerose acelerada, todavia, qualquer ocorrência de dispneia, dispepsia ou dor no ombro ou no braço merece consideração da possibilidade de doença cardíaca isquêmica.
O diagnóstico diferencial de lúpus envolve duas perguntas interligadas: o paciente tem doença reumática? E, se tiver, qual?
A síndrome de febre, citopenia, erupções e adenopatia sugere muitas infecções, entre as quais a infecção pelo HIV, citomegalovírus, mononucleose e endocardite bacteriana. Uma poliartrite aguda, erupções e citopenias podem ser produzidas por diversas infecções virais, como a hepatite, parvovirose e rubéola. A resolução destas síndromes ocorre de maneira espontânea, em algumas semanas. Esta síndrome também pode ocorrer em malignidades hematológicas, primariamente linfomas, leucemias e síndromes mielodisplásicas. Embora o FAN seja comum em muitas destas doenças, os anticorpos anti-DNA ou anti-Sm não são. Igualmente incomuns são as erupções específicas do lúpus, nefrite e manifestações vasculares, como a telangiectasia periungueal e as pápulas vasculíticas. A fotossensibilidade e a alopecia frontal também são características do lúpus que não ocorrem nestas outras doenças.
As causas de nefrite não relacionadas ao LES incluem a glomerulonefrite pós-estreptocócica, doença de Goodpasture, nefropatias genéticas e toxemia. A erupção em rosácea costuma ser confundida com a erupção lúpica. A PTI ou a anemia hemolítica autoimune podem ocorrer como doenças isoladas ou como parte do envolvimento multissistêmico lúpico. Nestas circunstâncias, uma completa avaliação clínica e sorológica para lúpus irá localizar os achados no contexto apropriado.
O lúpus pode ser parecido com outras doenças reumáticas [Tabela 1]. Estas incluem a artrite reumatoide e a síndrome de Sjögren [ver Artrite reumatoide], bem como o esclerodermia [ver Esclerodermia e doenças correlatas]. A apresentação poliartrítica do lúpus é bastante semelhante com a apresentação da artrite reumatoide, doença de Lyme e outras doenças reumáticas. A esclerodermia em fase inicial muitas vezes se manifesta como um edema bilateral nas mãos, que é confundido com a poliartrite.
A dermatomiosite [ver Miopatias inflamatórias idiopáticas] atinge o pico em três grupos de faixas etárias: 5 a 10 anos; final da adolescência e início da terceira década da vida; e acima dos 45 anos. A erupção da dermatomiosite é similar à erupção lúpica, porém com envolvimento ocular diferente: na dermatomiosite, a telangiectasia produz o conhecido aspecto de heliotropo nas pálpebras; no lúpus, as erupções envolvem as sobrancelhas, porém a erupção malar cessa abruptamente na órbita. Do mesmo modo, a erupção associada à dermatomiosite comumente preserva os canais auditivos que, por sua vez, com frequência estão envolvidos no lúpus. A telangiectasia periungueal é mais dramática na dermatomiosite do que no lúpus, ocorrendo também na esclerodermia. As erupções que surgem sobre as pequenas articulações das mãos são sugestivas de dermatomiosite, enquanto aquelas localizadas entre as articulações sugerem lúpus.
Em comparação ao LES, a artrite reumatoide ocorre em indivíduos de idade mais avançada (40 a 60 anos) e apresenta uma predominância feminina menor (2:1 a 3:1). Apesar da rigidez matinal, fadiga e perda de peso serem comuns em pacientes com artrite reumatoide, não é comum que ocorradoença multissistêmica visceral específica. A ocorrência de leucocitose, em vez de leucopenia, é característica da artrite reumatoide. A doença renal é bastante rara e, quando de fato ocorre, é atribuível à doença tubular ou à amiloidose, mas não decorre de uma glomerulonefrite. Pacientes com artrite reumatoide não têm febre alta. Os FANs estão presentes em 10 a 20% dos casos de artrite reumatoide, sendo que pequeno percentual de pacientes apresenta baixos níveis de anticorpos anti-DNA, enquanto uma minoria apresenta anticorpos anti-SS-A e anti-SS-B. Os níveis de complemento geralmente estão elevados. O fator reumatoide ou o anticorpo antipeptídeo citrulinado cíclico (anti-CCP) estão presentes em 80% dos pacientes com artrite reumatoide. O fator reumatoide é encontrado em 25% dos pacientes com LES, enquanto os anticorpos anti-CCP são menos comuns.
Alguns pacientes apresentam sintomas sugestivos de lúpus (mais comumente, artrite, dor pleurítica e citopenia), entretanto não atendem aos critérios diagnósticos específicos para lúpus (erupção em forma de asa de borboleta, glomerulonefrite e níveis altos de anticorpos anti-DNA ou anti-Sm). Outros pacientes apresentam sintomas do tipo lúpico aliados a achados sugestivos de artrite reumatoide, dermatomiosite ou esclerodermia. Os pacientes sem sorologia definível e quadro clínico indeterminado são definidos como portadores de doença indiferenciada do tecido conjuntivo (DITC). Há ainda outros pacientes que apresentam miosite inflamatória, fenômeno de Raynaud e esclerodactilia acompanhados de níveis bastante elevados de anticorpos contra o antígeno ribonucleoproteico (U1-RNP), mas não possuem anticorpos anti-DNA nem anti-Sm. Este conjunto de achados é definido como doença mista do tecido conectivo (DMTC).
