(Carregando Índice)... (Carregando Índice)... |
Última revisão: 23/05/2018
Comentários de assinantes: 0
|
Artigo original: Wells, M, MD. Clair, W ST, MD. Primary Sjögren Syndrome. SAM [The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2015 Decker Intellectual Properties Inc. All Rights Reserved.] Tradução: Paulo Henrique Machado. Revisão técnica: Dr. Lucas Santos Zambon.
|
E. William St. Clair, MD
Professor de Medicina e Imunologia na Divisão de Reumatologia e Imunologia da Duke University Medical Center (Durham, NC).
Melissa A. Wells, MD
Bolsista na Divisão de Reumatologia e Imunologia da Duke University Medical Center (Durham, NC).
Resumo
O foco principal desta revisão é a categoria primária da síndrome de Sjögren (SS), uma condição inflamatória crônica definida pela presença de olhos secos (ceratoconjuntivite seca) ou boca seca (xerostomia) na ausência de outras doenças reumatológicas. A SS primária também pode apresentar manifestações extraglandulares na forma de doenças pulmonares, renais, gastrintestinais e neurológicas que poderão produzir morbidade significativa e elevar o índice de mortalidade, sendo totalmente distinta de quaisquer outras doenças nos tecidos conjuntivos. Embora a etiologia da SS primária seja desconhecida, estudos de associação genômica ampla continuam a revelar que a suscetibilidade à doença se baseia na predisposição genética; pacientes com SS primária foram identificados com vários poliformismos genéticos do complexo de histocompatibilidade menor que estão estatisticamente associados ao aumento na suscetibilidade à doença. Um estudo recente demonstrou que as células sanguíneas T CD4+de pacientes com SS primária apresentavam padrões diferentes de metilação do DNA, em comparação com controles sadios e revelou que muitos genes envolvidos na ativação de linfócitos e na resposta imune estavam em estado de equilíbrio para transcrição. O tratamento de SS primária envolve principalmente alívio dos sintomas e prevenção das complicações da doença no longo prazo. Embora as terapias biológicas tenham sido estudadas com detalhes, os resultados foram negativos ou inconclusivos até o momento.
|
Síndrome Primária de Sjögren |
Definição da Doença
A síndrome de Sjögren (SS) é uma condição inflamatória crônica que afeta principalmente a função das glândulas lacrimais e salivares resultando na presença de olhos secos e boca seca. Entretanto, a SS pode também apresentar manifestações extraglandulares que causam morbidade significativa e aumentar a mortalidade.
Em 1933, o Dr. Henrik Sjögren, um oftalmologista sueco, descreveu em sua tese de doutoramento, Zur Kenntinis der Keratoconjunctivitis Sicca, casos de 19 mulheres com sintomas de olhos secos e boca seca e alterações patológicas na parte anterior dos olhos e nas glândulas lacrimais e salivares.1 O Dr. Sjögren criou o termo keratoconjunctivitis sicca (KCS) para designar essa condição ocular, distinguindo-a de xeroftalmia causada por hipovitaminose A.
O termo síndrome de Sjögren surgiu mais tarde e passou a ser utilizado em todo o mundo para se referir à tríade da KCS (olhos secos), xerostomia (boca seca) e artrite. Outras denominações na literatura médica para SS incluem: “doença de Gougerot”, “doença de Gougerot- Sjögren”, “doença de Mikulicz” e “epitelite autoimune”.
A SS se divide em duas categorias principais: SS primária e SS secundária. Define-se SS primária como a presença de olhos secos e boca seca na ausência de alguma doença associada nos tecidos conjuntivos. A SS secundária se refere à presença da tríade da KCS ou xerostomia no quadro de outra doença reumatológica, como, por exemplo, artrite reumatoide (AR), lúpus eritematoso sistêmico (LES), esclerose sistêmica ou polimiosite/dermatomiosite.
As manifestações extraglandulares possivelmente estejam associadas à SS primária ou secundária. O padrão de envolvimento extraglandular na SS primária é distinto de outras doenças nos tecidos conjuntivos, embora algumas de suas tipificidades se sobreponham às de AR e LES. Por outro lado, tipicamente, as características extraglandulares da SS secundária seguem as mesmas características associadas à condição reumatológica. O foco desta revisão é abordar a epidemiologia, a fisiopatologia e as características clínicas da SS primária.
Epidemiologia
Na maior parte dos estudos, a prevalência da SS primária varia de 0,2 a 1,6%.2 Esse tipo de doença afeta com mais frequência mulheres na meia idade (pico de incidência na quarta e quinta décadas de vida) com uma proporção de 9:1 entre mulheres e homens. No entanto, a SS primária pode ocorrer durante a infância e também em pessoas idosas. A prevalência da SS secundária em casos de AR, LES e esclerose sistêmica foi calculada em 17,1,3 8 a 20,4,5 e 14%,6 respectivamente.
Etiologia e Genética
Ainda não se conhece a etiologia da SS primária. Os efeitos cumulativos da suscetibilidade genética e das alterações epigenéticas, em combinação com desencadeadores ambientais, provavelmente iniciem a doença durante muitos anos, antes do surgimento das primeiras manifestações clínicas.
Foram feitas várias tentativas de relacionar a SS primária a uma etiologia viral, como, por exemplo, o vírus de Epstein-Barr, o herpes-vírus humano tipo 6 e o vírus linfotrópico de células T humanas tipo 1 (HTVL-1), assim como aos retrovírus. Entretanto, os resultados desses estudos não eram reproduzíveis e, consequentemente, não conseguiram confirmar o papel de qualquer agente infeccioso como deflagrador da doença.
A contribuição genética para a suscetibilidade à SS primária foi descoberta há várias décadas, embora somente agora tenha sido revelada por meio de estudos da associação ampla a genomas. A evidência para predisposição genética surgiu inicialmente nas observações de que a SS primária tinha uma prevalência aproximada de cinco vezes em parentes de primeiro grau de probandos afetados e uma concordância de 20 a 30% da doença em gêmeos monozigóticos.7
Pares de gêmeos com SS primária também compartilham fenótipos clínicos semelhantes. A SS primária, assim como a AR e o LES, aparenta ser um distúrbio poligênico complexo. Os genes que se localizam no lócus do complexo de histocompatibilidade principal (MHC), basicamente os alelos DR e DQ do antígeno leucocitário humano (HLA), estão entre os primeiros loci de suscetibilidade genética que foram estudados.
O Quadro 1 contém os critérios revisados da classificação internacional da SS.
Quadro 1
|
Critérios Revisados da Classificação Internacional da Síndrome de Sjögren |
|
I. Sintomas oculares: resposta positiva a, pelo menos, uma entre as seguintes perguntas: 1. Você teve olhos secos diariamente, de forma persistente e incômoda, por mais de 3 meses? 2. Você tem uma sensação recorrente de areia ou de cascalho nos olhos? 3. Você usa substitutos de lágrimas mais de 3 vezes ao dia? II. Sintomas orais: resposta positiva a, pelo menos, uma entre as seguintes perguntas: 1. Você teve a sensação de boca seca diariamente por mais de 3 meses? 2. Você teve, de forma recorrente ou persistente, glândulas salivares inchadas depois de adulto? 3. Você toma líquidos com frequência para ajudar a engolir alimentos secos? III. Sintomas oculares: ou seja, evidência objetiva de envolvimento ocular definido como resultado positivo em, pelo menos, um entre os seguintes testes: 1. Teste de Schimer I sem anestesia (<5mm em 5min). 2. Pontuação do corante rosa de Bengala ou pontuação de outro corante ocular (>4, de acordo com o sistema de pontuação de van Bijsterveld). IV. Histopatologia: nas glândulas salivares menores (obtenção através de mucosa com aparência normal), sialoadenite linfocitária focal, avaliada por um histopatologista experiente, com pontuação focal >1, definida como o número de focos linfocitários (adjacentes ao ácido mucoso com aparência normal e contendo mais de 50 linfócitos) por 4mm2 de tecido glandular. V. Envolvimento da glândula salivar: evidência objetiva de envolvimento da glândula salivar definido por um resultado positivo para, ao menos, um entre os seguintes testes diagnósticos: 1. Fluxo salivar total não estimulado (<1,5mL em 15min). 2. Sialografia da parótida mostrando a presença de sialectasias difusas (padrão pontilhado, cavitário ou destrutivo), sem evidência de obstrução nos ductos maiores. 3. Cintilografia salivar mostrando absorção tardia, concentração reduzida e/ou excreção tardia do radiomarcador. VI. Autoanticorpos: presença no soro dos seguintes autoanticorpos: 1. Antígenos dos anticorpos Ro (SS-A) ou La (SS-B), ou ambos. Regras revisadas para classificação: Para SS primária Em pacientes sem qualquer doença potencialmente associada à SS primária, poderá ser definida como segue: Presença de 4 entre os seguintes 6 itens, é uma indicação de SS primária, desde que o item IV (Histopatologia) ou VI (Sorologia) seja positivo. Presença de 3 entre os 4 itens dos critérios objetivos (p. ex., itens III, IV, V, VI). O procedimento da árvore classificatória representa um método alternativo válido para classificação, embora o uso mais adequado seja em pesquisas clínicas e epidemiológicas. Para SS secundária Em pacientes sem qualquer doença potencialmente associada (p. ex., outra doença bem definida no tecido conjuntivo), a presença do item I ou do item II mais 2 entre os itens III, IV e V pode ser considerada uma indicação de SS secundária. Critérios de exclusão Tratamento anterior na cabeça e pescoço com radiação Infecção por hepatite C Aids Linfoma pré-existente Sarcoidose DEVH Uso de medicamentos anticolinérgicos (desde que o tempo seja mais curto que 4 vezes a meia-vida do medicamento). |
Aids: síndrome da imunodeficiência adquirida; DEVH: doença do enxerto versus hospedeiro.
Os polimorfismos nos loci DRB1*0301 (DR3) e DRB1*1501 são responsáveis por, aproximadamente, 90% da associação com MHC em pacientes com SS primária.8 Os loci DQB1*0201 e HLA-DQA1*0501 também estão fortemente associados à suscetibilidade à doença. Esses marcadores de classe II do HLA aparentemente estão mais associados aos autoanticorpos relacionados à doença do que a própria doença, sugerindo que esses haplótipos particulares do MHC desempenham papel importante na indução de respostas específicas contra antígenos.9,10
Alguns estudos de associação ampla de genomas de pacientes com SS primária feitos nas populações dos EUA e da Europa identificaram vários polimorfismos genéticos não MHC que estão estatisticamente associados ao aumento na suscetibilidade à doença. Essas variações genéticas são polimorfismos de nucleotídeo único (SNPs) que ocorrem com uma frequência de mais de 1% na população em geral (variantes alélicas comuns).
Os SNPs não MHC estão muito menos associados à suscetibilidade à doença em comparação com os SNPs localizados na região do MHC. Essas variantes de risco não MHC incluem os SNPs nos genes IRF5 e STAT4, que estão envolvidos na via de sinalização do interferon tipo 1; o gene BLK, que está envolvido na sinalização de células B, e o CXCR5, que está envolvido na atração química das células B.11
As diferenças entre indivíduos induzidas pelo ambiente se refletem nas variações do epigenoma. Por exemplo, a metilação do DNA, que é uma modificação epigenética importante, regula a expressão genética por meio da alteração na acessibilidade das regiões reguladoras no interior dos genes na ligação dos fatores de transcrição. Em um estudo recente, as células T CD4+ eram diferentes nos respetivos padrões de metilação do DNA, em comparação com controles saudáveis, e demonstraram que muitos genes envolvidos na ativação de linfócitos e na resposta imune foram estabilizados para transcrição.12
Fisiopatologia e Patogênese
A patogênese da SS primária é o resultado de interações complexas entre o ambiente, a suscetibilidade genética e o sistema imune. Alguns estudos mostraram que o sistema imune na SS primária se caracteriza pela presença de distúrbios reguladores generalizados no braço adaptativo e no braço inato.
O Quadro 2 apresenta a classificação para SS proposta pela American College of Rheumatology (ACR) em 2012, e a Figura 1 contém a patogênese da SS primária.
Quadro 2
|
Classificação para a Síndrome de Sjögren Proposta pela American College Of Rheumatology em 2012 |
|
Os pacientes com sintomas que sugerem SS atendem aos critérios se apresentam, pelo menos, duas entre as três características descritas a seguir: Anti-SS-A/Ro e/ou anti-SS-B/La soropositivo ou (fator reumatoide positivo e titulação do AAN =1:320). Biópsia da glândula salivar labial mostrando sialadenite linfocitária focal com pontuação de foco =1 foco/4mm2. Ceratoconjuntivite seca com pontuação de coloração ocular =3 (presumindo-se que o indivíduo não esteja usando colírio para glaucoma e não tenha feito nenhuma cirurgia na córnea ou cirurgia cosmética na pálpebra nos últimos 5 anos).
Critérios de exclusão: Histórico de terapia com radiação na cabeça e pescoço Infecção por hepatite C Aids Sarcoidose Amiloidose DEVH Doença relacionada à IgG4 |
Adaptação de Shiboski SC et al.26
AAN: anticorpo antinuclear; Aids: síndrome da imunodeficiência adquirida; DEVH: doença do enxerto versus hospedeiro; SS: síndrome de Sjögren.
