(Carregando Índice)... (Carregando Índice)... |
Você está em:
Inicial  acp-medicine Pneumologia
acp-medicine Pneumologia
Última revisão: 01/06/2018
Comentários de assinantes: 0
|
Artigo original: Stephen MJ, MD. Cystic Fibrosis and Non-Cystic Fibrosis Bronchiectasis. SAM. [The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2015 Decker Intellectual Properties Inc. All Rights Reserved.] Tradução: Paulo Henrique Machado. Revisão técnica: Dr. Lucas Santos Zambon.
|
Michael J. Stephen, MD
Professor Assistente de Medicina na Drexel University School of Medicine (Philadelphia, PA)
Resumo
Fibrose cística (FC) é uma doença autossômica recessiva que se caracteriza por níveis elevados de cloreto no suor, bronquiectasia difusa e deficiência pancreática exócrina. FC é a doença letal hereditária mais comum em brancos. A maior parte dos pacientes se apresenta com a doença no nascimento ou na fase inicial da infância, embora os diagnósticos tardios sejam comuns. Limpeza das vias respiratórias e gerenciamento das infecções após a constatação de que a FC é uniformemente fatal no estágio inicial, mas que evolui na nutrição, resultaram na sobrevida média de 37 anos. O aspecto mais recente do tratamento foi o advento de moduladores proteicos, que podem aumentar ainda mais a expectativa de vida. Este artigo apresenta algumas discussões sobre epidemiologia, genética, fisiopatologia, patogênese, diagnóstico, diagnóstico diferencial e tratamento da síndrome de FC. Apresentamos também a definição, a epidemiologia, a etiologia, a patogênese, o diagnóstico, o gerenciamento e o prognóstico de bronquiectasia não cística. As figuras apresentam ilustrações sobre os reguladores da condutância transmembrana da FC (CFTR), hipótese de círculo vicioso da lesão pulmonar, taxas de germes respiratórios por idade, diagnóstico de FC, caminhos terapêuticos para FC, volume expiratório forçado em 1 segundo, função percentual prevista versus índice de massa corporal (IMC) e idade mediana de sobrevida prevista de pacientes com FC. Apresentamos também uma radiografia e uma varredura por tomografia computadorizada (TC) de uma FC. Os quadros e a tabela apresentam a descrição mais comum de mutações de FC em 2011, mutações de classe da FC, método mnemônico para exacerbações de FC, diagnóstico de diabetes relacionado à FC em um paciente estável, valor dos testes de suor e diagnóstico diferencial de bronquiectasia.
Fibrose Cística
Definição da Doença
Fibrose cística (FC) é uma doença autossômica recessiva que se caracteriza por níveis elevados de cloreto no suor, bronquiectasia difusa e deficiência pancreática exócrina. FC é a doença letal hereditária mais comum em brancos. A maior parte dos pacientes se apresenta com a doença no nascimento ou na fase inicial da infância, embora os diagnósticos tardios sejam comuns.
Limpeza das vias respiratórias e gerenciamento da infecção após a constatação de que a FC é uniformemente fatal no estágio inicial, mas evolui com a nutrição, levaram a uma sobrevida média de 37 anos. O aspecto mais recente do tratamento foi o advento de moduladores proteicos, que podem aumentar ainda mais a expectativa de vida.
Epidemiologia
Aproximadamente, 30.000 pacientes nos EUA e 60 mil em todo o mundo vivem, atualmente, com FC, sendo que a variação étnica é bastante ampla.1 Na população branca, um em cada 3.200 nascidos vivos é afetado pela doença, com um índice médio de transmissão de um em 26 casos.2 Outras taxas de incidência da doença por etnia incluem um em 9.200 hispânicos, um em 10.500 nativos norte-americanos, um em 15.000 afro-americanos e um em 30.000 asiáticos americanos.
Em termos mundiais, a prevalência é mais elevada na Europa e em países com descendência europeia e mais baixa na Ásia e na África. A título de exemplo, alguns estudos recentes estimaram uma prevalência de um em 70.000 na Índia e de um em 350.000 no Japão.3,4
Genética
O gene da FC, descoberto em 1989, se localiza no braço longo do cromossomo 7 e codifica a proteína de um canal de cloreto na superfície apical das células epiteliais.5 A delF508, uma deleção da fenilalanina na 508ª posição, foi a primeira e mais comum entre as mutações que foram descobertas. A partir de então, foram identificadas mais de 1.900 mutações genéticas. O órgão Cystic Fibrosis Mutation Database (http://www.genet.sickkids.on.ca/cftr/app) catalogou e mantém uma lista atualizada de mutações.
Entre as 1.900 mutações identificadas, apenas 10% causam FC, e apenas 22 alelos foram identificados com uma frequência mais comum que 0,1% de todos os alelos.6 Embora a significância das outras mutações não seja muito clara, trata-se de uma área de pesquisas em curso.
O site http://www.cftr2.org é um recurso bastante útil em que são apresentadas 160 das mutações mais comuns com dados para os resultados clínicos usuais (por exemplo, taxa prevista de declínio do volume expiratório forçado em 1 segundo [FEV1] e probabilidade de suficiência pancreática). No futuro, a grande meta será adicionar os dados dos resultados das mutações incomuns, na medida em que forem sendo coletados mais dados prospectivos.
A Tabela 1 contém as mutações mais comuns em FC.
Tabela 1
|
MUTAÇÕES MAIS COMUNS EM FIBROSE CÍSTICA, 2011 | |
|
Mutação |
Percentual de pessoas com uma ou duas mutações |
|
F508del |
86,8 |
|
G542X |
4,6 |
|
G551D |
4,4 |
|
R117H |
2,7 |
|
N1303K |
2,5 |
|
W1282X |
2,4 |
|
R553X |
1,8 |
|
621 + 1G->T |
1,7 |
|
1717- 1G->A |
1,6 |
|
3849 + 10kbC->T |
1,5 |
|
2789 5G->A |
1,3 |
|
3120- 1G->A |
1,0 |
Esta tabela foi adaptada com permissão do Cystic Fibrosis Foundation Patient Registry 2011 Annual Data Report, Bethesda, Maryland, © 2012 Cystic Fibrosis Foundation.1
A mutação delF508 corresponde a, aproximadamente, 87% dos alelos totais, sendo que nenhuma outra mutação chega a atingir 5% de alelos.7 Há uma heterogeneidade considerável para genes específicos, sendo que o rastreio genético do gene 23 abrange 90% das mutações em populações brancas e apenas 68% em hispânicos.8 Vários estudos enfatizaram a variação genética em diversos países. Por exemplo, na Itália, o perfil genético mostra apenas 49% com a mutação delF508, sendo que os genes N1303K e G542X têm 6% cada.9
O gene da FC codifica a proteína membranosa da superfície apical das células epiteliais que se denomina proteína do regulador da condutância transmembrana da FC (CFTR). Os diferentes genes da FC foram classificados com base no ponto em que a via celular da proteína encontra algum tipo de problema.
A mutação mais comum, delF508, é de classe II e encontra alguma dificuldade no estágio de tráfego proteico. As mutações das classes I-III (assim como as mutações da classe VI) geralmente produzem FC mais grave e foram associadas a taxas elevadas de mortalidade, FEV baixa, capacidade vital forçada (FVC) e redução na estatura e no peso.10
O Quadro 1 mostra as classes de mutações em FC.143
Quadro 1
|
CLASSES DE MUTAÇÕES EM FIBROSE CÍSTICA | ||
|
Classe |
Defeito molecular |
Exemplo de mutação |
|
I |
Gene FC com sinal de parada |
W1282X |
|
II |
Problemas no transporte de CFTR; degradação no retículo endoplasmático. |
delF508 |
|
III |
Sinal defeituoso para o funcionamento proteico na superfície apical; o canal não abre. |
G551D |
|
IV |
Falha na condutância do CFTR; diminuição no transporte de cloreto. |
R117H |
|
V |
Defeito na separação do CFTR com produção reduzida. |
3849 + 10kbC ->T |
|
VI |
Degradação mais rápida do CFTR. |
Q1412X |
FC: fibrose cística; CFTR: regulador da condutância transmembrana de FC.
Há uma ampla variação nos resultados clínicos, mesmo entre irmãos com o mesmo genótipo, e os genes modificadores passaram a ser o foco principal como área para pesquisas. Como exemplo, a lectina de ligação da manose (MBL), uma proteína envolvida na ativação de complementos, foi associada a uma redução substancial na função pulmonar em pacientes com FC e aos alelos da variante da MBL.11
Fisiopatologia e Patogênese
A proteína do CFTR se localiza basicamente na superfície apical das células epiteliais. Órgãos como pulmões, pâncreas e fígado são os mais afetados. Acredita-se que a causa principal de doença pulmonar fenotípica seja a interrupção no transporte de ferro.
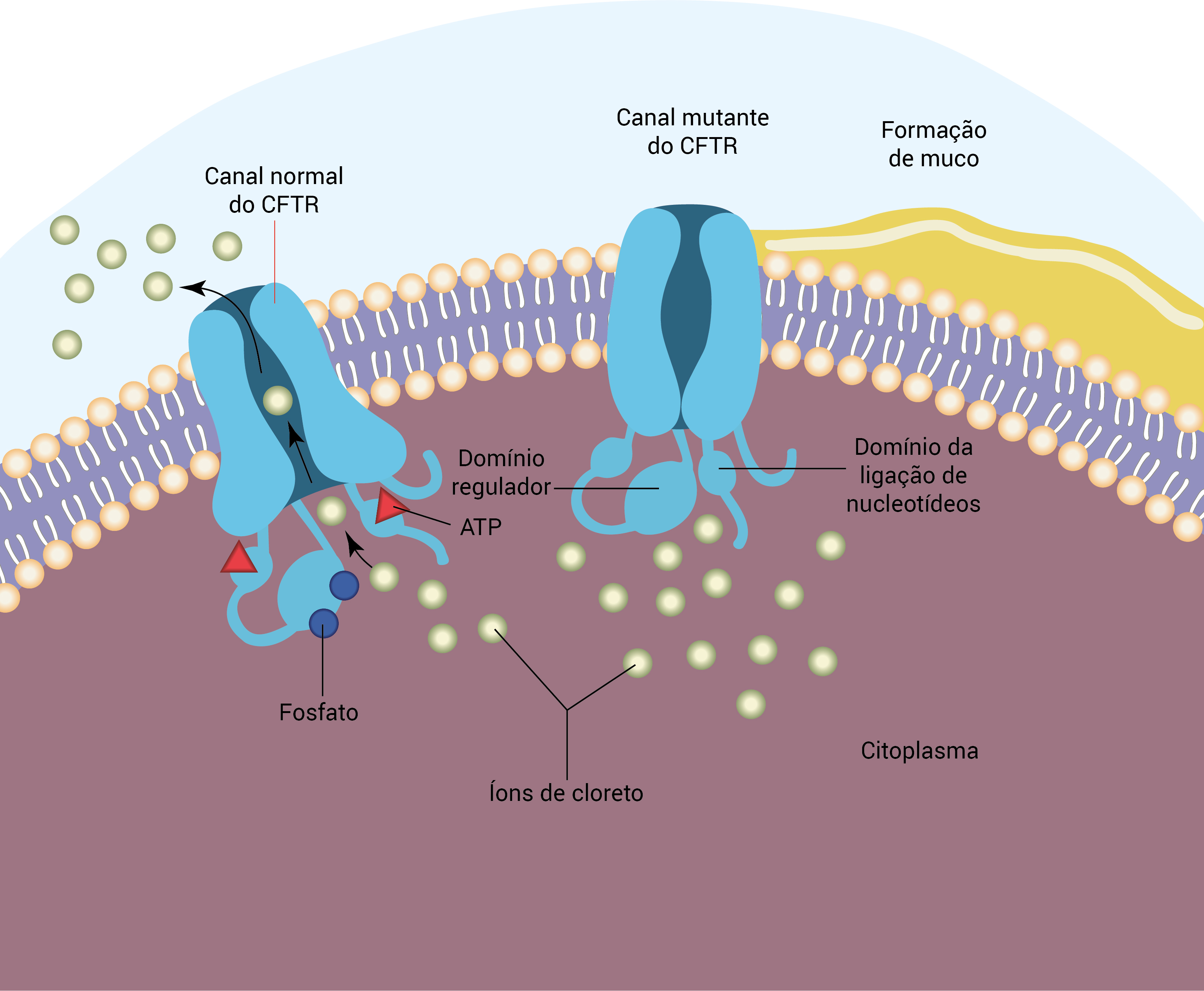
O cloreto não tem capacidade para deixar a célula de forma apropriada, levando, consequentemente, à reabsorção de sódio e água provenientes da camada superficial das células epiteliais, como mostra a Figura 1. O resultado final é a desidratação das superfícies das vias respiratórias e os problemas subsequentes com a limpeza mucociliar.12 Esse cenário cria o estágio para colonização bacteriana, inflamação com a liberação de citocinas e bronquiectasia em um ciclo repetitivo.13 A fisiopatologia das doenças hepáticas e pancreáticas pode envolver questões como hidratação de superfícies epiteliais alteradas e secreções inspissadas.

ATP: trifosfato de adenosina; CFTR: condutância transmembrana de FC.
Figura 1 - Regulador da CFTR normal e anormal.
Diagnóstico
Manifestações Clínicas
As manifestações clínicas de FC variam amplamente e dependem da gravidade da doença e da idade do paciente no momento do diagnóstico. Um dos princípios que se deve ter sempre em mente é que as complicações pulmonares são as principais fontes de morbidade e mortalidade.
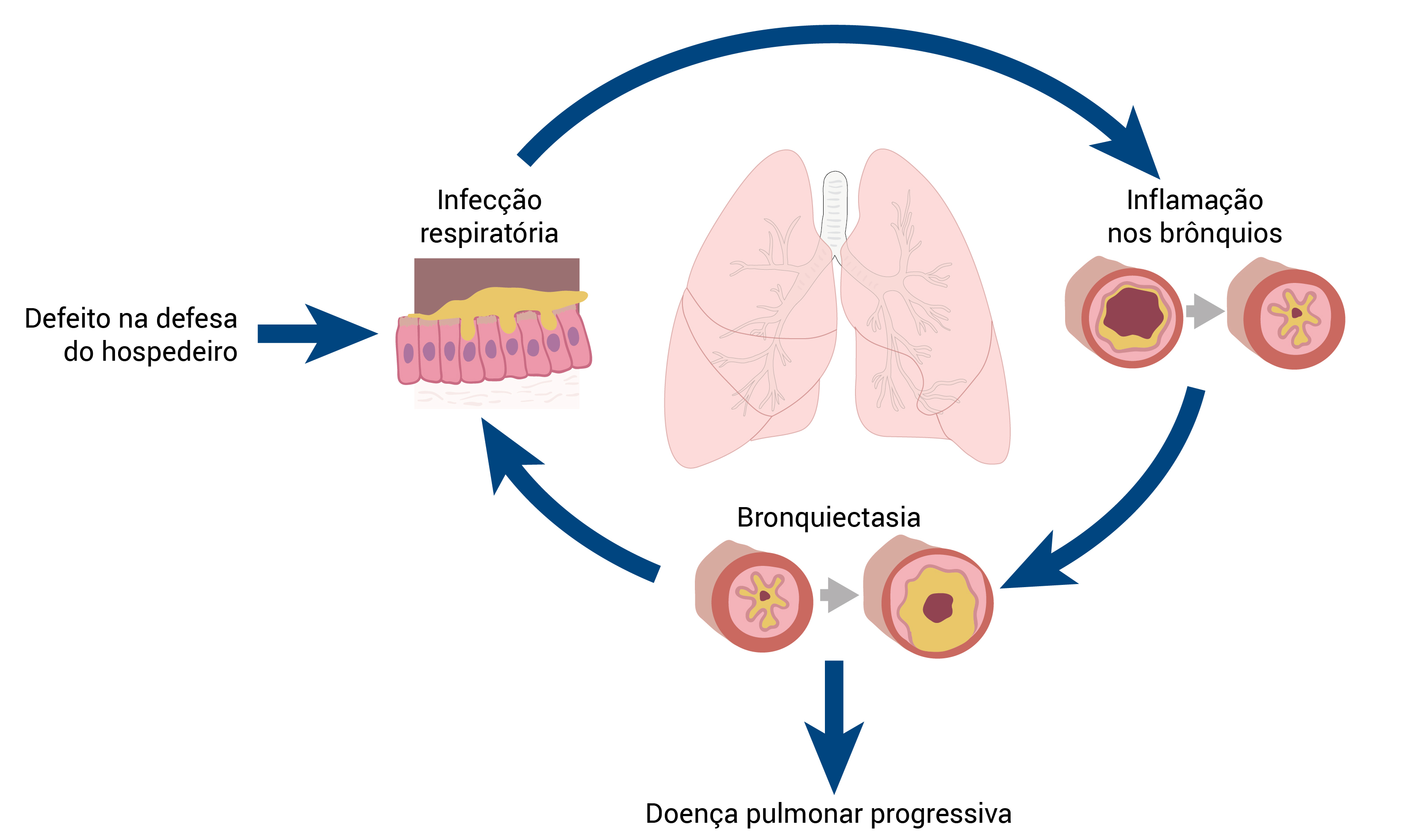
A Figura 2 mostra o círculo vicioso hipotético de uma lesão pulmonar.
