(Carregando Índice)... (Carregando Índice)... |
Última revisão: 20/02/2018
Comentários de assinantes: 0
|
Artigo original: Nieman, L, MD. Cushing Syndrome, SAM. [The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2015 Decker Intellectual Properties Inc. All Rights Reserved.] Tradução: Paulo Henrique Machado. Revisão técnica: Dr. Lucas Santos Zambon. |
Resumo
A síndrome de Cushing é uma condição com manifestações proteiformes causadas pela exposição crônica ao excesso de glicocorticoides. A causa mais comum da doença é o tratamento com doses suprafisiológicas de glicocorticoides. O hipercortisolismo patológico possivelmente seja resultado da produção adrenal autônoma ou da ação da produção excessiva do hormônio adrenocorticotrófico (ACTH) por algum tumor, estimulando a produção de cortisol adrenal. As formas adrenais primárias incluem carcinoma ou adenoma unilateral ou, mais raramente, hiperplasia bilateral e/ou nódulos. Este artigo apresenta detalhes sobre epidemiologia, etiologia, fisiopatologia e diagnóstico da síndrome de Cushing. As manifestações clínicas, as descobertas de exames físicos e os testes laboratoriais, incluindo exames de sangue e de outros líquidos corporais, estudos de imagens e biópsias, também serão discutidos neste artigo. Serão também descritos com detalhes o diagnóstico diferencial, as opções de tratamento, as complicações e o prognóstico. Os quadros apresentam fatores como características clínicas e causas da síndrome de Cushing, anormalidades associadas às causas adrenais primárias da doença, precisão diagnóstica dos testes de rastreamento, hipercortisolismo endógeno sem a síndrome de Cushing e terapia médica para a síndrome de Cushing. As figuras ilustram as causas da síndrome de Cushing e uma comparação do eixo hipotalâmico-hipofisário-adrenal em pacientes com síndrome de Cushing dependente do ACTH e em pacientes com síndrome de pseudo-Cushing. Os algoritmos mostram a avaliação de uma possível síndrome de Cushing e a avaliação das causas da síndrome. Este artigo apresenta também detalhes sobre os tratamentos de segunda linha para a síndrome de Cushing nas situações em que a cirurgia não tenha sido bem-sucedida ou nas situações em que não for viável.
Definição e Subclassificação da Doença
A síndrome de Cushing é uma condição com manifestações proteiformes causadas pela exposição crônica ao excesso de glicocorticoides.1 A causa mais comum da doença é o tratamento com doses suprafisiológicas de glicocorticoides; o uso de glicocorticoides exógenos, seja qual for a via de administração, é a melhor opção para pacientes portadores da síndrome de Cushing. Este artigo foca as causas endógenas de hipercortisolismo.
Provavelmente, o hipercortisolismo patológico seja o resultado da produção adrenal autônoma ou da ação da produção excessiva do hormônio adrenocorticotrófico (ACTH) por algum tumor, estimulando a produção de cortisol adrenal. As formas adrenais primárias incluem carcinoma ou adenoma unilateral ou, mais raramente, hiperplasia bilateral e/ou nódulos. É possível que o excesso de ACTH (conhecido como secreção ectópica de ACTH) tenha origem em tumores que se localizam nas células hipofisárias corticotrópicas. Enquanto os tumores primários são quase sempre benignos, os ectópicos podem ser malignos.
Epidemiologia
A síndrome de Cushing endógena é uma condição rara que, na Europa, ocorre em duas a três pessoas por ano em uma população de 1 milhão de adultos.2 A produção de ACTH ectópico é mais comum em homens, embora todas as outras formas do distúrbio sejam três vezes mais comuns em mulheres e ocorram com mais frequência durante os anos reprodutivos.3 As possíveis predileções geográficas, raciais ou étnicas, ainda não foram estudadas, exceto um efeito mãe sobre o câncer adrenal em famílias brasileiras com a síndrome de Li-Fraumeni4 e uma taxa de incidência de tumores adrenais além das expectativas no Japão.5
Etiologia e Genética
A maior parte das causas das formas de síndrome de Cushing dependentes de ACTH é desconhecida. Embora essas causas geralmente não sejam familiares, há relatos de uma grande variedade de mutações somáticas.6 Por outro lado, inúmeras causas adrenais familiares carregam mutações genéticas conhecidas.
Por exemplo, os pacientes com displasia suprarrenal nodular pigmentada primária (DSRNPP) apresentam mutações na via de sinalização da proteína quinase A (em inglês, protein kinase A [PKA]).7 Inúmeras anormalidades genéticas e vias moleculares disfuncionais foram descritas em casos de câncer adrenal; embora não sejam relevantes para os cuidados clínicos nesse momento, os pacientes poderão receber tratamentos individualizados no futuro.7
Fisiopatologia e Patogênese
Embora a patogênese dos tumores que produzem ACTH e a maior parte dos adenomas adrenais não seja muito clara, sua fisiopatologia é inerente à estratégia dos testes diagnósticos. Em indivíduos saudáveis, níveis elevados de cortisol inibem a produção do hormônio liberador de corticotrofina (em inglês, corticotropin-releasing hormone [CRH]) e a liberação de corticotrofos hipofisários que produzem ACTH, respectivamente.
Este feedback negativo é operacional nos casos de síndrome de Cushing, de modo que ocorre a supressão da função corticotrópica normal; em outras palavras, a secreção de ACTH é extremamente baixa. Como resultado, qualquer ACTH em circulação deriva da produção tumoral. Consequentemente, as causas da síndrome de Cushing dependentes e independentes do ACTH são distinguíveis pela presença de níveis inapropriadamente normais ou elevados de ACTH e por concentrações baixas ou não detectáveis de ACTH, respectivamente.
Os carcinoides pulmonares são os tumores ectópicos mais comuns (não hipofisários) que produzem ACTH; entretanto, a secreção ectópica também ocorre em outros carcinoides do intestino anterior (pâncreas, timo), feocromocitoma, câncer tireóideo medular e outros tipos de tumor.8 Em situações extremamente raras, algum tumor poderá produzir secreção de ACTH em excesso.
Embora a etiologia não seja conhecida, a maior parte dos adenomas benignos que produzem cortisol apresenta ativação anormal da via de sinalização da proteína-quinase A (PKA).7 Em células adrenais normais, a ligação do ACTH ativa a subunidade a estimuladora da proteína G, aumentando, consequentemente, o monofosfato cíclico de adenosina (em inglês, cyclic adenosine monophosphate [cAMP]).
O cAMP se liga às subunidades reguladoras da PKA, liberando-as para ativar a proteína de ligação com o elemento de resposta ao cAMP, e influencia a transcrição dos genes-alvo. As fosfodiestérases desativam o cAMP. Portanto, as mutações que ativam as subunidades reguladoras da PKA ou as mutações que desativam as fosfodiestérases possivelmente intensifiquem a atividade dessa via.