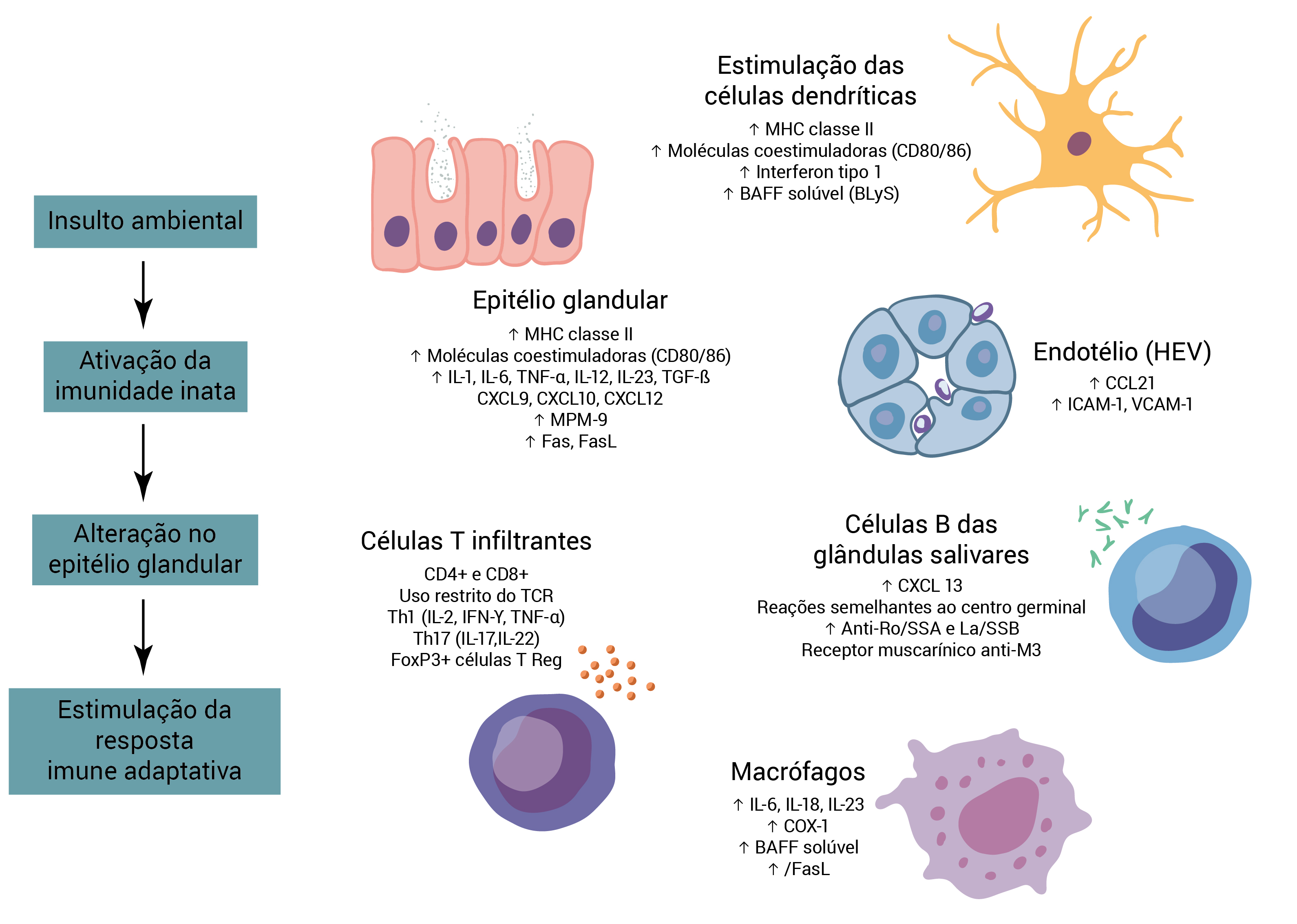
COX-1: cicloxigenase-1; ICAM-1: molécula de adesão intercelular 1; IFN-?: interferon y; MPM: metaloproteinases matriciais; TCR: receptor de células T; TGF-ß: fator de crescimento transformador ß; T Reg: regulador T; VCAM-1: molécula de adesão celular vascular-1.

Figura 1 - Patogênese da SS primária. Postula-se que as vias que iniciam a inflamação nas glândulas salivares sejam insultos ambientais como as infecções virais. Esses fatores ambientais disparam imunidade inata e determinam o estágio das inflamações glandulares persistentes. No caso de SS primária, as vias imunes inatas relevantes incluem as células epiteliais ativadas e as células dendríticas. As células epiteliais suprarregulam a expressão do complexo de histocompatibilidade principal (MHC) classe II e as moléculas coestimuladoras na superfície celular, habilitando-as a atuar como células acessórias apresentadoras de antígenos e a estimular a ativação das células T. As células epiteliais também são fontes ricas de citocinas pró-inflamatórias (fator de necrose tumoral [TNF], interleucina-1 [IL-1] e IL-6) e quimiocinas (CXCL9, CXCL10, CXCL12) cuja função é amplificar a resposta inflamatória. O aumento na expressão de Fas e do ligante Fas (FasL) na superfície das células epiteliais também contribui para sua morte, limitando a reserva secretora das glândulas. As células T ativadas percorrem desde a circulação até as vênulas endoteliais altas (HEVs) no interior do microambiente das glândulas salivares, onde estimulam outras células imunes. As células dendríticas plasmacitoides das glândulas salivares produzem quantidades abundantes de interferon tipo 1 e do fator de ativação de células B (BAFF). Em particular, o excesso de produção de BAFF estimula o desenvolvimento de estruturas que se assemelham ao centro germinal, que consiste de agregados de células B circundados por uma bainha de células T. A presença de células T auxiliares tipo 1 (Th1) e de células Th17 cria um estímulo inflamatório constante e ajuda as células B a se diferenciarem em células formadoras de anticorpos. Os macrófagos também segregam fatores pró-inflamatórios que amplificam a resposta inflamatória crônica.
As aberrações na imunidade adaptativa, que é geradora de memória imune, são inferidas pelas associações do MHC, pela presença de autoanticorpos relacionados à doença, por outras anormalidades que refletem uma miríade de distúrbios nas células B e pela importância das células T e B nos tecidos alvos (por exemplo, glândulas lacrimais e salivares).
Embora os deflagradores ambientais permaneçam indefinidos nesse tipo de doença, teoricamente eles poderão agir por meio do disparo de eventos sinalizadores posteriores nos receptores Toll-like da via das células imunes, que, por sua vez, estimulam as vias celulares pró-inflamatórias que fornecem combustível para o processo da doença. Os estímulos ambientais ativam também programas celulares indutores de apoptose (morte celular), que podem gerar autoimunidade, aumentando a exposição dos próprios antígenos ao sistema imune.
Pode-se questionar, posteriormente, que a operação dos loci de suscetibilidade genética não MHC diminui os limiares quantitativos da sinalização imune que estimulam estados de ativação imune crônica e a perda de controle regulador. Considerados conjuntamente, esses eventos conspiram para perpetuar as respostas inflamatórias crônicas.
A maior parte das células que infiltram no tecido gandular é formada por linfócitos T aß CD4+ e linfócitos B. Alguns estudos de biópsias da glândula salivar menor de pacientes com SS primária mostraram que os linfócitos T e B englobam mais de 90% das células mononucleares infiltrantes.13 A população remanescente consiste de números baixos de células T CD8+, células exterminadoras (killer cells) naturais CD56+, macrófagos CD68+ e células dendríticas (CDs).
Nas glândulas salivares, as células T e B têm a tendência de se agregar ao redor de ductos e vasos sanguíneos. Com frequência, esses agregados formam folículos consistindo de agrupamentos de células B circundados por uma bainha de células T CD3+. Nos casos de lesões mais graves, as células T e B se organizam em estruturas que se assemelham aos centros germinais.
As células apresentadoras de antígenos clássicas, em especial DCs e macrófagos, normalmente são encontradas nas proximidades do epitélio ductal, onde produzem hospedeiros de mediadores pró-inflamatórios como a interleucina-6 (IL-6), o fator a de necrose tumoral (TNF-a), IL-12 e IL-18, assim como a citocina anti-inflamatória IL-10.
Na SS primária, a maior parte das células T CD4+ que infiltram nas glândulas salivares menores expressa o antígeno CD45RO, que é o fenótipo das células T de memória, indicando experiência anterior com antígenos. Com embasamento no paradigma clássico de células T auxiliares tipo 1 (helper cells) (Th1) versus células T auxiliares tipo 2 (Th2), aparentemente as células T CD4+ das glândulas salivares produzem basicamente citocinas Th1 (IL-2 e interferon y).14
As glândulas salivares menores de pacientes com SS primária também contêm células T auxiliares tipo 17 (Th17).15 As células Th17 têm esse nome porque produzem a citocina IL-17; muitas células Th17 produzem também interferon y. As glândulas salivares são ricas no fator de crescimento transformador ß (TGF-ß), assim como em IL-6 e IL-23, criando um meio favorável para o desenvolvimento de células Th17.
Além dos efeitos promotores de Th17, o TGF-ß também tem papel importante na geração de células T reguladoras. Entretanto, vem se tornando cada vez mais evidente o fato de que os fenótipos diferentes das células T (por exemplo, Th1, Th2, Th17, células T reguladoras) são indicadores da medição de plasticidade.16 Portanto, nas situações em que as células podem alterar suas identidades de acordo com o ambiente, o paradigma tradicional da célula T auxiliar como modelo conceitual para a patogênese de SS primária pode não ter uma aplicação direta.
As células B também são importantes na fisiopatologia da SS primária. As proporções de subgrupos de células B sanguíneas importantes na SS primária diferem das proporções em controles saudáveis e mostram um aumento relativo no percentual de células B virgens CD27- e uma redução relativa no percentual de células B de memória CD27+.
A SS primária está associada a uma gama variada de autoanticorpos, que são produtos de células plasmáticas derivadas de células B. Entre esses autoanticorpos, os anticorpos anti-SS-A/Ro e anti-SS-B/La são marcadores significativos para diagnóstico e prognóstico da SS primária.
Entre outros autoanticorpos associados à SS primária, estão aqueles direcionados contra o receptor muscarínico (MR). A família de MRs faz a mediação dos efeitos autonômicos pré-gangliônicos e parassimpáticos pós-gangliônicos da acetilcolina nas glândulas salivares, que operam in tandem com o sistema nervoso simpático para controlar o fluxo salivar.
Entre os cinco subtipos de receptor muscarínico (M1R a M3R), o subtipo M3R representa o MR predominante expresso nas glândulas salivares e faz a mediação da maior parte dos efeitos pós-funcionais que estimulam a contração dos músculos lisos e a secreção das glândulas acinares.17 Especula-se que os anticorpos contra o receptor M3R bloqueiam a secreção glandular. Entretanto, a detecção de anticorpos do receptor anti-M3R em pacientes com SS primária produziu resultados variáveis no ensaio,18 de modo que a relevância patogênica desses autoanticorpos não é muito clara.
As células do sistema imune inato, tais como as células dendríticas plasmacitoides (pDCs), macrófagos e células epiteliais ductais, são igualmente importantes na fisiopatologia de SS primária. Embora os espécimes das glândulas salivares menores com SS primária contenham um pequeno percentual de pDCs, elas são as principais produtoras de interferons tipo 1 (interferon a e interferon ß) e das células responsáveis pela assinatura de um interferon tipo 1 robusto (genes cuja expressão é induzida pelo interferon tipo 1).19
As células mononucleares do sangue de pacientes com SS primária também expressam genes induzíveis por interferons.20 As células pDCs expressam TLR7 e TLR9, os principais sensores de ácidos nucleicos estranhos de patógenos virais de RNA e DNA, respectivamente, que desencadeiam inflamações e promovem a liberação de fatores antivirais como os interferons.
Tanto o interferon 1 como o interferon y aumentam a expressão do fator ativador de células B (BAFF), outro regulador crucial de respostas imunes inatas.21 Em modelos animais, o excesso de BAFFs resulta no desenvolvimento de doença autoimune que se assemelha à SS e, em pacientes com SS primária, níveis elevados de BAFFs são detectados nos soro.22
As principais fontes de BAFF nas glândulas salivares são as células mononucleares e as células epiteliais ductais.23 Nas interações com seus receptores, as BAFFs dão suporte à ativação e à sobrevida das células B e contribuem para o início e resolução de reações no centro germinal, para troca de isotipos e para a sobrevida de células plasmáticas. Consequentemente, pode-se postular que o círculo vicioso de estimulação do receptor Toll-like, produção excessiva de interferon tipo 1 e expressão suprarregulada do BAFF reforça, pelo menos, alguns dos distúrbios locais e sistêmicos das células B nesse tipo de doença.
Várias linhas de evidências mostram que as células epiteliais glandulares têm um papel central na estimulação de respostas inflamatórias crônicas nos tecidos alvos. Primeiramente, os infiltrados glandulares de células T têm distribuição periductal, sugerindo que há uma ligação entre o epitélio ductal e a resposta aberrante das células T.