Figura 2 - Círculo vicioso hipotético de uma lesão pulmonar.
Manifestações Típicas por Idade
Aproximadamente, 10% de pacientes com FC são neonatos com íleo meconial, que é o bloqueio das primeiras fezes. De um modo geral, essa situação quase sempre é uma indicação da presença de FC. Outras manifestações comuns em neonatos incluem icterícia prolongada e atresia intestinal.
As manifestações gastrintestinais são comuns na primeira infância e na infância, tendo em vista que a insuficiência pancreática poderá se apresentar precocemente. A perda de enzimas digestivas apropriadas cria um retardo na progressão do crescimento em combinação com diarreia crônica e esteatorreia.
Flatulência, fezes gordurosas e distensão abdominal são outros sintomas de insuficiência pancreática.14 Condições como pancreatite crônica ou recorrente e sinusite, com ou sem pólipos nasais, também são observadas nessa fase da vida. O sintoma pulmonar inicial na infância é tosse seguida da produção de esputo.
A partir do momento em que os pacientes com FC atingem a adolescência ou a vida adulta, as manifestações pulmonares se tornam dispositivas em combinação com a inflamação em curso e a destruição tecidual. Finalmente, ocorre o desenvolvimento de bronquiectasia nos lobos superiores.
O fluxo expiratório forçado reduzido de 25 a 75% e o aumento na proporção entre volume residual/capacidade pulmonar total (RV/TLC) possivelmente sejam indicadores precoces de doença pulmonar, cujo padrão obstrutivo óbvio é indicado pela redução na proporção FEV1/FVC que ocorre mais tarde.15 Outras alterações evidenciadas pelo teste da função pulmonar são aprisionamento de ar com volume residual elevado e hiperinsuflação dos pulmões aumentando a capacidade pulmonar total.
Microbiologia das Vias Respiratórias com Fibrose Cística
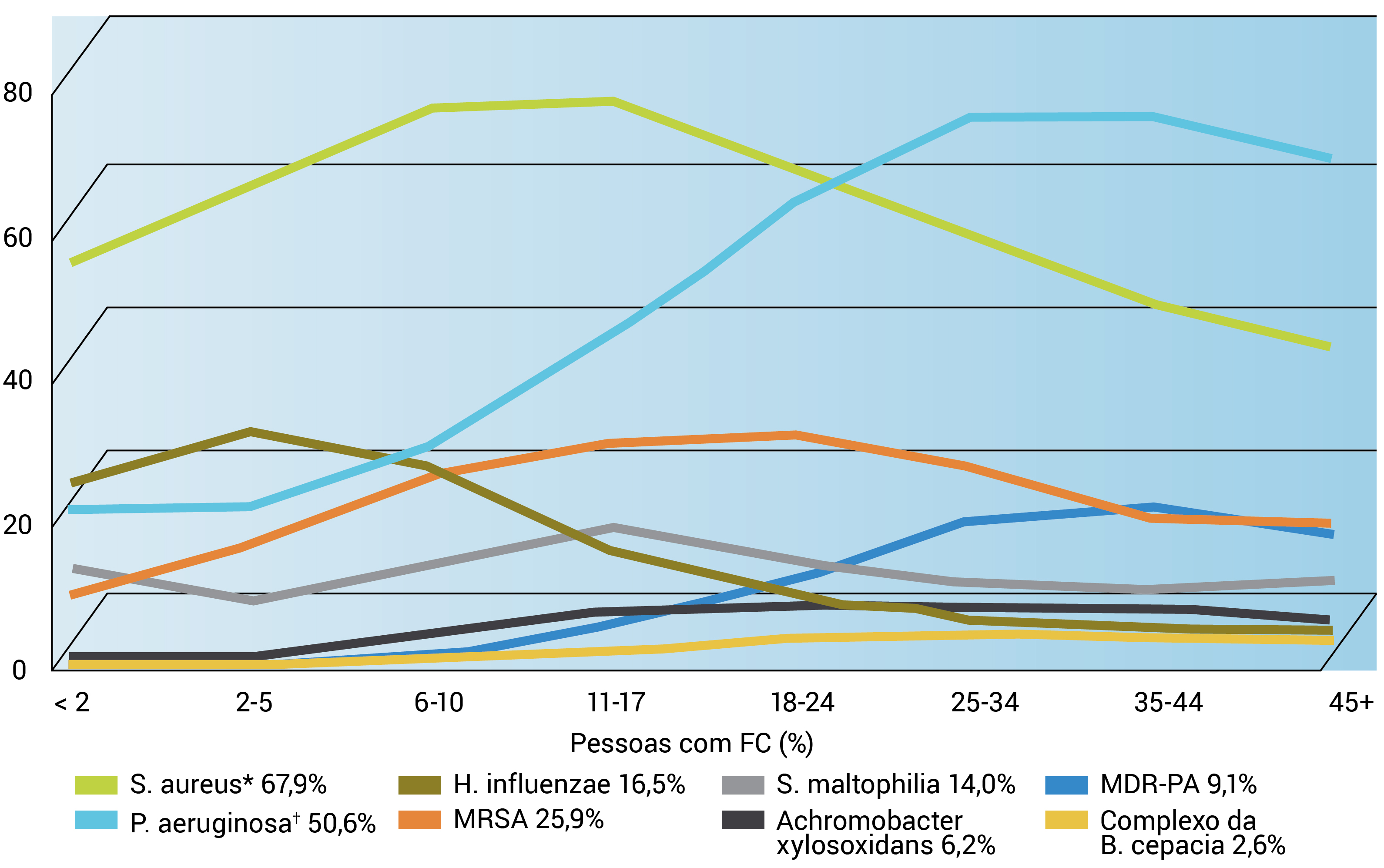
Na fase inicial da doença, é muito comum que as crianças com FC sejam colonizadas pelos organismos Staphylococcus aureus ou Haemophilus influenzae, embora, mais tarde, o Pseudomonas aeruginosa se torne o organismo dominante. As infecções pseudomonais são causadas inicialmente por um fenótipo não mucoide, que progride para fenótipo mucoide com a produção de alginato e uma camada subsequente de biofilme.16 A Figura 3 apresenta um gráfico do fator tempo e do percentual de organismos por idade em casos de FC.
Figura 3 - Germes respiratórios por idade. Este gráfico foi adaptado com permissão do Cystic Fibrosis Foundation Patient Registry, 2011 Annual Data Report, Bethesda, Maryland, © Cystic Fibrosis Foundation.1 *Staphylococcus aureus inclui pessoas com S. aureus resistente à meticilina (MRSA).
?Pseudomonas aeruginosa inclui pessoas com P. aeruginosa resistente a diversos medicamentos (MDF-PA).
Conforme mostra a Figura 3, a colonização de Pseudomonas em pulmões com FC ocorre em 60% de pacientes com idade entre 18 e 24 anos e em 80% de pacientes na faixa etária de 25 a 28 anos. A penetração da função imune e dos antibióticos na camada de biofilme produzida pelo fenótipo mucoide é extremamente difícil.
Os pacientes com colonização pelo Pseudomonas apresentam perda acelerada da função pulmonar em comparação com indivíduos sem esse organismo, assim como uma tendência para aumento na mortalidade, particularmente em pacientes que adquirirem o fenótipo mucoide.17,18
Conforme mencionado anteriormente, o S. aureus é um colonizador precoce. O organismo Staphylococcus aureus foi associado ao aumento nos marcadores inflamatórios nos pulmões, enquanto que o Staphylococcus aureus resistente à meticilina (MRSA) foi associado ao declínio na função pulmonar.19,20 O Cystic Fibrosis Patient Registry registrou um aumento dramático nas culturas positivas de esputo em casos de FC no período de 1996 a 2011 (de 2,1 para 25%).1
Outros organismos que são frequentemente cultivados a partir das vias respiratórias nos casos de FC incluem Achromobacter, Stenotrophomonas, Aspergillus, a espécie Burkholderia e microbactérias atípicas. Excetuando-se as espécies Burkholderia, não está suficientemente claro se o organismo cultivado é um colonizador ou um contribuinte ativo para doenças pulmonares. O julgamento clínico e os testes de suporte geralmente ajudam a tomar a decisão de tratar pessoas com um microrganismo individual.
A taxa de colonização do organismo Stenotrophomonas varia de 7 a 40%, sendo que um estudo mostrou que não ocorreram resultados adversos nesses pacientes.21,22 A colonização do Achromobacter xylosoxidans ocorre em menos de 10% de pacientes e, aparentemente, não acelera o declínio da função pulmonar.23 Recentemente, a frequência da colonização com a espécie Aspergillus foi calculada em 14%.21
As culturas positivas são indicações para a existência de aspergilose broncopulmonar alérgica (ABPA), definida como uma reação de hipersensibilidade à espécie Aspergillus. Respiração ofegante e presença de infiltrados pulmonares são ocorrências típicas. Os resultados dos testes laboratoriais de suporte incluem nível de IgE total acima de 500IU/mL, teste cutâneo positivo e nível elevado específico para imunoglobulina G (IgG) ou nível de imunoglobulina E (IgE) para a espécie Aspergillus.24 Ainda não está suficientemente claro se a espécie Aspergillus é um patógeno autêntico na ausência de ABPA.
A taxa de culturas de espécies microbacterianas atípicas é de 13%, sendo que 72% desses isolados correspondem ao complexo Mycobacterium avium (MAC) e 16% ao Mycobacterium abscessus.25 Há uma clara associação entre M. abscessus e o declínio na função pulmonar, ao passo que no curto prazo o MCA afeta a FEV1.26 As varreduras por tomografia computadorizada (TC) do tórax ajudam a decidir sobre a necessidade de terapia, sendo que uma das descobertas usuais é a formação de agrupamentos de nódulos inflamatórios.
O complexo Burkholderia é um conjunto de nove espécies distintas (genomovares), sendo que os isolados mais comuns são os organismos Burkholderia multivorans e Burkholderia cenocepacia. Considerados conjuntamente, esses dois isolados correspondem a, aproximadamente, 60 a 85% do total, dependendo do país em que foi realizada a pesquisa.27,28
A espécie Burkholderia provoca doenças graves com o declínio acelerado da função pulmonar e aumenta a mortalidade em comparação com pacientes com FC que não foram infectados por esse tipo de organismo.29 Um subgrupo de pacientes infectados pela espécie Burkholderia também desenvolve início rápido no declínio dos pulmões, acompanhado por pneumonia necrosante e sepse devastadora, que se denomina síndrome por cepacia.30
A teoria de que os pulmões são colonizados por vários organismos diferentes, ao invés de um único organismo dominante, se tornou o foco das pesquisas mais recentes. Novas técnicas moleculares conhecidas por microbioma pulmonar comprovaram que os pulmões com FC podem abrigar mais de 60 gêneros diferentes de bactérias.31 Um estudo documentou que, além dos organismos Pseudomonas e S. aureus, o Streptococcus anaeróbico, Rothia e Prevotella também eram comuns.32
O acompanhamento de pacientes ao longo do tempo mostrou que, aparentemente, a diversidade das espécies se perde na medida em que os indivíduos envelhecem e perdem a função pulmonar, sugerindo que a manutenção da homeostase de muitas espécies diferentes é um aspecto importante para manter saudáveis os pulmões com FC.33
Sintomas Pulmonares
A tosse que caracteriza a FC na infância evolui para bronquiectasia e produção crônica de esputo, que geralmente ocorrem no início da vida adulta. Juntamente com os sinais obstrutivos observados nos testes da função pulmonar, a doença progressiva cria uma incompatibilidade na ventilação-perfusão e, finalmente, condições como hipercarbia e hipoxemia.6
Observa-se, com frequência, respiração ofegante e reversibilidade com uso de broncodilatadores. Esses sintomas imitam asma e sugerem que a remodelagem dos músculos lisos tem papel importante nesse processo.34 Um estudo longitudinal de cerca de 1 ano demonstrou que quase a metade dos pacientes apresentou responsividade extrema, pelo menos, uma vez, com mais frequência no inverno do que no verão.35
Inflamação nas vias respiratórias e colonização bacteriana resultam em um aumento na vascularidade e na quantidade de vasos sanguíneos frágeis, sendo que a hemoptise poderá se tornar um grande problema. Assim como na maior parte dos casos, a hemoptise tem origem na circulação da artéria dos brônquios e poderá colocar os pacientes em risco de vida. A colonização crônica dos cistos pulmonares pela espécie Aspergillus é uma ocorrência provável (conhecida por aspergiloma) que poderá precipitar a incidência de hemoptise.
A hemoptise pode se tornar maciça e é definida como mais de 240mL em um período de 24 horas ou mais de 100mL/dia durante vários dias. No entanto, isso poderá colocar em risco a vida dos pacientes como decorrência da perda das vias respiratórias e da asfixia. Em um estudo retrospectivo, a hemoptise maciça ocorreu em 0,87% de pacientes por ano e em 4,1% de todos os pacientes no curso de 10 anos.36 Os riscos da doença incluíram idade avançada e função pulmonar mais baixa.
Pneumotórax espontâneo, assim como a hemoptise, é outra complicação em pacientes mais velhos com função pulmonar mais baixa. A explicação tradicional é o rompimento das bolhas subpleurais na pleura visceral, embora estudos mais recentes indiquem que elevação na pressão alveolar, rompimento causado pelo tamponamento mucoso e inflamação, possivelmente sejam os fatores mais importantes.37
Uma revisão retrospectiva que cobriu um período de 10 anos e envolveu pacientes com FC e pneumotórax mostrou uma incidência anual de 0,64% e uma incidência cumulativa de 3,4% durante todo o período.38 A idade média foi de 22 anos, sendo que 75% dos casos tinham FEV1 abaixo da previsão de 40%. Os pacientes com pneumotórax permaneceram mais tempo no hospital e apresentaram taxa de mortalidade mais elevada depois de 2 anos.