Muitos pacientes com hiperplasia adrenal macronodular bilateral (HAMB) apresentam expressão aberrante dos receptores acoplados da proteína G.6 A demonstração recente da expressão e secreção do ACTH em células esteroidogênicas adrenais sugere que o ACTH intranodular possivelmente intensifique a ação dos receptores expressos de forma aberrante.9
Diagnóstico
Manifestações Clínicas
O Quadro 1 apresenta uma lista de sinais e sintomas clínicos da síndrome de Cushing.1,11,45,46
Quadro 1
|
CARACTERÍSTICAS CLÍNICAS DA síndrome de Cushing | ||||
|
Características clínicas |
Sensibilidade (%) |
Especificidade (%) |
Razão de probabilidade | |
|
Resultado positivo |
Resultado negativo | |||
|
Fadiga intensa |
100 |
? |
? |
?
|
|
Redução na libido |
33?100 |
? |
? |
?
|
|
Ganho de peso |
79?97 |
33?100 |
? |
?
|
|
Irritabilidade, labilidade emocional |
40?86 |
? |
? |
?
|
|
Insônia |
69 |
? |
? |
?
|
|
Baixa concentração |
66 |
? |
? |
?
|
|
Problemas de memória no curto prazo |
83 |
? |
? |
?
|
|
Alterações no apetite |
54 |
? |
? |
?
|
|
Letargia, depressão |
40?67 |
57 |
? |
?
|
|
Alterações na menstruação |
35?86 |
49?74 |
0,7?1,7 |
1,3?0,3
|
|
Osteopenia ou fratura recente |
48?83 |
90?94 |
8,0?13,8 |
0,6?0,2
|
|
Cefaleia |
47?58 |
63 |
1,3?1,6 |
0,7?0,8
|
|
Dor nas costas |
39?83 |
? |
? |
?
|
|
Intolerância à glicose |
13?35 |
83 |
? |
?
|
|
Infecções recorrentes |
14?25 |
? |
? |
?
|
|
Obesidade ou ganho de peso generalizado |
51?90 |
71 |
1,8?3,1 |
0,1?0,7
|
|
Obesidade no tronco |
3?97 |
38 |
0,05?1,6 |
0,1?2,6
|
|
Pletora |
78?94 |
69 |
2,5?3,0 |
0,1?0,3
|
|
Rosto arredondado |
88?92 |
? |
? |
?
|
|
Hirsutismo |
64?84 |
48?61 |
2,2?2,9 |
0,3?0,6
|
|
SOP |
8 |
58 |
? |
?
|
|
Hipertensão |
74?90 |
52?83 |
4,4?5,3 |
0,1?0,3
|
|
Equimose |
36?68 |
90?94 |
10,0?11,3 |
0,3?0,4
|
|
Estrias com mais de 1cm de largura e de cor roxa
|
50?64 |
61?78 |
2,7?2,9 |
0,5?0,6 |
|
Fraqueza |
28?90 |
70?93 |
8,0?12,6 |
0,1?0,7
|
|
Distribuição anormal de gorduras (centrípeta, dorsocervical, supraclavicular e/ou temporal)
|
34?67 |
? |
? |
? |
|
Edema |
48?66 |
83 |
2,8?3,9 |
0,4?0,6
|
|
Pele fina e fraca |
84 |
? |
? |
?
|
|
Dor abdominal |
21 |
? |
? |
?
|
|
Acne |
21?82 |
61?76 |
0,9?3,4 |
0,2?1,0
|
|
Calvície feminina |
13?51 |
? |
? |
? |
SOP: síndrome do ovário policístico.
*Os traços indicam que não há dados disponíveis.
Muitos desses sinais e sintomas são comuns na população em geral. Características mais específicas da síndrome de Cushing (infecções não usuais, estrias roxas, fraqueza proximal) tendem a ocorrer nos casos de hipercortisolismo mais grave.
Como resultado, o diagnóstico talvez seja mais difícil nos casos mais leves que se apresentarem com sinais e sintomas menos específicos. As características apontam mais fortemente para a síndrome de Cushing nas situações que forem atípicas para a idade (por exemplo, osteoporose ou hipertensão em adultos jovens) ou quando forem cumulativas ao longo do tempo (ao contrário dos sinais e sintomas da SOP, que tendem a ser mais estáveis).10
Descobertas nos Exames Físicos
O exame físico procura localizar qualquer distribuição anormal de gorduras (aumento na adiposidade supraclavicular, na fossa temporal, dorsocervical e/ou abdominal), pele anormal para a idade (pletora facial, afinamento >1cm em estrias roxas largas, equimose, acne, infecções, edema), redução no enfraquecimento dos músculos proximais (a força é testada pela capacidade de erguer-se de uma cadeira sem usar os braços ou erguer-se da posição agachada), hirsutismo, hipertensão, obesidade, alterações na memória e na cognição (pelo histórico, setes seriais e lembrança de três objetos), e alterações no humor (por meio da observação e do histórico).
As descobertas mais úteis para o diagnóstico diferencial incluem lentigo e nevos azuis-escuros, que sugerem a presença de DSRNPP no contexto da síndrome de Carney. A probabilidade de doença antes dos testes é mais elevada nas situações em que houver múltiplos sinais e sintomas, quando se acumularem ao longo do tempo e quando forem atípicos para a idade.11
Por exemplo, fatores como aumento de peso, nova depressão, fratura e hipertensão sugerem a presença da síndrome de Cushing em homens com 24 anos de idade, porém não em mulheres com 64 anos. A probabilidade pré-teste é mais baixa nos casos de características estáveis; é comum na população e esperada para a idade do paciente.
Consequentemente, uma mulher com 32 anos de idade com oligomenorreia, acne e hirsutismo por 15 anos, sem quaisquer outras características, provavelmente tenha SOP, e é pouco provável que tenha síndrome de Cushing.
O Quadro 2 contém as causas da síndrome de Cushing e o Quadro 3, as anormalidades associadas às causas adrenais primárias da síndrome de Cushing.6,7
Quadro 2
|
CAUSAS DA síndrome de Cushing |
|
Formas independentes de ACTH (15%) Tumor unilateral Adenoma (60%) Câncer (40%) Tumor(es) ou hiperplasia (casos raros) DSRNPP, com ou sem as características do complexo de Carney HAMB SMA
Formas dependentes de ACTH (85%) Tumor corticotrópico (“doença de Cushing”, aproximadamente 80%) Tumor não hipofisário (denominado secreção ou síndrome ectópica de ACTH, cerca de 20%) Secreção ectópica tumoral de CRH (condição rara) |
ACTH: hormônio adrenocorticotrófico; CRH: hormônio liberador de corticotrofina; DSRNPP: doença suprarrenal nodular pigmentada primária; HAMB: hiperplasia adrenal macronodular bilateral; SMA: Síndrome de McCune-Albright.
Quadro 3
|
ANORMALIDADES ASSOCIADAS ÀS CAUSAS ADRENAIS PRIMÁRIAS DA síndrome de Cushing | |
|
Mutação |
Associação com causa de SC |
|
Mutações no gene da catenina-ß (CTNNB1) |
Adenoma adrenal esporádico (20%), carcinoma adrenal (20%).
|
|
Subunidade reguladora 1a do tipo somático das mutações da PKA dependente de cAMP (PRKAR1A)
|
Adenoma adrenal (20%), DSRNPP familiar e complexo de Carney. |
|
Deleção genética do lócus de PRKAR1A |
Adenoma adrenal (23%).
|
|
Variantes genéticas da fosfodiestérase somática 11A (PDE11A) |
Adenoma adrenal (19%), carcinomas adrenais (16%), HAMB (24%).
|
|
Mutações somáticas na subunidade catalítica de PKA (PRKACA) |
Adenoma adrenal (37%).
|
|
Mutações desativadoras da repetição de testes em tatus contendo 5 genes (ARMC5)
|
Famílias de HAMB, HAMB esporádica (50%). |
|
Mutações ativadoras da subunidade Gsa |
Síndrome de McCune-Albright.
|
cAMP: monofosfato cíclico de adenosina; DSRNPP: doença suprarrenal nodular pigmentada primária; HAMB: hiperplasia adrenal macronodular bilateral; PKA: proteína-quinase A; SC: síndrome de Cushing.
Testes Laboratoriais para Diagnóstico da síndrome de Cushing
As orientações mais recentes sugerem que são necessários dois entre três testes de rastreamento para fazer o diagnóstico da síndrome de Cushing.12 Esses testes incluem cortisol urinário livre, cortisol salivar noturno e supressão de dexametasona. Em uma metanálise que incluiu estudos nos quais havia uma grande proporção de pacientes com a síndrome de Cushing, cada teste apresentou uma alta precisão diagnóstica.13 Entretanto, ainda não se conhece o desempenho desses testes em ambientes fora das pesquisas e com um menor número de pacientes afetados.