Em segundo lugar, as células epiteliais ductais e acinares do tecido das glândulas salivares de pacientes com SS primária apesentam um fenótipo “ativado”, que poderá mediar o alojamento de células mononucleares, a apresentação de antígenos e a ampliação da imunidade adaptativa. Nas glândulas salivares inflamadas, as células epiteliais expressam níveis elevados de proteínas classes I e II no MHC, as moléculas co-estimuladoras CD80 e CD86 e as moléculas de adesão intercelular tipo 1 (ICAM-1), moléculas de adesão celular vascular tipo 1 (VCAM-1) e a seletina-E.23
Finalmente, nos casos de SS primária, as células epiteliais ductais dos tecidos de glândulas salivares inflamadas produzem citocinas pró-inflamatórias (IL-1, IL-6, IL-12 e o fator a de necrose tumoral [TNF-a], quimiocinas que atraem células T, quimiocinas que foram centros germinais e quimiocinas que atraem pDC, assim como o fator ativador de células B.23 Essas informações mostram porque a SS primária foi denominada “epitelite autoimune”.
Órgãos parenquimatosos como pulmões e rins também poderão ser afetados nos casos de SS primária. Nos pulmões, a histopatologia se assemelha à das glândulas salivares labiais, em especial a presença de infiltrados linfocitários peribronquiais e intersticiais.
O envolvimento renal se caracteriza pela presença de infiltrados linfocitários ao redor do epitélio tubular, que são responsáveis pela disfunção tubular observada com frequência em pacientes com esse tipo de doença. O envolvimento de outros sistemas de órgãos possivelmente surja a partir da deposição de complexos imunes na pele (púrpura), nas artérias menores que suprem os nervos periféricos (neuropatia periférica) e nos rins (glomerulonefrite [GN]).
Diagnóstico
O diagnóstico de SS primária se baseia na presença de olhos secos (KCS), xerostomia (boca seca), anticorpos anti-Ro/SS-A e anti-La/S-B e evidências histopatológicas de sialadenite crônica. O European Study Group estabeleceu inicialmente um grupo de critérios preliminares para classificação24 que foi posteriormente revisado pelo grupo de consenso Americano-Europeu.25
A presença de quatro entre os seis critérios foi considerada indicativa do diagnóstico de SS primária, com sensibilidade de 89,5% e especificidade de 95,2%. Mais recentemente, foram publicados os critérios preliminares do ACR para classificação da SS que, essencialmente, identificam os mesmos grupos de pacientes que os critérios revisados do American-European Consensus Group.26
Em termos práticos, o diagnóstico da maior parte dos pacientes com SS primária se fundamenta na presença dos sintomas de olhos secos, teste 1 de Schirmer e/ou coloração ocular anormal, sintomas de boca seca, fluxo salivar anormalmente baixo e biópsia positiva das glândulas salivares labiais ou anticorpos séricos anti-Ro/SS-A e/ou anti-La/SS-B.
Manifestações Clínicas
Doença da Glândula Exócrina
Envolvimento ocular. O fenômeno conhecido por olhos secos resulta de uma redução na produção de lágrimas aquosas nas glândulas lacrimais, causando danos no epitélio conjuntival e bulbar. Os pacientes possivelmente não se importam com a secura ocular propriamente dita, mas quase sempre se queixam de arenosidade ou da sensação da presença de algum corpo estranho, em combinação com outros sintomas de irritação, queimação, acúmulo de muco espesso nos cantos internos dos olhos (filamentos mucosos), fotofobia e fadiga ocular.
Com frequência, ocorre uma redução na produção de lágrimas que poderá ser medida pelo teste 1 de Schirmer. Tipicamente, o exame com lâmpada fendida revela uma redução nas lágrimas no saco conjuntival, vasos conjuntivais bulbares dilatados com injeção, filamentos mucosos e danos nas córneas confirmados pela coloração verde com lissamina (para coloração de células desvitalizadas) ou fluoresceína (para coloração de defeitos epiteliais). Essa condição se denomina KCS e, de um modo geral, está associada à blefarite (infecções nas pálpebras). Os casos graves poderão produzir abrasões e ulcerações corneais.
O diagnóstico diferencial para olhos secos poderá ser mais bem compreendido por meio da análise da composição das lágrimas. Qualquer função inadequada das pálpebras, anormalidade na anatomia do filme lacrimal ou redução no volume das lágrimas pode resultar em olhos secos e irritação ocular. O filme lacrimal se compõe de três camadas: camada lipídica externa, camada aquosa intermediária e camada mucínica interna.
As lágrimas aquosas são produzidas pelas glândulas lacrimais. A deficiência nessa camada é responsável pelos olhos secos da SS primária. Outras condições que podem afetar a camada aquosa incluem medicações como antidepressivos tricíclicos, anti-histamínicos e diuréticos. Fatores como envelhecimento e alterações hormonais também podem estar associados à redução na camada de lágrimas aquosas.
A camada lipídica e a camada mucínica estabilizam e distribuem as lágrimas com uniformidade sobre a superfície ocular. A camada mucínica tem origem nas células caliciformes da conjuntiva, em que, caso sejam danificadas, possivelmente ocorra distribuição irregular das lágrimas sobre a superfície dos olhos. Dois exemplos típicos de distúrbios associados à produção mucínica anormal são: deficiência de vitamina A e síndrome de Stevens-Johnson.
A camada lipídica é um produto das glândulas meibomianas e tem a função de proteger o filme de lágrimas contra evaporação rápida. A disfunção das glândulas meibomianas causada por blefarite poderá reduzir a camada lipídica, resultando na decomposição lacrimal. Outras causas de camadas lipídicas anormais incluem dermatite rosácea, seborreica e tabagismo.
Envolvimento orofaríngeo: xerostomia. A orofaringe é afetada de maneira uniforme nos casos de SS primária por alterações na quantidade e qualidade da secreção das glândulas salivares. Os sintomas de boca seca são comuns na população em geral, embora tenham a tendência de ser brandos e, com bastante frequência, são explicados pelo uso de medicações.
Entretanto, os pacientes com SS primária apresentam sintomas mais graves como queimação oral, deglutição difícil de alimentos secos (por exemplo, bolachas), alteração no paladar (metálico, salgado ou amargo), dificuldade para falar em períodos prolongados de tempo e problemas para usar dentaduras. De um modo geral, o exame revela a presença de mucosa oral seca, eritematosa e viscosa, embora a cavidade oral pareça normal em muitos casos.
Há duas consequências importantes da xerostomia grave. Em primeiro lugar, poderá produzir cáries dentárias incontroláveis, dentes rachados e obturações soltas. Em segundo lugar, poderá aumentar o risco de candidíase oral, fato que foi descrito em um estudo, com probabilidade de ocorrer em 74% de pacientes com SS primária.27
Tipicamente, a candidíase oral se manifesta como queilite angular (lesões secas e com rachaduras nos cantos da boca), eritema e atrofia das papilas filiformes da língua, e um exsudato branco fino na língua. Em geral, não se observa a aparência de “cândida” da candidíase oral nesse contexto.
Aproximadamente, um terço dos pacientes com SS primária desenvolve aumento de volume nas glândulas parótidas e submandibulares ao longo do curso da doença. Nos casos crônicos, geralmente, o inchaço é indolor, firme e difuso, ao passo que os episódios agudos de inchaço são transitórios, doloridos e sensíveis e, provavelmente, são atribuíveis à obstrução nos ductos das glândulas salivares por muco inspissado. O aumento no volume das glândulas assimétricas, que poderá ser rígido ou nodular, indica a presença de neoplasmas, como, por exemplo, um linfoma.
Outros envolvimentos das glândulas exócrinas. Praticamente, qualquer glândula exócrina do corpo poderá ser afetada nos casos de SS primária. Em particular, a secura poderá afetar também as passagens nasais, a vagina e a pele. As mulheres com secura vaginal geralmente se queixam de dispareunia, prurido ou irritação. A secura na garganta e na traqueia (xerotraqueia) poderá produzir tosse crônica.
Manifestações Extraglandulares
Fadiga. A presença de fadiga está entre as manifestações extraglandulares mais comuns de SS primária.28 Em um estudo norte-americano de uma coorte de SS primária, a fadiga foi o preditor dominante da saúde geral e da função física.29 Provavelmente, a causa da fadiga seja multifatorial e possa ser disparada por alguma disfunção imunológica ou por fatores como transtorno do sono e depressão.
Em um estudo de 94 indivíduos que atendiam aos critérios do American-European Consensus Group para SS primária, os fatores psicológicos foram responsáveis por apenas 62% da variabilidade na Fatigue Severity Scale.30 Embora a depressão se correlacione com a gravidade da fadiga, ela não foi a causa principal de fadiga nos casos de SS primária. A fadiga teve impacto significativo sobre o estado funcional dos pacientes com SS primária. Na maior parte dos casos, esse sintoma precisa de intervenção médica e pode exigir a aplicação de abordagens multidisciplinares.31
Fenômeno de Raynaud. Até 30% de indivíduos com SS primária têm o fenômeno de Raynaud. Aparentemente, os ataques de Raynaud são mais brandos em pacientes com SS primária, em comparação com outras doenças nos tecidos conjuntivos, como, por exemplo, esclerose sistêmica. Esses ataques ocorrem com mais frequência em pacientes com outras manifestações extraglandulares como poliartrite e vasculite cutânea, assim como na forma de teste positivo para os anticorpos séricos anti-Ro/SS-A e anti-La/SS-B.
Manifestações cutâneas. Xerose (pele seca) é a manifestação cutânea mais frequente de SS primária que, geralmente, produz prurido e queimação. Provavelmente, a xerose seja secundária a modificações na função protetora da epiderme causadas por alterações na função exócrina das glândulas sudoríparas e sebáceas. Eritema anular também é uma manifestação cutânea comum de SS primária observada com mais frequência em pacientes com anti-Ro/SS-A.
Ocasionalmente, os pacientes com SS primária poderão desenvolver lesões causadas por lúpus subagudo, que também foram associadas com a presença de anticorpos anti-Ro/SS-A e, não necessariamente, alteram o diagnóstico de LES.
Os pacientes com SS primária também poderão desenvolver púrpura causada por uma condição conhecida por “púrpura hipergamaglobulinêmica de Waldenström” ou por vasculite em vasos menores. Na realidade, aproximadamente 50% de pacientes com púrpura hipergamaglobulinêmica de Waldenström benigna secundária têm SS primária.32 Essa condição se apresenta com púrpura não palpável simétrica que se estende até a perna e sintomas de coceira e queimação nas proximidades das lesões. Em alguns casos, ela foi associada à neuropatia periférica sensorial.
A vasculite de vasos menores (por exemplo, vasculite leucocitoclástica) também é relativamente comum na SS primária e se manifesta como púrpura palpável ou, com menos frequência, como lesões urticárias doloridas. Tipicamente, as lesões urticárias produzidas por vasculite duram entre 3 a 4 dias, deixando feridas ou descolorações no local, fato que distingue esse tipo de lesão de urticária idiopática ou alérgica.
Manifestações musculoesqueléticas. Artralgia e artrite estão entre as queixas mais comuns de manifestações extraglandulares da SS primária. Os sintomas articulares precedem os sintomas de sicca em até um terço de casos. O envolvimento poliarticular e a distribuição simétrica da artrite se assemelham aos de LES e de AR.
Ao contrário da AR, a poliartrite tende a ser menos grave e intermitente e, tipicamente, não produz erosões articulares nas radiografias. Os sítios mais comuns de envolvimento articular são as articulações interfalângicas proximais, as articulações metacarpofalângicas e os punhos, em 33, 27 e 12% de casos, respectivamente.33
Mialgia, que lembra fibromialgia, é uma queixa comum entre indivíduos com SS primária. Embora, em muitos casos, as biópsias musculares mostrem evidências de inflamações leves, um número relativamente pequeno de pacientes desenvolve um quadro clínico de polimiosite com fraqueza muscular.34
Manifestações pulmonares. Sob o ponto de vista clínico, o envolvimento significativo dos pulmões ocorre em 9 a 24% de indivíduos com SS primária, embora 75% desses pacientes apresentem anormalidades nos testes da função pulmonar (TFPs) e nas varreduras por tomografia computadorizada (TC) no tórax, assim como na composição do líquido do lavado broncoalveolar (LBA).35 O reconhecimento do envolvimento pulmonar é muito importante porque está associado ao aumento de quatro vezes na mortalidade depois de 10 anos com a doença.
A SS primária envolve o trato respiratório superior e o inferior. As manifestações mais frequentes na via respiratória superior estão relacionadas à hipersecreção glandular e incluem epistaxe, sinusite recorrente, formação de crostas nasais, rouquidão e tosse irritante e seca. O envolvimento do trato respiratório inferior ocorre de várias formas. Talvez a forma mais comum seja um leve envolvimento da via respiratória menor em que os pacientes têm tosse seca e falta de ar.36
Os tipos de doença pulmonar intersticial (DPI) na SS primária incluem pneumonite intersticial linfoide (PIL), pneumonia intersticial inespecífica (PII), pneumonia intersticial usual (PIU), pneumonia em organização (PO) e doença pulmonar cística.35 A PII é a mais comum e seu prognóstico depende da extensão da fibrose. Normalmente, a PIU não responde tão bem quanto a PII ao tratamento com glicocorticoides e outros agentes imunossupressivos.
Tipicamente, a PO é tratada com sucesso com glicocorticoides. Ito e colaboradores estudaram 33 pacientes com SS primária e com doença pulmonar confirmada por biópsia do pulmão.37 Vinte pacientes (61%) tinham PII (1 celular, 19 fibrosantes), quatro tinham o tipo de linfoma não Hodgkin (LNH) no tecido linfoide associado à mucosa (MALT), quatro tinham bronquiolite difusa e dois tinham doença mailoide.
