(Carregando Índice)... (Carregando Índice)... |
Autor:
Jerusa Smid
Neurologista do Grupo de Neurologia Cognitiva e do Comportamento do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo
Neurologista do Instituto de Infectologia Emílio Ribas
Especialista em Neurologia Cognitiva e do Comportamento pela Faculdade de Medicina da USP
Última revisão: 10/11/2008
Comentários de assinantes: 0
A síndrome demencial é caracterizada por declínio cognitivo adquirido, cuja intensidade é capaz de interferir nas atividades profissionais e sociais da vida diária do indivíduo. Segundo a definição do DSM IV, o déficit cognitivo deve compreender alteração de memória associado à alteração em pelo menos um outro domínio cognitivo, como praxia (capacidade de realizar atividades motoras), linguagem, funções executivas ou gnosia (capacidade de reconhecer ou identificar objetos).
As demências ocorrem mais freqüentemente em indivíduos idosos, e a prevalência de demência dobra a cada cinco anos a partir dos 65 anos de idade, como observamos na tabela 1. O envelhecimento populacional é um processo mundial e, como resultado, as conseqüências socioeconômicas relacionadas aos quadros demenciais, que já são enormes, se tornarão ainda maiores em decorrência desse processo.
Tabela 1: Prevalência de demência nas diversas faixa etárias
|
Idade |
Demência (%) |
|
60-64 |
0,7 |
|
65-69 |
1,4 |
|
70-74 |
2,8 |
|
75-79 |
5,6 |
|
80-84 |
10,5 |
|
85-89 |
20,8 |
|
90-95 |
38,6 |
Jorm et al. 1998
As diferentes causas de demência podem estar relacionadas não apenas a quadros neurológicos primários, mas também a condição médica sistêmica, a efeitos persistentes de abuso de substâncias, ou à combinação desses fatores. Devemos lembrar que o diagnóstico de demência não deve ser feito na vigência de delirium (situação clínica em que há agudamente um déficit global da atenção, geralmente causado por doenças clínicas).
A doença de Alzheimer (DA) e demência vascular (DV) são as causas mais comuns de demência, seguidas pela demência com corpos de Lewy (DCL). No entanto, muitas outras causas podem ser responsáveis por esse processo. Segundo Mesulam, podemos dividir as diferentes etiologias das demências em dois grupos: o das doenças que costumam se apresentar como demência pura e o das doenças que apresentam sintomas sensitivo-motores mais proeminentes associados à demência (tabela 2).
Tabela 2: Etiologia das síndromes demenciais
|
Doenças que podem se apresentar como demência pura |
Doenças degenerativas |
doença de Alzheimer Degeneração lobar frontotemporal demência com corpos de Lewy |
|
Doença vascular
|
Múltiplos infartos Doença de Binswanger Arterites CADASIL | |
|
Lesões com efeito de massa |
Neoplasias Hidrocefalia de pressão normal Hematoma subdural Cistos parasitários, abscessos cerebrais | |
|
Doenças infecciosas
|
HIV Sífilis Herpes simples Doença de Lyme Doença de Whipple | |
|
Nutricional-tóxico-metabólico
|
Doença de Wernicke-Korsakoff Alcoolismo crônico Deficiência de vitamina B12 Hipotiroidismo severo Pelagra Exposição a solventes orgânicos Intoxicação por metais pesados Encefalopatia hepática | |