Os resultados dos testes próximos do ponto de corte devem ser encarados com ceticismo, sendo muito importante considerar outros estados hipercortisolêmicos. Os pacientes com resultados discordantes poderão fazer novos testes periódicos nos casos em que houver forte suspeita clínica. Os com resultados negativos não precisam fazer testes adicionais, a menos que a suspeita clínica seja muito elevada e, além disso, haja suspeita clínica de síndrome de Cushing cíclica.
Há no mercado um nomograma que incorpora a probabilidade pré-teste na interpretação dos resultados.13 A escolha do teste de rastreamento deve ser individualizada para cada paciente com base no potencial para resultados falsos, na conveniência e na preferência do paciente. Os testes utilizados para determinar a causa da síndrome de Cushing não devem ser usados para fins diagnósticos.
Testes de sangue
O teste de supressão da dexametasona avalia a presença de inibição do feedback negativo normal do eixo hipotalâmico-hipofisário-adrenal (HPA). Em indivíduos saudáveis, os glicocorticoides inibem a secreção do ACTH por corticotropos, de modo que a administração de 1mg de dexametasona entre 11h e meia-noite resulta na supressão de cortisol (<1,8µg/dL, 50mmol/L) na manhã seguinte entre 8h e 9h.
Uma segunda forma de teste, o teste com 2mg por 2 dias (às vezes, chamado de teste de dose baixa), não é usado com muita frequência nos EUA. Esse tipo de teste consiste na administração de 0,5mg de dexametasona a cada 6 horas, iniciando às 9h, no total de 8 doses, e medição do nível de cortisol às 9h, 6 horas após a última dose. Valores inferiores a 1,8µg/dL são considerados normais.14
Embora os corticotropos tumorais mantenham alguma sensibilidade ao feedback de glicocorticoides, o procedimento deverá ser reinicializado depois de níveis mais elevados e, em geral, exige doses mais altas de dexametasona para fins de supressão. As outras causas da síndrome de Cushing não mantêm a supressão de dexametasona.
Há alguns avisos de alerta que influenciam a escolha e a interpretação dos resultados desse teste. Em primeiro lugar, o nível de corte no cortisol para fins de interpretação do teste está próximo do limite funcional de detecção de alguns ensaios, de modo que a variabilidade possivelmente leve a um resultado normal ou anormal. Em segundo lugar, o metabolismo interpessoal da dexametasona é variável e afetado pelo uso de medicações e por problemas hepáticos e renais.
A dexametasona é metabolizada através do complexo CYP3A4. As medicações podem intensificar (por exemplo, rifampicina, fenobarbital) ou inibir (ritonavir, cetoconazol) sua eliminação, levando a respostas falsamente anormais ou normais, respectivamente. O site da Indiana University School of Medicine15 fornece uma lista extensa de medicações com grande potencial de interação. Por causa dessas interações, talvez esse tipo de teste não seja boa escolha para indivíduos que tomam muitas medicações.16
O metabolismo anormal pode ser avaliado por meio da medição do nível de dexametasona em comparação com o nível de cortisol, levando-se em conta que os laboratórios comerciais determinaram as concentrações esperadas de dexametasona.
Para finalizar, quase todos os níveis séricos de cortisol se ligam à globulina ligadora do cortisol (CBG, do inglês cortisol-binding globulin), cujo nível poderá ser elevado em mulheres que tomam estrogênios orais, levando a resultados falsamente elevados.17 Para evitar esse tipo de inconveniência, o uso de estrogênios orais deve ser interrompido por 4 a 6 semanas antes do início dos testes. Como alternativa, a medição da CBG possivelmente evite esse tipo de preocupação.
Testes de outros líquidos corporais
A medição do nível noturno de cortisol salivar antes de deitar, ou em alguns estudos feitos à meia-noite, pode identificar com grande precisão pacientes com a síndrome de Cushing.18 Esse tipo de medição aproveita a vantagem do nadir fisiológico do cortisol, que ocorre logo após o início do sono e se perde em pacientes com a síndrome de Cushing.
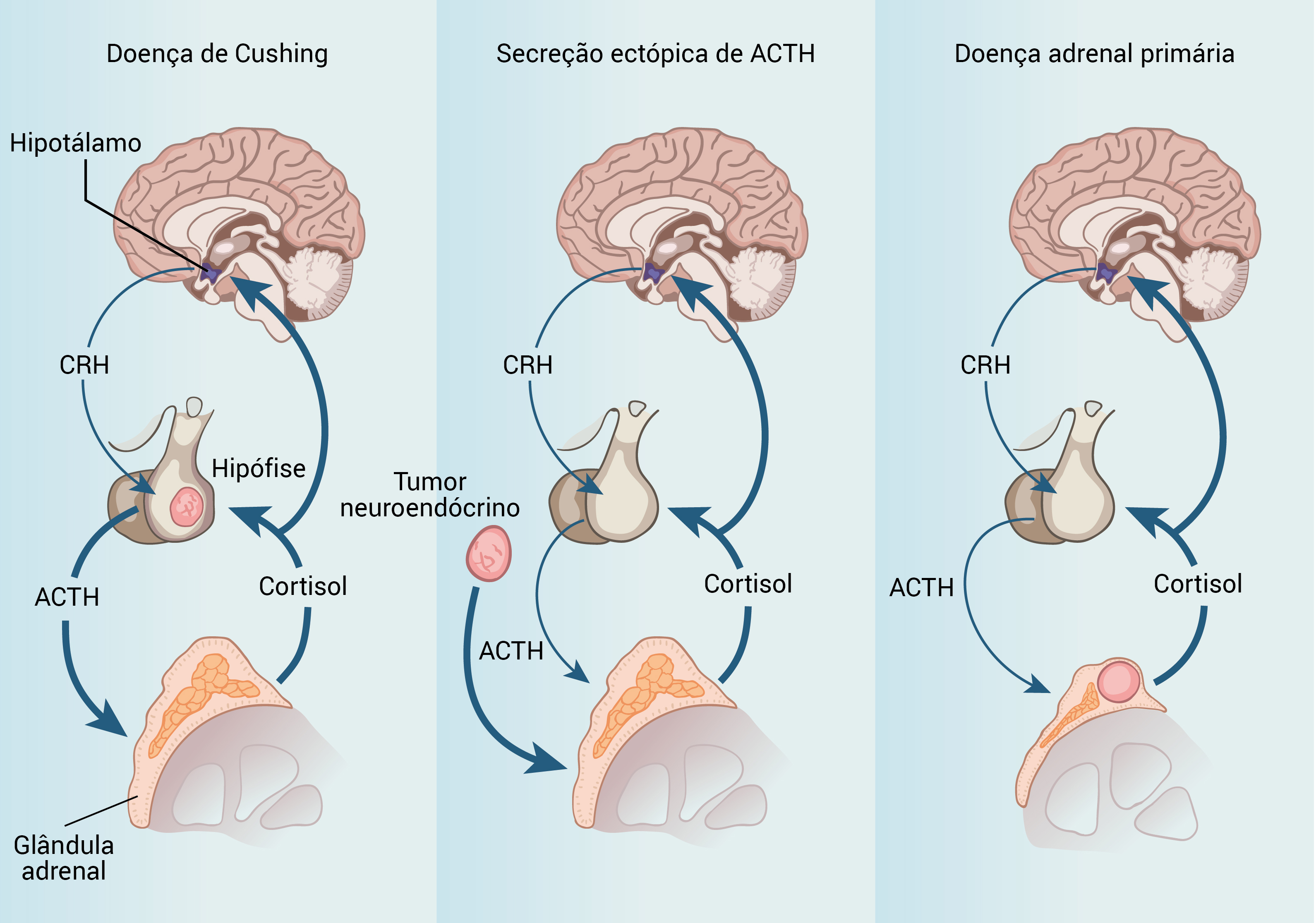
A Figura 1 mostra as causas da síndrome de Cushing.

