(Carregando Índice)... (Carregando Índice)... |
Autores:
Fernando de Paula Machado
Médico pela Faculdade de Medicina da Universidade de São Paulo (FMUSP). Residência em Clínica Médica no Hospital das Clínicas da FMUSP (HC-FMUSP). Residência em Cardiologia pelo Instituto do Coração (InCor) do HC-FMUSP. Médico Diarista do Pronto-Atendimento do Hospital Sírio-Libânes.
Leonardo Vieira da Rosa
Médico Cardiologista pelo Instituto do Coração do Hospital das Clínicas da Faculdade de Medicina da USP. Médico Assistente da Unidade de Terapia Intensiva do Instituto do Coração do Hospital das Clínicas da Faculdade de Medicina da USP. Doutorando em Cardiologia do InCor-HC-FMUSP. Médico Cardiologista da Unidade Coronariana do Hospital Sírio Libanês.
Última revisão: 01/04/2019
Comentários de assinantes: 0
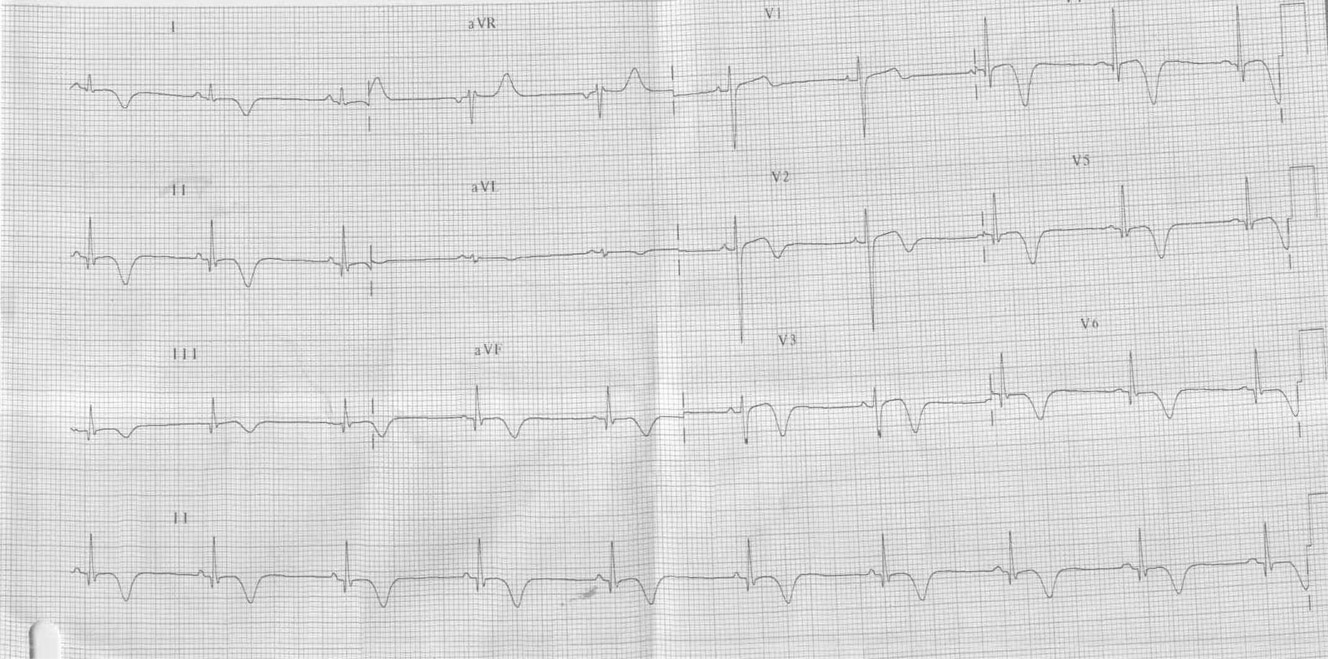
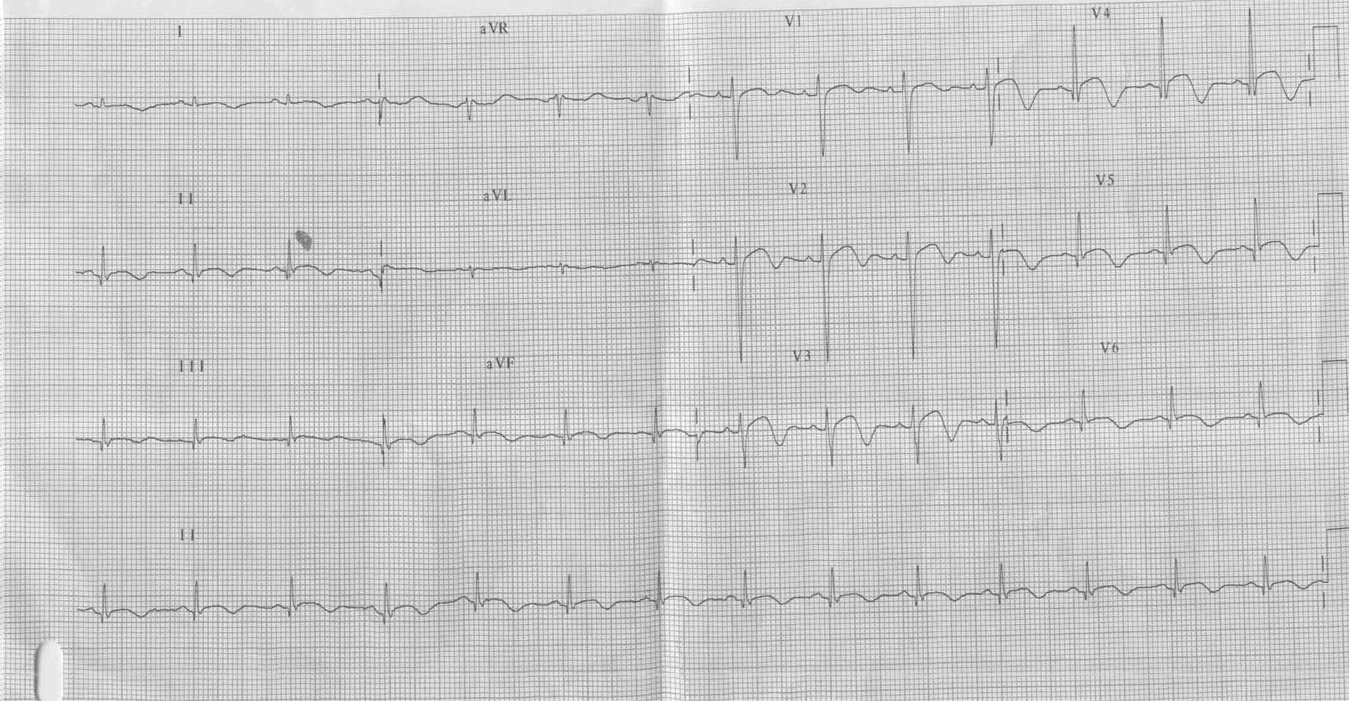
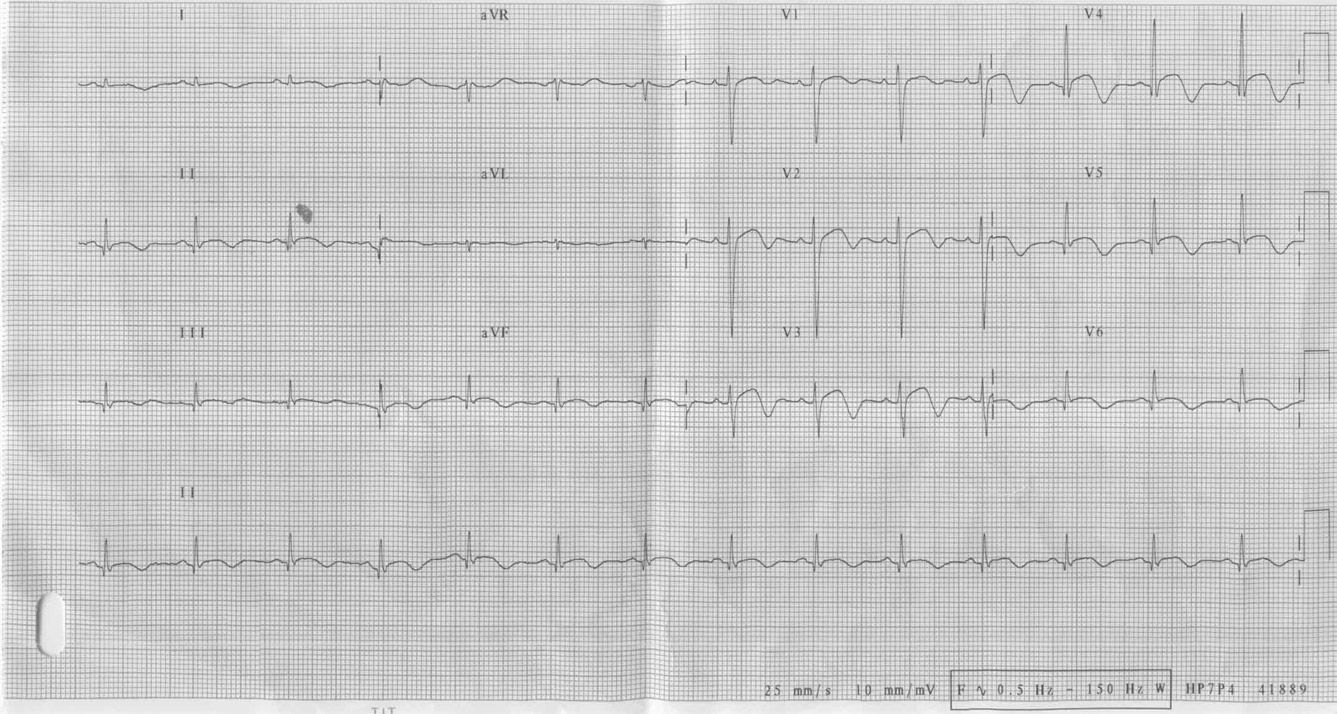
Paciente de 15 anos do sexo masculino, apresentou PCR em fibrilação ventricular durante internação para investigação de síncope

Ver diagnóstico abaixo
Síndrome do QT longo congênito. O que mais chama atenção nesses traçados não é o aumento do QTc e sim as ondas T. Elas se apresentam com alternância elétrica, ou seja, sendo ora positivas ora negativas ou isodifásicas e com variabilidade em sua amplitude. Poderiam estar associadas a momentos de estresse físico ou emocional, precedendo uma taquicardia ventricular polimórfica. Esse achado eletrocardiográfico é incomum; trata-se, porém, de um dos critérios diagnósticos maiores para o diagnóstico dessa enfermidade.