|
Imune/Inflamatório |
Encefalite límbica paraneoplásica Lúpus eritematoso sistêmico Cerebrite associada a doenças imunes do colágeno | |
|
Doenças de depósito |
Leucodistrofia metacromática Doença de Kufs | |
|
Doença de Creutzfeldt-Jakob | ||
|
Doenças que apresentam sintomas sensitivo-motores mais proeminentes associados à demência |
Doença de Parkinson Paralisia supranuclear progressiva Degeneração córtico-basal Doença de Huntington Doença de Wilson Doença de Hallervoden-Spatz Ataxia espino-cerebelar Esclerose lateral amiotrófica Esclerose múltipla Doença de Gerstmann-Sträussler-Scheinker | |
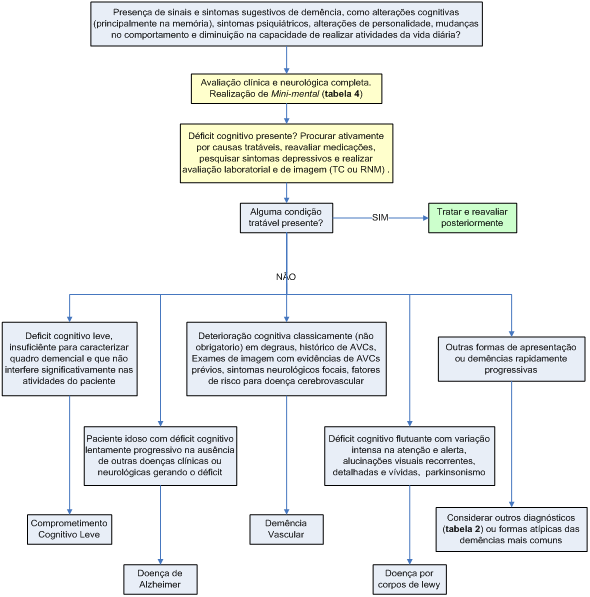
Devemos suspeitar de quadro demencial quando o paciente apresentar alterações cognitivas (principalmente perda de memória), sintomas psiquiátricos, alterações de personalidade, mudanças no comportamento ou diminuição na capacidade de realizar atividades da vida diária (tabela 3).
Tabela 3: Sinais e sintomas que podem indicar avaliação para quadro demencial
|
Alterações cognitivas: diminuição de memória, dificuldade em compreender comunicação escrita ou verbal, dificuldade em encontrar as palavras, esquecimento de fatos de conhecimento comum (por exemplo, nome do presidente) |
|
Sintomas psiquiátricos: apatia, depressão, ansiedade, insônia, desconfiança, delírios, paranóia, alucinações |
|
Alterações de personalidade: comportamentos inapropriados, desinteresse, isolamento social, ataques explosivos, frustração excessiva |
|
Mudanças no comportamento: agitação, inquietude, deambulação durante a noite |
|
Diminuição de capacidade de realizar atividades da vida diária: dificuldade em dirigir, se perder constantemente, dificuldade em cozinhar, cuidado pessoal ruim, problemas com compras e no trabalho. |
Interrogar familiares e amigos é importante, pois o paciente muitas vezes não tem percepção ou esconde suas dificuldades. Devem-se questionar alterações de memória, orientação, capacidade de realizar atividades diárias, incluindo trabalho, questões financeiras, compras e cuidados pessoais. Pesquisa de sintomas depressivos precedendo ou simultaneamente ao quadro cognitivo é fundamental, pois muitas vezes depressão pode simular quadros demenciais.
Testes padronizados para avaliar as diferentes funções cognitivas estão disponíveis e constituem bons métodos de rastreamento e acompanhamento de quadros demenciais. Apresentam algumas limitações, como o fato de serem influenciadas pela idade e escolaridade.
Na prática clínica, o teste mais utilizado é o Miniexame do Estado Mental (Mini-mental) (tabela 4). Nesse exame é considerado um resultado normal um escore acima de 23, lembrando novamente que os resultados são influenciados pela idade e escolaridade. Se houver dúvida após avaliação clínica e Mini-mental pode-se solicitar uma avaliação neuropsicológica para um psicólogo especializado, o que ajudará a definir com precisão o tamanho do déficit e qual a função cognitiva com maior comprometimento.