ACTH: hormônio adrenocorticotrófico; CRH: hormônio liberador de corticotrofina.
Figura 1 - Causas da síndrome de Cushing. Um dos conceitos fisiológicos importantes é o efeito do excesso de cortisol para suprimir a secreção do CRH e do ACTH por corticotropos normais. Como resultado, qualquer ACTH em circulação representa uma secreção tumoral, permitindo fazer a distinção entre doença adrenal primária (independente do ACTH) e causas que dependem do ACTH.
Embora o teste noturno do cortisol salivar tenha sensibilidade e especificidade de 95%, sua interpretação depende do ensaio que foi utilizado. De um modo geral, o limite superior da normalidade é utilizado como critério de corte.
Ainda que esse tipo de teste tenha apresentado bom desempenho em estudos de pesquisas que incluíram basicamente pacientes em idade reprodutiva, talvez não seja o teste ideal em indivíduos mais velhos com diabetes melito e/ou hipertensão. Em um dos estudos, de 10 a 43% desses indivíduos, dos quais nenhum tinha a síndrome de Cushing, apresentaram resultados anormais.19
O Quadro 4 apresenta a precisão diagnóstica dos testes de rastreamento para a síndrome de Cushing.13
Quadro 4
|
PRECISÃO DIAGNÓSTICA DOS TESTES DE RASTREAMENTO PARA A síndrome de Cushing | |||
|
Teste de rastreamento |
Número de estudos |
Razão de probabilidade de um teste positivo (IC 95%) |
Razão de probabilidade de um teste negativo (IC 95%) |
|
UFC |
14 |
10,6 (5,5?20,5) |
0,16 (0,08?0,33)
|
|
Cortisol salivar à meia-noite |
4 |
8,8 (3,5?21,8) |
0,07 (0?1,2)
|
|
1mg de DST durante a noite |
14 |
16,4 (9,3?28,8) |
0,06 (0,03?0,14)
|
|
Estratégias de testes combinados (p. ex., resultado positivo no UFC e em 1mg de DST durante a noite)
|
3 |
15,4 (0,7?358) |
0,11 (0,007?1,57) |
DST: teste de supressão de dexametasona; UFC: cortisol urinário livre.
Levando-se em conta que os resultados normais dependem de padrões normais de sono-vigília, os pacientes que trabalham em regime de turnos não são bons candidatos para o teste e, além do mais, o momento de aplicação do teste deve ser ajustado nos indivíduos que, cronicamente, irão se deitar em horários fora do padrão. Embora ainda não tenha sido documentado, há uma preocupação teórica de que a contaminação com sangue possa levar a resultados anormais.
A contaminação, presumivelmente causada pelo uso de agentes tópicos, é sugerida pela ocorrência de resultados bastante elevados e poderá ser confirmada por meio da documentação de uma proporção muito elevada de cortisol e cortisona determinada através da espirometria de massa.
A medição da excreção de cortisol de 24 horas (cortisol urinário livre [UFC]) permite fazer uma avaliação integrada da secreção de cortisol durante o dia. Esse tipo de teste teve boa sensibilidade no passado quando se utilizavam imunoensaios; estudos recentes de pequeno porte sugerem que a precisão diagnóstica é menor nas situações em que se utilizam ensaios estruturais específicos como a espectrometria de massa em série.20 Esse fato se explica pela reatividade cruzada de metabólitos não cortisóis nos imunoensaios, porém não em ensaios estruturais.
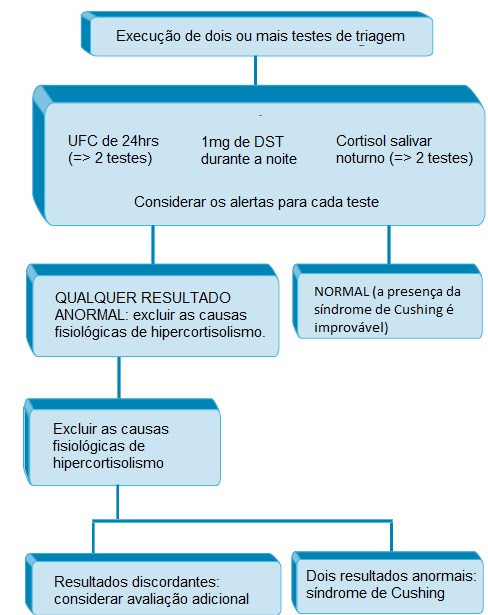
A Figura 2 ilustra o algoritmo para avaliação de possível síndrome de Cushing.
DST: teste de supressão da dexametasona; UFC: cortisol urinário livre.
Figura 2 - Algoritmo para avaliação de possível síndrome de Cushing. Adaptação de um trabalho realizado por Nieman et al.12
A coleta precisa de 24 horas é essencial para estimar o nível de cortisol urinário livre. Normalmente, a coleta inicia após a primeira urina da manhã e continua até a primeira urina da segunda manhã. Os resultados das medições do volume urinário e da secreção de creatinina ajudam a avaliar a integralidade dos dados da coleta. Volumes ou valores de creatinina muito baixos ou muito elevados são suspeitos nos casos de coleta excessiva ou de coleta insuficiente.
Os valores intraindividuais de creatinina variam relativamente pouco, de modo que diferenças diárias iguais ou superiores a 15% provavelmente representem coletas imprecisas. Levando-se em consideração que a secreção de cortisol declina com a insuficiência renal, a medição do UFC não tem muita utilidade em pacientes com taxas de filtração glomerular menores que 30 a 40mL/min.
Testes Laboratoriais para Determinar a Causa da síndrome de Cushing
Testes de sangue
A medição plasmática do ACTH é a melhor forma de fazer a distinção entre as formas de síndrome de Cushing dependentes e as independentes do ACTH, com valores não detectáveis ou valores baixos, normais ou elevados, respectivamente. Após ter sido feita essa distinção, torna-se necessário fazer testes adicionais para cada categoria, conforme descrevemos a seguir.
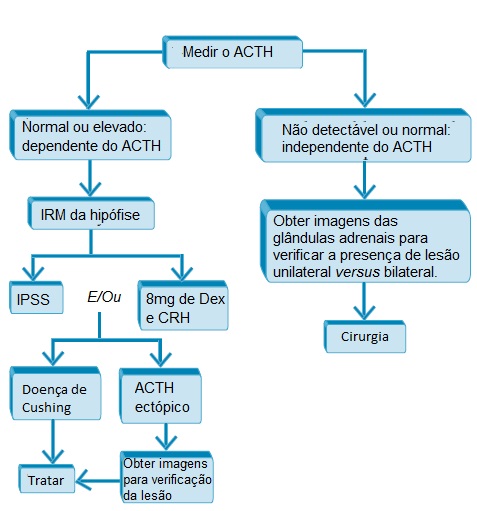
A Figura 3 apresenta um resumo dos testes laboratoriais usados para determinar a causa da síndrome de Cushing.
ACTH: hormônio adrenocorticotrófico; CRH: hormônio liberador de corticotrofinas; Dex: dexametasona; IPSS: amostragem do seio petroso inferior; IRM: imagens por ressonância magnética.
Figura 3 - Algoritmo para avaliação das causas da síndrome de Cushing.