O índice de sobrevida depois de 5 anos foi 83% para os indivíduos com PII. A pneumonite linfocitária é o resultado da proliferação de tecido linfoide associado aos brônquios (TLAB). Seu curso clínico é bastante variável, desde remissão sem tratamento a insuficiência respiratória e morte. Em, aproximadamente, 5% de indivíduos, as lesões por TLAB evoluem para linfoma.38
Outras manifestações de doença pulmonar na SS primária incluem hipertensão arterial pulmonar (HAP). HAP é uma complicação séria e potencialmente fatal da SS primária. Os pacientes podem ser assintomáticos ou apresentar sintomas leves.39 Tipicamente, a tomografia computadorizada por emissão de pósitrons (PET) e a ecocardiografia transtorácica (ETT) são usadas nos procedimentos de rastreamento.
O Quadro 3 contém as manifestações no trato respiratório em SS primária.
Quadro 3
|
Manifestações no Trato Respiratório em Síndrome de Sjögren Primária |
|
Via respiratória superior Sintomas: formação de crostas nasais, epistaxe, sinusite crônica, rouquidão, tosse seca. Causa: envolvimento das glândulas exócrinas que revestem o trato respiratório superior. Prevalência: variável; entretanto, formação de crostas nasais e epistaxe são as mais comuns. Doença distal nas vias respiratórias Bronquite folicular Sintomas: semelhantes aos sintomas de asma (tosse, respiração ofegante, dispneia). Prevalência: comum. Causa: folículos linfoides hiperplásicos com centros germinais reativos encontrados ao longo dos feixes bronquiovasculares. Avaliação: os TFPs mostram a presença de doença restritiva com DLCO reduzida. Prognóstico: bom. DPI Existem vários tipos de DPI que produzem sintomas de SS primária. Causa mais comum de DPI: PII. Outros tipos de DPI: pneumonia intersticial linfocitária (PIL), pneumonia intersticial usual (PIU), PO, doença pulmonar cística. A avaliação deve incluir TFPs e TC torácica. Após a suspeita de DPI, recomenda-se encaminhar o paciente para um pneumologista. O tipo de DPI subjacente tem prognóstico importante e implicações no tratamento. Linfoma pulmonar Muito raro Amiloidose pulmonar Muito rara Hipertensão na artéria pulmonar Complicação séria de doença e potencialmente fatal. Considerar o diagnóstico se o paciente tiver dispneia com esforço físico, ortopneia ou dispneia noturna paroxísmica. A avaliação inicial consiste de TFP e ETT. Considerar o encaminhamento para um especialista em artérias pulmonares se a DLCO for reduzida no TFP ou a PVDS for elevada no ETT. |
Adaptação de Stojan et al.35
DLCO: capacidade difusora do dióxido de carbono; DPI: doença pulmonar intersticial; ETT: ecocardiograma transtorácico; PII: pneumonia intersticial inespecífica; PO: pneumonia em organização; PVDS = pressão ventricular direita sistólica; TFP: teste da função pulmonar; TC: tomografia computadorizada; SS: síndrome de Sjögren.
Manifestações renais. A presença de doença renal clinicamente significativa não é frequente nos casos de SS primária. Um estudo constatou que até 4,9% de pacientes com SS primária tinham envolvimento renal.40 Condições como nefrite tubular intersticial (NTI), acidose renal tubular, doença glomerular e diabetes insípido nefrogênico estão associadas à SS primária e precedem o início dos sintomas de sicca. Normalmente, a NTI é assintomática e, em casos raros, evolui para doença renal em estágio final.
A GN é mais comum que a NIT. Diferentes formas de GN foram associadas à SS primária, incluindo GN membranoproliferativa, GN mesangial proliferativa, GN foca crescente e nefropatia membranosa.41?44 As crioglobulinas e níveis baixos de complementos são mais comuns na GN do que em casos de SS primária em geral. Em uma série, crioglobulinemia monoclonal mista e idade em faixas etárias mais elevadas foram dois preditores independentes de GN.41
Manifestações gastrintestinais. O trato gastrintestinal pode ser afetado desde a boca, o intestino delgado e o intestino grosso até o fígado e o pâncreas. Os sintomas mais comuns incluem disfagia, dor epigástrica e náusea. A disfagia é multifatorial e provavelmente esteja relacionada à ausência de saliva e ao ressecamento subsequente da mucosa, falta de motilidade esofágica e redes esofágicas superiores. A presença de gastrite atrófica crônica é comum em indivíduos com SS primária.
Valores anormais das enzimas hepáticas são relativamente comuns em pacientes com SS primária, sendo que foram encontradas em 49,1% de casos em um estudo.45 Embora a doença hepática autoimune possivelmente ocorra em associação com a SS, os valores anormais das enzimas do fígado nunca chegaram a ser explicados na maior parte dos casos.
Manifestações neurológicas. Condições como neuropatia periférica, envolvimento de nervos cranianos e, com menor frequência, doença no sistema nervoso central (SNC) ocorrem na SS primária. Neuropatia periférica, a forma mais comum de envolvimento, se apresenta como neuropatia atáxica sensorial, neuropatia sensorial dolorida sem ataxia sensorial, mononeuropatias múltiplas, neuropatia craniana múltipla, neuropatia do trigêmeo, neuropatia autonômica ou radiculoneuropatia.46
A prevalência de doença no SNC é discutível e varia amplamente nos estudos, desde não existente a comum. De acordo com esses estudos, o espectro da doença pode afetar o cérebro, a medula espinal e os nervos cranianos. A doença no SNC sutil cria problemas de memória e transtornos cognitivos. Outros sinais e sintomas de doença no SNC incluem hemiparesia, disartria, ataxia, convulsões, distúrbios de movimento como distonia, meningite asséptica, encefalopatia, demência, ansiedade e depressão.47 Ataques agudos e transitórios imitando esclerose múltipla também foram descritos em alguns casos.48
Manifestações reprodutivas. As informações sobre a gravidez de mulheres com SS primária são muito limitadas. Um estudo de pequeno porte de 34 gestações e SS primária constatou que houve um ligeiro aumento no risco de aborto espontâneo (índice de 30%) em comparação com a população em geral.49 A presença de anti-Ro/SS-A aumenta o risco de lactentes com bloqueio cardíaco fetal nas mulheres em idade reprodutiva.
Em um estudo de pequeno porte, a maior parte das gestações foi saudável; entretanto, 5% dos nascimentos tinham bloqueio cardíaco congênito.50 No mesmo estudo, 10% dos pacientes tiveram crises no primeiro ano após o nascimento. As mulheres grávidas com SS primária ou que estiverem planejando ter filhos, em especial as pacientes com anticorpos anti-Ro/SS-A e/ou anti-La/SS-B, devem ser encaminhadas para um obstetra com experiência em gestações de alto risco.
Vasculite sistêmica. Vasculite sistêmica (excetuando-se os casos de púrpura) aparentemente ocorre em casos raros no contexto de SS primária. Os casos que afetam predominantemente os vasos menores foram descritos no ambiente de crioglobulinemia (sem evidências de infecção pelo vírus da hepatite C [VHC]), sendo que o envolvimento dos vasos de porte médio provavelmente leve a uma apresentação clínica semelhante à poliarterite nodosa.
Doenças Associadas
Doença tireóidea. A presença de disfunção tireóidea é comum entre indivíduos com SS primária e entre parentes de primeiro grau.51 Os estudos iniciais sugeriam uma forte correlação entre SS primária e doença tireóidea. Por exemplo, um dos estudos registrou uma prevalência de 30% de doença tireóidea em pacientes com SS primária versus 4% no grupo de controle.52
Um estudo recente mais amplo comparou a prevalência de doença tireóidea em 160 pacientes com controles de 75 anos de idade e com controles na mesma faixa etária. Embora a doença tireóidea tenha sido mais comum sob o ponto de vista numérico em pacientes com SS primária, em comparação com os controles (36% versus 27%), a diferença total entre os dois grupos não chegou a ser estatisticamente significativa.53
Doença hepática autoimune. Os pacientes com SS primária podem ter cirrose biliar primária (CBP) coexistente, hepatite autoimune (HAI), colangite esclerosante ou hiperplasia nodular regenerativa.45 CPB e HAI ocorrem com mais frequência em pacientes com SS primária. Embora sua prevalência exata não seja conhecida, estima-se que fique ao redor de 5%.54
Doença celíaca. A doença celíaca também foi descrita em pacientes com SS primária. Um estudo húngaro encontrou uma frequência 10 vezes mais elevada de doença celíaca em pacientes com SS primária em comparação com os controles.55
Linfoma. LNH é uma complicação importante da SS primária. Inicialmente, a frequência de LNH foi 44 vezes maior em pacientes com esse tipo de doença em comparação com a população em geral.56 Uma prevalência estimada de 4,3% foi registrada em um estudo populacional posterior, em que o tempo mediano entre o diagnóstico de SS primária até o desenvolvimento do linfoma foi de 7,5 anos.57
Há registros de diversas variações histológicas de LNH, incluindo linfoma de células foliculares, linfoma de células B grandes e difusas, linfoma linfoplasmacitoide e linfoma de células B na zona marginal. Os linfomas de células B na zona marginal, incluindo o linfoma MALT, são os tipos mais comuns na SS primária.57,58
Embora, geralmente, se desenvolvam nas glândulas salivares, os linfomas MALT podem surgir a partir de outros sítios extranodais, como, por exemplo, estômago, nasofaringe, pele, fígado, rins e pulmões. A presença de linfadenopatia e/ou glândulas parótidas inchadas deve levantar a suspeita de linfoma.
Neutropenia, crioglobulinemia, esplenomegalia, linfadenopatia, componente M no soro ou na urina, nível elevado de microglobulina-ß2, neuropatia periférica e nível baixo de C4 estão entre os fatores de risco para o desenvolvimento de linfoma;59 a presença de qualquer um desses fatores representa um aumento de cinco vezes no risco de linfoma, em comparação com indivíduos sem fatores de risco.58 O grau histológico (intermediário ou elevado) do linfoma, o tamanho do tumor (>7cm) e a presença de sintomas B (febre, suor noturno e perda de peso) são determinantes de aumento na mortalidade.57
Exame Físico
O exame físico deve ser bastante amplo, com atenção especial nos olhos e na mucosa oral, glândulas lacrimais e salivares e outros tecidos com glândulas exócrinas como pele, mucosa nasal e vagina, levando-se em consideração que a SS primária é uma doença multissistêmica. O aumento no volume das glândulas lacrimais não é uma característica usual da SS primária.
Na sua presença, deve-se considerar um diagnóstico alternativo como sarcoidose, doença relacionada à IgG4 (IgG4-RD) ou linfoma. A despeito da redução na secreção de saliva, a mucosa oral não tem aparência anormal, a menos que o defeito secretor seja grave e produza uma aparência opaca nas membranas mucosas (em oposição à aparência brilhante) com ausência de um pool salivar sublingual. Os dentes podem apresentar sinais de perda de esmalte (descoloração), quebras e cavidades.
A descoberta de queilite angular, membranas mucosas avermelhadas, revestimento da língua ou atrofia das papilas da língua levanta suspeitas de candidíase oral. O exame da cabeça e do pescoço é importante para detectar qualquer aumento nas glândulas parótidas e submandibulares, assim como qualquer aumento nos linfonodos cervicais. Tipicamente, o volume da glândula parótida aumenta e diminui ao longo do curso da doença. Entretanto, o inchaço nessa glândula durante muitos meses deve levantar a suspeita de linfoma.