Tabela 4: Miniexame do Estado Mental (Mini-mental)
|
Área cognitiva avaliada |
Comandos de avaliação |
Escore | |
|
Orientação temporal |
Perguntar qual o(a): ANO – ESTAÇÃO – MÊS – DIA – DIA DA SEMANA. (um ponto para cada) |
| |
|
Orientação espacial |
Perguntar qual o(a): ESTADO – RUA – CIDADE – LOCAL – ANDAR. (um ponto para cada) |
| |
|
Registro |
O examinador nomeia 3 palavras comuns (por exemplo, carro, vaso, bola). Em seguida, pede-se que o paciente repita as 3 palavras. O paciente receberá um ponto por cada acerto. Permita 5 tentativas até o paciente aprender as 3 palavras, mas pontuar apenas a primeira |
| |
|
Atenção e cálculo |
Peça para subtrair 7 de 100 sucessivamente (5 vezes): 100 – 93 – 86 – 79 – 72 - 65 Dar um ponto para cada acerto. Se não atingir o escore máximo, peça para que soletre a palavra MUNDO. Corrija os erros de soletração e então peça para que soletre a palavra MUNDO de trás para frente. Dar um ponto para cada letra na posição correta. Considerar o maior resultado obtido no cálculo ou na soletração da palavra |
| |
|
Memória de evocação |
Peça para o paciente repetir as 3 palavras aprendidas anteriormente. (um ponto para cada acerto) |
| |
|
Linguagem |
1o teste |
Apontar o lápis e perguntar o que é. Fazer o mesmo com o relógio. (um ponto para cada acerto) |
|
|
2o teste |
Pedir para repetir a seguinte frase: NEM AQUI, NEM ALÍ NEM LÁ. (um ponto se acertar) |
| |
|
3o teste |
Peça para que execute a seguinte tarefa: PEGUE ESTE PAPEL COM A MÃO DIREITA (pausa), COM AS DUAS MÃOS DOBRE-O AO MEIO UMA VEZ (pausa) E EM SEGUIDA JOGUE-O NO CHÃO. Dar um ponto para o acerto em cada comando |
| |
|
4o teste |
Escrever em uma folha de papel o seguinte comando: FECHE OS OLHOS. Peça para o paciente ler e obedecer ao comando. (um ponto se acertar) |
| |
|
5o teste |
Peça para o paciente escrever uma frase completa. (um ponto se conseguir) |
| |
|
6o teste |
Peça para que copie o seguinte desenho:
|
| |
|
|
|
|
Escore total de |
Pesquisar com os familiares o tempo de evolução do declínio cognitivo, assim como sintomas associados, doenças clínicas ou neurológicas concomitantes, medicações utilizadas (muitas podem comprometer as funções cognitivas). Causas clínicas ou neurológicas tratáveis de demência devem ser ativamente pesquisadas.
Da mesma forma, o exame clínico e neurológico devem ser completos, mais uma vez, em busca da etiologia do quadro demencial e de causas tratáveis. Déficit neurológico focal associado a quadro demencial, por exemplo, pode sugerir DV.
No Brasil, os exames necessários para investigação de síndrome demencial em faixa etária senil são hemograma, função renal (dosagem de eletrólitos, uréia e creatinina), hepática e tiroidiana, dosagem sérica de vitamina B12 e ácido fólico, sorologia para sífilis, glicemia de jejum e exame de neuroimagem (tomografia computadorizada [TC] de crânio ou ressonância magnética [RM] de encéfalo).
A análise do líquido céfalo-raquidiano (LCR), sorologia para HIV e eletroencefalograma (EEG) são solicitados em casos de síndrome demencial em idade pré-senil ou com evolução atípica (por exemplo, demência rapidamente progressiva).
Em casos em que o déficit cognitivo instalou-se de forma aguda, devemos pensar primeiramente em delirium (situação clínica em que há agudamente um déficit global da atenção, geralmente causado por doenças clínicas) e realizar exames para afastar problemas clínicos como infecção urinária (ou outras infecções), desidratação, isquemia miocárdica, hipóxia, distúrbios hidroeletrolíticos ou outros problemas clínicos, pois geralmente a alteração cognitiva é revertida com o tratamento adequado.