Testes Laboratoriais para o Diagnóstico Diferencial de síndrome de Cushing Independente do Acth
Estudos de Imagens
Os estudos de imagens das glândulas adrenais com tomografia computadorizada (TC) ou imagens por ressonância magnética (IRM) são as melhores opções para fazer a distinção entre distúrbios unilaterais e bilaterais. As características das imagens das lesões unilaterais sugerem a presença de carcinoma (bordas irregulares, heterogeneidade, unidades de Housfield >25) ou de adenoma (liso, homogêneo, unidades de Housfield <20).21
As lesões bilaterais incluem nódulos grandes acima de 1cm, com ou sem hiperplasia, ou nódulos pequenos, sugerindo a presença de HAMB e DSRNPP, respectivamente. É importante observar que a hipersecreção hormonal deve ser excluída em pacientes com massa adrenal cuja descoberta for incidental, assim como os resultados obtidos após uma varredura abdominal por TC, para avaliação de dor.22
Esse tipo de avaliação inclui varredura para detectar a presença de feocromocitoma, síndrome de Cushing e hiperaldosteronismo (em pacientes hipertensos). De um modo geral, os pacientes com incidentaloma apresentam desregulação do eixo HPA nos testes bioquímicos, que normalmente se manifesta como insuficiência de supressão de dexametasona.23
Com frequência, o cortisol urinário livre é normal. O cortisol salivar ainda não foi avaliado extensivamente na população. Além disso, esses pacientes possivelmente tenham poucas características clássicas da síndrome de Cushing e têm mais chances de se apresentar com características da síndrome metabólica.24
Como resultado, há uma grande controvérsia em relação aos melhores testes para estabelecer o diagnóstico de síndrome de Cushing (ou “diagnóstico subclínico”, isto é, síndrome de Cushing leve) e, da mesma forma, não há nenhum consenso sobre qual seria o melhor tratamento para esses pacientes.
Testes de sangue
Em pacientes com valores indeterminados do ACTH, a medição do sulfato de desidroepiandrosterona (em inglês, dehydroepiandrosterone sulfate [DHEA]) possivelmente ajude a identificar pacientes com a síndrome de Cushing independente do ACTH, em que ela tem a tendência de ser subnormal (com base na idade). Isso ocorre porque o nível de ACTH, que normalmente estimula a produção do DHEA, é baixo.
Testes Laboratoriais para o Diagnóstico Diferencial de síndrome de Cushing Dependente do Acth
Conforme já mencionado, os testes para o diagnóstico diferencial de síndrome de Cushing dependente do ACTH exigem supressão do corticotropo hipofisário normal do hipercortisolismo para possibilitar a remoção das respostas hipofisárias normais dos resultados observados. Por essa razão, possivelmente os resultados dos testes sejam incorretos em pacientes com síndrome de Cushing cíclica ou leve. A situação ideal é que os testes sejam feitos depois de um período em que o hipercortisolismo sustentado (isto é, UFC >duas vezes o nível normal) tenha sido confirmado documentalmente por 6 a 8 semanas.
Testes de Sangue
Os pacientes portadores da doença de Cushing tendem a manter as características de corticotropos normais, incluindo elevação nos níveis de ACTH e de cortisol após a administração de CRH, supressão de ACTH (e de cortisol) depois de doses elevadas de dexametasona (8mg) e secreção de ACTH pela hipófise. Por outro lado, os pacientes com secreção ectópica de ACTH geralmente não respondem ao hormônio liberador de corticotrofinas (CRH) ou à dexametasona e não apresentam evidências de secreção de ACTH pela hipófise.
Atualmente, há inúmeros protocolos que realizam e interpretam esses testes.25 O teste de estimulação do CRH tem maior precisão diagnóstica25 que o teste de supressão de dexametasona.26 Nas situações em que ambos os testes são positivos, observam-se resultados discordantes ou negativos em qualquer forma de doença dependente do ACTH.
A amostragem do seio petroso inferior (em inglês, inferior petrosal sinus sampling [IPSS]) é o melhor teste para distinguir entre causas dependentes do ACTH da síndrome de Cushing. Isso envolve a colocação de cateteres no seio petroso para drenar a hipófise. O sangue é retirado medindo-se o ACTH e uma veia periférica em pontos de dois tempos antes e 3, 5 e 10 minutos após a administração de CRH (1µg/kg ou 100µg).
Os pacientes com a doença de Cushing apresentam uma etapa variando de petrosa a periférica acima do ACTH (> duas vezes na linha de base ou > três vezes após a administração de CRH) em um ou mais pontos de tempo. Um gradiente lado a lado de mais de 1,4 se localiza no lado do tumor em 69% de pacientes com tumor lateral.27 Por outro lado, os pacientes com secreção ectópica do ACTH não apresentam essa quantidade de gradiente.
Embora a IPSS tenha uma precisão diagnóstica de 95%, é necessária a experiência técnica de um radiologista intervencionista; o resultado possivelmente seja falso negativo se a cateterização não for bem-sucedida ou a anatomia venosa for anormal.28,29 Os marcadores tumorais (calcitonina, catecolaminas, ácido 5-hidroxi-indolacético [5-HIAA]) podem sugerir uma fonte de ACTH ectópico.
Estudos de Imagens
Os pacientes portadores de doença dependente de ACTH geralmente fazem a IRM da hipófise como primeiro teste; a finalidade desse teste é avaliar tumores acima de 6mm (nesse caso, a IPSS provavelmente não seja necessária). Apenas 50% de pacientes com a doença de Cushing apresentam IRM positiva e até 10% da população saudável têm lesão com 6mm ou menos.30 Logo, a presença de lesões pequenas não é suficiente para estabelecer o diagnóstico de doença de Cushing.
A obtenção de imagens ajuda a identificar a presença de tumores logo após a suspeita do diagnóstico de secreção ectópica de ACTH através de testes bioquímicos ou IPSS. Levando-se em consideração que, com frequência, esses tumores ocorrem no tórax, o passo inicial é a TC de corte fino; IRM e varredura de octreotídeos são estratégias imagiológicas adjuvantes bastante úteis.8
O tumor causador pode permanecer oculto em até 40% de pacientes após os testes de imagens iniciais. Esses pacientes devem ser tratados com meios médicos ou com adrenalectomia bilateral; estudos de imagens e ressecção em intervalos são muito importantes por causa do risco de malignidades.
Biópsia
Em pacientes com lesões metastáticas, a biópsia (em geral, no fígado) ajuda a determinar a melhor opção de tratamento.
Diagnóstico Diferencial
Níveis muito elevados de cortisol urinário (>4 vezes o nível normal) geralmente são observados apenas na síndrome de Cushing. O tratamento das condições associadas (como depressão, por exemplo) poderá normalizar os resultados bioquímicos anormais. A observação ao longo do tempo (por exemplo, 6 a 12 meses) ajuda a identificar a presença de progressão, sugestiva da síndrome de Cushing, ou de remissão da doença (contra o diagnóstico).31
O Quadro 5 mostra os distúrbios que se sobrepõem à síndrome de Cushing em termos de características clínicas e bioquímicas (situação conhecida por síndrome de pseudo-Cushing).
Quadro 5
|
DIAGNÓSTICO DIFERENCIAL ? HIPERCORTISOLISMO ENDÓGENO SEM síndrome de Cushing |
|
Algumas das características clínicas da síndrome de Cushing: Gravidez Depressão e outras condições psiquiátricas Dependência de álcool Resistência aos glicocorticoides Obesidade mórbida Diabetes melito sem controle Improbabilidade de ter algumas características clínicas da síndrome de Cushing: Estresse físico (hospitalização, cirurgia, dor) Má nutrição, anorexia nervosa Exercício físico crônico intenso Amenorreia hipotalâmica Excesso de CBG (nível sérico elevado, mas não UFC) |
CBG: globulina ligadora do cortisol; UFC: cortisol urinário livre.
Tratamento
O tratamento ideal da síndrome de Cushing procura normalizar os sinais e sintomas associados enquanto mantém normal a função hipofisária e adrenal.32 De um modo geral, o tratamento de primeira linha envolve ressecção do(s) tumor(es) causativo(s). Na ausência de qualquer malignidade, esses resultados na cura (evidenciados por hipocortisolismo pós-operatório que reflete a supressão do eixo HPA normal) de todos os tumores que produzem ACTH adrenal e ectópico.