A diferenciação do LES em relação à DITC, DMTC e síndrome de Sjögren depende da extensão e do padrão de envolvimento de órgãos diferentes (a glomerulonefrite é rara em todos estes distúrbios, exceto no lúpus), bem como das anormalidades sorológicas associadas. Níveis elevados de anticorpos anti-DNA ou anti-Sm geralmente indicam lúpus. Níveis altos de anticorpos anti-RNP sem positividade para outros anticorpos indicam DMTC. A detecção de anticorpos anti-SS-A e anti-SS-B é consistente com a síndrome de Sjögren, mas também ocorre no lúpus e na artrite reumatoide. Ocasionalmente, os pacientes apresentam uma artrite reumatoide destrutiva característica, nódulos subcutâneos e níveis elevados de fator reumatoide e anticorpos anti-DNA. Estes pacientes, bem como aqueles com outros aspectos sobrepostos, devem ser tratados como se tivessem as duas doenças.
O prognóstico tende a ser mais benigno em casos de pacientes com DITC do que para pacientes com LES. Os pacientes com DMTC não desenvolvem glomerulonefrite, porém apresentam um prognóstico a longo prazo que é agravado pelo desenvolvimento gradual de hipertensão pulmonar.
As recomendações para o tratamento dos sintomas agudos do lúpus dependem do grau de severidade e dos sistemas orgânicos envolvidos. As manifestações não prejudiciais à vida, como uma artrite mínima, artralgias, mal-estar, mialgias, serosite e febre baixa, muitas vezes podem ser controladas com a administração de doses plenas de AINH. Não há uma preferência específica entre os AINH, no entanto os pacientes com lúpus são inusitadamente suscetíveis a toxicidades hepáticas e renais, que devem ser monitoradas. Do mesmo modo, em casos raros, os pacientes lúpicos desenvolvem uma febre alta abrupta e meningite após tomarem ibuprofeno e outros fármacos similares. Os pacientes irresponsivos aos AINH usualmente respondem a baixas doses (5 a 10 mg/dia) de prednisona. Como regra, os pacientes que apresentam erupções inflamatórias, bem como aqueles sob tratamento com duração prevista de vários meses ou anos, devem receber terapia antimalárica com hidroxicloroquina (200 mg, 2 vezes/dia). Ao longo de um curso de 3 meses ou mais, a hidroxicloroquina diminui a artralgia, mialgia, erupção, fadiga, mal-estar e outros sintomas semelhantes.65 Os pacientes sob tratamento com corticosteroides, cuja duração prevista seja superior a algumas semanas, devem considerar firmemente a adição de medidas de proteção óssea [Osteoporose, adiante]. Estes pacientes também devem cogitar a adição de um agente redutor de lipídios, (p. ex., estatina) ao regime. As erupções faciais, especialmente as lesões eritematosas com edema ou telangiectasia, podem responder à terapia tópica com cremes à base de corticosteroide.
A recuperação da alopecia pode ocorrer de forma espontânea, mas de outro modo não costuma ser facilmente atenuada com terapias. Perucas e apliques ou extensores de cabelo são úteis. Uma maquiagem bem-feita é capaz de cobrir a maior parte das alterações pigmentares causadas pelo lúpus discoide.
A terapia com baixas doses de corticosteroide pode ser apropriada para casos leves de trombocitopenia ou anemia. A leucopenia usualmente dispensa tratamentos. Uma terapia com altas doses de corticosteroide (60 mg de prednisona/dia) é administrada aos pacientes que apresentam sintomas sistêmicos graves, doença renal ou outra doença visceral potencialmente prejudicial à vida. O tratamento deve ser iniciado com doses divididas ao longo do dia, mantido por 4 a 6 semanas e, então, diminuído gradativamente. Em geral, uma redução da dose muito precocemente resulta no reaparecimento da atividade da doença. Quando há previsão da instituição de um tratamento prolongado à base de corticosteroide, o paciente apresenta vasculite ou outra condição prejudicial à vida, ou a toxicidade produzida pelo corticosteroide é inaceitável, em geral recomenda-se adicionar uma terapia imunossupressora. Os agentes imunossupressores utilizados em casos de lúpus incluem a ciclofosfamida administrada por via oral ou endovenosa; azatioprina e micofenolato de mofetil. Um regime padrão para casos de nefrite lúpica ativa inclui uma alta dose de corticosteroide e ciclofosfamida IV. A ciclofosfamida é administrada a uma dosagem de 1 g/m2 mensalmente, durante 6 meses, passando então para uma dose a cada 3 meses, durante mais 2 anos. Outros regimes terapêuticos estão sendo testados.
Os sintomas cerebrais agudos (diferentes do AVC) usualmente são tratados com altas doses de corticosteroides. Alucinações e outros sintomas psicóticos respondem às medicações antipsicóticas, como haloperidol, olanzapina ou outros fármacos similares, muitas vezes administrados com os corticosteroides. Como a psicose também pode advir do uso de uma dose alta de corticosteroide isoladamente, pode ser necessário suspender o tratamento com corticosteroide em alguns casos. É bastante difícil distinguir a psicose por esteroides da psicose lúpica. Não há nenhum conjunto de critérios que permita distinguir estas duas condições. A evidência da existência de lúpus ativo em outros sistemas orgânicos constitui uma indicação para que o paciente receba tratamento para psicose lúpica.