Uma das causas frequentes de mortalidade em pacientes com FC é o agravamento subagudo dos sintomas pulmonares, também conhecido por exacerbação aguda. Fatores como tosse, aumento no volume de esputo, perda de peso e queda na FEV1 são frequentes, embora ainda não exista uma definição prospectiva válida de exacerbação aguda. Vários sistemas diferentes de pontuação foram utilizados em testes clínicos, sendo que os sintomas podem ser mais importantes que os sinais objetivos.39
A frequência da exacerbação aguda aumenta com o avanço da idade e com FEV1 mais baixa, e, ao redor dos 18 anos de idade, aproximadamente 50% de pacientes apresentam uma ou mais exacerbações por ano que exigem terapia antibiótica intravenosa.40
O Quadro 2 apresenta um método mnemônico bastante útil.
Quadro 2
|
MÉTODO MNEMÔNICO PARA EXACERBAÇÕES AGUDAS DEFIBROSE CÍSTICA: CF PÂNCREAS |
|
C = Cough/Tosse (aumento na gravidade ou frequência) |
|
F – Fever/Febre (usualmente de grau baixo) |
|
P = Pulmonary function decline/Declínio na função pulmonar |
|
A = Appetite/Apetite (reduzido) |
|
N = Nutrition/Nutrição (perda de peso) |
|
C = Complete blood count/Hemograma completo (leucocitose) |
|
R = Radiograph/Radiografia (aumento na bainha peribronquial, tamponamento com muco, excesso de aeração) |
|
E = Examination/Exame (estalidos, respiração ofegante, uso de músculos acessórios) |
|
A = Activity/Atividade (tolerância reduzida a exercícios) |
|
S = Sputum/Esputo (mudança de cor, consistência, quantidade) |
Este quadro foi adaptado com permissão de Schidlow DV.14
Sintomas Gastrintestinais
O espectro amplo das doenças gastrintestinais tem uma grande prevalência na população com FC, incluindo doença pancreática, intestinal e hepatobiliar.
Doença pancreática. Insuficiência pancreática talvez seja o sinal inicial da doença e, antes da introdução das enzimas pancreáticas, foi a causa principal de mortalidade em crianças com FC. A má absorção de gorduras e enzimas é proeminente e resulta no retardo da progressão do crescimento. As deficiências das vitaminas A, D, E e K também são ocorrências prováveis e poderão resultar em anemia, neuropatia, cegueira noturna e diáteses hemorrágicas. Normalmente, a incidência de pancreatite aguda é uma possibilidade em pacientes com mutações mais brandas.41
Doença gastrintestinal. Os problemas gastrintestinais provavelmente sejam comuns nos casos de FC e incluem doença por refluxo gastroesofágico (DRGE), constipação, síndrome da obstrução intestinal distal (Soid), prolapso retal, crescimento excessivo do intestino delgado e intussuscepção.
Aparentemente, a incidência de DRGE é muito mais elevada em pacientes com FC do que na população em geral, sendo que um estudo pediátrico revelou a incidência de 26,5% versus 5,6% em controles compatíveis.42 Tradicionalmente, se considera a SOID como o equivalente adulto ao íleo paralítico e ela se manifesta como início rápido de dor abdominal e obstrução total ou parcial por fezes na válvula ileocecal.43 Vale a pena fazer a distinção entre início lento de dor abdominal e enchimento do colo, observados nos casos de constipação, cuja incidência também é maior em pacientes com FC.
Embora os valores absolutos sejam baixos, parece haver um aumento na incidência de cânceres intestinais, em especial no intestino delgado, no colo e no trato biliar. Esse quadro é mais pronunciado em pacientes que tenham feito transplante de pulmão.44
Colonopatia fibrosante é uma complicação intestinal final em casos de FC que foi observada originalmente em 1990 após o advento das enzimas entéricas pancreáticas revestidas. A apresentação usual se caracteriza pela presença de dor abdominal e constipação, que poderá confundi-la com Soid.45
Em estudos de enemas com contraste, sob o ponto de vista radiográfico, a colonopatia fibrosante se assemelha à doença de Crohn e, sob o ponto de vista patológico, há um espessamento submucoso produzido por tecido conjuntivo fibroso sem inflamação. A colonopatia fibrosante foi associada ao uso de doses elevadas de enzimas pancreáticas.46
Doença hepatobiliar. O espectro das doenças hepáticas observado em pacientes com FC varia de esteatose hepática a cirrose biliar focal, cirrose biliar multilobular e colelitíase. Acredita-se que a etiologia de doença hepática nos casos de FC seja a obstrução causada por bile espessa e inspissada, assim como malnutrição e inflamação sistêmica.47
Até 72% de pacientes com FC, com idade aproximada de 24 anos, apresentam alterações hepáticas patológicas após a morte, sendo que uma pequena minoria apresenta elevações no nível de fosfatase alcalina ou de aspartato aminotransferase.48 A significância desse fato não é muito clara e a incidência de doença hepática clinicamente significativa é muito mais baixa.
O universo de 1 a 2% de pacientes que desenvolvem cirrose e suas complicações geralmente é formado por pacientes pediátricos com idade média de 10 anos.49 Cabe observar que, mesmo em indivíduos cirróticos, a função hepática poderá ser preservada por muitos anos. Não é comum ver função pulmonar bem preservada em pacientes com doença hepática grave.
Problemas metabólicos
Os problemas metabólicos associados com mais frequência à FC são diabetes melito relacionado à FC (CFRD, do inglês cystic fibrosis-related diabetes), doença óssea e nefrolitíase.
CFRD. As modificações estruturais no pâncreas iniciam precocemente em pacientes com FC, sendo que o percentual de pacientes com insuficiência pancreática antes dos 5 anos de idade atinge 75%; as anormalidades são demonstradas por ultrassonografia.50 Ao longo do tempo, ocorre a destruição das células de ilhotas, assim como a reposição constante das células que produzem insulina, na presença de amiloide e fibrose.51
Esse quadro abaixa os níveis insulínicos normais e eleva os níveis de glicose. Aparentemente, a autoimunidade não tem papel importante nesse processo, embora o mecanismo exato da perda das células de ilhotas não seja muito claro.52
A incidência de CFRD aumenta com o avanço da idade e as estimativas mostram que, aproximadamente, 50% de pacientes se apresentam com a doença ao redor dos 30 anos de idade.53 Os pacientes com FC apresentam os sintomas típicos de poliúria, polidipsia e perda de peso durante um terço do tempo. Outros sintomas são declínio inexplicável na função pulmonar e redução na velocidade de crescimento.54
O Quadro 3 contém o diagnóstico de diabetes melito relacionado à FC em pacientes estáveis.55
Quadro 3
|
DIAGNÓSTICO DE DIABETES MELITO RELACIONADO A FIBROSE CÍSTICA EM PACIENTES ESTÁVEIS |
|
1. Teste oral de tolerância à glicose ao nível de 2 horas ? diabetes melito se =200mg/dL* 2. Teste de confirmação com um entre os seguintes testes: Hemoglobina A1C =6,5% Glicose em jejum =126mg/dL Glicose aleatória =200mg/dL com poliúria ou polidipsia |
FC: fibrose cística.
*Nos casos em que o resultado for anormal, este teste deverá ser repetido em dias diferentes.
Uma orientação consensual recente recomenda a realização de varreduras anuais, iniciando na idade de 10 anos, com testes de tolerância à glicose e medições da hemoglobina glicosilada (hemoglobina A1C).55 O teste oral de tolerância à glicose é o padrão para fazer o diagnóstico, embora níveis elevados em jejum ou da hemoglobina A1C devam ser usados como testes de confirmação. Cabe observar que a hemoglobina A1C pode ser falsamente baixa em pacientes com FC, de modo que não é confiável quando for considerado isoladamente.
Alguns centros estão usando rotineiramente o monitoramento contínuo da glicemia, que é um indicador mais moderno e potencialmente mais sensível do status do diabetes melito. Esse método monitora a glicemia a cada 5 minutos durante todo o dia e também à noite. Um estudo que utilizou o monitoramento contínuo conseguiu traçar um paralelo entre peso e declínio da função pulmonar e níveis elevados de açúcar no sangue de mais de 4,5% do tempo total por um dia.56
Doença óssea. A doença óssea observada nos casos de FC provavelmente seja multifatorial. Deficiência das vitaminas D e K, malnutrição e aumento de reabsorção durante episódios infecciosos têm papel importante nesse processo.57 Essas deficiências aumentam o risco de ocorrência de fraturas vertebrais e não vertebrais, sendo que um estudo envolvendo uma população de adultos mostrou a prevalência de 14%.58
Não chega a causar nenhuma surpresa o fato de que as pontuações da densidade mineral óssea total em pacientes com FC também sejam baixas, e em 23% dos casos ficam abaixo da linha de corte para osteoporose.59 O uso de glicocorticoides foi relacionado a densidades minerais ósseas ainda mais baixas, sendo que essa situação é exacerbada por transplantes de pulmão.60
A artropatia por FC (AFC) é uma condição que inicia na idade média de 15 anos e afeta entre 2 e 8% de pacientes com FC.61 Descreve-se AFC como um curso recidivante de monoartrite ou poliartrite que afeta as grandes articulações dos joelhos, tornozelos, ombros, punhos e cotovelos.
De um modo geral, as exacerbações têm 5 a 7 dias de duração, sendo que os resultados das radiografias e dos testes com anticorpos são negativos.62 A AFC possivelmente esteja associada a erupções cutâneas semelhantes a eritema nodoso ou febre, porém, aparentemente, não está associada à função pulmonar mais baixa ou a exacerbações agudas.63
Nefrolitíase. O risco de cálculos renais aumenta em pacientes com FC. Embora os estudos não tenham utilizado controles, a média de seis estudos realizados ao longo dos anos mostrou a incidência de 4,1% (51 entre 1.231 pacientes), sendo que a maioria dos casos ocorreu na fase final da adolescência e no início da vida adulta (idade média entre 13 e 27 anos).64 A incidência foi de 1% em um estudo de grupos de controle por idade envolvendo indivíduos saudáveis na faixa de 20 a 29 anos.65
Acredita-se que o mecanismo exato da formação de cálculos tenha sido multifatorial, em que a excreção urinária alterada de oxalato e de ácido úrico desempenhou papel importante nesse processo. Houve também grande interesse na ausência da bactéria Oxalobacter formigenes que decompõe o oxalato no colo, aumentando sua absorção e a secreção através da urina.66
Sintomas Reprodutivos
A fertilidade é reduzida tanto em homens como em mulheres com FC, ainda que essa situação seja muito mais acentuada nos homens. Aproximadamente, 98% dos homens com FC são estéreis por causa da ausência bilateral congênita do ducto deferente, que não pode ser corrigida por meios cirúrgicos.67 De um modo geral, os 2% de homens com ducto deferente normal são pacientes com mutações mais brandas ou atípicas.68
A análise dos espermatozoides é o primeiro passo na avaliação de problemas reprodutivos em pacientes do sexo masculino com FC; a finalidade desse tipo de teste é confirmar a presença de azoospermia. No caso de confirmação da azoospermia, os espermatozoides poderão ser retirados dos testículos por aspiração percutânea de espermatozoides do epidídimo (em inglês, percutaneous epididymal sperm aspiration [Pesa]) ou por aspiração testicular de espermatozoides (em inglês, testicular sperm aspiration [Tesa]) com anestesia local. Tipicamente, as taxas de recuperação de espermatozoides são excelentes.69
Após a colheita, faz-se a inseminação artificial ou a injeção intracitoplasmática do espermatozoide. Em um estudo, as taxas de fertilização de óvulos por injeção intracitoplasmática de espermatozoides foram de 60% e os índices de gravidez foram de 40% por ciclo.70 Outro estudo apresentou um índice de gravidez semelhante em quatro de oito casais, resultando no nascimento de três conjuntos de gêmeos e um nascimento simples.71
Embora cerca de 20% de mulheres com FC tenham problema de fertilidade, as causas são diferentes, levando-se em conta que elas têm anatomia estruturalmente normal. Observaram-se algumas alterações no muco cervical, sendo que os fatores contribuintes incluíam desidratação na linha de base e ausência de diluição na ovulação.72 Além disso, condições como malnutrição e doença em estado avançado estão associadas à infertilidade pela predisposição a ciclos anovulatórios e amenorreia secundária.
O efeito da gravidez em pacientes com FC e os resultados fetais também foram objetos de várias investigações. Embora os registros iniciais tenham sido desfavoráveis em ambos os aspectos, a gravidez na FC se tornou relativamente comum e segura. Entretanto, ainda se recomenda prudência, sendo que as mulheres com exacerbações frequentes, ou com outras comorbidades ou estressores, devem levar em consideração a carga adicional da gravidez e de um recém-nascido.
Um estudo recente de pequeno porte registrou os casos de oito mulheres com FC que haviam passado por 11 gestações. Em 10 dos 11 casos, a paciente apresentou função pulmonar estável durante a gravidez, sendo que 4 entre as 11 apresentaram FEV1 inferior à previsão de 50%.73 Na Cidade de Toronto, no Canadá, um estudo de maior porte apresentou os resultados de 92 gestações em 54 mulheres entre 1961 e 1998.74
Na realidade, aparentemente, a gravidez não afetou a função pulmonar, e a mortalidade depois de 11 anos não foi diferente em comparação com a população em geral com FC. Nesse estudo, duas mulheres apresentaram FEV1 abaixo de 35% e se saíram muito bem durante e depois da gravidez, de modo que é difícil estabelecer um ponto de corte para gestações bem-sucedidas.
Os resultados neonatais nos dois estudos mencionados anteriormente foram semelhantes aos da população em geral, com uma ligeira tendência para partos prematuros, em especial nas mulheres com função pulmonar mais baixa. Observou-se um aumento na incidência de diabetes melito gestacional, sugerindo a necessidade de varreduras mais frequentes.75
Problemas Psiquiátricos
A frequência das comorbidades médicas é cada vez maior, levando-se em consideração que os indivíduos com FC estão vivendo mais. A depressão não é uma exceção à regra, sendo que há um aumento de incidência na população com FC, com início na adolescência e na vida adulta.
No entanto, não está muito claro se a depressão aumenta em pacientes com FC em comparação com a população em geral, sendo que um estudo registrou um aumento de 30% em uma população de adultos, e outro estudo, um aumento de 11% em um grupo com faixa etária entre 21 e 30 anos.76,77
Não chegou a causar nenhuma surpresa o fato de que alguns pacientes com função pulmonar mais baixa, complicação médica recente ou dor significativa tenham apresentado taxas de depressão mais elevadas.78 É extremamente importante verificar a presença de um desses fatores de risco ou se surgiram outros estressores.
Ainda não se estudou com o rigor necessário a prevalência de abuso de substâncias em casos de FC. Um estudo mais antigo mostrou que, em média, os adolescentes com FC abusam menos de tabaco, álcool e maconha em comparação com seus pares.79 Um estudo recente de uma pesquisa envolvendo 100 pacientes revelou uma taxa de abuso de bebidas alcoólicas de 16% e uma taxa de abuso de drogas de 4%.80
Considerando que o mix de enfermidade crônica e abuso de substâncias não é sustentável, vale a pena considerar a avaliação de pacientes com comportamentos de risco e oferecer orientação o quanto antes.
Exame Físico
O exame físico nos casos de FC varia de acordo com a gravidade da doença e do órgão afetado. Com frequência, os seios nasais são afetados por pólipos e rinite. O exame dos pulmões possivelmente revele a presença de hiper-ressonância, acompanhada de respiração ofegante e roncos.