O sucesso da ressecção do tumor corticotrópico inicial é mais baixo, 67 a 97%, e depende da experiência e das técnicas utilizadas pelo cirurgião, do tamanho do tumor e da presença de invasão dural.33 A exploração hipofisária subsequente é possível, embora o índice de sucesso seja mais baixo, e melhora se houver suspeita de localização do tumor na IPSS, na IRM ou na ressecção parcial.
Após a adrenalectomia bilateral, os pacientes precisam fazer reposição de glicocorticoides e de mineralocorticoides por toda a vida; todos os outros pacientes que forem curados precisam fazer reposição de glicocorticoides (por exemplo, 12 a 15mg/m2/dia de hidrocortisona) até a recuperação do eixo HPA, o que geralmente ocorre dentro de um período de 6 a 18 meses.
A terapia médica é o tratamento de segunda linha nas situações em que a cirurgia não for curativa ou possível. Essas opções incluem inibidores da esteroidogênese, que agem ao nível da glândula adrenal, uso de medicamentos direcionados para tumores (para as causas dependentes do ACTH) e uso de qualquer agente que antagonize a ação do cortisol. O efeito desses medicamentos é curto, de modo que apenas o tratamento médico é um comprometimento para toda a vida com os riscos apresentados no Quadro 6.
Para pacientes com doença de Cushing persistente ou recorrente ou que não sejam candidatos cirúrgicos, a radiação hipofisária, inicialmente com terapia médica, é outra opção. A radiação poderá ser direcionada para toda a hipófise por meio de radioterapia fracionada convencional ou por radiocirurgia. A radiocirurgia poderá ter também como alvo uma parte específica da glândula que supostamente contenha o tumor, na expectativa de preservar o tecido e a função normal.
Em uma série de grande porte, 53% dos pacientes conseguiram a remissão com tratamento com faca gama.34 Possivelmente, essa modalidade não seja uma opção para tumores muito próximos do(s) nervo(s) ótico(s) por causa do grande potencial para causar danos. Além do hipopituitarismo, a radioterapia poderá causar danos ocasionais no nervo craniano, colocando os pacientes em um grande risco de meningioma tardio e, possivelmente, acidente cerebrovascular.35
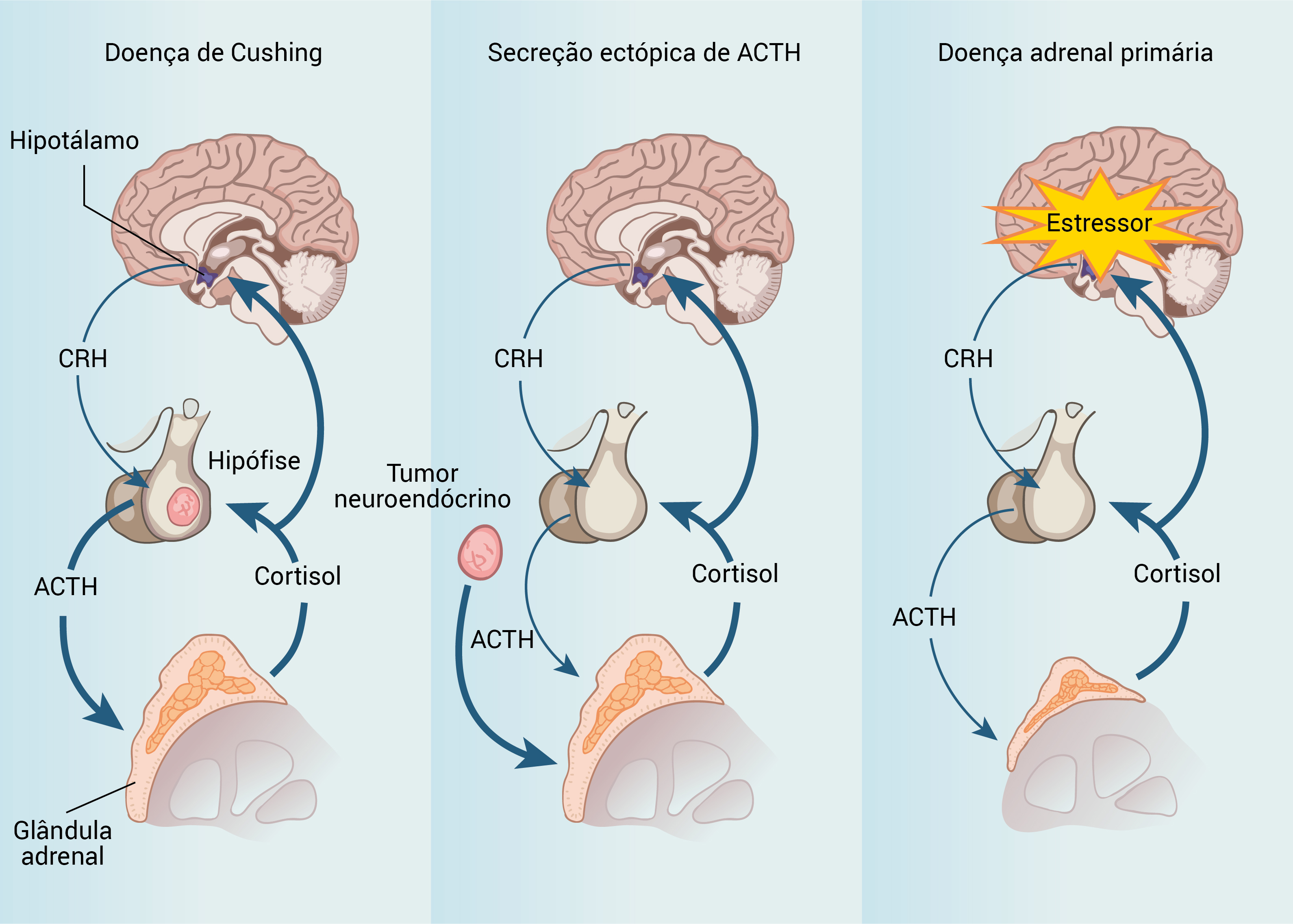
A Figura 4 mostra a comparação do eixo hipotalâmico-hipofisário-adrenal em pacientes com síndrome de Cushing dependente do ACTH e em pacientes com a síndrome de pseudo-Cushing, e a Figura 5, os tratamentos de segunda linha para a síndrome de Cushing.
ACTH: hormônio adrenocorticotrófico; CRH: hormônio liberador de corticotrofinas.
Figura 4 - Comparação do eixo hipotalâmico-hipofisário-adrenal em pacientes com síndrome de Cushing dependente do ACTH e em pacientes com a síndrome de pseudo-Cushing. Curiosamente, a secreção excessiva de cortisol inibe a secreção do CRH e do ACTH e, dessa forma, retém o hipocortisolismo da síndrome de pseudo-Cushing.
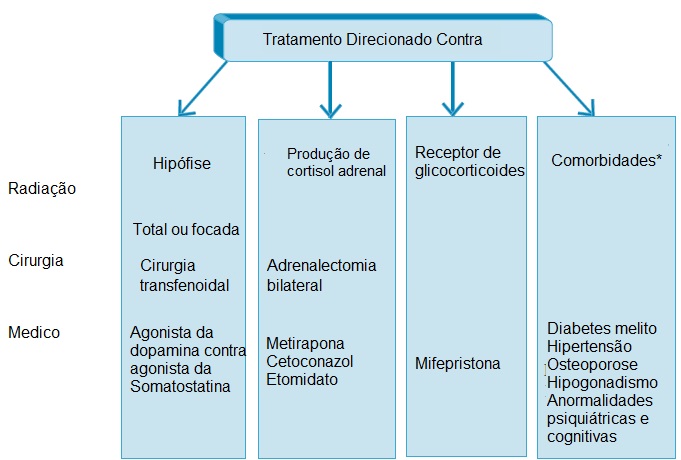
Figura 5 - Tratamentos de segunda linha para a síndrome de Cushing nas situações em que a cirurgia não for possível, a cirurgia for falha e houver recorrência.