Os episódios de lúpus agudo frequentemente são tratados com pulsoterapia com corticosteroide (em geral, 1.000 mg de metilprednisolona administradas via infusão IV rápida [1 hora], 1 vez/dia, durante 3 dias). Poucos estudos formais sustentam esta prática, no entanto a experiência clínica sugere sua eficácia e relativa segurança. O tratamento com pulso de corticosteroide pode produzir elevações abruptas da pressão arterial, vasoespasmo agudo com consequente AVC, infarto cardíaco ou infarto intestinal. O paciente também pode apresentar oligúria transiente e aumento dos níveis séricos de creatinina. Para prevenir estas complicações, a terapia em pulso deve ser monitorada intensivamente e, quando não for possível controlar a hipertensão, suspendida. O acesso à terapia de reposição renal deve estar disponível, para o caso de o paciente apresentar diminuição da função renal.
Muitas terapias novas têm sido investigadas. Estas são, em grande parte, terapias biológicas e incluem o uso de fármacos dirigidos contra receptores de células imunoativadoras ou células de reconhecimento, e o uso de moduladores da resposta imune, como CD154, CTLA-4 e anti-C5b. A remoção de anticorpos por meio da passagem do plasma do paciente em uma coluna de absorção também está sendo estudada. A inibição do fator de necrose tumoral-alfa – que no passado era considerada perigosa para pacientes com LES – tem sido revista. As tentativas ao nível da manipulação hormonal, como no caso da desidroepiandrosterona, têm alcançado um sucesso apenas modesto.
Nenhum teste isolado informa aos médicos se o tratamento para LES será ou não bem-sucedido. Em vez disso, é necessário monitorar todo o quadro clínico, incluindo os sintomas e os resultados do exame físico, exames laboratoriais de rotina e estudos de função imune.
Exacerbação. Uma intensificação da inflamação em qualquer sistema orgânico afetado pelo LES é denominada exacerbação, que constitui uma variável contínua, em vez de dicotômica. Em um certo paciente, a exacerbação pode ocorrer em diferentes sistemas orgânicos, a diferentes taxas e intensidades. Exemplificando, uma erupção pode se tornar severa, enquanto uma nefrite permanece estável. Por isso, foram criados vários esquemas para medir as exacerbações. Estes esquemas diferem entre si, por atribuírem diferentes pesos a medidas individuais da atividade da doença (uma nova nefrite conta mais ou menos do que uma nova erupção?) e no que diz respeito às medidas sorológicas (título de FAN, anticorpos anti-DNA, complemento) contarem ou não para a determinação. Os índices disponíveis – SLE Disease Activity Index (SLEDAI), Systemic Lupus Activity Measure (SLAM) e British Isles Lupus Assessment Group (BILAG)66 – geralmente concordam quanto à identificação das exacerbações nas populações de pacientes, mas com frequência são discordantes em aspectos específicos. Há pouco consenso no que se refere à distinção da variação da atividade da doença de um dia para o outro e uma exacerbação definida. Vários componentes dos índices (p. ex., quantificação da erupção ou da artrite) são suficientemente subjetivos a ponto de obrigarem os pesquisadores, nos estudos clínicos, a se submeterem a um treinamento de padronização para poderem validar seus escores para pacientes individuais.
Exacerbação na gravidez. Proteinúria, trombocitopenia e outros eventos relacionados à gestação que ocorrem na ausência de lúpus invalidam a maioria dos sistemas de pontuação para pacientes grávidas. Um instrumento específico – o SLE Disease Activity Index (SLEDAI) – foi projetado para uso em casos de pacientes grávidas.67
Inflamação recorrente e obstrução vascular induzem cicatrização irreversível e déficits permanentes, tais como AVC, catarata, adelgaçamento da pele, osteoporose, osteonecrose e insuficiência renal. É prática comum, portanto, atribuir escores aos pacientes com LES de acordo com seus índices de atividade (exacerbação) e índices de dano. O índice de dano mais amplamente utilizado é o Systemic Lupus International Collaborating Clinics (SLICC).68
A gravidez em pacientes com LES, antigamente considerada contraindicada, hoje constitui um evento de rotina. As complicações da gestação, nestas pacientes, estão relacionadas a três aspectos principais: função renal anormal, anticorpos antifosfolípide e anticorpos anti-SS-A e anti-SS-B. Ainda se discute se o lúpus é exacerbado pela gravidez, porém atualmente existe um consenso de que as pacientes grávidas com LES não necessitam de aumentos profiláticos na terapia de corticosteroide. Em vez disso, elas devem ser tratadas do mesmo modo que as pacientes não grávidas, a não ser pelo fato de que os fármacos com toxicidade fetal não devem ser administrados, sendo necessário realizar alguns ajustes de dose considerando o aumento de peso e metabolismo.
A doença renal, em particular a insuficiência renal, predispõe fortemente ao desenvolvimento de toxemia da gravidez. Hipertensão, diminuição da depuração da creatinina e LES ativo são todos prejudiciais à evolução do feto. Ao contrário, as mulheres que engravidam sem apresentar doença renal nem hipertensão usualmente permanecem bem. As perdas recorrentes de gestação, particularmente durante o segundo trimestre, constituem umas principais manifestações clínicas da síndrome antifosfolípide. Para as pacientes com positividade para anticorpos antifosfolipídios, o período periparto impõe alto risco de desenvolvimento de doença tromboembólica. Mulheres que possuem anticorpos contra os antígenos SS-A ou SS-B correm risco de dar à luz a crianças com síndrome lúpica neonatal.