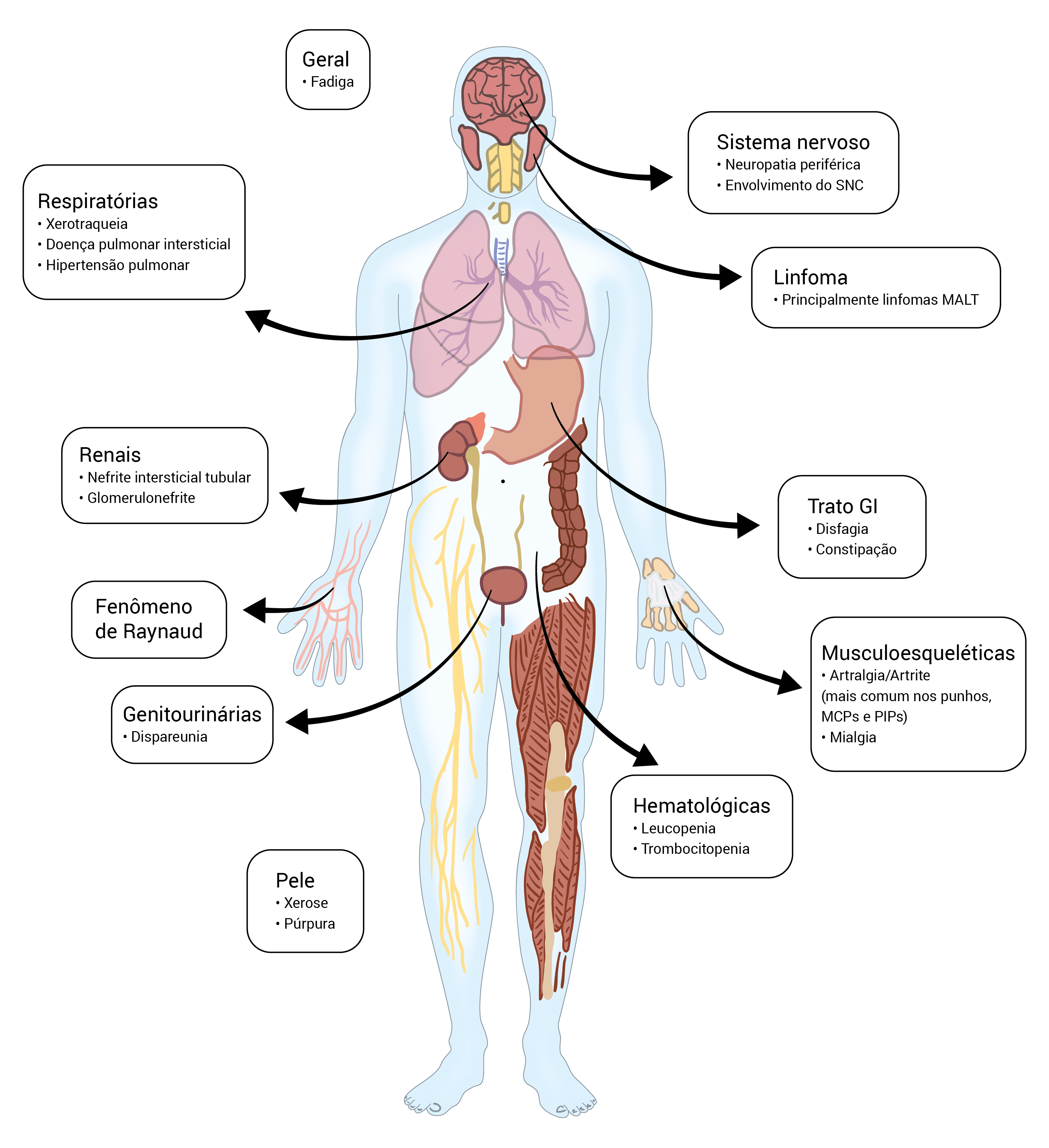
A Figura 2 apresenta um resumo das manifestações extraglandulares.
IFP: interfalângico proximal; MALT: tecido linfoide associado à mucosa; MCP: metacarpofalange.
Figura 2 - Manifestações clínicas da SS primária. As manifestações exócrinas envolvem os olhos, a orofaringe e os sistemas respiratório, cutâneo e reprodutivo. Outras características clínicas incluem manifestações constitucionais e o envolvimento do trato respiratório, o trato gastrintestinal, sistema musculoesquelético, a pele e o sistema hematológico, assim como aumento no risco de desenvolvimento de linfoma.
A utilidade do exame físico para diagnosticar SS primária ainda não foi investigada. Entretanto, a sensibilidade e a especificidade de algumas características clínicas relacionadas à doença foram observadas nos estudos realizados pelo grupo Sjögren’s International Collaborative Clinical Alliance Cohort, que desenvolveu os critérios mais recentes de classificação.26
Nesse estudo, o teste de Schimer I anormal (=5mm/5min) apresentou uma sensibilidade de menos de 50% para classificar corretamente os pacientes com SS primária. Sabe-se também que olhos secos são altamente prevalentes na população em geral, em especial nos grupos de idade mais avançada. Por essa razão, os valores preditivos positivos e negativos de um teste de Schimer I anormal poderão ser relativamente baixos para SS primária, mesmo em clínicas reumatológicas.
Da mesma forma, os sintomas de boca seca isoladamente não são muito úteis para fazer o diagnóstico de SS primária por causa da alta prevalência dessas queixas na população em geral. O aumento da glândula parótida ocorre em menos de um terço de pacientes com SS primária; portanto, sua ausência não exclui de forma confiável o diagnóstico.
Testes Laboratoriais
A esmagadora maioria de pacientes com SS primária apresenta resultados positivos nos anticorpos antinucleares (AANs) séricos. Em uma grande coorte de pacientes com SS primária, 85% tiveram AAN positivo, 52% tiveram anticorpo RO/SS-A positivo e 34% apresentaram teste positivo para anticorpos anti-La/SS-B.60
Outros autoanticorpos também podem ser encontrados na SS primária e podem estar associados a fenótipos clínicos diferentes. No mesmo estudo, 9% de pacientes com SS primária apresentaram níveis séricos baixos de complementos (C3 ou C4) e 28% tiveram várias citopenias, incluindo 16% com leucopenia e 13% com trombocitopenia. Normalmente, as citopenias não são graves ou não precisam de intervenção.
O Quadro 4 apresenta os autoanticorpos na SS primária, prevalência e fenótipos clínicos.89
Quadro 4
|
Autoanticorpos na Síndrome de Sjögren Primária, Prevalência e Fenótipos Clínicos | ||
|
Caraterísticas sorológicas |
Prevalência (%) |
Fenótipo clínico/associações |
|
FR |
40?50 |
Idade mais jovem, manifestações extraglandulares.
|
|
Crioglobulina |
10?15 |
Idade mais jovem, manifestações extraglandulares, mau prognóstico.
|
|
AAC
|
4?17 |
Fenômeno de Raynaud, menor probabilidade de anti-Ro/SS-A e anti-La/SS-HB, FR positivo, leucocitopenia e hipergamaglobulinemia.
|
|
AAM |
5?6,5 |
CBP
|
|
ASMAs
|
6,5?62 |
Hepatite autoimune tipo 1 |
|
anti-CCP
|
7?10 |
Sinovite não erosiva |
|
anti-CA
|
12,5?20,8 |
Acidose tubular renal distal |
AAC: anticorpo anticentrômero; AAM: anticorpo antimitocondrial; anti-CA: anticorpo II antianidrase carbônica; anti-CCP: anticorpo antipeptídeo citrulinado cíclico; ASMA: anticorpo antimúsculo liso; CBP: Cirrose biliar primária; FR: fator reumatoide; pSS: síndrome de Sjögren primária.
Testes Oculares
Evidências objetivas do envolvimento ocular podem ser obtidas com auxílio do teste de Schimer I e coloração da córnea e da conjuntiva. O teste de Schimer I mede a quantidade de produção de lágrimas no olho não anestesiado. Para fazer esse tipo de teste, coloca-se um pedaço de papel de filtro estéril (com, aproximadamente, 35mm de comprimento) no meio do terço lateral da pálpebra inferior e, em seguida, faz-se a medição da distância que as lágrimas percorrem em 5 minutos.
Distâncias de 5mm ou menos são consideradas anormais e indicam produção reduzida de lágrimas. O teste com o corante lissamina verde é utilizado para avaliar a integridade da superfície conjuntival. O rosa de Bengala, que anteriormente era usado para colorir a córnea, é um corante à base de anilina que colore células mortas ou degeneradas em áreas sem lágrimas.
O uso desse corante é limitado pelo desconforto ocular e, por essa razão, seu uso foi descontinuado. O corante lissamina verde adere nas superfícies epiteliais com deficiência de mucina. O corante fluoresceína também costuma ser usado na avaliação de olhos secos e colore áreas da córnea e da conjuntiva onde ocorre o rompimento da membrana celular.
Fluxo da Glândula Salivar e Estudos de Imagens
Existem vários métodos diferentes para avaliar os casos de xerostomia. Sialometria é um tipo de medição não invasiva da velocidade do fluxo salivar usado para quantificar as velocidades de fluxo de glândulas diferentes (parótida, submandibular ou sublingual) ou na boca como um todo. As velocidades não estimuladas anormais do fluxo salivar total são de 1,5mL por 15 minutos ou menos.24
A sialografia é uma opção para identificar padrões ductais e áreas de obstrução no ducto salivar que permitem distinguir processos inflamatórios crônicos de neoplasmas localizados. A aplicação dessa técnica possibilita injetar contraste radiográfico no interior do ducto salivar, permitindo obter radiografias seriais para visualizar o corante.
É extremamente importante usar contraste à base de água em comparação com contraste à base de óleo, considerando que este último poderá danificar o tecido salivar adjacente. A sialografia é um procedimento invasivo com algumas complicações, incluindo rompimento do ducto, dor e infecção e, de um modo geral, não é necessário para fazer diagnósticos.
Cintilografia é uma técnica moderadamente sensível, porém não é específica para medir a função das glândulas salivares nesse tipo de doença. Para fazer esse teste, injeta-se pertecnetato de sódio marcado com tecnécio (99mTc) no sangue e quantifica-se a velocidade de absorção do material radioativo pela glândula salivar e a posterior secreção na boca. Para esse tipo de teste, estima-se que a sensibilidade e a especificidade diagnósticas sejam de 75 e 78%, respectivamente.61
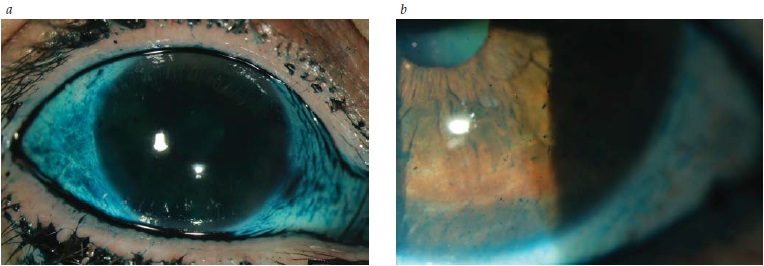
A Figura 3 mostra a coloração de um olho seco com lissamina conforme mostra o exame com lâmpada fendida.
Figura 3 - Coloração de um olho seco com lissamina conforme mostra o exame com lâmpada fendida: (A) Coloração difusa da conjuntiva; (B) Coloração pontilhada da córnea.
Técnicas avançadas de imagens radiográficas, tais como ultrassonografia, imagens por ressonância magnética (IRMs) e sialografia por ressonância magnética são muito usadas para avaliar pacientes nesse contexto. A ultrassonografia das glândulas salivares (USGS) é um teste diagnóstico recente usado na avaliação de SS primária. A USGS aumenta a sensibilidade do diagnóstico.62
Entretanto, a experiência com essas técnicas avançadas de obtenção de imagens tem alguma limitação e exige validação ulterior no caso de serem adotadas na prática clínica. Em um dos estudos, a sialografia por ressonância nuclear magnética foi a mais sensível entre as três técnicas para detectar alterações glandulares, ao passo que as IRMs foram a melhor opção para avaliar alterações parenquimatosas.63
Biópsia
A biópsia nas glândulas salivares labiais ainda é o padrão de ouro para o diagnóstico de SS primária. Trata-se de um procedimento invasivo, embora relativamente benigno, em que se removem ao menos quatro lóbulos das glândulas salivares por meio de uma pequena incisão na parte interna do lábio e que são analisados por histopatologia. A biópsia é considerada positiva se a pontuação do foco for 1 ou maior que 4mm2 de tecido, onde se define foco como um agrupamento de 50 ou mais linfócitos.
Na prática, as biópsias nas glândulas salivares labiais são reservadas para aquelas poucas situações em que o diagnóstico não é suficientemente claro (por exemplo, teste negativo para AAN, diagnóstico alternativo como sarcoidose). Alguns estudos recentes mostraram que a interpretação dos resultados das biópsias está sujeita a uma considerável variabilidade, dependendo da experiência do analista.64
Diagnóstico Diferencial
Existem vários distúrbios que podem imitar pelo menos algumas das características clínicas da SS primária. Algumas dessas condições, em especial aquelas que causam infiltração linfocitária nas glândulas exócrinas, poderão dificultar a distinção de SS primária.65 As infecções virais crônicas são consideradas no diagnóstico diferencial dessa doença.
Os pacientes com infecção produzida pelo VHC possivelmente apresentam sintomas de sicca, teste de Schimer I anormal e biópsia positiva da glândula salivar labial. Entretanto, a infecção por VHC acomete os homens com mais frequência do que as mulheres e está predominantemente associada a doenças hepáticas, embora não esteja associada aos anticorpos anti-Ro/SS-A e anti-La/SS-B.66-68
O HTLV-1 foi reconhecido como a causa de leucemia de células T/linfoma e de uma síndrome denominada “mielopatia/paraparesia espástica tropical”; esse vírus produz características clínicas e histopatológicas que não podem ser distinguidas daquelas da SS primária.69
A síndrome da linfocitose infiltrativa difusa (SLID) também imita a SS primária no quadro de infecção por HIV. A SLID ocorre em qualquer estágio da doença por HIV e se caracteriza por olhos secos, boca seca e aumento das glândulas salivares. A maior parte dos pacientes com essa condição é formada por homens sem os anticorpos anti-Ro/SS-A e anti-La/SS-B.
Recentemente, os estudiosos descreveram uma nova entidade clínica denominada IgG4-RD.70,71 A IgG4-RD possui uma distribuição típica dos órgãos envolvidos que se assemelha à distribuição da SS primária, embora seja diferente em vários aspectos sob a ótica patológica e clínica.
Em comparação com a SS primária, a entidade IgG4-RD está associada a uma frequência mais baixa de olhos secos, boca seca, artralgia e positividade AAN, assim como a uma taxa mais elevada de pancreatite autoimune. Além disso, os pacientes com IgG4-RD apresentam glândulas lacrimais aumentadas, que é uma descoberta usual nos casos de SS primária
Os sintomas de sicca são queixas comuns em pacientes com doença do enxerto versus hospedeiro (DEVH) após o transplante alogênico de medula óssea. Em um estudo, as biópsias das glândulas salivares de 90% dos pacientes com DEVH revelaram a presença de linfócitos T infiltrantes.72 Os linfomas primários das glândulas salivares também produzem sintomas de sicca e aumento da glândula parótida e, consequentemente, poderão ser considerados no diagnóstico diferencial de SS primária no contexto clínico apropriado.