A adrenalectomia bilateral tem a vantagem de produzir remissão imediata. Talvez seja a melhor escolha em mulheres jovens que desejarem fertilidade futura (evitando hipopituitarismo depois de radiação) ou em pacientes portadores de doença metastática ou rosada.36 Além do risco cirúrgico, há necessidade de terapia de reposição hormonal por toda a vida.
Entretanto, uma metanálise de grande porte constatou que 46% dos pacientes morreram dentro do período de 1 ano após a adrenalectomia bilateral;37 quase metade dos pacientes apresentou secreção ectópica do ACTH, e muitos dos estudos haviam sido feitos antes que a cirurgia transesfenoidal estivesse disponível. A mortalidade recente para a doença de Cushing foi mais baixa.38?40
Possivelmente, os tumores malignos precisem de tratamento adicional (quimioterapia, radioterapia, embolização) dependendo do tipo de tumor. A confirmação histológica e a consulta oncológica são bastante úteis.
O Quadro 6 apresenta a terapia médica para a síndrome de Cushing.47?49
Quadro 6
|
TERAPIA MÉDICA PARA A síndrome de Cushing | ||||
|
Tipo de medicamento |
Medicamento* |
Prós |
Contras |
Dose |
|
Inibidores da esteroidogênese |
Cetoconazol |
Início rápido da ação |
Efeitos adversos: GI, discrasia hepática (morte); precisa de ácido para a atividade biológica.
|
400?1.600mg/dia |
|
|
Metirapona |
Início rápido da ação |
Efeitos adversos: GI, hirsutismo, HT; difícil de obter.
|
500mg ? 2g/dia |
|
|
Mitotano |
Adrenolítico; usado no câncer adrenal. |
Ação de início lento: teratogênico, efeitos adversos no trato GI e no SNC.
|
5.008g/dia |
|
|
Etomidato |
Administração intravenosa, início rápido da ação.
|
Precisa de monitoramento. |
Aplicação em bolo e titulada. |
|
Direcionado para a hipófise |
Cabergolina |
Agente oral sem toxicidade hepática. |
Efeitos adversos: astenia, GI, tontura.
|
1?7mg/semana |
|
|
Pasireotida? |
Para pacientes com doença de Cushing que continuarem não respondendo à cirurgia anterior ou que não forem candidatos à cirurgia.
|
Efeitos adversos: diarreia, náusea, colelitíase. |
600?900µg b.i.d. |
|
Direcionado para o receptor de glicocorticoides |
mifepristona |
Para pacientes com diabetes melito tipo 2 ou intolerância à glicose que continuarem não respondendo à cirurgia anterior ou que não forem candidatos à cirurgia. |
Difícil de titular; abortivo. |
300?600mg/dia |
SNC: sistema nervoso central; GI: gastrintestinal; HT: hipertensão.
* O medicamento pasireotida foi aprovado pela Food and Drug Administration (FDA) para tratamento de pacientes com doença de Cushing que não são candidatos cirúrgicos ou cuja cirurgia não tenha sido bem-sucedida. A mifepristona foi aprovada pela FDA para tratamento de hiperglicemia em pacientes com síndrome de Cushing que não são candidatos cirúrgicos ou cuja cirurgia não tenha sido bem-sucedida. O uso de outros agentes listados nesse Quadro representa o uso de medicamentos sem prescrição médica para tratamento da síndrome de Cushing.
? O medicamento pasireotida deve ser administrado por via subcutânea; os outros agentes, exceto o etomidato, são administrados por via oral.
Complicações
Todos os pacientes portadores da síndrome de Cushing correm grande risco de doença cardiovascular e de trombose venosa; embora provavelmente melhorem esses riscos, eles permanecem em até 5 anos e 1 ano após a cura cirúrgica.41,42 Fatores como ganho persistente de peso, hiperlipidemia, hipertensão e diabetes melito provavelmente contribuam para o aumento no risco cardiovascular e, portanto, devem ser tratados de forma agressiva.
Ainda não há consenso sobre a administração de anticoagulação profilática. Os pacientes com síndrome de Cushing ativa, em particular os casos graves, correm grande risco de infecção, incluindo patógenos incomuns geralmente associados com estados imunocomprometidos.32
A osteoporose induzida por glicocorticoides melhora após a cura, embora não normalize completamente; ela deve ser tratada como em pacientes sem síndrome de Cushing. Em comparação com a população em geral, a qualidade de vida é reduzida em pacientes com todas as formas de síndrome de Cushing, antes e depois de muitos anos após a cura, a despeito das melhoras.41
Prognóstico
O índice de mortalidade é elevado em pacientes com síndrome de Cushing ativa, e continua elevado após a cura em alguns estudos, mas não em todos eles.38?40 O prognóstico de pacientes com câncer adrenal não é bom, sendo que o prognóstico de pacientes com secreção ectópica de ACTH depende do tipo de tumor e se é maligno ou não.
Conforme já mencionado, a má qualidade de vida persiste durante o período de remissão, e problemas psicológicos como depressão, apatia, problemas cognitivos, ansiedade e fobia social são muito comuns nessa população de pacientes, em comparação com os indivíduos de controle.43
Alguns estudos fazem referência ao prognóstico de longo prazo em pacientes que foram curados da síndrome de Cushing; a ausência desses dados, assim como a atenção especial ao tratamento de comorbidades para neutralizar seus efeitos danosos, é extremamente importante.44
Referências
1. Newell-Price J, Trainer P, Besser M, et al. The diagnosis and differential diagnosis of Cushing syndrome and pseudo-Cushing states. Endocr Rev 1998;19:647–72.
2. Steffensen C, Bak AM, Rubeck KZ, et al. Epidemiology of Cushing syndrome. Neuroendocrinology 2010;92 Suppl 1:1–5.
3. Valassi E, Santos A, Yaneva M, and ERCUSYN Study Group. The European Registry on Cushing syndrome: 2-year experience. Baseline demographic and clinical characteristics. Eur J Endocrinol 2011;165:383–92.
4. Garritano S, Gemignani F, Palmero EI, et al. Detailed haplotype analysis at the TP53 locus in p.R337H mutation carriers in the population of Southern Brazil: evidence for a founder effect. Hum Mutat 2010;31:143–50.
5. Shizume K, Harada Y, Ibayashi H, et al. Survey studies on pituitary diseases in Japan. Endocrinol Jpn 1977;24:139–47.
6. Lacroix A, Feelders R, Stratakis CS, et al. Cushing syndrome. Lancet 2014. [In press]
7. Lerario AM, Moraitis A, Hammer GD. Genetics and epigenetics of adrenocortical tumors. Mol Cell Endocrinol 2014;386:67–84.
8. Alexandraki KI, Grossman AB. The ectopic ACTH syndrome. Rev Endocr Metab Disord 2010;11:117–26.
9. Louiset E, Duparc C, Young J, et al. Intraadrenal production of corticotropin in maronodular bilateral adrenal hyperplasia causing Cushing syndrome. N Engl J Med 2013;369:2115–25.
10. Boscaro M, Arnaldi G. Approach to the patient with possible Cushing syndrome. J Clin Endocrinol Metab 2009;94:3121–31.
11. Schneider HJ, Dimopoulou C, Stalla GK, et al. Discriminatory value of signs and symptoms in Cushing syndrome revisited: what has changed in 30 years? Clin Endocrinol (Oxf) 2013;78:153–4.
12. Nieman LK, Biller BM, Findling JW, et al. The diagnosis of Cushing syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2008;93:1526–40.
13. Elamin MB, Murad MH, Mullan R, et al. Accuracy of diagnostic tests for Cushing syndrome: a systematic review and metaanalyses. J Clin Endocrinol Metab 2008;93:1553–62.