O aspecto mais importante do tratamento para os pacientes com lúpus de longa duração é a prevenção ou supervisão do dano às artérias, rins, ossos e cérebro, ao invés do controle da resposta imune e da inflamação. O médico deve prever os efeitos crônicos tanto da doença como da terapia.
Durante o tratamento com altas doses de corticosteroides, com ou sem agentes imunossupressores, a prevenção de infecções constitui uma preocupação básica. Herpes-zóster, tuberculose e infecções bacterianas são as ameaças primárias. A infecção por Pneumocystis jiroveci (antigo Pneumocystis carinii) é observada relativamente com pouca frequência. A maioria dos reumatologistas não sugere uma profilaxia de rotina contra este organismo. As complicações da terapia prolongada com corticosteroides incluem osteoporose (ver adiante) e cataratas, estrias cutâneas, hemorragia cutânea, diabetes e candidíase oral e vaginal. Em pacientes com doença de longa duração, estas complicações produzem uma morbidade equivalente àquela associada à própria doença.
O tratamento da síndrome antifosfolípide consiste na anticoagulação, tendo como alvo uma relação normalizada internacional de 2 a 3. A varfarina e baixas doses de aspirina são utilizadas para prevenir as manifestações da síndrome. A heparina ou a heparina de baixo peso molecular são utilizadas em casos de pacientes grávidas.69,70 Dados recentes sugerem que a adição de fármacos à base de estatina, para inibir ativação endotelial, também pode ser benéfica.71
A aterosclerose de início precoce constitui um problema comum entre pacientes com LES de longa duração; manifesta-se mais comumente como obstrução arterial cerebral e coronariana, mas também como obstrução vascular periférica. Embora a causa seja desconhecida, também foram implicados a inflamação crônica, terapia com corticosteroides, hipertensão descontrolada, diabetes e tabagismo, entre outros fatores. A maioria dos especialistas desta área recomenda a instituição rápida e vigorosa do tratamento dos fatores de risco conhecidos, para todos os casos de pacientes lúpicos. A aterosclerose lúpica é supervisionada do mesmo modo que a aterosclerose associada a outras condições, exceto quanto ao fato de as intervenções vasculares serem arriscadas para pacientes com síndrome antifosfolípide.
Embora o mecanismo da osteonecrose não seja totalmente conhecido, muitos autores acreditam que um aumento esteroide-induzido do volume dos lipócitos promove aumento da pressão na medula óssea, bloqueando o fluxo sanguíneo para as áreas vulneráveis. Em consequência, quando a osteonecrose é identificada antes do colapso articular (usualmente, por cintilografia ou RM), recomenda-se a trepanação do osso para redução da pressão intraóssea (conhecida como descompressão central). Entretanto, a validade desta teoria e a eficácia do tratamento continuam sem comprovação. Em geral, a substituição articular eventualmente é necessária.72
A osteoporose sucede a terapia prolongada à base de corticosteroide com uma frequência suficiente para justificar que todos os pacientes submetidos a esta terapia recebam profilaxia contra o desenvolvimento desta complicação. A administração por via oral de uma dose alta de cálcio (1.500 mg/dia), vitamina D, fármaco bisfosfonato e paratormônio constitui a medida preventiva inicial. A reposição de estrogênio pode ser considerada durante os primeiros anos de pós-menopausa para mulheres que não possuem anticorpos antifosfolipídio. A prática de exercícios com sustentação de carga deve ser estimulada. Outras medidas profiláticas, incluindo a administração de calcitonina e paratormônio, podem ser convenientes. Como os pacientes com lúpus são fotossensíveis, é imprudente aumentar a exposição solar para prevenir a osteoporose. Os bifosfonatos não devem ser utilizados por mulheres possivelmente grávidas.
A miocardiopatia inflamatória, ao contrário da isquêmica, responde aos corticosteroides. As insuficiências valvares e o tromboembolismo são complicações tardias da doença cardíaca lúpica. A endocardite bacteriana raramente complica esta anomalia. A valvulite geralmente é irresponsiva ao tratamento, embora alguns relatos de casos ocasionais tenham sugerido que a valvulite aguda responde à terapia com corticosteroides.73 Um número pequeno de pacientes necessita de substituição de valva, usualmente a valva aórtica ou a mitral. O mecanismo da valvopatia no LES é desconhecido, assim como os métodos preventivos.
Um erro comum no tratamento do LES é considerar que uma determinada queixa reflete uma doença inflamatória em andamento ao invés de um dano irreversível, e que é possível controlá-la por meio da terapia anti-inflamatória e imunossupressora, em vez de usar métodos paliativos. Alguns exemplos destes sintomas são as convulsões, demência e outras síndromes neurológicas associadas a infartos cerebrais; úlceras cutâneas causadas por uma insuficiência vascular prolongada; fenômeno embólico decorrente de aterosclerose; insuficiência respiratória por fibrose pulmonar ou hipertensão pulmonar; artrite decorrente de osteonecrose, artrite erosiva do tipo reumatoide, ou tendinose; e insuficiência renal progressiva decorrente de arterionefroesclerose. Para estes casos, indica-se uma terapia paliativa no lugar de aumento nos corticosteroides.