A síndrome do QT longo caracteriza-se pelo prolongamento do intervalo QT no traçado eletrocardiográfico associado à síncope, morte cardíaca súbita e arritmias ventriculares malignas, como o torsades de pointes. Pode, de acordo com sua etiologia, ser classificada em congênita ou adquirida.
Os mecanismos fisiopatológicos envolvidos são caracterizados por mutações nos genes que codificam os canais de sódio e potássio da membrana plasmática das células miocárdicas. Inicialmente duas formas de QT longo congênito foram descritas: a síndrome de Jervell e Lange-Nielsen e a síndrome de Romano-Ward. Pacientes acometidos pela síndrome de Jervell e Lange-Nielsen caracterizam-se por apresentarem intervalo QT longo associado a surdez congênita, episódios de síncope ou morte súbita. A transmissão hereditária se dá através de herança autossômica e recessiva. A síndrome de Romano-Ward, mais freqüente que a anterior, apresenta características clínicas semelhante, porém não acompanhada de surdez congênita, e possui herança autossômica dominante. Atualmente, sete genótipos (LQT1, LQT2, LQT3, LQT4, LQT5, LQT6 e LQT7) estão relacionados a essa síndrome.
A síndrome freqüentemente se manifesta antes dos 40 anos de idade, principalmente durante a infância e adolescência. Aproximadamente dois terços dos indivíduos com a síndrome do QT longo congênito são diagnosticados mediante a realização de um eletrocardiograma de triagem após a avaliação de um parente de primeiro grau acometido. Pacientes sintomáticos podem apresentar palpitações, síncope, tonturas e parada cardíaca. A arritmia ventricular geralmente verificada é o torsades de pointes.
O diagnóstico da síndrome depende basicamente da história clínica e familiar do paciente, associados aos achados eletrocardiográficos sugestivos. Um sistema de escore baseado em alterações no eletrocardiograma e em dados clínicos e familiares dos pacientes pode auxiliar no diagnóstico da síndrome. A probabilidade de a síndrome do QT longo congênito estar presente é então subdividida em baixa (escore = 1 ponto), intermediária (2-3 pontos) e alta probabilidade (escore = 4 pontos).
Tabela 1: Critérios diagnósticos para síndrome do QT longo
|
Características do paciente |
Pontos | ||
|
História clínica |
Síncope |
Com estresse |
2 |
|
Sem estresse |
1 | ||
|
Surdez congênita |
|
0,5 | |
|
História familiar |
Membros com QT longo |
1 | |
|
Morte súbita cardíaca em parentes < 30 anos |
0,5 | ||
|
Achados eletrocardiográficos |
QT corrigido |
> 480 ms |
3 |
|
460-470 ms |
2 | ||
|
450 ms (em mulheres) |
1 | ||
|
Torsades de pointes |
2 | ||
|
Alternância da onda T |
1 | ||
|
Onda T tocada em tr6es derivações |
1 | ||
|
Baixa frequência cardíaca para idade (abaixo do segundo percentil) |
0,5 | ||
|
Probabilidade de síndrome do QT longo congênito: baixa (escore = 1 ponto), intermediária (2-3 pontos) e alta probabilidade (escore = 4 pontos). | |||
A terapêutica da síndrome do QT longo é baseada principalmente na interrupção do estímulo simpático ao miocárdio (betabloqueador / simpatectomia). Outras possíveis terapias incluem a implantação de marcapasso definitivo e cardiodesfibrilador implantável.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.