14. Martin NM, Dhillo WS, Banerjee A, et al. Comparison of the dexamethasone-suppressed corticotropin-releasing hormone test and low-dose dexamethasone suppression test in the diagnosis of Cushing syndrome. J Clin Endocrinol Metab 2006;91:2582–6.
15. Flockhart DA. Drug interactions: cytochrome P450 drug interaction table. Indiana University School of Medicine (2007). Available at: http://medicine.iupui.edu/clinpharm/ddis/clinical-table/ (accessed March 10, 2014).
16. Valassi E, Swearingen B, Lee H, et al. Concomitant medication use can confound interpretation of the combined dexamethasone-corticotropin releasing hormone test in Cushing syndrome. J Clin Endocrinol Metab 2009;94:4851–9.
17. Manetti L, Rossi G, Grasso L, et al. Usefulness of salivary cortisol in the diagnosis of hypercortisolism: comparison with serum and urinary cortisol. Eur J Endocrinol 2013;168:315–21.
18. Raff H. Cushing syndrome: diagnosis and surveillance using salivary cortisol. Pituitary 2012;15:64–70.
19. Liu H, Bravata DM, Cabaccan J, et al. Elevated late-night salivary cortisol levels in elderly male type 2 diabetic veterans. Clin Endocrinol (Oxf) 2005;63:642–9.
20. Alexandraki KI, Grossman AB. Is urinary free cortisol of value in the diagnosis of Cushing syndrome? Curr Opin Endocrinol Diabetes Obes 2011;18:259–63.
21. Hamrahian AH, Ioachimescu AG, Remer EM, et al. Clinical utility of noncontrast computed tomography attenuation value (Hounsfield units) to differentiate adrenal adenomas/hyperplasias from nonadenomas: Cleveland Clinic experience. J Clin Endocrinol Metab 2005;90:871–7.
22. Young WF Jr. Clinical practice. The incidentally discovered adrenal mass. N Engl J Med 2007;356:601–10.
23. Chiodini I. Clinical review: Diagnosis and treatment of subclinical hypercortisolism. J Clin Endocrinol Metab 2011;96:1223–36.
24. Eller-Vainicher C, Morelli V, Salcuni AS, et al. Accuracy of several parameters of hypothalamic-pituitary-adrenal axis activity in predicting before surgery the metabolic effects of the removal of an adrenal incidentaloma. Eur J Endocrinol 2010;163:925–35.
25. Kola B, Grossman AB. Dynamic testing in Cushing syndrome. Pituitary 2008;11:155–62.
26. Aron DC, Raff H, Findling JW. Effectiveness versus efficacy: the limited value in clinical practice of high dose dexamethasone suppression testing in the differential diagnosis of adrenocorticotropin-dependent Cushing syndrome. J Clin Endocrinol Metab 1997;82:1780–5.
27. Wind JJ, Lonser RR, Nieman LK, et al. The lateralization accuracy of inferior petrosal sinus sampling in 501 patients with Cushing disease. J Clin Endocrinol Metab 2013;98:2285–93.
28. Mulligan GB, Eray E, Faiman C, et al. Reduction of false-negative results in inferior petrosal sinus sampling with simultaneous prolactin and corticotropin measurement. Endocr Pract 2011;17:33–40.
29. Lad SP, Patil CG, Laws ER Jr, et al. The role of inferior petrosal sinus sampling in the diagnostic localization of Cushing disease. Neurosurg Focus 2007;23:E2.
30. Hall WA, Luciano MG, Doppman JL, et al. Pituitary magnetic resonance imaging in normal human volunteers: occult adenomas in the general population. Ann Intern Med 1994;120:817–20.
31. Bertagna X, Guignat L, Groussin L, et al. Cushing disease. Best Pract Res Clin Endocrinol Metab 2009;23:607–23.
32. Biller BM, Grossman AB, Stewart PM, et al. Treatment of adrenocorticotropin-dependent Cushing syndrome: a consensus statement. J Clin Endocrinol Metab 2008;93:2454–62.
33. Sheth SA, Bourne SK, Tritos NA, et al. Neurosurgical treatment of Cushing disease. Neurosurg Clin N Am 2012;23:639–51.
34. Jagannathan J, Yen CP, Pouratian N, et al. Stereotactic radiosurgery for pituitary adenomas: a comprehensive review of indications, techniques and long-term results using the gamma knife. J Neurooncol 2009;92:345–56.
35. Sattler MG, Vroomen PC, Sluiter WJ, et al. Incidence, causative mechanisms, and anatomic localization of stroke in pituitary adenoma patients treated with postoperative radiation therapy versus surgery alone. Int J Radiat Oncol Biol Phys 2013;87:53–9.
36. Morris LF, Harris RS, Milton DR, et al. Impact and timing of bilateral adrenalectomy for refractory adrenocorticotropic hormone-dependent Cushing syndrome. Surgery 2013;154:1174–83; discussion 1183–4.
37. Ritzel K, Beuschlein F, Mickisch A, et al. Clinical review: Outcome of bilateral adrenalectomy in Cushing syndrome: a systematic review. J Clin Endocrinol Metab 2013;98:3939–48.
38. Ntali G, Asimakopoulou A, Siamatras T, et al. Mortality in Cushing syndrome: systematic analysis of a large series with prolonged follow-up. Eur J Endocrinol 2013;169:715–23.
39. Clayton RN, Raskauskiene D, Reulen RC, et al. Mortality and morbidity in Cushing disease over 50 years in Stoke-on-Trent, UK: audit and meta-analysis of literature. J Clin Endocrinol Metab 2011;96:632–42.
40. Dekkers OM, Horváth-Puhó E, Jørgensen JO, et al. Multisystem morbidity and mortality in Cushing syndrome: a cohort study. J Clin Endocrinol Metab 2013;98:2277–84.
41. Barahona MJ, Resmini E, Sucunza N, et al. Diagnosis of cure in Cushing syndrome: lessons from long-term follow-up. Front Horm Res 2010;38:152–7.
42. van der Pas R, Leebeek FW, Hofland LJ, et al. Hypercoagulability in Cushing syndrome: prevalence, pathogenesis and treatment. Clin Endocrinol (Oxf) 2013;78:481–8.
43. Andela CD, van der Werff SJ, Pannekoek JN, et al. Smaller grey matter volumes in the anterior cingulate cortex and greater cerebellar volumes in patients with long-term remission of Cushing disease: a case-control study. Eur J Endocrinol 2013;169:811–9.
44. Ragnarsson O, Johannsson G. Cushing syndrome: a structured short- and long-term management plan for patients in remission. Eur J Endocrinol 2013;169:R139–52.
45. Starkman MN, Schteingart DE. Neuropsychiatric manifestations of patients with Cushing syndrome. Relationship to cortisol and adrenocorticotropic hormone levels. Arch Intern Med 1981;141:215–9.
46. Pecori Giraldi F, Pivonello R, Ambrogio AG, et al. The dexamethasone-suppressed corticotropin-releasing hormone stimulation test and the desmopressin test to distinguish Cushing syndrome from pseudo-Cushing states. Clin Endocrinol (Oxf) 2007;66:251–7.
47. Feelders RA, Hofland LJ, de Herder WW. Medical treatment of Cushing syndrome: adrenal-blocking drugs and ketoconazole. Neuroendocrinology. 2010;92 Suppl 1:111–5.
48. Fleseriu M, Biller BM, Findling JW, et al; SEISMIC Study Investigators. Mifepristone, a glucocorticoid receptor antagonist, produces clinical and metabolic benefits in patients with Cushing syndrome. J Clin Endocrinol Metab 2012;97:2039–49.
49. Colao A, Petersenn S, Newell-Price J, et al; Pasireotide B2305 Study Group. A 12-month phase 3 study of pasireotide in Cushing disease. N Engl J Med 2012;366:914–24.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.