Há certa prevalência de desconforto respiratório e do uso de músculos acessórios para aumentar a inspiração, principalmente nos casos de exacerbações agudas. Nos quadrantes inferiores, é possível palpar a hepatoesplenomegalia e a impactação fecal. Hipocratismo digital nos dedos das mãos e dos pés é uma ocorrência comum. O enrugamento aquagênico das palmas das mãos é um sinal físico raro associado exclusivamente à FC.
O surgimento de placas ou pápulas esbranquiçadas é muito comum após a imersão total das mãos em água.81 Possivelmente, o enrugamento aquagênico esteja associado a dores ou queimaduras, e acredita-se que seja decorrência do teor elevado de sal na pele de pacientes com FC.
Testes Laboratoriais
Tradicionalmente, a FC sempre foi diagnosticada com base na combinação de sintomas clínicos ou danos em órgãos e evidências biológicas de disfunção no regulador da CFTR. Define-se disfunção no CFTR como teste positivo de suor, que se caracteriza pela varredura genética que mostra duas mutações causadoras da doença ou uma diferença nasal potencialmente positiva.
Entretanto, os dados sugerem que diagnosticar FC no nascimento versus aguardar por sintomas clínicos é uma situação benéfica sob o ponto de vista nutricional e sob a perspectiva da função pulmonar e da mortalidade, o que levou à instituição de varreduras em recém-nascidos.82,83
A partir de 2010, todos os Estados norte-americanos instituíram a realização de varredura obrigatória em recém-nascidos, sendo que, aproximadamente, 60% dos diagnósticos de FC nos EUA em 2011 foram feitos com embasamento nas varreduras em recém-nascidos.1 Trata-se de um aumento dramático desde 2004, quando apenas 10% de casos foram diagnosticados a partir do rastreamento.
Os detalhes sobre o momento em que cada Estado começou a fazer o rastreamento e o protocolo exato que foi utilizado poderão ser encontrados no site http://www.cff.org/AboutCF/Testing/NewbornScreening/ScreeningforCF/, incluindo os links com o site de cada Estado.
As varreduras para FC que são feitas na maior parte dos estados iniciam primeiramente com a verificação de níveis elevados de triptogênio imunorreativo (TIR), seguida por análise do DNA para verificar a eventual presença de mutações na FC nos casos em que o nível do TIR for muito elevado.
O TIR é um precursor de enzimas pancreáticas cujo nível é elevado em recém-nascidos com FC; trata-se de um teste sensível, porém inespecífico. Todos os recém-nascidos com rastreamento positivo para FC pela análise TIR/DNA são avaliados com o teste de cloreto no suor para confirmar o diagnóstico.84
O diagnóstico de FC é uma área que vem sendo debatida com bastante frequência. Aproximadamente, 90% dos pacientes se apresentam com os sinais e sintomas clássicos, nível elevado de cloreto no suor e duas mutações causadoras da doença. Nos 10% de pacientes remanescentes, o valor do teste de cloreto no suor poderá ser obtido imediatamente, e o teste genético geralmente apresenta uma ou duas mutações, cuja significância não é muito clara.
Em reação a toda essa confusão, a North American Cystic Fibrosis Foundation (NACFF) decidiu publicar uma declaração de consenso.85 Essa declaração reconhece que os pacientes com FC se apresentam em idades variáveis e, de um modo geral, com envolvimento de sistemas de órgãos diferentes, desde doença sinusal isolada a patologia isolada do fígado ou do pulmão.
A NACFF reconhece que a obtenção rápida do diagnóstico de FC é importante para os resultados e que o encaminhamento dos pacientes a um centro acreditado de FC é uma abordagem razoável nos casos em que o diagnóstico não for suficientemente claro.
O Quadro 4 contém os valores do teste de suor (mmol/L).
Quadro 4
|
VALORES DO TESTE DE SUOR (MMOL/L) | ||
|
|
<6 meses de idade |
=6 meses de idade |
|
Normal |
<30 |
<40
|
|
Intermediário |
30?59 |
50?59
|
|
Anormal |
=60 |
=60
|
O teste de cloreto no suor ainda é o mais importante para detectar evidências de disfunção no regulador de CFTR. O teste de suor dever ser feito em locais acreditados que seguem os protocolos dos métodos de coleta. O teste de cloreto no suor tem algumas limitações, considerando que um dos estudos mostrou que 13,5% dos pacientes com FC apresentaram resultados inferiores a 60mmol/L e, em um estudo de pacientes com suficiência pancreática, 21% apresentaram níveis inferiores a 60mmol/L.86,87
Os testes genéticos são mais úteis nas situações em que houver valores intermediários de cloreto no suor. Entretanto, os valores apresentados no teste de cloreto no suor também não são absolutos, sendo que, nos casos de FC, apenas 9,7% de pacientes apresentaram uma das mutações que haviam sido identificadas.40
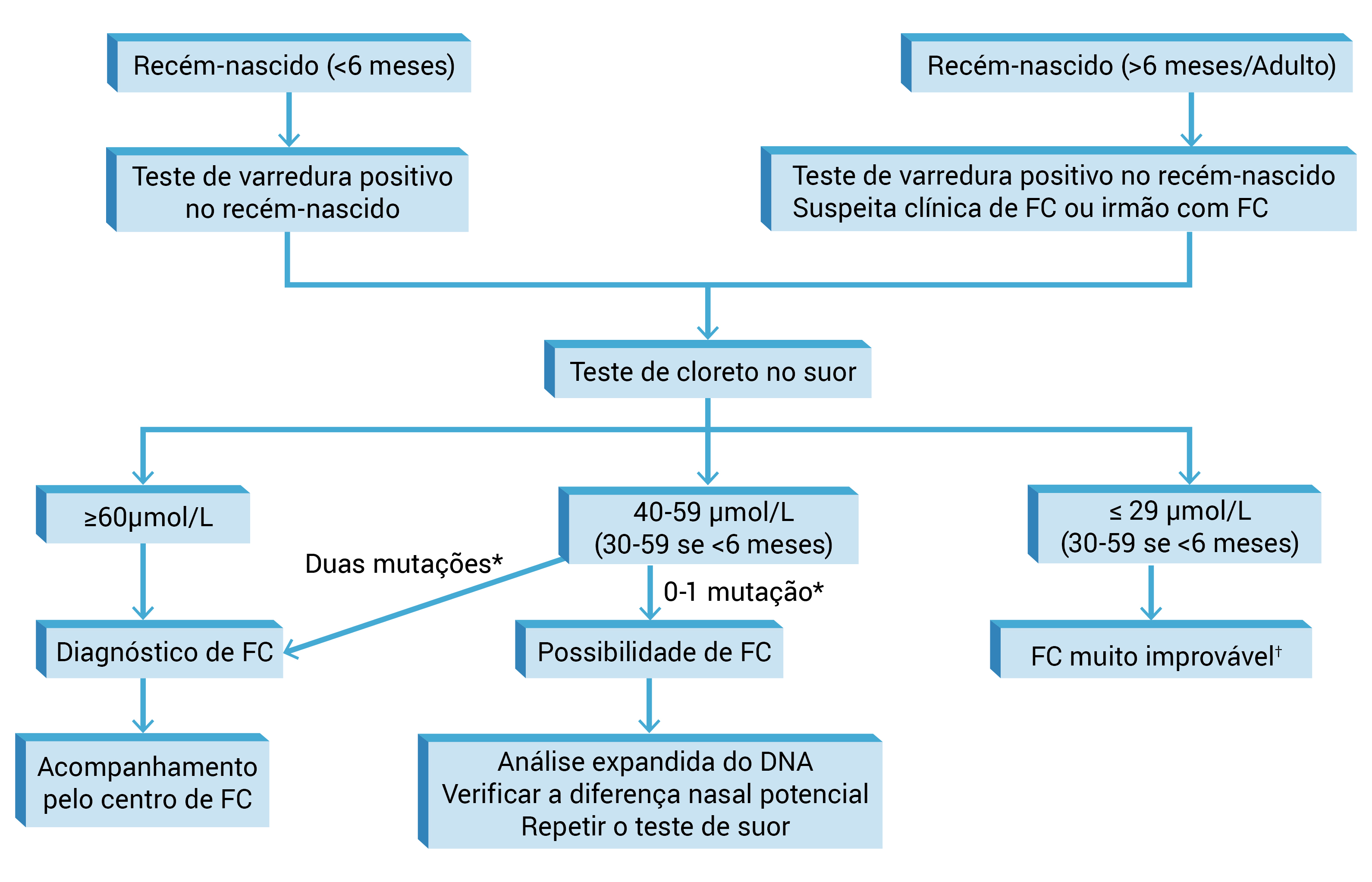
A Figura 4 apresenta um algoritmo resumido das orientações da declaração recente de consenso para fins diagnósticos.
DNA: ácido desoxirribonucleico; FC: fibrose cística.
*As mutações devem causar reconhecidamente FC fenotípica.
?O diagnóstico poderá ser feito na presença de dois genes.
Figura 4 - Diagnóstico de FC.85
Há um grande esforço em curso para aprimorar a classificação do significado dos resultados anormais do teste de suor e do rastreamento genético em todas as faixas, assim como estão em andamento alguns estudos longitudinais de resultados e de identificação de genes modificadores. Há um consenso de que a maior parte dos desafios diagnósticos poderá se concentrar no cenário de recém-nascidos, tendo em vista que um percentual maior de rastreamentos é feito nessa população.
Um grupo único de pacientes que não haviam sido classificados como portadores de FC, mas que corriam o risco de contrair a doença, foi designado como portador de doença relacionada ao CFTR. Esses pacientes têm disfunção em um único órgão isolado como, por exemplo, azoospermia, pancreatite ou bronquiectasia e, possivelmente, apresentem valores intermediários no teste de cloreto no suor.
Essa população deve ser acompanhada de perto para verificar a hipótese de conversão potencial para FC manifesta. Os lactentes que atenderem aos critérios diagnósticos após uma varredura suspeita inicial também devem ser acompanhados longitudinalmente e, atualmente, são classificados como portadores de síndrome metabólica relacionada ao CFTR.
Imagens em Fibrose Cística
Embora os estudos radiológicos não façam parte dos critérios principais para o diagnóstico de FC, as imagens podem ser muito úteis como testes auxiliares, em especial no que diz respeito ao diagnóstico em questão. Em pacientes pediátricos, a hiperinsuflação é o primeiro sinal de alteração nas radiografias, que poderá ser observada nos diafragmas planos, no aumento do espaço intercostal das costelas e no aumento do ar retroesternal.88
Os sinais tardios incluem evidências de inflamações com espessamento peribronquial e nódulos espalhados, seguidos pelo desenvolvimento de bronquiectasia. Normalmente, a bronquiectasia se localiza nos lobos superiores.89
As varreduras por TC apresentam visões mais detalhadas do parênquima pulmonar em comparação com as radiografias torácicas, além de serem mais sensíveis para o diagnóstico de bronquiectasia.90 Nas varreduras por TC, define-se bronquiectasia como uma via respiratória maior que o vaso adjacente.
As varreduras por TC mostram também espessamento peribronquial, nódulos espalhados devido à impactação mucoide dos bronquíolos e ao aprisionamento de ar. O aprisionamento de ar pode ser mais bem avaliado comparando-se as imagens expiratórias com as imagens inspiratórias.
Diagnóstico Diferencial
O diagnóstico diferencial de doença pulmonar causada por FC geralmente inclui os distúrbios que produzem bronquiectasia difusa. Esses tipos de distúrbios incluem discinesia ciliar primária (DCP), imunodeficiência, doenças autoimunes, doença intestinal inflamatória e ABPA.
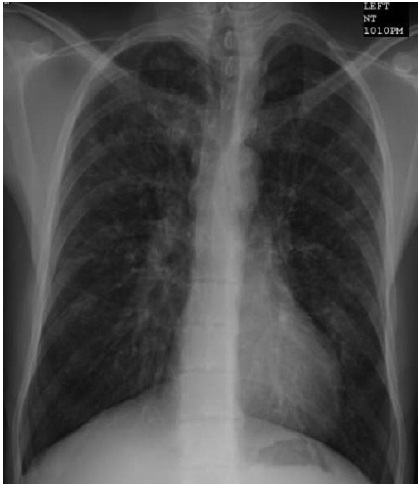
A Figura 5 ilustra a radiografia torácica de FC.
FC: fibrose cística.
Figura 5 - Radiografia torácica de FC. Observe a hiperinsuflação e a doença cística predominante no lobo superior bilateral.
Levando-se em consideração que a FC poderá se apresentar com disfunção de órgãos isolados, o diagnóstico diferencial depende do órgão especifico que estiver envolvido no processo da doença. Tipicamente, inclui-se a FC nas discussões sobre doença sinusal isolada, pancreatite, insuficiência hepática e infertilidade (especialmente em homens).
O diagnóstico diferencial para disfunção de órgãos em cada um desses sítios está fora do escopo deste artigo, embora seja interessante observar que doença sinusal e infertilidade estão associadas a estados de doença sem FC que também produzem bronquiectasia.
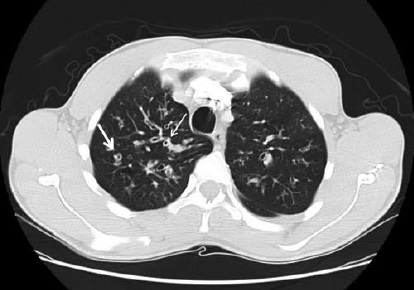
A Figura 6 ilustra a varredura por TC de FC.
FC: fibrose cística; TC: tomografia computadorizada.
Figura 6 - Varredura por TC de FC. Evidência de anel tipo sinete (seta grossa) e espessamento peribronquial (seta fina).
O Quadro 5 mostra o diagnóstico diferencial de bronquiectasia.113
Quadro 5
|
DIAGNÓSTICO DIFERENCIAL DE BRONQUIECTASIA |
|
Causas pós-infecciosas: espécies de micobatérias (Aspergillus), vírus (HIV, influenza, adenovírus), bactérias (Pseudomonas), aspiração.
|
|
Imunodeficiência: hipogamaglobulinemia, leucemia linfocitária crônica, leucemia linfocitária aguda, pós-transplante de células-tonco.
|
|
Doença autoimune: artrite reumatoide, síndrome de Sjögren, doença intestinal inflamatória.
|
|
Condições congênitas: DCP, FC, deficiência de a1-antitripsina, traqueobroncomegalia (síndrome de Mounier-Kuhn), deficiência de cartilagem (síndrome de Williams-Campbell).
|
|
Outras causas: corpo estranho, síndrome de Young, síndrome da unha amarela, síndrome de Job. |
DCP: discinesia ciliar primária; HIV: vírus da imunodeficiência adquirida.
Tratamento
Doença Pulmonar
Os pesquisadores de FC e a Cystic Fibrosis Foundation têm sido bastante agressivos na busca de novas terapias. Informações mais atualizadas sobre os estudos em curso, a fase em que se encontram e o que já foi aprovado estão à disposição no site da Cystic Fibrosis Foundation (http://www.cff.org/research/DrugDevelopmentPipeline/).
A Cystic Fibrosis Foundation classificou os componentes do tratamento nas respectivas classes. Tradicionalmente, os pacientes com FC fazem várias terapias uma ou mais vezes ao dia. O comprometimento com o tempo necessário para essas terapias é bastante significativo e, em geral, a adesão às terapias é um problema comum.
Limpeza das Vias Respiratórias
Uma das terapias mais antigas e mais recomendadas é a limpeza das vias respiratórias. A meta é mobilizar as secreções crônicas e melhorar a função pulmonar e o bem-estar dos pacientes. Existem vários métodos disponíveis no mercado, incluindo o ciclo respiratório ativo, dispositivos de oscilação da parede torácica de alta frequência (incluindo o colete vibratório), fisioterapia manual convencional no tórax e dispositivos de pressão positiva, entre outros.