A doença neurológica crônica, muitas vezes sob a forma de demência, constitui uma sequela do LES a longo prazo. Algumas de suas causas são o AVC (aterosclerótico, hipertensivo ou trombótico, associado à presença de anticorpos antifosfolipídios), ataque de autoanticorpos a alvos cerebrais específicos, doença obstrutiva de pequenos vasos e fármacos. Ocasionalmente, os pacientes apresentam incapacitação severa. Não existe nenhuma profilaxia efetiva conhecida.
Pacientes com doença renal frequentemente precisam de inibidores da enzima conversora de angiotensina para tratar a proteinúria, anti-hipertensivos para a hipertensão, eritropoietina para a anemia, e diuréticos para o edema. No lúpus, a insuficiência renal pode ocorrer de três modos. No primeiro, a nefrite inflamatória aguda caracteriza-se por urinálises distintamente anormais e uma doença com atividade clínica e sorologicamente evidente. Pacientes com este tipo de quadro renal apresentam uma rápida elevação dos níveis séricos de creatinina e desenvolvem insuficiência renal logo após serem diagnosticados. Quando tratada de maneira agressiva, com altas doses de corticosteroides e fármacos imunossupressores, a insuficiência renal é passível de reversão em cerca de 1/3 destes pacientes.74 No segundo modo de insuficiência, que ocorre apenas ocasionalmente, os pacientes desenvolvem insuficiência renal por toxicidade farmacológica – usualmente AINH – ou em decorrência de necrose tubular aguda. Outras manifestações do LES são modificadas com a uremia: a erupção é menos proeminente, enquanto a febre, caquexia, úlceras mucosas e citopenias são mais proeminentes.
No terceiro modo, que é o mais comum, os pacientes lúpicos desenvolvem insuficiência renal lentamente, após sofrerem da doença por muitos anos. Caracteristicamente, no momento em que a insuficiência se manifesta pela primeira vez, o paciente apresenta uma doença sistêmica de pequenas proporções e sofre há meses ou anos de uma insuficiência renal modesta (com uma depuração de creatinina de 10 a 30 mL/min e níveis séricos abaixo de 3,5 mg/dL), com elevação lenta dos níveis séricos de creatinina, sedimentos urinários relativamente não inflamatórios e anemia progressiva. Então, no decorrer de alguns meses, o paciente desenvolve hipertensão e retenção de líquidos, com ou sem insuficiência cardíaca. Uma dor abdominal frequentemente está presente. O ultrassom mostra rins fibróticos e pequenos ou córtices renais delgados e com cicatriz. A insuficiência renal é apenas transientemente reversível nestes pacientes. Uma terapia imunossupressora agressiva não tem utilidade neste estágio, podendo, ao contrário, acelerar a deterioração renal e complicar a iniciação da diálise.
Menos comumente, a insuficiência renal é causada por uma microangiopatia trombótica associada aos anticorpos antifosfolípides. Nestes casos, a manifestação consiste numa modesta proteinúria com sedimentos urinários brandos e elevação lenta dos níveis séricos de creatinina, frequentemente acompanhada de hipertensão moderada. A vasculite lúpica com envolvimento dos rins tende a surgir de forma abrupta, com urinálise severamente anormal, hipertensão severa e insuficiência renal de progressão rápida. Esta complicação é tratada com altas doses de corticosteroides e imunossupressão. Contudo, a recuperação total é incomum.
Os pacientes lúpicos, em particular aqueles que desenvolvem insuficiência renal lentamente, toleram bem a diálise e o transplante renal. Por outro lado, um dano cardíaco, cerebral e osteoarticular preexistente pode atuar como fator limitante. Os pacientes que desenvolvem insuficiência renal de forma aguda muitas vezes apresentam outra doença sistêmica ativa, com convulsões e citopenias sendo as condições mais comuns. A crença de que o lúpus se torna inativo na insuficiência renal provavelmente é falsa. Em vez disto, na maioria dos pacientes que seguem para a diálise, o lúpus já está sistemicamente inativo e permanece neste estado. É provável que a insuficiência renal não resulte da doença contínua e sim da cicatrização progressiva. Em poucos pacientes que desenvolvem insuficiência renal durante uma exacerbação sistêmica aguda, usualmente no início do curso da doença, a doença sistêmica ativa tende a continuar e representa uma relativa contraindicação ao transplante renal. Pacientes com LES ativo que se submetem à diálise geralmente possuem altos níveis de anticorpo anti-DNA e baixos níveis de complemento. Estes pacientes respondem à terapia com corticosteroide, em geral com doses menores do que as doses administradas aos pacientes que não fazem diálise.
No LES, há quatro elementos prognósticos: os imediatos para a vida e para sistemas orgânicos individuais, e a longo prazo para sistemas orgânicos e para a vida.
Durante as fases iniciais do lúpus, espera-se alcançar a reversão completa de quase todas as manifestações (erupção, artrite, febre, citopenias e nefrite) com uma terapia agressiva. As exceções incluem as erupções discoides com cicatriz, os infartos cerebrais ou de medula espinal, e a nefrose ou nefrite severa. Em casos raros (< 5%), os pacientes apresentam a doença severa mesmo com o tratamento e progridem rapidamente para a morte, dentro de um período de 2 anos. Como a maioria dos pacientes recém-diagnosticados responde à terapia, a sobrevida em 5 anos é de 80 a 90%, enquanto a de 10 anos é de 70 a 90% e a de 20 anos chega quase a 70%.6 Os determinantes são a idade, doença renal e etnia, sendo que os afro-americanos apresentam taxas de sobrevida geral menores do que os brancos.75
Um sistema orgânico (rins ou plaquetas) pode falhar em responder integralmente ao tratamento, sem contudo ameaçar diretamente a vida do paciente. Estes pacientes podem ser monitorados sem intervenção. Um paciente pode desenvolver insuficiência renal crônica (creatinina sérica igual a 3 mg/dL) ou apresentar trombocitopenia persistente (contagem de plaquetas igual a 30.000/mm3) e ser considerado em estado de remissão e sem necessidade de tratamento.