O Quadro 5 contém o diagnóstico diferencial da SS primária.
Quadro 5
|
Diagnóstico Diferencial da Síindrome de Sjögren Primária | ||||
|
Doença |
Epidemiologia |
Características clínicas |
Biópsia da glândula salivar |
Outros |
|
VHC |
Homens > mulheres |
Sintomas de sicca; testes oculares anormais; frequência de aumento na glândula parótida menor do que na SS. |
Infiltrado linfocitário focal CD3+ CD4+ com predominância de células B em alguns casos; a inflamação tende a ser mais leve que a SS primária.
|
Ausência de anticorpos anti-Ro/SS-A e anti-La/SS-B no soro. |
|
Vírus linfotrópico de células T humanas tipo 1 (HTLV-1)
|
Endêmico em áreas da África, América do Sul, América Central, sudoeste do Japão e Caribe. |
Sintomas de sicca; testes oculares anormais. |
Infiltrado linfocitário CD3+ CD4+ que talvez não possa ser distinguido de SS primária.
|
Causas de leucemia/linfoma e mielopatia/paraparesia espástica tropical em adultos. |
|
SLID
|
Homens > mulheres |
Sintomas de sicca; testes oculares anormais; aumento nas glândulas salivares (usualmente bilateral); cistos na parótida.
|
Infiltrado linfocitário focal CD3+ com predominância de células T CD8+ sobre as células CD4+.
|
Aumento no risco de linfoma. |
|
Tumor de Warthin (cistadenoma papilar linfomatoso) |
Segundo tumor mais comum nas glândulas salivares; os fumantes têm risco oito vezes mais elevado de desenvolver esse tipo de tumor em comparação com a população em geral. |
O tumor afeta com mais frequência a cauda da glândula parótida e pode ser unilateral ou bilateral. |
Papilas múltiplas compostas de uma camada dupla de células epiteliais que se projetam no interior dos cistos; o estroma linfoide abaixo do epitélio geralmente contém centros germinais.
|
- |
|
Doença relacionada à IgG4 |
Prevalência mais alta em homens em comparação com SS primária. |
Com frequência apresenta aumento na glândula parótida, submandibular e/ou na glândula lacrimal; prevalência de sicca mais baixa do que na SS primária. |
Infiltração mononuclear proeminente nas glândulas salivares e lacrimais com formação de folículos linfoides, infiltração de células plasmáticas IgG4+ e fibrose; infiltração linfocitária nos ductos (lesões linfoepiteliais) não é comum.
|
Ausência de anticorpos séricos anti-Ro/SS-A e anti-La/SS-B; aumento nos níveis séricos de IgG4; rinite alérgica e pancreatite são mais comuns nesta condição do que na SS primária. |
|
DEVH |
Complicação do transplante alogênico de medula óssea.
|
Sintomas de sicca, principalmente com envolvimento ocular. |
Infiltrado linfocitário CD3+. |
- |
|
Linfoma nas glândulas salivares |
2,5% de todos os tumores primários das glândulas salivares; a maior parte dos pacientes com linfomas nas glândulas salivares não tem SS primária.
|
Sintomas de sicca em apenas uma minoria de pacientes; aumento na glândula parótida com massa firme, usualmente sem dor. |
Proliferação clonal de células B. |
Linfomas MALT > linfomas foliculares de células B > linfomas difusos de células B grandes; linfomas de células T são raros. |
|
Sarcoidose |
Rara; prevalência mais elevada em afro-americanos. |
Sintomas de sicca; testes oculares anormais; aumento nas glândulas salivares e lacrimais. |
Infiltração granulomatosa não caseante. |
Possivelmente ocorra adenopatia hilar, uveíte, eritema nodoso ou outras características de sarcoidose; ausência de anticorpos anti-Ro/SS-A e anti-La/SS-B no soro.
|
|
Amiloidose |
Rara |
Sintomas de sicca; aumento nas glândulas salivares; outras características de amiloidose como inchaço na língua, artropatia, proteinuria com insuficiência renal.
|
Infiltração amiloide com coloração vermelha do Congo positiva; sem infiltrado linfocitário ou infiltrado mínimo. |
- |
|
Hipertrigliceridemia |
Rara |
Aumento indolor na glândula parótida (pode ser bilateral) com ou sem sintomas de sicca. |
Infiltração gordurosa de lóbulos glandulares com perda de células acinares; sem infiltrado linfocitário.
|
- |
|
Hemocromatose |
Rara |
Sintomas de sicca. |
Deposição de hemosiderina; sem infiltrado linfocitário.
|
- |
|
Diabetes; alcoolismo; bulimia; má nutrição crônica. |
- |
Sintomas de sicca; aumento da glândula parótida leve e difuso, usualmente sem dor. |
Sialadenose com infiltração gordurosa e aumento no tamanho da célula acinar. |
- |
DEVH: doença do enxerto versus hospedeiro; MALT: tecido linfoide associado à mucosa; SLID: síndrome da linfocitose infiltrativa difusa; SS: síndrome de Sjögren; VHC: vírus da hepatite C.
A sarcoidose pode produzir sintomas de sicca com aumento nas glândulas lacrimais e salivares e, portanto, pode ser confundida com SS primária. A sarcoidose pode ser distinguida de SS primária por meios histopatológicos pela presença de inflamação granulomatosa. Além disso, os pacientes com sarcoidose são sorologicamente negativos para os anticorpos anti-Ro/SS-A e anti-La/SS-B.
Outros estudos, como as radiografias torácicas, são bastante úteis na busca de evidências de sarcoidose em outros órgãos ou tecidos. Raramente, a sarcoidose e a SS primária ocorrem no mesmo paciente. Amiloidose, hipertrigliceridemia e hemocromatose podem imitar SS primária ao aumentar o volume das glândulas exócrinas por meio da infiltração tecidual com amiloides, triglicérides e ferro, respectivamente.65 Os sintomas de sicca também são comuns entre indivíduos com diabetes melito.
Gerenciamento
A meta do tratamento consiste basicamente em aliviar os sintomas e prevenir as complicações da doença no longo prazo. A Figura 4 apresenta uma abordagem por etapas ao gerenciamento dos sintomas. Há uma miríade de opções de tratamento para olho seco. Essas opções incluem evitar condições que possam agravar os olhos secos (aquecimento central desumidificado e ar-condicionado, ambientes expostos ao vento e medicações que diminuam ainda mais a produção de lágrimas) e usar suplementos lacrimais e secretagogos, assim como obstruir os ductos lacrimais (punctae).
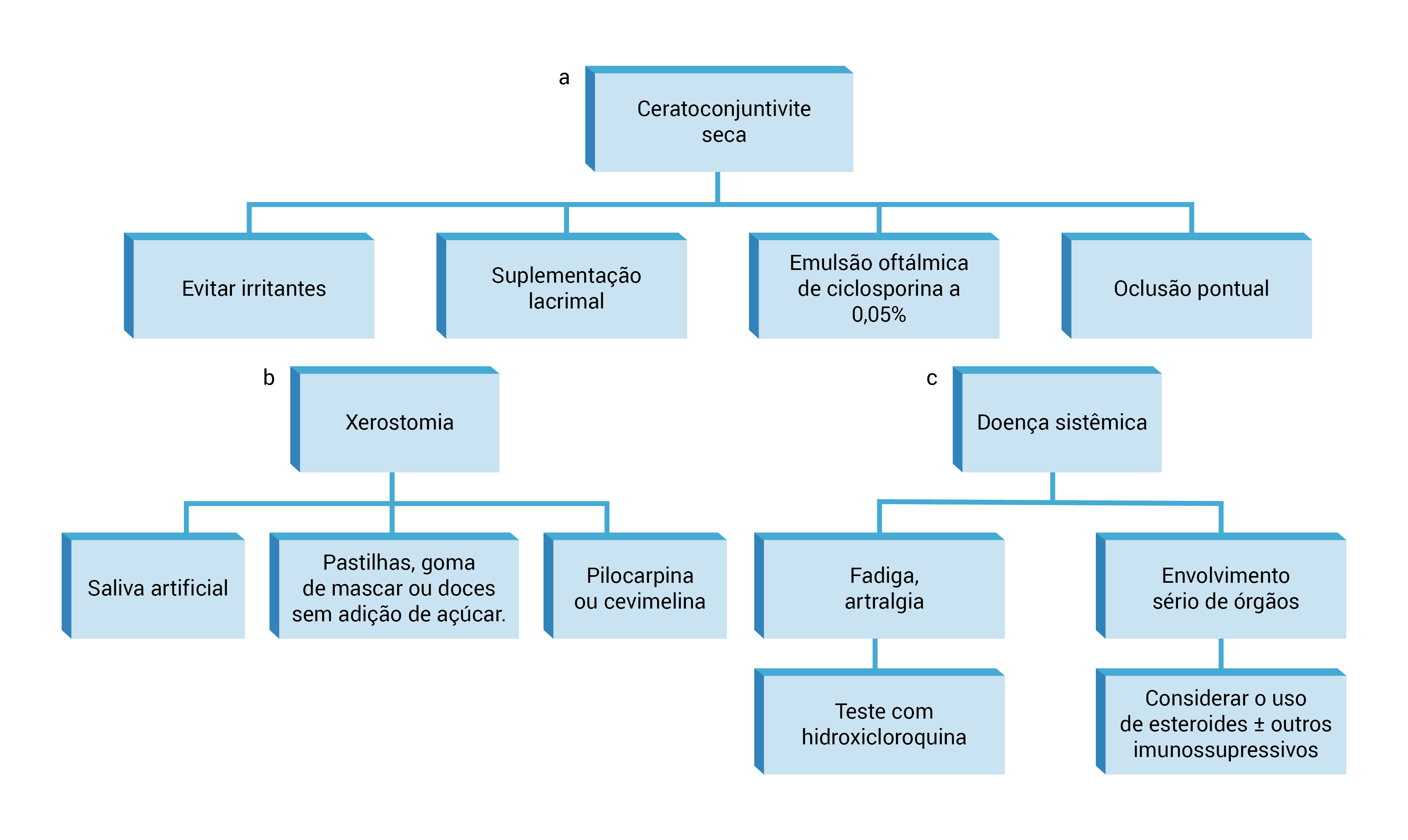
DPI: doença pulmonar intersticial; GN: glomerulonefrite; NTI: nefrite tubular intersticial.
Figura 4 - Gerenciamento da SS primária. (A) O gerenciamento inicial de ceratoconjuntivite consiste em evitar irritantes potenciais, suplementos lacrimais e emulsão oftálmica de ciclosporina a 0,05%. Oclusão pontual, temporária ou permanente, pode ser usada para bloquear os canais de drenagem e preservar os efeitos lubrificantes das lágrimas nos olhos por períodos mais longos de tempo. (B) Para os casos de xerostomia, o tratamento se enquadra em uma de duas categorias: estimulação do fluxo existente da glândula salivar ou terapia de reposição. Os secretagogos orais, pilocarpina e cevimelina, foram aprovados pela Food and Drug Administration (FDA) para tratamento de boca seca e podem ser bastante eficazes no gerenciamento desses sintomas. (C) Fadiga, artralgia e mialgia podem ser tratadas empiricamente com um teste da terapia com hidroxicloroquina por 6 a 12 meses (indicação não aprovada pela FDA). O envolvimento mais grave de sistemas de órgãos, como DPI, NTI, GN e vasculite sistêmica, geralmente exige tratamento com corticosteroides e/ou outros agentes imunossupressivos.
As lágrimas artificiais são comercializadas sem prescrição médica em uma grande variedade de formulações e com diferentes viscosidades. Quanto mais baixa a viscosidade, maior será a frequência de administração da solução. Soluções com viscosidade mais espessa são muito úteis na hora de dormir porque têm longa duração; talvez exijam o uso de esfoliador de pálpebras todas as manhãs para impedir o desenvolvimento de blefarite.
Existem, atualmente, no mercado dois secretagogos orais que estimulam a produção de lágrimas. A pilocarpina (Salagen) é um agente muscarínico oral que estimula o receptor M3R. O uso dessa medicação na dose de 5mg, 3 a 4x/dia, melhora significativamente as avaliações globais subjetivas de olhos secos73,74 e as medições subjetivas da integridade da superfície conjuntival.73
Os efeitos colaterais possíveis da pilocarpina incluem ruborização, sudorese, aumento na frequência de micção e desconforto abdominal. A cevimelina (Evoxac) também estimula os MRs (principalmente os receptores M1 e M3) nas glândulas salivares e lacrimais. Esse medicamento deve ser administrado na dose de 30mg, 3x/dia.
Os resultados de testes randomizados controlados mostraram que o tratamento à base de cevimelina nessa dosagem estava associado a uma melhora estatisticamente significativa na avaliação subjetiva global de olhos secos e boca seca e a uma melhora objetiva nas velocidades de fluxo salivares e lacrimais.75
Possivelmente, o uso de cevimelina esteja associado a cefaleia, sudorese, rinite, náusea e diarreia, assim como a distúrbios visuais. Por causa de suas propriedades farmacológicas, as terapias à base de pilocarpina e de cevimelina são contraindicadas para pacientes com irite, glaucoma com ângulo estreito e asma de moderada a grave.