Uma revisão recente feita por Cochrane não mostrou nenhuma evidência de que um método seja superior ao outro.91 Existem poucos dados de longo prazo documentando a eficácia desses métodos, embora os dados de curto prazo mostrem que há uma redução no volume de esputo.92
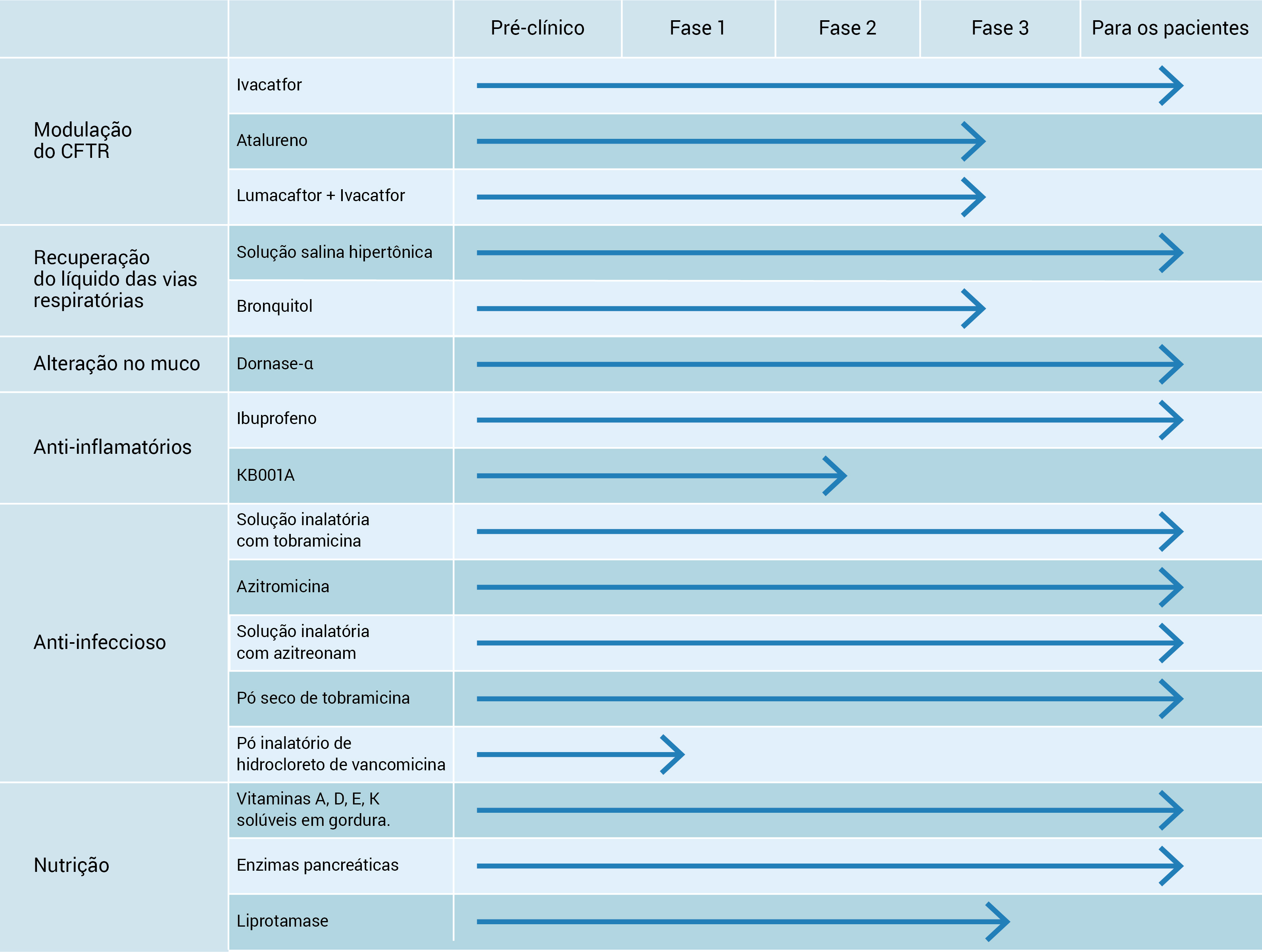
A Figura 7 mostra as etapas terapêuticas da FC.
FC: fibrose cística.
Figura 7 - Etapas terapêuticas da FC.
Esta figura foi adaptada com permissão da Cystic Fibrosis Foundation. Etapas do desenvolvimento farmacológico. Disponível no site http://www.cff.org/research/DrugDevelopmentPipeline/ (acesso em 4 de abril de 2013).
Mobilização de Secreções
As duas terapias aprovadas para melhorar a mobilização de secreções são a dornase-a (DNAse I) e a solução salina hipertônica. A dornase-a é um agente mucolítico que age por meio da clivagem do DNA liberado degenerando os neutrófilos. Diversos estudos mostraram os benefícios, sendo que o maior deles comprovou que houve melhoras de 5,8% na FEV1 em 900 pacientes com doença pulmonar moderada durante período de 6 meses.93
A solução salina hipertônica também produziu alguns benefícios, sendo que um estudo envolvendo 164 pacientes usando uma solução a 7% demonstrou que houve uma melhora modesta na FEV1. Além disso, esses pacientes apresentaram taxas reduzidas de exacerbação e um maior comparecimento ao trabalho.94
Antibióticos
Não se recomenda o uso regular de antibióticos, embora os antibióticos inalatórios sejam os grandes pilares do tratamento de determinados tipos de pacientes.92 Um estudo pioneiro de 1997 sobre o uso de tobramicina em pacientes com colonização crônica pela espécie Pseudomonas demonstrou que houve uma melhora significativa na FEV1 e uma redução na taxa de hospitalização depois de 24 semanas em comparação com placebo.95
Um estudo complementar mostrou que houve uma melhora persistente depois de 2 anos, com ligeiro aumento na resistência, que não teve nenhuma correlação com aumento no uso de antibióticos.96 O uso de aztreonam inalatório é uma alternativa para a tobramicina; recentemente, foi publicado um teste randomizado que revelou a ocorrência de melhoras na função pulmonar e nos sintomas respiratórios.97
Embora os dados sejam escassos, a colistina é uma terceira opção de antibiótico inalatório preferida por alguns pacientes cuja ação é bastante satisfatória. Tradicionalmente, os antibióticos inalatórios são aplicados como solução nebulizada e exigem tempo substancial para a administração. Em 2013, nos EUA, foi aprovada uma formulação de pó seco de tobramicina inalatória em um teste randomizado de resistência com 6 meses de duração,98 que representou uma economia significativa de tempo para os pacientes.
Broncodilatadores
Os broncodilatadores, normalmente ß-agonistas de ação prolongada, são utilizados com bastante frequência nos casos de FC. Muitos pacientes com FC têm asma ou reatividade brônquica extrema, sendo que um estudo envolvendo 113 pacientes mostrou que 51% apresentaram reação positiva à metacolina.99
Excetuando-se a asma, não houve nenhuma correlação com os testes cutâneos ou com rinite alérgica. Vários estudos de curto prazo também mostraram que houve melhoras na FEV1 com uso de ß-agonistas.100 Recomenda-se o uso diário de ß-agonistas em pacientes com resposta aos broncodilatadores ou com hiper-reatividade brônquica. Os dados sobre a utilização de agentes anticolinérgicos não são tão convincentes; as orientações mais recentes da Cystic Fibrosis Foundation indicam que as evidências não são suficientes para recomendar o uso desse medicamento.92
Agentes Anti-Inflamatórios
Acredita-se que as respostas inflamatórias intensas às bactérias sejam a principal causa de bronquiectasia e de agravamento da função pulmonar.101 O uso crônico de antibióticos macrolídeos, com possível efeito anti-inflamatório, melhorou ligeiramente a função pulmonar, porém não de forma significativa.102 O nível de evidências é mais marcante em pacientes com colonização crônica do organismo Pseudomonas.
Os dados positivos dão suporte ao uso de ibuprofeno, embora a maior parte das melhorias no teste maior tenha ocorrido em pacientes com idade inferior a 13 anos.103 As orientações mais recentes para tratamento de FC recomendam o uso de ibuprofeno apenas em pacientes na faixa etária de 6 a 17 anos.92 Recomenda-se fazer o monitoramento rigoroso nos níveis do medicamento.
Diversos outros compostos também foram testados, tais como esteroides orais, esteroides inalatórios, cromolina e modificadores do leucotrieno. Considerando a qualidade variável de evidências, não se recomenda nenhum desses medicamentos para uso rotineiro Uma exceção aos esteroides ou aos modificadores do leucotrieno seria o diagnóstico concomitante de ABPA ou asma.
Moduladores dos Reguladores da Condutância Transmembrana da Fibrose Cística
A classe de medicamentos moduladores de proteínas foi a primeira tentativa para corrigir o defeito básico da FC ao invés de tratar os danos posteriores. Até o presente momento, o ivacaftor foi o primeiro medicamento dessa classe a ser aprovado. A Food and Drug Administration o aprovou em janeiro de 2012 para uso em pacientes com FC com genótipo incluindo, pelo menos, uma cópia da mutação G551D (mutação de classe III com prevalência de 4,4%).
Um teste randomizado controlado envolvendo 167 pacientes constatou que houve melhoras significativas na FEV1, peso, necessidade de antibióticos, nível de cloreto no suor e na pontuação de bem-estar.104 A função pulmonar melhorou depois de 2 semanas e as mudanças permaneceram depois de 48 semanas.
A meta atual é estender o uso de ivacaftor para outras mutações de classe III e desenvolver outros moduladores de proteínas com foco em classes diferentes de mutações. O mais importante entre esses estudos atualmente em curso é uma combinação de ivacaftor e lumacaftor em pacientes homozigotos para a mutação IF508.
Se os resultados forem positivos, esse medicamento poderá abranger uma porção significativa da população com FC. Embora não se acredite que esses medicamentos curem a FC, a grande expectativa é que ajudem a retardar bastante o declínio pulmonar e se tornem parte importante do tratamento.
Gerenciamento de Exacerbações Agudas
Os principais focos do tratamento de exacerbações agudas são adesão às terapias de longo prazo, intensificação na limpeza das vias respiratórias e uso oral ou intravenoso de antibióticos. A decisão sobre a via de administração de antibióticos depende do julgamento clínico, sendo que os principais fatores são sintomas, nível de queda da FEV1 e experiência anterior do paciente. A escolha do antibiótico se fundamenta nas culturas anteriores e no sucesso dos tratamentos prévios.
Um documento orientador publicado pela Cystic Fybrosis Foundation fez uma revisão dos dados mais recentes.105 Recomenda-se o uso de dois medicamentos, normalmente um aminoglicosídeo e um ß-lactâmico, nos casos de pacientes com colonização do organismo Pseudomonas que precisam de administração intravenosa de antibióticos. Os testes de suscetibilidade e de sinergia não apresentaram nenhum benefício útil sob o ponto de vista clínico.106
Nas situações em que o organismo S. aureus resistente à meticilina também estiver presente, o procedimento padrão é adicionar vancomicina ou linezolida. Nos casos em que houver suporte apropriado, a terapia doméstica pode ser tão eficaz quanto a hospitalar.
Transplante de Pulmão
As orientações de encaminhamento para avaliação de transplante de pulmão foram publicadas pela International Society of Heart and Lung Transplantation (ISHLT) e incluem FEV1 abaixo de 30%, exacerbações frequentes e recorrência de pneumotórax ou hemoptise.107 O benefício da sobrevida foi bastante evidente nos casos de pacientes com FEV1 abaixo de 30%.108
De acordo com os casos mais recentes publicados pela ISHLT, a sobrevida de pacientes com FC que receberam transplante de pulmão é de 7,5 anos; a rejeição crônica é o fator que mais contribui para a mortalidade.109 Trata-se de um resultado melhor em comparação com outras categorias de doença.
Colonização crônica pela espécie Burkholderia é uma contraindicação relativa específica para FC. Levando-se em consideração que o transplante de pulmão não é uma cura definitiva, a decisão de prosseguir com esse tipo de procedimento é uma escolha pessoal de alguns pacientes.
Nutrição
Os cuidados com a nutrição foram identificados como o grande pilar do tratamento ideal. Uma declaração de consenso da Cystic Fibrosis Foundation recomenda acompanhar regularmente o crescimento dos pacientes pediátricos e fazer a intervenção o mais rapidamente possível, caso ela seja necessária.110 Em adultos, recomenda-se um índice de massa corporal (IMC) mínimo de 22 para mulheres e 23 para homens.
Estados nutricionais subótimos foram associados a uma função pulmonar mais baixa e a piores resultados.111 Dois terços dos pacientes com FC nascem com insuficiência pancreática e 85% poderão ter insuficiência pancreática em algum momento na vida.112 Esses pacientes devem ser tratados com enzimas de reposição. Recomenda-se fazer um estudo de elastase fecal nos casos em que houver alguma dúvida sobre a insuficiência pancreática. A ingestão de calorias e de gorduras não deve ser restrita a não ser nos casos de obesidade.
O monitoramento anual dos biomarcadores nutricionais é extremamente importante. Isso inclui níveis de monitoramento das vitaminas solúveis em gorduras A, D, E e K, juntamente com o teste anual de tolerância à glicose e o nível de hemoglobina glicosilada.
Quaisquer problemas nutricionais identificados devem ser suplementados o mais rapidamente possível. Recomenda-se fazer uma varredura anual com absortometria por raios X de dupla energia para verificar a saúde dos ossos, sendo que a suplementação de cálcio e de vitamina D é o tratamento de primeira linha.
Prognóstico
Nos dias atuais, o prognóstico para pacientes com FC é bem melhor do que em qualquer outro momento na história, sendo que a sobrevida média é de 37 anos.1 A melhora nos resultados resultou principalmente do melhor gerenciamento da função pulmonar e da nutrição. O advento dos modificadores da proteína do CFTR é um dos desenvolvimentos recentes mais importantes no mundo das pesquisas e promete melhorar ainda mais a expectativa de vida dos pacientes.
Bronquiectasia não Fibrose Cística
Definição da Doença
As doenças pulmonares supurativas além da FC englobam uma ampla gama de entidades, cada uma delas com uma patogênese exclusiva. Essas doenças são relacionadas pela característica morfológica dos brônquios dilatados de forma permanente por inflamações e infecções crônicas, em combinação com as características clínicas de tosse crônica e produção de esputo.
As exacerbações agudas são comuns e geralmente exigem intervenção com antibióticos. Os testes da função pulmonar revelam a presença de doença obstrutiva com queda na proporção FEV1/CVF. As radiografias torácicas mostram evidências de doença pulmonar cística, embora as varreduras torácicas por TC tenham se tornado o padrão para diagnosticar e quantificar a extensão da doença.