Revisaremos a seguir as causas mais importantes de demência.
A DA é a principal causa de demência, e é responsável por mais de 80% dos casos de demência após os 65 anos de idade. Descrita por Alois Alzheimer em 1906, representa atualmente a sétima causa de morte nos Estados Unidos. Era considerada uma forma rara de demência até o início da década de 1970, por ser diagnosticada apenas em casos de demência pré-senis (antes dos 65 anos de idade). Atualmente sabemos que essa doença é mais freqüente após os 65 anos e que o principal fator de risco é a idade.
Em cerca de 5% dos casos, a doença tem herança autossômica dominante, sendo na maioria das vezes de ocorrência esporádica. Além da idade, outros fatores de risco para a DA são sexo feminino, história familiar de DA em parentes de primeiro grau, história de traumatismo cranioencefálico e presença do alelo E4 da apolipoproteína E.
A DA é uma doença degenerativa, sem fator etiológico determinado. Os principais achados anatomopatológicos são as placas amilóides, depósitos insolúveis de proteína beta-amilóide, e os emaranhados neurofibrilares, constituídos por proteína tau fosforilada. O diagnóstico pós-mortem é determinado pela distribuição e densidade desses achados. Outras características patológicas são perda neuronal, diminuição da densidade sináptica e gliose.
As alterações mais precoces ocorrem em estruturas do sistema límbico, como hipocampo, córtex entorrinal e transentorrinal, núcleo basal de Meynert, amígdala e o córtex têmporo-polar. Posteriormente há acometimento dos córtex associativos parietal e frontal, e, no estágios avançados da doença, comprometimento de áreas sensitivo-motoras primárias.
Ocorre perda de neurônios colinérgicos do núcleo basal de Meynert precocemente na DA, que é responsável pela perda de inervação colinérgica cortical. Esse é um dos mecanismos fisiopatológicos da doença, e o principal alvo terapêutico até o momento. Outros mecanismos fisiopatológicos envolvidos na DA são inflamação e estresse oxidativo.
A DA ocorre de forma insidiosa e com progressão lenta dos sintomas. A sobrevida de doença pode chegar a 20 anos, mas é em média de oito anos. A principal e mais precoce característica clínica da DA é o déficit de memória. O paciente tem dificuldade em memorizar novas informações, experiências e eventos recentes. Dificuldade em nomear é comum e o paciente torna-se repetitivo. Os sintomas podem flutuar em intensidade, e a presença de anosognosia (incapacidade de reconhecimento da doença pelo paciente) é comum. No início do quadro, como o déficit de memória é leve, as atividades do dia-a-dia ainda podem ser realizadas com relativa independência. O paciente realiza atividades como dirigir, cuidar da casa, fazer compras e participar de eventos sociais, porém de forma mais ineficiente e com menos interesse.
Nos estágios intermediários, associam-se ao déficit de memória déficits mais intensos nos outros domínios cognitivos, como linguagem, praxia, atenção, funções executivas e orientação espacial. A afasia na DA geralmente é fluente. O paciente torna-se mais dependente para suas atividades, necessitando de ajuda para dirigir, cuidar da casa, pagar contas, e pode apresentar também dificuldade nas atividades de cuidado pessoal. Nesta fase, as alterações do ciclo sono-vigília são comuns, podendo haver piora dos sintomas cognitivos e comportamentais ao entardecer (fenômeno do pôr-do-sol). Os sintomas psiquiátricos são comuns, e é freqüente a presença de delírios. Os mais comuns são delírios de ciúmes (do cônjuge) e de roubo (ao tentar encontrar objeto guardado em local não usual). O paciente também pode apresentar alucinações, agitação, apatia e sintomas depressivos. A necessidade de cuidados de terceiros é crescente.