O prognóstico a longo prazo é uma função do dano orgânico causado pelo LES ou decorrente de seu tratamento. A fibrose e a hipertensão pulmonares são pouco responsivas à terapia, embora novos protocolos (p. ex., bosentan ou infusão de prostaciclina) possam ser úteis no caso da hipertensão pulmonar. Os pacientes com insuficiência cardiopulmonar que apresentam doenças limitantes em outros sistemas são candidatos à transplante de coração e de pulmão. Os pacientes lúpicos com insuficiência renal são candidatos à diálise e a transplante renal.
O autor não possui relações comerciais com os fabricantes de produtos ou prestadores de serviços mencionados neste capítulo.
1. Zonana-Nacach A, Barr SG, Magder LS, et al. Damage in systemic lupus erythematosus and its association with corticosteroids. Arthritis Rheum 2000;43:1801.
2. Mimouni D, Nousari CH. Systemic lupus erythematosus and the skin. In: Lahita RG, editor. Systemic lupus erythematosus. 4th ed. San Diego: Academic Press; 2004. p. 855.
3. Mongey A-B, Hess EV. Drug and environmental lupus: clinical manifestations and differences. In: Lahita RG, editor. Systemic lupus erythematosus. 4th ed. San Diego: Academic Press; 2004. p. 1121.
4. Lockshin MD. Antiphospholipid syndrome. In: Ruddy S, Harris ED Jr, Sledge C, editors. Kelley’s textbook of rheumatology. Philadelphia: WB Saunders; 2001. p. 1145.
5. Buyon JP. Neonatal lupus syndromes. In: Lahita RG, editor. Systemic lupus erythematosus. 4th ed. San Diego: Academic Press; 2004. p. 449.
6. Gladman DD. Epidemiology of systemic lupus erythematosus. In: Lahita RG, editor. Systemic lupus erythematosus. 4th ed. San Diego: Academic Press; 2004. p. 697.
7. Karlson EW, Daltroy LH, Lew RA, et al. The relationship of socioeconomic status, race, and modifi able risk factors to outcomes in patients with systemic lupus erythematosus. Arthritis Rheum 1997;40:47.
8. Hahn BH. Antibodies to DNA. N Engl J Med 1998; 338:1359.
9. Balow JE, Boumpas DT, Austin HA III. Systemic lupus erythematosus and the kidney. In: Lahita RG, editor. Systemic lupus erythematosus. 3rd ed. San Diego: Academic Press; 1999. p. 657.
10. Bonfa E, Golombek SJ, Kaufman LD, et al. Association between lupus psychosis and anti–ribosomal P protein antibodies. N Engl J Med 1987;317:265.
11. Kozora E, Hanly JG, Lapteva L, Filley CM. Cognitive dysfunction in systemic lupus erythematosus. Arthritis Rheum 2008. [In press].
12. Harrison MJ, Ravdin LD, Lockshin MD. Relationship between serum NR2a antibodies and cognitive dysfunction in systemic lupus erythematosus. Arthritis Rheum 2006; 54:2515–22.
13. Walport MJ. Lupus, DNAse, and defective disposal of cellular debris. Nat Genet 2000;25:135.
14. Blanco P, Palucka AK, Gill M, et al. Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science 2001;294:1540.
15. Davies KA, Robson MG, Peters AM, et al. Defective Fc-dependent processing of immune complexes in patients with systemic lupus erythematosus. Arthritis Rheum 2002; 46:1028.
16. Marrack P, Kappler J, Kotzin BL. Autoimmune disease: why and where it occurs. Nat Med 2001;7:899.
17. Davidson A, Diamond B. Autoimmune diseases. N Engl J Med 2001;345:340.
18. Crow MK, Kirou KA. Interferon-alpha in systemic lupus erythematosus. Curr Opin Rheumatol 2004;16:541.
19. Kobayashi S, Ikari K, Kaneko H, et al. Association of STAT4 with susceptibility to rheumatoid arthritis and systemic lupus erythematosus in the Japanese population. Arthritis Rheum 2008;58:1940–6.
20. Taylor KE, Remmers EF, Lee AT, et al. Specifi city of the STAT4 genetic association for severe disease manifestations of systemic lupus erythematosus. PLoS Genet 2008;4(5): e1000084.
21. Dieguez-Gonzalez R, Calaza M, Perez-Pampin E, et al. Association of interferon regulatory factor 5 haplotypes, similar to that found in systemic lupus erythematosus, in a large subgroup of patients with rheumatoid arthritis. Arthritis Rheum 2008;58:1264–74.
22. Kawasaki A, Kyogoku C, Ohashi J, et al. Association of IRF5 polymorphisms with systemic lupus erythematosus in a Japanese population: support for a crucial role of intron 1 polymorphisms. Arthritis Rheum 2008;58:826–34.