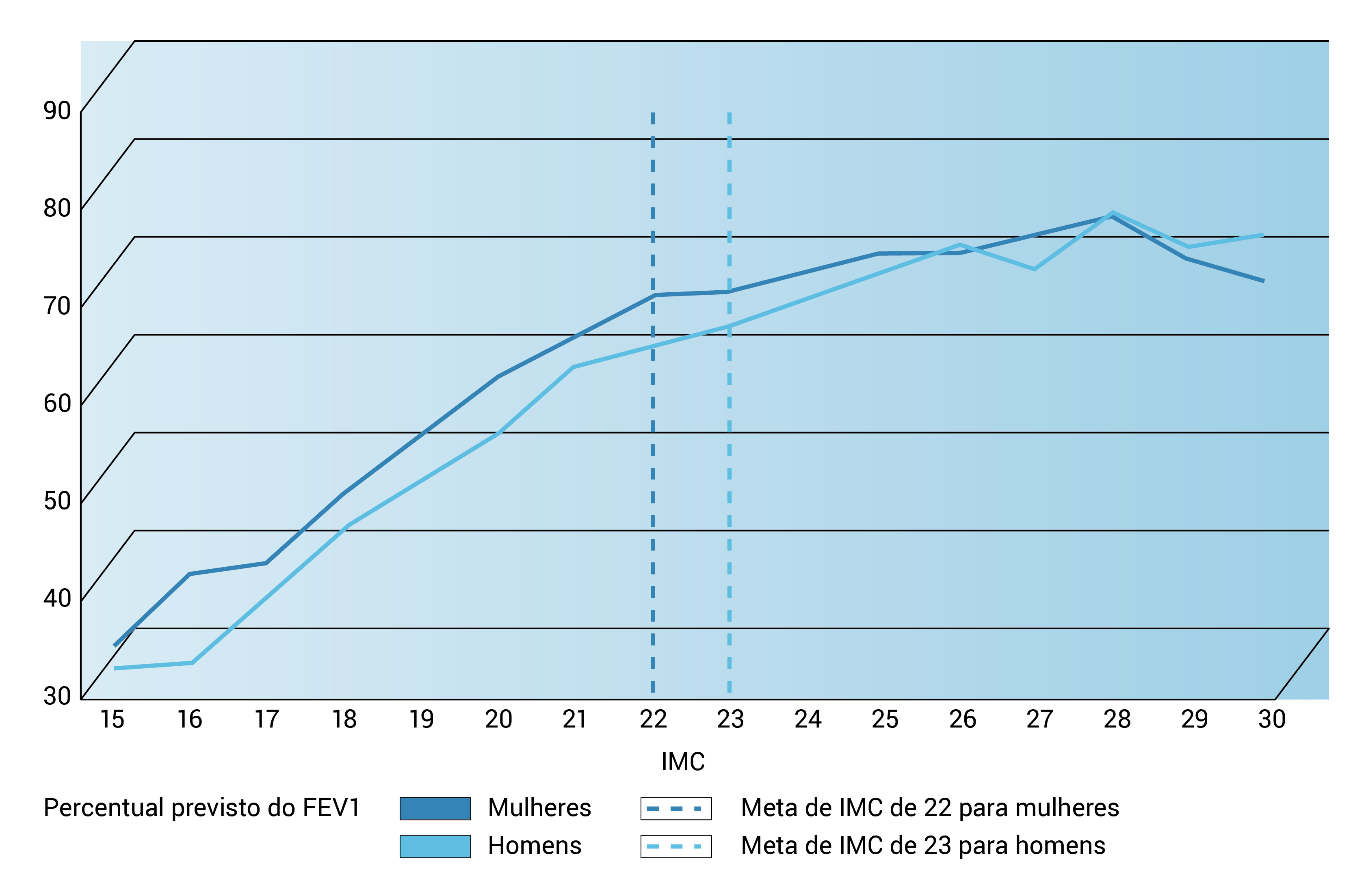
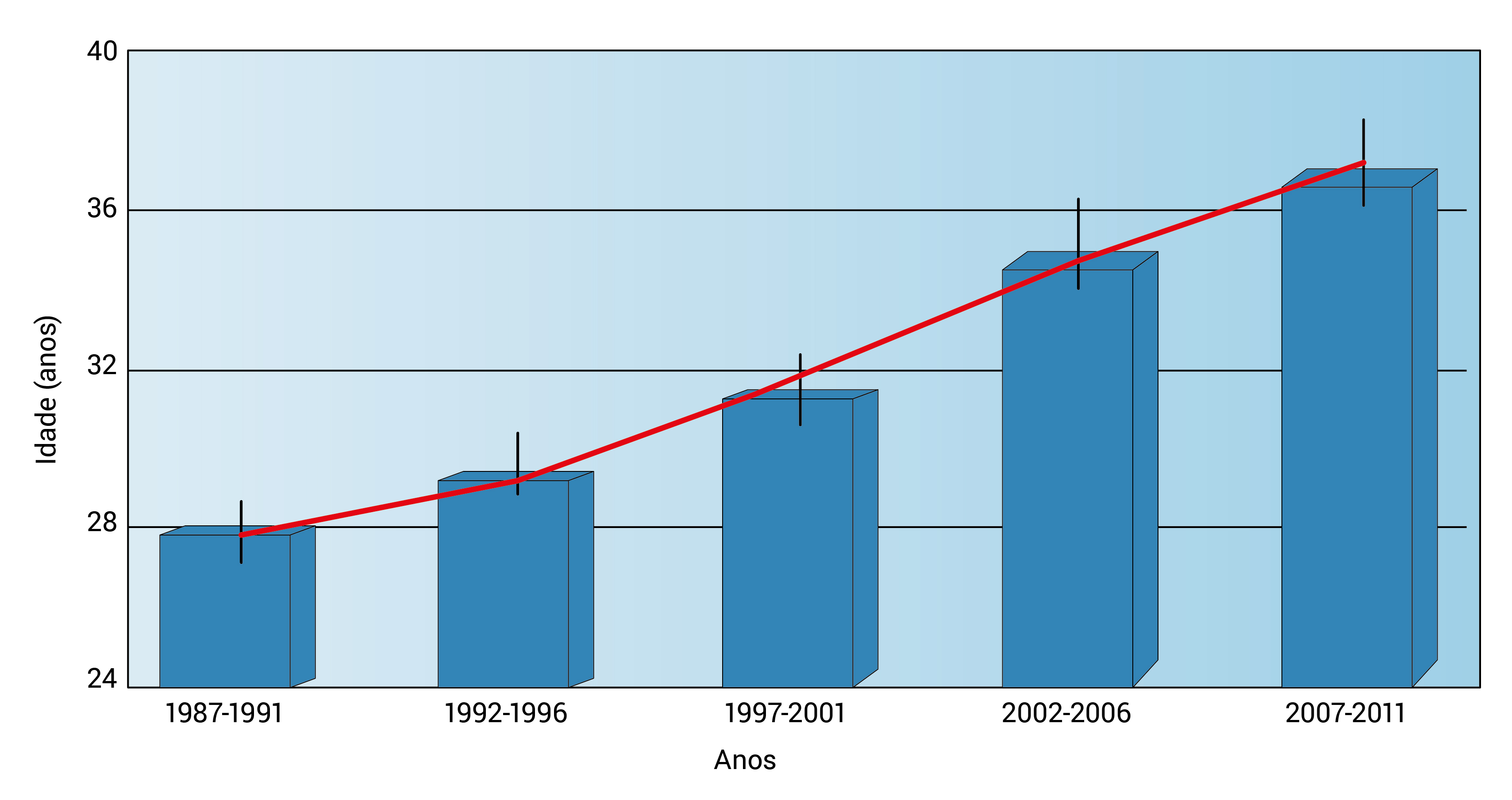
A Figura 8 mostra o FEV1 em relação ao percentual previsto da função pulmonar versus DCP e a Figura 9, a idade mediana de sobrevida prevista em pacientes com FC por períodos de 5 anos, de 1987 a 2011.
FEV1: volume expiratório forçado em 1 segundo; IMC: índice de massa corporal.
Figura 8 - FEV1 em relação ao percentual previsto da função pulmonar versus IMC para adultos de 20 a 40 anos de idade, 2011. Esta figura foi adaptada com permissão da Cystic Fibrosis Foundation Patient Registry 2011 Annual Data Report, Bethesda, Maryland, © 2012 Cystic Fibrosis Foundation.1
Figura 9 - Idade mediana de sobrevida prevista em pacientes com FC por períodos de 5 anos, de 1987 a 2011. Esta figura foi adaptada com permissão da Cystic Fibrosis Foundation Patient Registry 2011 Annual Data Report , Bethesda, Maryland, © 2012 Cystic Fibrosis Foundation.1
Epidemiologia
A prevalência de bronquiectasia é difícil de ser quantificada levando-se em conta que não há dados prospectivos. Este tema está sendo avaliado pelo Bronchiectasis Research Consortium, que, a partir de 2008, começou a cadastrar pacientes com base nos dados recebidos de 11 locais nos EUA. Informações adicionais sobre esse consórcio poderão ser obtidas no site http://www.cscc.unc.edu/bron/.
Foram feitas várias estimativas usando dados retrospectivos da saúde. O primeiro desses estudos se baseou em uma coorte durante o período de 1999 a 2001 e estimou uma prevalência média anual de 52,3 casos por 100 mil adultos.113 A prevalência da doença aumentou com a idade, juntamente com aumento na participação feminina em todas as faixas etárias.
Um segundo estudo de 8 anos, de 2000 a 2007, também fez uma revisão de dados e mostrou uma prevalência anual de 138 casos por 100 mil adultos.114 Uma prevalência mais elevada foi observada em mulheres, em pessoas mais velhas e em asiáticos.
É interessante observar que a prevalência de bronquiectasia aumentou uma média de 8,7% por ano e pode também ser uma indicação do aumento no número de diagnósticos e de uma maior consciência da doença ou de um aumento na incidência. A intensificação no uso de varreduras por TC possivelmente tenha um papel importante nessa tendência de aumento na prevalência desde os estudos mais antigos aos estudos mais recentes.
Etiologia
As causas de bronquiectasia são classificadas de acordo com a área focal ou difusa da doença observada nas imagens. As causas focais da doença incluem área prévia de pneumonia, presença de algum corpo estranho ou compressão extrínseca de um grande linfonodo. As principais causas difusas de bronquiectasia são DCP, deficiências de imunoglobulina, doença autoimune, ABPA, deficiência de a1-antitripsina e síndrome de Mounier-Khun (traqueobroncomegalia).115
A causa exata da doença em pacientes específicos depende da idade e da localização geográfica. Um estudo pediátrico realizado no Reino Unido mostrou que, em um grupo de 136 pacientes (idade média de 12,1 anos), que haviam sido examinados extensivamente, 67% tinham imunodeficiência, aspiração ou DCP.116
No geral, a etiologia foi identificada em 74% e, em 57% dos casos, o exame completo resultou em alteração no gerenciamento. Um estudo recente feito com 106 pacientes revelou a etiologia em 93% de casos, sendo que os diagnósticos mais comuns foram doença autoimune (33 pacientes), deficiência de imunoglobulina (17), malignidade hematológica (15), aspiração (12), deficiência de a1-antitripsina (12) e doença micobacteriana não tuberculosa (10).117
Patogênese
Causas Infecciosas
O desenvolvimento de pneumonia na infância pode predispor as pessoas para bronquiectasia mais tarde. Um estudo encontrou uma associação entre pneumonia antes da idade de 7 anos e redução na função pulmonar na quarta década de vida, evidenciada por obstrução nos testes da função pulmonar.118
A doença micobacteriana não tuberculosa vem cada vez mais sendo reconhecida como causa de bronquiectasia, geralmente em mulheres mais velhas e mais altas e magras em comparação com os controles na mesma faixa etária.119 Normalmente, o Aspergillus, na forma de ABPA, é precedido por asma, e acredita-se que represente uma ação imune contra várias espécies deste organismo.
Discinesia Ciliar Primária
A DCP é uma doença de disfunção ciliar e de limpeza mucociliar inapropriada, geralmente herdada em um padrão autossômico recessivo.120 As características clínicas incluem bronquiectasia difusa, juntamente com rinossinusite crônica, otite crônica, infertilidade masculina e, na metade dos casos, situs inversus. A DCP recebe a denominação de síndrome de Kartagener quando associada aos situs inversus.
Deficiência de ?1-Antitripsina
Embora a deficiência de a1-antitripsina esteja tipicamente associada à presença de enfisema, um estudo avaliou as varreduras por TC de 74 pacientes e encontrou evidências de bronquiectasia em 70 casos (94%).121 Vinte pacientes (27%) apresentaram evidências de bronquiectasia difusa e de produção crônica de esputo.
Traqueomegalia
Traqueias com aumento de volume poderão resultar em condições congênitas como a síndrome de Mounier-Khun, ou a traqueomegalia poderá ser causada por inflamações crônicas induzidas pelo tabagismo ou por uma traqueostomia prévia. A síndrome de Mounier-Khun é um distúrbio raro causado por uma redução nas fibras musculares elásticas e lisas na traqueia e nos brônquios maiores.122
Doença Autoimune
A artrite reumatoide (AR) e a síndrome de Sjögren estão associadas ao desenvolvimento de bronquiectasia. Nos casos de AR, a bronquiectasia poderá se apresentar antes do desenvolvimento de artropatia inflamatória, assim como em pacientes com doença sistêmica em estado avançado.123,124
O mecanismo provável de ambas as condições é alguma alteração no sistema imune que permite o desenvolvimento de infecções pulmonares crônicas. As doenças intestinais inflamatórias também foram mencionadas como causas pouco frequentes, geralmente associadas mais à colite ulcerativa do que à doença de Crohn.125
Defesa Deficiente
Hipogamaglobulinemia e malignidades hematológicas são duas condições que predispõem as pessoas para bronquiectasia, provavelmente como resultado de alguma alteração no sistema imune. A imunodeficiência variável comum (IDVC) é a doença hipogamaglobulínica mais prevalente. A IDVC se caracteriza por níveis baixos de imunoglobulina A (IgA) ou imunoglobulina M (IgM), em combinação com baixa resposta a imunizações.126
Infecções sinopulmonares e gastrintestinais são ocorrências comuns. Aproximadamente, 11% dos pacientes desenvolvem bronquiectasia.127 Vale a pena verificar subclasses específicas de IgG, considerando que alguns pacientes se apresentam com nível normal da IgG. Um estudo revelou que as malignidades hematológicas associadas com mais frequência à bronquiectasia eram leucemia linfocitária crônica e aguda, sendo que a maior dos pacientes apresentava histórico de transplante de células-tronco.116
Diagnóstico
Manifestações Clínicas
O diagnóstico de bronquiectasia deve focar inicialmente a obtenção de históricos adequados que indiquem o momento do início dos sintomas, incluindo enfermidades ou episódios de infecções na infância. A quantificação da quantidade diária de esputo é bastante útil para avaliar a gravidade da doença. A perda do olfato, resultando em sinusite crônica, também é uma ocorrência comum.128
Em um estudo de 103 pacientes que apresentaram diagnóstico inicial de bronquiectasia, os sintomas incluíam 90% de casos com tosse, 87% com esputo quase todos os dias da semana, 73% com rinossinusite, 73% com fadiga, 60% com dispneia e 26% com hemoptse.129 No exame físico do mesmo estudo, 73% tinham estertores crepitantes e 21%, respiração ofegante.
O fato mais interessante é que 80% das pessoas tiveram enfermidades na infância, incluindo tosse, respiração ofegante, asma e infecções no trato respiratório superior e inferior. Nesse grupo, a função pulmonar apresentou leve obstrução com uma previsão média do VEF1 de 76%, embora outros estudos tenham apresentado função pulmonar mais baixa com o percentual de VEF1 na faixa de 50 a 60%.130
Testes Laboratoriais
Os exames completos para etiologia de bronquiectasia devem incluir hemograma completo com diferencial; medição dos níveis de IgG (incluindo subclasses), IgA e IgM; e cultura de esputo para micobactérias, fungos e bactérias.