A emulsão oftálmica de ciclosporina a 0,05% (Restasis) foi aprovada em 2003 pela FDA para tratamento de KCS. Em pacientes com olhos secos, a ciclosporina diminui os marcadores superficiais das células de linfócitos T ativados e aumenta a densidade das células calciformes.76
Em testes clínicos, essa preparação oftalmológica de ciclosporina melhorou as queixas subjetivas de olhos secos e os valores nos teste de Schimer I, além de diminuir o uso de lágrimas artificiais complementares.77 O efeito colateral conhecido mais comum é a sensação de queimação nos olhos, que poderão limitar seu uso em alguns casos.
Oclusões pontuais temporárias ou permanentes podem aliviar os sintomas por meio do bloqueio dos canais nasolacrimais de drenagem e da preservação das lágrimas. Nos casos de oclusões pontuais temporárias, colocam-se inicialmente tampões nas duas extremidades inferiores, levando-se em consideração que a drenagem lacrimal ocorre através desses canais.
Ocasionalmente, para melhorar o nível de benefícios, esses tampões poderão ser colocados nas duas extremidades superiores. O uso de tampões de colágeno no curto prazo é uma opção para avaliar as respostas dos pacientes às oclusões pontuais. Os tampões de longo prazo são uma opção semipermanente ou permanente nos casos em que as oclusões pontuais temporárias forem bem toleradas pelos pacientes. Normalmente, conseguem-se oclusões pontuais permanentes com cautério ou cirurgia a lêiser. A produção excessiva de lágrimas é a complicação mais significativa das oclusões pontuais.
Os pacientes com olhos secos geralmente se queixam de irritação nos olhos apesar da instilação de lágrimas artificiais ou da aplicação de outras medidas de tratamento. Às vezes, essas queixas resultam dos efeitos dos preservativos contidos nas soluções lubrificantes. Os lubrificantes sem preservativos são comercializados em embalagens únicas e somente deverão ser administrados se as lágrimas forem instiladas mais de três vezes ao dia.
A blefarite também pode exacerbar os sintomas subjacentes de olhos secos, que poderão ser gerenciados com compressões mornas, esfoliantes para as pálpebras como o limpador de sobrancelhas Johnson’s Baby Shampoo misturado com água na proporção de 1:1 ou terapia antibiótica. Os pacientes com filamentos mucosos nos cantos internos dos olhos poderão ter algum benefício com a instilação de uma solução oftálmica de acetilcisteína (agente mucolítico), que pode ser manipulada em farmácias para uso nesse contexto.
De um modo geral, costuma-se abordar a xerostomia ou boca seca de duas maneiras: estimulação do fluxo salivar existente e reposição da saliva. Com frequência, os indivíduos percebem que pastilhas de açúcar, doces e goma de mascar sem adição de açúcar ajudam a estimular o fluxo de saliva. O uso de produtos sem adição de açúcar é extremamente importante, levando-se em consideração o aumento no risco de cáries dentárias resultante da disfunção das glândulas salivares.
Assim como no tratamento de olhos secos, os medicamentos pilocarpina e cevimelina também estimulam o fluxo salivar. Os resultados de alguns testes clínicos mostraram que o tratamento com 5mg de pilocarpina, 3x/dia, melhora as condições dos pacientes, a avaliação global de boca seca pelos médicos, bem como aumenta a velocidade do fluxo salivar.74,78
Da mesma forma, doses de 30mg de cevimelina, 3x/dia, produzem benefícios clínicos semelhantes.75,79 Nos dias atuais, as preparações de saliva artificial contendo hipromelose ou metilcelulose são comercializadas regularmente e poderão ser úteis em alguns casos.
Os pacientes com SS primária e xerostomia grave correm grande risco de degradação rápida dos dentes. Recomendam-se uso rotineiro de fio dental, visitas frequentes ao dentista, aplicações regulares de fluoreto dental e modificações na dieta para manter a saúde oral. Candidíase oral é uma das complicações mais comuns da SS primária. De um modo geral, os pacientes com xerostomia percebem a sensação de queimação ou de lesões na boca.
Além disso, esses pacientes desenvolvem queilite angular que, provavelmente, tenha sido causada por alguma infecção oral por leveduras. Medicamentos como elixir de nistatina, pastilhas de clotrimazol e fluconazol são usados no tratamento de candidíase oral. O creme tópico de clotrimazol usado em combinação com clotrimazol oral por 1 a 2 semanas geralmente é suficiente para eliminar a queilite angular.
Lábios, pele e passagens nasais secas, assim como a secura vaginal, também são sintomas comuns na SS primária. A secura nessas áreas pode ser tratada com uso regular de agentes umidificadores, bálsamo labial, spray nasal salino e umidificadores vaginais. Em determinados casos, o creme de estrogênio vaginal para uso tópico é eficaz no tratamento da dispareunia produzida pela secura na mucosa vaginal.
Condições como fadiga, artralgia e mialgia respondem bem à terapia com hidroxicloroquina, embora essa abordagem não tenha o suporte de evidências produzidas por testes randomizados controlados. Um teste randomizado duplo-cego e controlado por placebo da terapia com hidroxicloroquina envolvendo 122 pacientes com SS primária não conseguiu atingir o desfecho primário.80
Todavia, parece razoável tratar pacientes com hidroxicloroquina como teste, considerando que seu perfil de segurança é favorável e o custo é relativamente baixo, embora esse medicamento ainda não tenha sido aprovado pela FDA para essa indicação. Entretanto, o uso de hidroxicloroquina deverá ser interrompido depois de 6 a 12 meses de terapia contínua, na ausência de respostas sintomáticas.
Recomenda-se tomar muito cuidado para evitar doses acima 6,5mg/kg/dia por causa do aumento no risco de toxicidade retinal com doses mais elevadas de hidroxicloroquina. Geralmente, recomenda-se fazer exame dos olhos em intervalos de 12 meses para monitorar quaisquer sinais de toxicidade retinal.
Normalmente, o tratamento à base de glicocorticoides e de outros agentes imunossupressivos deve ser reservado para pacientes com envolvimento sério do sistema de órgãos, como PII ou PIL, nefrite intersticial ou neuropatia. Ainda não há medicamentos aprovados pela FDA para tratamento dessas manifestações extraglandulares da doença.
Vários estudos recentes investigaram o uso de terapias biológicas para SS primária. Até o presente momento, os resultados foram negativos ou inconclusivos. Os bloqueadores do TNF-a etanercept e infliximabe não produziram benefícios clínicos em testes randomizados controlados.81,82 As descobertas de testes clínicos de pequeno porte envolvendo o rituximabe (anticorpo monoclonal anti-CD20) e o epratuzumab (anticorpo monoclonal anti-CD22) produziram resultados mistos.83?85
Recentemente, um teste randomizado, duplo cego, controlado por placebo, que avaliou a terapia com rituximabe em 120 pacientes com SS primária não confirmou seus benefícios na doença,86 ao passo que os resultados de outro teste clínico com desenho semelhante ainda não tinham sido divulgados até o momento da revisão deste texto.
Prognóstico
Dois grandes estudos de coorte analisaram os resultados da SS primária no longo prazo. Em um estudo de coorte realizado na Grécia envolvendo 723 pacientes, Ioannidis e colaboradores constataram que a mortalidade padrão em indivíduos com SS primária era semelhante à da população em geral.87
Os autores concluíram que os indivíduos com SS primária poderiam ser classificados em dois grupos com base no risco do desenvolvimento de algum distúrbio linfoproliferativo. Os indivíduos que foram classificados como tipo I (aproximadamente, 20%) tinham púrpura palpável e níveis baixos de C4 na apresentação e risco mais elevado de complicações no longo prazo e de morte. Os classificados como tipo II tinham curso descomplicado da doença e deviam ser tranquilizados quanto ao risco de morte, que não é mais alto que na população em geral.
Na Suécia, Theander e colaboradores acompanharam 484 pacientes e chegaram a conclusões semelhantes.88 Não houve aumento na mortalidade por todas as causas entre indivíduos com SS primária em comparação com a população em geral. Nesse estudo, nível baixo de C3 e C4 no momento do diagnóstico foram os preditores mais fortes de resultados desfavoráveis.
Referências
1. Sjögren H. Zur Kenntnis der Keratoconjunctivitis Sicca (Keratitis Filiformis bei Hypofunktion der Tranendrusen). Acta Ophthalmol (Kbh) 1933;11 Suppl 2:1–151.
2. Gabriel SE, Michaud K. Epidemiological studies in incidence, prevalence, mortality, and comorbidity of the rheumatic diseases. Arthritis Res Ther 2009;11:229.
3. Turesson C, O’Fallon WM, Crowson CS, et al. Occurrence of extraarticular disease manifestations is associated with excess mortality in a community based cohort of patients with rheumatoid arthritis. J Rheumatol 2002;29:62–7.
4. Andonopoulos AP, Skopouli FN, Dimou GS, et al. Sjögren’s syndrome in systemic lupus erythematosus. J Rheumatol 1990;17:201–4.
5. Nossent JC, Swaak AJ. Systemic lupus erythematosus VII: frequency and impact of secondary Sjögren’s syndrome. Lupus 1998;7:231–4.
6. Avouac J, Sordet C, Depinay C, et al. Systemic sclerosis-associated Sjögren’s syndrome and relationship to the limited cutaneous subtype: results of a prospective study of sicca syndrome in 133 consecutive patients. Arthritis Rheum 2006;54:2243–9.
7. Bolstad AI, Jonsson R. Genetic aspects of Sjögren’s syndrome. Arthritis Res 2002;4:353–9.
8. Cobb BL, Lessard CJ, Harley JB, Moser KL. Genes and Sjögren’s syndrome. Rheum Dis Clin North Am 2008;34:847–68.
9. Harley JB, Reichlin M, Arnett FC, et al. Gene interaction at HLA-DQ enhances autoantibody production in primary Sjögren’s syndrome. Science 1986;232:1145–7.
10. Gottenberg JE, Busson M, Loiseau P, et al. In primary Sjögren’s syndrome, HLA class II is associated exclusively with autoantibody production and spreading of the autoimmune response. Arthritis Rheum 2003;48:2240–5.
11. Lessard CJ, Li He, Adrianto I, et al. Variants at multiple loci implicated in both innate and adaptive immune responses are associated with Sjögren’s syndrome. Nat Genet 2013;45:1284–92.
12. Altorok N, Coit P, Hughes T, et al. Genome-wide DNA methylation patterns in naïve CD4+ T cells from patients with primary Sjögren’s syndrome. Arthritis Rheum 2014;66:713–39.
13. Christodoulou MI, Kapsogeorgou EK, Moutsopoulos HM. Characteristics of the minor salivary gland infiltrates in Sjögren’s syndrome. J Autoimmun 2009. DOI: 10.1016/j.jaut.2009.10.004.
14. Mavragani CP, Moutsopoulos HM. The geoepidemiology of Sjögren’s syndrome. Autoimmun Rev 2009. DOI: 10.1016/j.autrev.2009.11.004.
15. Katsifis GE, Rekka S, Moutsopoulos NM, et al. Systemic and local interleukin-17 and linked cytokines associated with Sjögren’s syndrome immunopathogenesis. Am J Pathol 2009;175:1167–77.
16. Palmer MT, Weaver CT. Autoimmunity: increasing suspects in the CD4+ T cell lineup. Nat Immunol 2010;11:36–40.
17. Tobin G, Giglio D, Lundgren O. Muscarinic receptor subtypes in the alimentary tract. J Physiol Pharmacol 2009;60:3–21.
18. Routsias JG, Tzioufas AG. Sjögren’s syndrome—study of autoantigens and autoantibodies. Clin Rev Allergy Immunol 2007;32:238–51.
19. Gottenberg JE, Cagnard N, Lucchesi C, et al. Activation of IFN pathways and plasmacytoid dendritic cell recruitment in target organs of primary Sjögren’s syndrome. Proc Natl Acad Sci U S A 2006;103:2770–5.
20. Emamian ES, Leon JM, Lessard CJ, et al. Peripheral blood gene expression profiling in Sjögren’s syndrome. Genes Immun 2009;10:285–96.
21. Moisini I, Davidson A. BAFF: a local and systemic target in autoimmune diseases. Clin Exp Immunol 2009;158:155–63.
22. Groom J, Kalled SL, Cutler AH, et al. Association of BAFF/BLyS overexpression and altered B cell differentiation with Sjögren’s syndrome. J Clin Invest 2002;109:59–68.
23. Manoussakis MN, Kapsogeorgou EK. The role of epithelial cells in the pathogenesis of Sjögren’s syndrome. Clin Rev Allergy Immunol 2007;32:225–30.
24. Vitali C, Bombardieri S, Moutsopoulos HM, et al. Preliminary criteria for the classification of Sjögren’s syndrome. Results of a prospective concerted action supported by the European Community. Arthritis Rheum 1993;36:340–7.
25. Vitali C, Bombardieri S, Jonsson R, et al. Classification criteria for Sjögren’s syndrome: a revised version of the European criteria proposed by the American-European Consensus Group. Ann Rheum Dis 2002;61:554–8.
26. Shiboski SC, Shiboski CH, Criswell LA, et al. American College of Rheumatology classification criteria for Sjögren’s syndrome: a data-driven, expert consensus approach in the Sjögren’s International Collaborative Clinical Alliance Cohort. Arthritis Care Res 2012;64:475–87.