23. Nath SK, Han S, Kim-Howard X, et al. A nonsynonymous functional variant in integrin-alpha(M) (encoded by ITGAM) is associated with systemic lupus erythematosus. Nat Genet 2008;40:152–4. Epub 2008 Jan 20.
24. International Consortium for Systemic Lupus Erythematosus Genetics (SLEGEN), Harley JB, Alarcón-Riquelme ME, Criswell LA, et al. Genome-wide association scan in women with systemic lupus erythematosus identifi es susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet 2008;40:204–10. Epub 2008 Jan 20.
25. Hom G, Graham RR, Modrek B, et al. Association of systemic lupus erythematosus with C8orf13-BLK and ITGAM-ITGAX. N Engl J Med 2008;358:900–9. Epub 2008 Jan 20.
26. Reveille JD. Major histocompatibility complex class II and non–major histocompatibility complex genes in the pathogenesis of systemic erythematosus. In: Lahita RG, editor. Systemic lupus erythematosus. 3rd ed. San Diego: Academic Press; 1999. p. 67.
27. Harley JB, Moser KL, Gaffney PM, et al. The genetics of human systemic lupus erythematosus. Curr Opin Immunol 1998;10:690.
28. Abel G, Agnello V. Complement deficiency and systemic lupus erythematosus. In: Lahita RG, editor. Systemic lupus erythematosus. 4th ed. San Diego: Academic Press; 2004. p. 173.
29. Zuniga R, Ng J, Peterson MG, et al. Low binding alleles of Fc gamma receptor types Iia and IIIa are inherited independently and are associated with systemic lupus erythematosus in Hispanic patients. Arthritis Rheum 2001;44:361.
30. James JA, Kaufman KM, Farris AD, et al. An increased prevalence of Epstein-Barr virus infection in young patients suggests a possible etiology for systemic lupus erythematosus. J Clin Invest 1997;100:3019.
31. Arbuckle MR, McClain MT, Rubertone MV, et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med 2003;349:1526.
32. Petri M, Robinson C. Review: oral contraceptives and systemic lupus erythematosus. Arthritis Rheum 1997;40: 797.
33. Lockshin MD, Sammaritano LR, Schwartzman S. Lupus pregnancy. In: Lahita RG, editor. Systemic lupus erythematosus. 4th ed. San Diego: Academic Press; 2004. p. 659.
34. Ruiz-Irastorza G, Lima F, Alves J, et al. Increased rate of lupus flare during pregnancy and the puerperium: a prospective study of 78 pregnancies. Br J Rheumatol 1996; 35:133.
35. Bynoe MS, Grimaldi CM, Diamond B. Estrogen up-regulates Bcl-2 and blocks tolerance induction of naive B cells. Proc Natl Acad Sci U S A 2000;97:2703.
36. Cooper GS, Dooley MA, Treadwell EL, et al. Hormonal, environmental, and infectious risk factors for developing systemic lupus erythematosus. Arthritis Rheum 1998;41: 1714.
37. Alarcon GS, McGwin G Jr, Bartolucci AA, et al. Systemic lupus erythematosus in three ethnic groups: IX. Differences in damage accrual. LUMINA Study Group. Arthritis Rheum 2001;44:2797.
38. Salmon JE, Roman MJ. Accelerated atherosclerosis in systemic lupus erythematosus: implications for patient management. Curr Opin Rheumatol 2001;13:341.
39. Hochberg MC. Updating the American College of Rheumatology revised criteria for the classifi cation of systemic lupus erythematosus [letter]. Arthritis Rheum 1997;40: 1725.
40. Lockshin MD, Sammaritano LR, Schwartzman S. Brief report: validation of the Sapporo Criteria for antiphospholipid antibody syndrome. Arthritis Rheum 2000;43:440.
41. Lahita RG. The clinical presentation of systemic lupus erythematosus in adults. In: Lahita RG, editor. Systemic lupus erythematosus. 4th ed. San Diego: Academic Press; 2004. p. 435.
42. Lawrence EC. Systemic lupus erythematosus and the lung. In: Lahita RG, editor. Systemic lupus erythematosus. 4th ed. San Diego: Academic Press; 2004. p. 961.
43. Roldan CA, Shively BK, Crawford MH. Echocardiographic study of valvular heart disease associated with lupus erythematosus. N Engl J Med 1996;335:1424.
44. Farzaneh-Far A, Roman MJ, Lockshin MD, et al. Relationship of antiphospholipid antibodies to cardiovascular manifestations of systemic lupus erythematosus. Arthritis Rheum 2006;54:3918–25.
45. Lockshin MD, Salmon JE, Roman MJ. Atherosclerosis and lupus: a work in progress [editorial]. Arthritis Rheum 2001;44:2215.
46. Roman MJ, Shanker BA, Davis A, et al. Prevalence and correlates of accelerated atherosclerosis in systemic lupus erythematosus. N Engl J Med 2003;349:2399.
47. Bywaters EGL. Jaccoud’s syndrome: a sequel to the joint involvement of systemic lupus erythematosus. Clin Rheum Dis 1975;1:125.
48. Mont MA, Glueck CJ, Pacheco IH, et al. Risk factors for osteonecrosis in systemic lupus erythematosus. J Rheumatol 1997;24:645.
49. Nagasawa K, Tsukamoto H, Tada Y, et al. Imaging study on the mode of development and changes in avascular necrosis of the femoral head in systemic lupus erythematosus: long-term observations. Br J Rheumatol 1994;33:343.