Nas situações em que o histórico indicar, os testes adicionais deverão incluir teste para HIV, medição do óxido nítrico exalado e biópsia nasal para DCP, IgE e níveis de anticorpos contra o organismo Aspergillus para ABPA, nível de cloreto no suor para FC, fator reumatoide para artrite reumatoide, e medição dos níveis de a1-antitripsina. Além da coleta de culturas para excluir a hipótese de obstrução por corpos estranhos ou massa, a broncoscopia é uma excelente opção nas situações em que as imagens revelarem que a bronquiectasia é focal.
Imagens
As descobertas das radiografias torácicas foram anormais em 91% de casos em um estudo envolvendo 104 pacientes que se apresentaram pela primeira vez com bronquiectasia, embora essas descobertas tenham sido inespecíficas e, com frequência, indicam a presença de infiltrados com aparência crônica.129 A TC de alta resolução do tórax se tornou o padrão para diagnóstico da doença e para quantificar sua gravidade.131
Via respiratória maior que os vasos que a acompanham é o sinal clássico, também conhecido por “sinal em anel de sinete”. Outros sinais indiretos são espessamento da parede brônquica ou vias respiratórias cheias de muco. A gravidade das descobertas feitas por TC são preditoras do declínio da função pulmonar.132
A localização da bronquiectasia nas varreduras por TC fornecem dicas para o diagnóstico, sendo que, tipicamente, a ABPA é uma indicação da presença de condições como bronquiectasia central, doença lobular superior por FC e doença lobular inferior por DCP.133
A presença de doença apical unilateral é típica da infecções causadas pelo organismo Mycobacterium tuberculosis, sendo que as doenças micobacterianas atípicas poderão se apresentar com doença apical unilateral, com doença em língula ou com doença no lobo médio direito.89
Gerenciamento
O gerenciamento de bronquiectasia crônica não FC se fundamenta na mobilização de secreções, terapia anti-inflamatória, agentes anti-infecciosos e cirurgia, que são os grandes pilares do tratamento.134 Existem algumas semelhanças com o tratamento de FC, assim como diferenças importantes. Além do gerenciamento geral, a terapia com foco específico é indicada nos casos em que forem encontradas etiologias específicas.
A higiene dos brônquios ajuda a limpar as secreções e, geralmente, é recomendada com ciclos ativos de respiração, terapia percussiva manual ou com dispositivos compressivos vibratórios, embora ainda não tenham sido realizados estudos que mostrem o benefício sobre a tosse não tratada.135
Os estudos sobre agentes mucolíticos em casos de bronquiectasia não FC apresentaram resultados mistos. Um estudo sobre DNAs em 349 pacientes com bronquiectasia idiopática não apresentou melhora no VEF1 e mostrou que houve mais exacerbações no grupo de tratamento.136 No início, a administração de solução salina era uma grande promessa, porém um estudo randomizado em 40 pacientes demonstrou que não houve nenhuma diferença na função pulmonar, na qualidade de vida ou nas exacerbações.137
A administração de antibióticos inalatórios no longo prazo também é uma boa opção, embora estudos mais amplos que utilizaram a tobramicina inalatória em 74 pacientes com colonização crônica com Pseudomonas não apresentaram nenhuma alteração no VEF1. O grupo de tratamento apresentou redução na carga bacteriana, ainda que tenha apresentado mais efeitos colaterais relacionados a tosse, dispneia, respiração ofegante e dor torácica não cardíaca.
Os medicamentos anti-inflamatórios testados incluem esteroides e antibióticos macrolídeos. De um modo geral, não se recomenda o uso de esteroides orais como terapia de manutenção devido aos efeitos colaterais potenciais e à falta de dados controlados. Os testes de esteroides inalatórios são muito pequenos, sendo que um estudo de 12 meses mostrou que não houve nenhuma diferença na função pulmonar ou na frequência das exacerbações.139
O subgrupo de pacientes que foram colonizados com Pseudomonas apresentou redução no volume de esputo e um índice reduzido de exacerbações. Em um teste randomizado recente, os antibióticos macrolídeos diminuíram os índices de exacerbações (0,59 versus 1,57 exacerbações médias em um período de 6 meses), embora não tenha produzido diferenças na função pulmonar ou nas pontuações dos sintomas.140
O tratamento cirúrgico contra bronquiectasia deve ser reservado para doença localizada em áreas focais ou para hemoptise recorrente ou com risco de vida. O transplante de pulmão é uma opção para uso em pacientes selecionados. As indicações para encaminhamento para transplante são as mesmas que para os casos de FC e incluem VEF1 inferior a 30%, exacerbações frequentes e recorrência de hemoptise ou pneumotórax.
Prognóstico
Levando-se em consideração fatores como heterogeneidade da doença, idade de apresentação e carga da doença, é extremamente difícil generalizar um prognóstico geral para bronquiectasia. Resultados significativos em pacientes com bronquiectasia incluem taxa de declínio da função pulmonar, frequência de hospitalizações para exacerbações agudas e mortalidade.
A função pulmonar declina a taxas mais rápidas que em indivíduos normais, sendo que um estudo retrospectivo mostrou um declínio médio de 53mL/ano versus 25mL/ano em indivíduos saudáveis.141 O declínio do VEF1 normal foi pior em pessoas com colonização crônica por Pseudomonas, exacerbações frequentes e evidências de inflamação sistêmica. O número anual de exacerbações agudas foi de 1,5 em um estudo multicentro de 349 pacientes.136 A taxa de mortalidade é mais elevada em pacientes com hipoxemia, hipercapnia, dispneia subjetiva e doença radiográfica mais grave.142
Referências
1. Cystic Fibrosis Foundation Patient Registry. 2011 annual data report. Bethesda (MD): Cystic Fibrosis Foundation; 2012.
2. Bobadilla JL, Macek M Jr, Fine JP, et al. Cystic fibrosis: a worldwide analysis of CFTR mutations—correlation with incidence data and application to screening. Hum Mutat 2002;19:575–606.
3. Kapoor V, Shastri SS, Kabra M, et al. Carrier frequency of F508del mutation of cystic fibrosis in Indian population. J Cyst Fibros 2006;5:43–6.
4. Yamashiro Y, Shimizu T, Oguchi S, et al. The estimated incidence of cystic fibrosis in Japan. J Pediatr Gastroenterol Nutr 1997;24:544–7.
5. Riordan JR, Rommens JM, Kerem B, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 1989;245:1066–72.
6. Ramsey Gibson RL, Burns JL, Ramsey BW. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am J Respir Crit Care Med 2003;168:918–51.
7. Drumm ML, Ziady AG, Davis PB. Genetic variation and clinical heterogeneity in cystic fibrosis. Annu Rev Pathol 2012;7:267–82.
8. Wong LJ, Wang J, Zhang YH, et al. Improved detection of CFTR mutations in Southern California Hispanic CF patients. Hum Mutat 2001;18:296–307.
9. Bossi A, Casazza G, Padoan R, et al. What is the incidence of cystic fibrosis in Italy? Data from the National Registry (1988-2001). Hum Biol 2004;76:455–67.
10. McKone EF, Emerson SS, Edwards KL, Aitken ML. Effect of genotype on phenotype and mortality in cystic fibrosis: a retrospective cohort study. Lancet 2003;361:1671–6.
11. Slieker MG, Sanders EA, Rijkers GT, et al. Disease modifying genes in cystic fibrosis. J Cyst Fibros 2005;4 Suppl 2:S7–13.
12. Coakley R, Boucher R. Pathophysiology: epithelial cell biology and ion channel function in the lung, sweat gland and pancreas. In: Hodson ME, Bush A, Geddes DM, editors. Cystic fibrosis. 3rd ed. London: Taylor and Francis; 2006. p. 59–68.
13. Cole PJ. Inflammation: a two-edged sword – the model of bronchiectasis. Eur J Respir Dis 1986;147:6–15.
14. Schidlow DV. Cystic fibrosis. In: Schidlow DV, Smith DS, editors. A practical guide to pediatric respiratory diseases. Philadelphia: Hanley and Belfus; 1994. p. 75–80.
15. Corey M, Edwards L, Levison H, Knowles M. Longitudinal analysis of pulmonary function decline in patients with cystic fibrosis. J Pediatr 1997;131:809–14.
16. Song Z, Wu H, Ciofu O, et al. Pseudomonas aeruginosa alginate is refractory to Th1 immune response and impedes host immune clearance in a mouse model of acute lung infection. Med Microbiol 2003;52:731–40.
17. Konstan MW, Morgan WJ, Butler SM, et al. Risk factors for rate of decline in forced expiratory volume in one second in children and adolescents with cystic fibrosis. J Pediatr 2007;151:134–9.
18. Henry RL, Mellis CM, Petrovic L. Mucoid Pseudomonas aeruginosa is a marker of poor survival in cystic fibrosis. Pediatr Pulmonol 1992;12:158–61.
19. Sagel SD, Gibson RL, Emerson J, et al. Impact of Pseudomonas and Staphylococcus infection on inflammation and clinical status in young children with cystic fibrosis. J Pediatr 2009;154:183.
20. Dasenbrook EC, Merlo CA, Diener-West M, et al. Persistent methicillin-resistant Staphylococcus aureus and rate of FEV1 decline in cystic fibrosis. Am J Respir Crit Care Med 2008;178:814–21.
21. Elborn JS. Identification and management of unusual pathogens in cystic fibrosis. J R Soc Med 2008;101 Suppl 1:S2–5.
22. Goss CH, Otto K, Aitken ML,et al. Detecting Stenotrophomonas maltophilia does not reduce survival of patients with cystic fibrosis. Am J Respir Crit Care Med 2002;166:356–61.
23. Rønne Hansen C, Pressler T, Høiby N, Gormsen M. Chronic infection with Achromobacter xylosoxidans in cystic fibrosis patients; a retrospective case control study. J Cyst Fibros 2006;5:245–51.
24. Stevens DA, Moss RB, Kurup VP, et al. Allergic bronchopulmonary aspergillosis in cystic fibrosis—state of the art: Cystic Fibrosis Foundation Consensus Conference. Clin Infect Dis 2003;37 Suppl 3:S225–64.
25. Olivier KN, Weber DJ, Wallace RJ, et al. Non-tuberculous mycobacteria. I: Multicenter prevalence study in cystic fibrosis. Am J Respir Crit Care Med 2003;167:828–34.
26. Olivier KN, Weber DJ, Lee JH, et al. Non-tuberculous mycobacteria. II: Nested-cohort study of impact on cystic fibrosis lung disease. Am J Respir Crit Care Med 2003;167:835–40.
27. Reik R, Spilker T, Lipuma JJ. Distribution of Burkholderia cepacia complex species among isolates recovered from persons with or without cystic ?brosis. J Clin Microbiol 2005;43:2926–8.
28. Kidd TJ, Bell SC, Coulter C. Genomovar diversity amongst Burkholderia cepacia complex isolates from an Australian adult cystic ?brosis unit. Eur J Clin Microbiol Infect Dis 2003;22:434–7.
29. Kalish LA, Waltz DA, Dovey M, et al. Impact of Burkholderia dolosa on lung function and survival in cystic fibrosis. Am J Respir Crit Care Med 2006;173:421.
30. Speert DP. Advances in Burkholderia cepacia complex. Paediatr Respir Rev 2002;3:230–5.
31. Zemanick ET, Sagel SD, Harris JK. The airway microbiome in cystic fibrosis and implications for treatment. Curr Opin Pediatr 2011;23:319–24.
32. Guss AM, Roeselers G, Newton IL, et al. Phylogenetic and metabolic diversity of bacteria associated with cystic fibrosis. ISME J 2011;5:20–9.
33. Cox MJ, Allgaier M, Taylor B, et al. Airway microbiota and pathogen abundance in age-stratified cystic fibrosis patients. PLoS One 2010; 5:e11044.
34. Hays SR, Ferrando RE, Carter R, et al. Structural changes to airway smooth muscle in cystic fibrosis. Thorax 2005;60:226–8.
35. Hordvik NL, König P, Morris D, et al. A longitudinal study of bronchodilator responsiveness in cystic fibrosis. Am Rev Respir Dis 1985;131:889–93.
36. Flume PA, Yankaskas JR, Ebeling M, et al. Massive hemoptysis in cystic fibrosis. Chest 2005;128:729–38.
37. Flume PA. Pneumothorax in cystic fibrosis. Chest 2003;123:217–21.
38. Flume PA, Strange C, Ye X, et al. Pneumothorax in cystic fibrosis. Chest 2005;128:720–8.
39. Rosenfeld M, Emerson J, Williams-Warren J, et al. Defining a pulmonary exacerbation in cystic fibrosis. J Pediatr 2001;139:359–65.
40. Cystic Fibrosis Foundation Patient Registry. 2004 Annual data report to the center directors. Bethesda (MD): Cystic Fibrosis Foundation; 2005.
41. De Boeck K, Weren M, Proesmans M, et al. Pancreatitis among patients with cystic fibrosis: correlation with pancreatic status and genotype. Pediatrics 2005;115:463–9.
42. Scott RB, O’Loughlin EV, Gall DG. Gastroesophageal reflux in patients with cystic fibrosis. J Pediatr 1985;106:223.
43. Houwen RH, van der Doef HP, Sermet I, et al. Defining DIOS and constipation in cystic fibrosis with a multicentre study on the incidence, characteristics, and treatment of DIOS. J Pediatr Gastroenterol Nutr 2010;50:38–42.
44. Maisonneuve P, Marshall BC, Knapp EA, Lowenfels AB. Cancer risk in cystic fibrosis: a 20-year nationwide study from the United States. J Natl Cancer Inst 2013;105:122–9.
45. Smyth RL. Fibrosing colonopathy in cystic fibrosis. Arch Dis Child 1996;74:464–8.
46. FitzSimmons SC, Burkhart GA, Borowitz D, et al. High-dose pancreatic-enzyme supplements and fibrosing colonopathy in children with cystic fibrosis. N Engl J Med 1997;336:1283–9.
47. Moyer K, Balistreri W. Hepatobiliary disease in patients with cystic ?brosis. Curr Opin Gastroenterol 2009;25:272–8.
48. Vawter GF, Shwachman H. Cystic fibrosis in adults: an autopsy study. Pathol Annu 1979;14:357–82.
49. Wilschanski M. Patterns of gastrointestinal disease associated with mutations of CFTR. Curr Gastroenterol Rep 2008;10:316–23.
50. Wilson-Sharp RC, Irving HC, Brown RC, et al. Ultrasonography of the pancreas, liver, and biliary system in cystic ?brosis. Arch Dis Child 1984;59:923–6.
51. Couce M, O’Brien TD, Moran A, et al. Diabetes mellitus in cystic ?brosis is characterized by islet amyloidosis. J Clin Endocrinol Metab 1996;81:1267–72.
52. Minicucci L, Cotellessa M, Pittaluga L, et al. Beta-cell autoantibodies and diabetes mellitus family history in cystic ?brosis. J Pediatr Endocrinol Metab 2005;18:755–60.
53. Lanng S. Glucose intolerance in cystic ?brosis patients. Paediatr Respir Rev 2001;2:253–9.
54. Lanng S, Hansen A, Thorsteinsson B, et al. Glucose tolerance in patients with cystic ?brosis: ?ve year prospective study. BMJ 1995;311:655–9.
55. Moran A, Brunzell C, Cohen RC, et al. Clinical Care Guidelines for Cystic Fibrosis-Related Diabetes: a position statement of the American Diabetes Association and a clinical practice guideline of the Cystic Fibrosis Foundation, endorsed by the Pediatric Endocrine Society. Diabetes Care 2010;33:2697–708.
56. Hameed S, Morton JR, Jaffe A, et al. Early glucose abnormalities in cystic ?brosis are preceded by poor weight gain. Diabetes Care 2010;33:221–6.