27. Soto-Rojas AE, Villa AR, Sifuentes-Osornio J, et al. Oral manifestations in patients with Sjögren’s syndrome. J Rheumatol 1998;25:906–10.
28. Thomas E, Hay EM, Hajeer A, Silman AJ. Sjögren’s syndrome: a community-based study of prevalence and impact. Br J Rheumatol 1998;37:1069–76.
29. Segal B, Bowman SJ, Fox PC, et al. Primary Sjögren’s syndrome: health experiences and predictors of health quality among patients in the United States. Health Qual Life Outcomes 2009;7:46.
30. Segal B, Thomas W, Rogers T, et al. Prevalence, severity, and predictors of fatigue in subjects with primary Sjögren’s syndrome. Arthritis Rheum 2008;59:1780–7.
31. Hackett KL, Newton JL, Frith J, et al. Impaired functional status in primary Sjögren’s syndrome. Arthritis Care Res 2012;64:1760–4.
32. Ferreiro JE, Pasarin G, Quesada R, Gould E. Benign hypergammaglobulinemic purpura of Waldenstrom associated with Sjögren’s syndrome. Case report and review of immunologic aspects. Am J Med 1986;81:734–40.
33. Pease CT, Shattles W, Barrett NK, Maini RN. The arthropathy of Sjögren’s syndrome. Br J Rheumatol 1993;32:609–13.
34. Lindvall B, Bengtsson A, Ernerudh J, Eriksson P. Subclinical myositis is common in primary Sjögren’s syndrome and is not related to muscle pain. J Rheumatol 2002;29:717–25.
35. Stojan G, Bae AN, Danoff SK. Pulmonary manifestation of Sjögren’s syndrome. Curr Allergy Asthma Rep 2013;13:354–60.
36. Papiris SA, Maniati M, Constantopoulos SH, et al. Lung involvement in primary Sjögren’s syndrome is mainly related to the small airway disease. Ann Rheum Dis 1999;58:61–4.
37. Ito I, Nagai S, Kitaichi M, et al. Pulmonary manifestations of primary Sjögren’s syndrome: a clinical, radiologic, and pathologic study. Am J Respir Crit Care Med 2005;171:632–8.
38. Swigris JJ, Berry GJ, Raffin TA, Kuschner WG. Lymphoid interstitial pneumonia: a narrative review. Chest 2002;122:2150–64.
39. Kobak S, Kalkan S, Kirilmaz B, et al. Pulmonary arterial hypertension in patients with primary Sjögren’s syndrome. Autoimmune Dis 2014. DOI: 10.1155/2014/710401.
40. Goules AV, Tatouli IP, Moutsopoulos HM, Tzioufas AG. Clinically significant renal involvement in primary Sjögren’s syndrome. Arthritis Rheum 2013;65:2945–53.
41. Goules A, Masouridi S, Tzioufas AG, et al. Clinically significant and biopsy-documented renal involvement in primary Sjögren syndrome. Medicine (Baltimore) 2000;79:241–9.
42. Bossini N, Savoldi S, Franceschini F, et al. Clinical and morphological features of kidney involvement in primary Sjögren’s syndrome. Nephrol Dial Transplant 2001;16:2328–36.
43. Maripuri S, Grande JP, Osborn TG, et al. Renal involvement in primary Sjögren’s syndrome: a clinicopathologic study. Clin J Am Soc Nephrol 2009;4:1423–31.
44. Ren H, Wang WM, Chen XN, et al. Renal involvement and follow-up of 130 patients with primary Sjögren’s syndrome. J Rheumatol 2008;35:278–84.
45. Kaplan MJ, Ike RW. The liver is a common non-exocrine target in primary Sjögren’s syndrome: a retrospective review. BMC Gastroenterol 2002;2:21.
46. Mori K, Iijima M, Koike H, et al. The wide spectrum of clinical manifestations in Sjögren’s syndrome-associated neuropathy. Brain 2005;128:2518–34.
47. Govoni M, Padovan M, Rizzo N, Trotta F. CNS involvement in primary Sjögren’s syndrome: prevalence, clinical aspects, diagnostic assessment and therapeutic approach. CNS Drugs 2001;15:597–607.
48. de Seze J, Devos D, Castelnovo G, et al. The prevalence of Sjögren syndrome in patients with primary progressive multiple sclerosis. Neurology 2001;57:1359–63.
49. De Carolis S, Salvi S, Botta A, et al. The impact of primary Sjögren’s syndrome on pregnancy outcome: our series and review of the literature. Autoimmun Rev 2014;13:103–7.
50. Priori R, Gattamelata A, Modesti M, et al. Outcomes of pregnancy in Italian patients with primary Sjögren’s syndrome. J Rheumatol 2013;40:1143–7.
51. Anaya JM, Tobon GJ, Vega P, Castiblanco J. Autoimmune disease aggregation in families with primary Sjögren’s syndrome. J Rheumatol 2006;33:2227–34.
52. D’Arbonneau F, Ansart S, Le Berre R, et al. Thyroid dysfunction in primary Sjögren’s syndrome: a long-term followup study. Arthritis Rheum 2003;49:804–9.
53. Ramos-Casals M, Garcia-Carrasco M, Cervera R, et al. Thyroid disease in primary Sjögren syndrome. Study in a series of 160 patients. Medicine (Baltimore) 2000;79:103–8.
54. Abraham S, Begum S, Isenberg D. Hepatic manifestations of autoimmune rheumatic diseases. Ann Rheum Dis 2004;63:123–9.
55. Szodoray P, Barta Z, Lakos G, et al. Coeliac disease in Sjögren’s syndrome—a study of 111 Hungarian patients. Rheumatol Int 2004;24:278–82.
56. Kassan SS, Thomas TL, Moutsopoulos HM, et al. Increased risk of lymphoma in sicca syndrome. Ann Intern Med 1978;89:888–92.
57. Voulgarelis M, Dafni UG, Isenberg DA, Moutsopoulos HM. Malignant lymphoma in primary Sjögren’s syndrome: a multicenter, retrospective, clinical study by the European Concerted Action on Sjögren’s Syndrome. Arthritis Rheum 1999;42:1765–72.
58. Baimpa E, Dahabreh IJ, Voulgarelis M, Moutsopoulos HM. Hematologic manifestations and predictors of lymphoma development in primary Sjögren syndrome: clinical and pathophysiologic aspects. Medicine (Baltimore) 2009;88:284–93.
59. Jonsson MV, Theander E, Jonsson R. Predictors for the development of non-Hodgkin lymphoma in primary Sjögren’s syndrome. Presse Med 2012;41:e511–6.
60. Ramos-Casals M, Solans R, Rosas J, et al. Primary Sjögren syndrome in Spain: clinical and immunologic expression in 1010 patients. Medicine (Baltimore) 2008;87:210–9.
61. Vinagre F, Santos MJ, Prata A, et al. Assessment of salivary gland function in Sjögren’s syndrome: the role of salivary gland scintigraphy. Autoimmun Rev 2009;8:672–6.
62. Cornec D, Jouse-Joulin S, Marhadour R. Salivary gland ultrasonography improves the diagnostic performance of the 2012 American College of Rheumatology classification criteria for Sjögren’s syndrome. Rheumatology (Oxford) 2014. DOI: 10.1093/rheumatology/keu037.
63. Niemela RK, Takalo R, Paakko E, et al. Ultrasonography of salivary glands in primary Sjögren’s syndrome. A comparison with magnetic resonance imaging and magnetic resonance sialography of parotid glands. Rheumatology (Oxford) 2004;43:875–9.
64. Stewart CM, Bhattacharyya I, Berg K, et al. Labial salivary gland biopsies in Sjögren’s syndrome: still the gold standard? Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2008;106:392–402.
65. Ramos-Casals M, Brito-Zeron P, Font J. Lessons from diseases mimicking Sjögren’s syndrome. Clin Rev Allergy Immunol 2007;32:275–83.
66. Ramos-Casals M, Font J. Extrahepatic manifestations in patients with chronic hepatitis C virus infection. Curr Opin Rheumatol 2005;17:447–55.
67. Ramos-Casals M, Loustaud-Ratti V, De Vita S, et al. Sjögren syndrome associated with hepatitis C virus: a multicenter analysis of 137 cases. Medicine (Baltimore) 2005;84:81–9.
68. Ceribelli A, Cavazzana I, Cattaneo R, Franceschini F. Hepatitis C virus infection and primary Sjögren’s syndrome: a clinical and serologic description of 9 patients. Autoimmun Rev 2008;8:92–4.
69. Merle H, Cabre P, Smadja D, et al. Sicca syndrome and HTLV-I-associated myelopathy/tropical spastic paraparesis. Jpn J Ophthalmol 1999;43:509–12.
70. Masaki Y, Dong L, Kurose N, et al. Proposal for a new clinical entity, IgG4-positive multiorgan lymphoproliferative syndrome: analysis of 64 cases of IgG4-related disorders. Ann Rheum Dis 2009;68:1310–5.
71. Stone JH, Khosroshahi A, Deshpande V, et al. Recommendations for the nomenclature of IgG4-related disease and its individual organ system manifestations. Arthritis Rheum 2012;64:3061–7.
72. Rouquette-Gally AM, Boyeldieu D, Gluckman E, et al. Autoimmunity in 28 patients after allogeneic bone marrow transplantation: comparison with Sjögren syndrome and scleroderma. Br J Haematol 1987;66:45–7.
73. Tsifetaki N, Kitsos G, Paschides CA, et al. Oral pilocarpine for the treatment of ocular symptoms in patients with Sjögren’s syndrome: a randomised 12 week controlled study. Ann Rheum Dis 2003;62:1204–7.
74. Vivino FB, Al-Hashimi I, Khan Z, et al. Pilocarpine tablets for the treatment of dry mouth and dry eye symptoms in patients with Sjögren syndrome: a randomized, placebo-controlled, fixed-dose, multicenter trial. P92-01 Study Group. Arch Intern Med 1999;159:174–81.
75. Petrone D, Condemi JJ, Fife R, et al. A double-blind, randomized, placebo-controlled study of cevimeline in Sjögren’s syndrome patients with xerostomia and keratoconjunctivitis sicca. Arthritis Rheum 2002;46:748–54.
76. Donnenfeld E, Pflugfelder SC. Topical ophthalmic cyclosporine: pharmacology and clinical uses. Surv Ophthalmol 2009;54:321–38.
77. Sall K, Stevenson OD, Mundorf TK, Reis BL. Two multicenter, randomized studies of the efficacy and safety of cyclosporine ophthalmic emulsion in moderate to severe dry eye disease. CsA Phase 3 Study Group. Ophthalmology 2000;107:631–9.
78. Nusair S, Rubinow A. The use of oral pilocarpine in xerostomia and Sjögren’s syndrome. Semin Arthritis Rheum 1999;28:360–7.
79. Fife RS, Chase WF, Dore RK, et al. Cevimeline for the treatment of xerostomia in patients with Sjögren syndrome: a randomized trial. Arch Intern Med 2002;162:1293–300.
80. Gottenberg JE, Ravaud P, Puechal X, et al. Inefficacy of hydroxychloroquine in primary Sjögren’s syndrome: results at 12 months of the randomized placebo-controlled trial of plaquenil in primary Sjögren’s syndrome. Ann Rheum Dis 2013;72 Suppl 3:90.
81. Sankar V, Brennan MT, Kok MR, et al. Etanercept in Sjögren’s syndrome: a twelve-week randomized, double-blind, placebo-controlled pilot clinical trial. Arthritis Rheum 2004;50:2240–5.
82. Mariette X, Ravaud P, Steinfeld S, et al. Inefficacy of infliximab in primary Sjögren’s syndrome: results of the randomized, controlled Trial of Remicade in Primary Sjogren’s Syndrome (TRIPSS). Arthritis Rheum 2004;50:1270–6.
83. Steinfeld SD, Tant L, Burmester GR, et al. Epratuzumab (humanised anti-CD22 antibody) in primary Sjögren’s syndrome: an open-label phase I/II study. Arthritis Res Ther 2006;8:R129.
84. Dass S, Bowman SJ, Vital EM, et al. Reduction of fatigue in Sjögren syndrome with rituximab: results of a randomised, double-blind, placebo-controlled pilot study. Ann Rheum Dis 2008;67:1541–4.
85. Devauchelle-Pensec V, Pennec Y, Morvan J, et al. Improvement of Sjögren’s syndrome after two infusions of rituximab (anti-CD20). Arthritis Rheum 2007;57:310–7.
86. Devauchelle-Pensec V, Mariette X, Jousse-Joulin S, et al. Treatment of primary Sjögren syndrome with rituximab: a randomized trial. Ann Intern Med 2014;160:233–42.
87. Ioannidis JP, Vassiliou VA, Moutsopoulos HM. Long-term risk of mortality and lymphoproliferative disease and predictive classification of primary Sjögren’s syndrome. Arthritis Rheum 2002;46:741–7.
88. Theander E, Manthorpe R, Jacobsson LT. Mortality and causes of death in primary Sjögren’s syndrome: a prospective cohort study. Arthritis Rheum 2004;50:1262–9.
89. Tzioufas AG, Tatouli IP, Moutsopoulos HM. Autoantibodies in Sjögren’s syndrome: clinical presentation and regulatory mechanisms. Presse Med 2012;41:e451–60.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.