50. Dotan I, Mayer L. Nonhepatic gastrointestinal manifestations of systemic lupus erythematosus. In: Lahita RG, editor. Systemic lupus erythematosus. 4th ed. San Diego: Academic Press; 2004. p. 975.
51. Mackay IR. Hepatic disease and systemic lupus erythematosus: coincidence or convergence. In: Lahita RG, editor. Systemic lupus erythematosus. 4th ed. San Diego: Academic Press; 2004. p. 993.
52. The American College of Rheumatology Nomenclature and Case Defi nitions for Neuropsychiatric Lupus Syndromes. ACR Ad Hoc Committee on Neuropsychiatric Lupus Nomenclature. Arthritis Rheum 1999;42:599.
53. Hanly JG. Evaluation of patients with CNS involvement in SLE. Baillieres Clin Rheumatol 1998;12:415.
54. Harrison MJ, Gershengorn J. Despite low rate of global impairment in SLE patients, cognitive performance is less than expected [abstract]. Arthritis Rheum 2001;44:S197.
55. Kozora E, Thompson LL, West SG, et al. Analysis of cognitive and psychological defi cits in systemic lupus erythematosus patients without overt central nervous system disease. Arthritis Rheum 1996;39:2035.
56. Sanna G, Piga M, Terryberry JW, et al. Central nervous system involvement in systemic lupus erythematosus: cerebral imaging and serological profi le in patients with and without overt neuropsychiatric manifestations. Lupus 2000;9:573.
57. Gladman DD, Urowitz MB, Slonim D, et al. Evaluation of predictive factors for neurocognitive dysfunction in patients with inactive systemic lupus erythematosus. J Rheumatol 2000;27:2367.
58. Weening JJ, D’Agati VD, Schwartz MM, et al. The classifi cation of glomerulonephritis in systemic lupus erythematosus revisited. Kidney Int 2004;65:521.
59. Clancy RM, Kapur RP, Molad Y, et al. Immunohistologic evidence supports apoptosis, IgG deposition, and novel macrophage/fi broblast crosstalk in the pathologic cascade leading to congenital heart block. Arthritis Rheum 2004; 50:173.
60. Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classifi cation criteria for defi nite antiphospholipid syndrome (APS). J Thromb Haemost 2006;4:295–306.
61. Salmon JE, Girardi G, Lockshin MD. The antiphospholipid syndrome as a disorder initiated by infl ammation: implications for the therapy of pregnant patients. Nat Clin Pract Rheumatol 2007;3:140–7.
62. Kozora E, West SG, Kotzin BL, et al. Magnetic resonance imaging abnormalities and cognitive defi cits in systemic lupus erythematosus patients without overt central nervous system disease. Arthritis Rheum 1998;41:41.
63. Sibbitt WL Jr, Sibbitt RR, Brooks W. Neuroimaging in neuropsychiatric systemic lupus erythematosus. Arthritis Rheum 1999;42:2026.
64. West SG, Emlen W, Wener MH, et al. Neuropsychiatric lupus erythematosus: a ten-year prospective study on the value of diagnostic tests. Am J Med 1995;99:153.
65. A randomized study of the effect of withdrawing hydroxychloroquine sulfate in systemic lupus erythematosus. Canadian Hydroxychloroquine Study Group. N Engl J Med 1991;324:150.
66. Liang MH, Socher SA, Roberts WN, et al. Measurement of systemic lupus erythematosus activity in clinical research. Arthritis Rheum 1988;31:817.
67. Buyon JP, Kalunian KC, Ramsey-Goldman R, et al. Assessing disease activity in SLE patients during pregnancy. Lupus 1999;8:677.
68. Gladman DD, Goldsmith CH, Urowitz MB, et al. The Systemic Lupus International Collaborating Clinics/American College of Rheumatology (SLICC/ACR) Damage Index for systemic lupus erythematosus international comparison. J Rheumatol 2000;27:373.
69. Crowther MA, Ginsberg JS, Julian J, et al. A comparison of two intensities of warfarin for the prevention of recurrent thrombosis in patients with the antiphospholipid antibody syndrome. N Engl J Med 2003;349:1133–8.
70. Finazzi G, Marchioli R, Brancaccio V, et al. A randomized clinical trial of high-intensity warfarin vs. conventional antithrombotic therapy for the prevention of recurrent thrombosis in patients with the antiphospholipid syndrome (WAPS). J Thromb Haemost 2005;3:848–53.
71. Meroni PL, Raschi E, Testoni C, et al. Statins prevent endothelial activation induced by antiphospholipid (antibeta2-glycoprotein I) antibodies: effect on the proadhesive and proinfl ammatory phenotype. Arthritis Rheum 2001;44: 2862.
72. Mont MA, Fairbank AC, Petri M, et al. Core decompression for osteonecrosis of the femoral head in systemic lupus erythematosus. Clin Orthop 1997;334:91.
73. Nesher G, Ilany J, Rosenmann D, et al. Valvular dysfunction in antiphospholipid syndrome: prevalence, clinical features and treatment. Semin Arthritis Rheum 1997;27:27.
74. Kimberly RP, Lockshin MD, Sherman RL, et al. Reversible “end-stage” lupus nephritis: analysis of patients able to discontinue dialysis. Am J Med 1983;74:361.
75. Trends in deaths from systemic lupus erythematosus— United States, 1979–1998. MMWR Morb Mortal Wkly Rep 2002;51:371.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.