57. Grey V, Atkinson S, Drury D, et al. Prevalence of low bone mass and deficiencies of vitamins D and K in pediatric patients with cystic fibrosis from 3 Canadian centers. Pediatrics 2008;122:1014–20.
58. Paccou J, Zeboulon N, Combescure C, et al. The prevalence of osteoporosis, osteopenia, and fractures among adults with cystic fibrosis: a systematic literature review with meta-analysis. Calcif Tissue Int 2010;86:1–7.
59. Javier RM, Jacquot J. Bone disease in cystic fibrosis: what’s new? Joint Bone Spine 2011;78:445–50.
60. Aris RM, Renner JB, Winders AD, et al. Increased rate of fractures and severe kyphosis: sequelae of living into adulthood with cystic fibrosis. Ann Intern Med 1998;128:186–93.
61. Thornton J, Rangaraj S. Pharmacological agents (anti-inflammatory and analgesic) for managing symptoms in people with cystic fibrosis-related arthritis. Cochrane Database Syst Rev 2008;(1):CD006838.
62. Merkel PA. Rheumatic disease and cystic fibrosis. Arthritis Rheum 1999;42:1563–71.
63. Turner MA, Baildam E, Patel L, David TJ. Joint disorders in cystic ?brosis. J R Soc Med 1997;90 Suppl 31:S13–20.
64. Gibney EM, Goldfarb DS. The association of nephrolithiasis with cystic fibrosis. Am J Kidney Dis 2003;42:1–11.
65. Hiatt RA, Dales LG, Friedman GD, Hunkeler EM. Frequency of urolithiasis in a prepaid medical care program. Am J Epidemiol 1982;115:255–65.
66. Sidhu H, Hoppe B, Hesse A, et al. Absence of Oxalobacter formigenes in cystic fibrosis patients: a risk factor for hyperoxaluria. Lancet 1998;352:1026–9.
67. Holsclaw DS, Perlmutter AD, Jockin H, et al. Genital abnormalities in male patients with cystic fibrosis. J Urol 1971;106:568–74.
68. Dreyfus DH, Bethel R, Gelfand EW. Cystic fibrosis 3849+10kb C > T mutation associated with severe pulmonary disease and male fertility. Am J Respir Crit Care Med 1996;153:858–60.
69. Dohle GR, Ramos L, Pieters M, et al. Surgical sperm retrieval and intracytoplasmic sperm injection as treatment of obstructive azoospermia. Hum Reprod 1998;13:620–3.
70. Hubert D, Patrat C, Guibert J, et al. Results of assisted reproductive technique in men with cystic fibrosis. Hum Reprod 2006;21:1232–6.
71. McCallum TJ, Milunsky JM, Cunningham DL, et al. Fertility in men with cystic fibrosis: an update on current surgical practices and outcomes. Chest 2000;118:1059–62.
72. Kopito LE, Kosasky HJ, Shwachman H. Water and electrolytes in cervical mucus from patients with cystic fibrosis. Fertil Steril 1973;24:512–6.
73. Barak A, Dulitzki M, Efrati O, et al. Pregnancies and outcome in women with cystic fibrosis. Isr Med Assoc J 2005;7:95–8.
74. Gilljam M, Antoniou M, Shin J, et al. Pregnancy in cystic fibrosis. Fetal and maternal outcome. Chest 2000;118:85–91.
75. Moran A, Hardin D, Rodman D, et al. Diagnosis, screening and management of cystic fibrosis related diabetes mellitus: a consensus conference report. Diabetes Res Clin Pract 1999;45:61–73.
76. Riekert KA, Bartlett SJ, Boyle MP, et al. The association between depression, lung function, and health-related quality of life among adults with cystic fibrosis. Chest 2007;132:231–7.
77. Goldbeck L, Besier T, Hinz A, et al. Prevalence of symptoms of anxiety and depression in German patients with cystic fibrosis. Chest 2010;138:929–36.
78. Quon BS, Aitken ML. Cystic fibrosis: what to expect now in the early adult years. Paediatr Respir Rev 2012;13:206–14.
79. Britto MT, Garrett JM, Dugliss MA, et al. Risky behavior in teens with cystic fibrosis or sickle cell disease: a multicenter study. Pediatrics 1998;101:250–6.
80. Malhotra S, Brager N, Rabin HR, et al. The prevalence of substance abuse in cystic ?brosis. J Cystic Fibrosis 2012;11 Suppl 1:S27.
81. Katz KA, Yan AC, Turner ML. Aquagenic wrinkling of the palms in patients with cystic fibrosis homozygous for the ?F508 CFTR mutation. Arch Dermatol 2005;141:621–4.
82. Farrell PM, Kosorok MR, Rock MJ, et al. Early diagnosis of cystic fibrosis through neonatal screening prevents severe malnutrition and improves long-term growth. Wisconsin Cystic Fibrosis Neonatal Screening Study Group. Pediatrics 2001;107:1–13.
83. Grosse SD, Rosenfeld M, Devine OJ, et al. Potential impact of newborn screening for cystic fibrosis on child survival: a systematic review and analysis. J Pediatr 2006;149:362–6.
84. Wagener JS, Zemanick ET, Sontag MK. Newborn screening for cystic fibrosis. Curr Opin Pediatr 2012;24:329–35.
85. Farrell PM, Rosenstein BJ, White TB, et al. Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic Fibrosis Foundation consensus report. J Pediatr 2008;153 Suppl 2:S4–14.
86. Rock M, Makholm L, Lai H, Cheng Y. Distribution of sweat chloride values in patients with cystic fibrosis. Pediatr Pulmonol 2003;36 Suppl 25:337.
87. Wilschanski M, Dupuis A, Ellis L, et al. Mutations in the cystic fibrosis transmembrane regulator gene and in vivo transepithelial potentials. Am J Respir Crit Care Med 2006;174:787–94.
88. Ruzal-Shapiro C. Cystic fibrosis. An overview. Radiol Clin North Am 1998;36:143–61.
89. Miller WT Jr. Diagnostic thoracic radiology. 1st ed. New York: McGraw-Hill; 2006.
90. Tiddens HA. Chest computed tomography scans should be considered as a routine investigation in cystic fibrosis. Paediatr Respir Rev 2006;7:202–8.
91. McKoy NA, Saldanha IJ, Odelola OA, Robinson KA. Active cycle of breathing technique for cystic fibrosis. Cochrane Database Syst Rev 2012;(12):CD007862.
92. Mogayzel PJ, Naurekas ET, Robinson KA, et al. Cystic fibrosis pulmonary guidelines: chronic medications for maintenance of lung health. Am J Respir Crit Care Med 2013;187:680–9.
93. Fuchs, HJ. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. N Engl J Med 1994;331:637–42.
94. Elkins MR, Robinson M, Rose BR, et al. A controlled trial of long-term inhaled hypertonic saline in patients with cystic fibrosis. N Engl J Med 2006;354:229–40.
95. Ramsey BW, Pepe MS, Quan JM, et al. Intermittent administration of inhaled tobramycin in patients with cystic fibrosis. Cystic Fibrosis Inhaled Tobramycin Study Group. N Engl J Med 1999;340:23–30.
96. Moss RB. Long-term benefits of inhaled tobramycin in adolescent patients with cystic fibrosis. Chest 2002;121:55–63.
97. McCoy KS, Quittner AL, Oermann CM, et al. Inhaled aztreonam lysine for chronic airway Pseudomonas aeruginosa in cystic fibrosis. Am J Respir Crit Care Med 2008;178:92.
98. Konstan MW, Geller DE, Minic P, et al. Tobramycin inhalation powder for P. aeruginosa infection in cystic fibrosis: the EVOLVE trial. Pediatr Pulmonol 2011;46:230–8.
99. Mitchell I, Corey M, Woenne R, et al. Bronchial hyperreactivity in cystic fibrosis and asthma. J Pediatr 1978;93:744–8.
100. Halfhide C, Evans HJ, Couriel J. Inhaled bronchodilators for cystic fibrosis. Cochrane Database Syst Rev 2005;(4):CD003428.
101. Chmiel JF, Konstan MW. Inflammation and anti-inflammatory therapies for cystic fibrosis. Clin Chest Med 2007;28:331.
102. Southern KW, Barker PM, Solis A. Macrolide antibiotics for cystic fibrosis. Cochrane Database Syst Rev 2011;(12):CD002203.
103. Konstan MW, Byard PJ, Hoppel CL, Davis PB. Effect of high-dose ibuprofen in patients with cystic fibrosis. N Engl J Med 1995;332:848–54.
104. Ramsey BW, Davies J, McElvaney NG, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med 2011;365:1663–72.
105. Flume PA, Mogayzel PJ Jr, Robinson KA, et al. Cystic fibrosis pulmonary guidelines: treatment of pulmonary exacerbations. Am J Respir Crit Care Med 2009;180:802–8.
106. Smith AL, Fiel SB, Mayer-Hamblett N, et al. Susceptibility testing of Pseudomonas aeruginosa isolates and clinical response to parenteral antibiotic administration: lack of association in cystic fibrosis. Chest 2003;123:1495–502.
107. Orens JB, Estenne M, Arcasoy S, et al. International guidelines for the selection of lung transplant candidates: 2006 update—a consensus report from the Pulmonary Scientific Council of the International Society for Heart and Lung Transplantation. J Heart Lung Transplant 2006;25:745–55.
108. Liou TG, Adler FR, Cahill BC, et al. Survival effect of lung transplantation among patients with cystic fibrosis. JAMA 2001;286:2683.
109. The Registry of the International Society for Heart and Lung Transplantation: 29th Adult Lung and Heart-Lung Transplant Report—2012. J Heart Lung Transplant 2012; 31:1073–86.
110. Borowitz D, Baker RD, Stallings V. Consensus report on nutrition for pediatric patients with cystic fibrosis. J Pediatr Gastroenterol Nutr 2002;35:246–59.
111. Steinkamp G, Wiedemann B. Relationship between nutritional status and lung function in cystic fibrosis: cross sectional and longitudinal analyses from the German CF quality assurance (CFQA) project. Thorax 2002;57:596.
112. Nousia-Arvanitakis S. Cystic fibrosis and the pancreas: recent scientific advances. J Clin Gastroenterol 1999;29:138–42.
113. Weycker D, Edelsberg J, Oster G, Tino G. Prevalence and economic burden of bronchiectasis. Clin Pulm Med 2005;12:205–9.
114. Seitz AE, Olivier KN, Adjemian J, et al. Trends in bronchiectasis among Medicare beneficiaries in the United States, 2000 to 2007. Chest 2012;142:432–9.
115. Barker AF. Bronchiectasis. N Engl J Med 2002;346:1383–93.
116. Li AM, Sonnappa S, Lex C, et al. Non-CF bronchiectasis: does knowing the aetiology lead to changes in management? Eur Respir J 2005;26:8–14.
117. McShane PJ, Naureckas ET, Strek ME. Bronchiectasis in a diverse US population: effects of ethnicity on etiology and sputum culture. Chest 2012;142:159.
118. Johnston ID, Strachan DP, Anderson HR. Effect of pneumonia and whooping cough in childhood on adult lung function. N Engl J Med 1998;338:581–7.
119. Kim RD, Greenberg DE, Ehrmantraut ME, et al. Pulmonary nontuberculous mycobacterial disease: prospective study of a distinct preexisting syndrome. Am J Respir Crit Care Med 2008;178:1066–74.
120. Leigh MW, Pittman JE, Carson JL, et al. Clinical and genetic aspects of primary ciliary dyskinesia/Kartagener syndrome. Genet Med 2009;11:473–87.
121. Parr DG, Guest PG, Reynolds JH, et al. Prevalence and impact of bronchiectasis in alpha1-antitrypsin deficiency. Am J Respir Crit Care Med 2007;176:1215–21.
122. Menon B, Aggarwal B, Iqbal A. Mounier-Kuhn syndrome: report of 8 cases of tracheobronchomegaly with associated complications. South Med J 2008;101:83–7.
123. Demoruelle MK, Weisman MH, Simonian PL, et al. Brief report: airways abnormalities and rheumatoid arthritis-related autoantibodies in subjects without arthritis: early injury or initiating site of autoimmunity? Arthritis Rheum 2012;64:1756–61.
124. Lieberman-Maran L, Orzano IM, Passero MA, Lally EV. Bronchiectasis in rheumatoid arthritis: report of four cases and a review of the literature—implications for management with biologic response modifiers. Semin Arthritis Rheum 2006;35:379–87.
125. Mahadeva R, Walsh G, Flower CD, Shneerson JM. Clinical and radiological characteristics of lung disease in inflammatory bowel disease. Eur Respir J 2000;15:41–8.
126. Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. Representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clin Immunol 1999;93:190–7.
127. Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood 2012;119:1650–7.
128. Guilemany JM, Mariño-Sánchez FS, Angrill J, et al. The importance of smell in patients with bronchiectasis. Respir Med 2011;105:44.
129. King PT, Holdsworth SR, Freezer NJ, et al. Characterisation of the onset and presenting clinical features of adult bronchiectasis. Respir Med 2006;100:2183–9.
130. Nicotra MB, Rivera M, Dale AM, et al. Clinical, pathophysiologic, and microbiologic characterization of bronchiectasis in an aging cohort. Chest 1995;108:955–61.
131. Bonavita J, Naidich DP. Imaging of bronchiectasis. Clin Chest Med 2012;33:233–48.
132. Sheehan RE, Wells AU, Copley SJ. A comparison of serial computed tomography and functional change in bronchiectasis. Eur Respir J 2002;20:581–7.
133. Cartier Y, Kavanagh PV, Johkoh T, et al. Bronchiectasis: accuracy of high-resolution CT in the differentiation of specific diseases. AJR Am J Roentgenol 1999;173:47.
134. O’Donnell AE. Bronchiectasis. Chest 2008;134:815–23.
135. McCool FD, Rosen MJ. Nonpharmacologic airway clearance therapies: ACCP evidence-based clinical practice guidelines. Chest 2006;129 Suppl 1:S250–9.
136. O’Donnell AE, Barker AF, Ilowite JS, Fick RB. Treatment of idiopathic bronchiectasis with aerosolized recombinant human DNase I. rhDNase Study Group. Chest 1998;113:1329–34.
137. Nicolson CH, Stirling RG, Borg BM. The long term effect of inhaled hypertonic saline 6% in non-cystic fibrosis bronchiectasis. Respir Med 2012;106:661–7.
138. Barker AF, Couch L, Fiel SB, et al. Tobramycin solution for inhalation reduces sputum Pseudomonas aeruginosa density in bronchiectasis. Am J Respir Crit Care Med 2000;162:481.
139. Tsang K, Tan K, Ho P, et al. Inhaled fluticasone in bronchiectasis: a 12 month study. Thorax 2005;60:239–43.
140. Wong C, Jayaram L, Karalus N, et al. Azithromycin for prevention of exacerbations in non-cystic fibrosis bronchiectasis (EMBRACE): a randomised, double-blind, placebo-controlled trial. Lancet 2012;380:660–7.
141. Martínez-García MA, Soler-Cataluña JJ, Perpiñá-Tordera M, et al. Factors associated with lung function decline in adult patients with stable non-cystic fibrosis bronchiectasis. Chest 2007;132:1565.
142. Onen ZP, Gulbay BE, Sen E, et al. Analysis of the factors related to mortality in patients with bronchiectasis. Respir Med 2007;101:1390–7.
143. Cystic Fibrosis Foundation. Targeting mutations that cause cystic fibrosis. Available at: www.cff.org/aboutCFFoundation/Publications/connections/archive/March2010/Targeting-Mutations-that-Cause-Cystic-Fibrosis.cfm (accessed April 3, 2013).
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.