No estágio final da doença, o indivíduo é totalmente dependente. Há incontinência vesical e fecal, incapacidade de reconhecer os familiares, dificuldade em alimentar-se e locomover-se. Todas as funções cognitivas e comportamentais são afetadas com a evolução da doença. Mais tardiamente, há comprometimento extrapiramidal, com ocorrência de mioclonias, rigidez, e instabilidade à marcha. Progressivamente, o indivíduo perde a capacidade de andar e fica restrito ao leito nas fases mais avançadas. A morte ocorre por complicações cardiopulmonares ou por infecção.
O exame neurológico é normal no início da doença; pode haver apraxia leve (perda da habilidade para executar movimentos e gestos precisos). Com a evolução do quadro, sinais extrapiramidais podem ser encontrados e o quadro apráxico se intensifica.
O diagnóstico da DA é feito a partir de história clínica e exame neurológico sugestivos. O diagnóstico clínico tem acurácia de 90%. Exames complementares devem ser solicitados conforme sugerido previamente, de forma a afastar causas de demência secundária a outras doenças clínicas ou neurológicas.
Os exames de neuroimagem estrutural (TC e RM) freqüentemente são normais em estágios iniciais da doença. A atrofia hipocampal evidente ao exame de RM pode estar presente já em fase inicial. Com a evolução da doença, a atrofia hipocampal torna-se mais evidente, e associa-se atrofia cortical com a evolução do quadro. Os exames funcionais (SPECT e PET) não são solicitados rotineiramente para o diagnóstico de DA, estando seu uso restrito a casos em que há dúvida diagnóstica após a investigação inicial. Nos casos de DA, a alteração mais freqüentemente encontrada é o hipometabolismo ou hipofluxo em região têmporo-parietal bilateral.
As dosagens de proteína tau fosforilada e de proteína beta-amilóide no LCR têm sido estudadas para o diagnóstico de DA e podem ter utilidade clínica para o diagnóstico de DA inicial no futuro.
O diagnóstico diferencial da DA envolve as outras causas de síndrome demencial apontadas na tabela 2.
A DA não tem tratamento específico. Os sintomas cognitivos são tratados com inibidores da acetilcolinesterase e memantina. Os inibidores da acetilcolinesterase (donepezil, galantamina e rivastigmina) constituem o tratamento inicial da doença. Representam a primeira linha terapêutica nos casos leves e moderados da doença. Nos casos moderados, a memantina deve ser associada ao tratamento inicial, e nos casos graves deve ser utilizada como monoterapia, embora estudos recentes apontem para a continuidade do uso dos inibidores da acetilcolinesterase nessa fase.
Os sintomas neuropsiquiátricos são tratados inicialmente com medidas não farmacológicas. As medicações utilizadas para o tratamento desses sintomas são inibidores da acetilcolinesterase, antidepressivos, estabilizadores de humor ou neurolépticos, a depender da qualidade e gravidade. Todos os estudos publicados até o momento mostram eficácia mínima ou duvidosa para o tratamento desses sintomas.
A DV é a segunda causa de demência na população idosa. Os principais fatores de risco são hipertensão arterial sistêmica, diabetes e a presença do alelo E4 da apolipoproteína E. Em algumas populações orientais, a DV é a principal causa de demência.
A DV ocorre secundariamente a insultos vasculares ao sistema nervoso central e pode ser dividida em demência por múltiplos infartos, demência isquêmica subcortical e demência por infarto estratégico.
A demência por múltiplos infartos está associada à recorrência clínica de acidentes vasculares cerebrais, geralmente associada à doença tromboembólica, hipertensão arterial sistêmica e evidência de doença aterosclerótica extracerebral.
A demência isquêmica subcortical está associada a doença de pequenos vasos cerebrais, em pacientes com hipertensão arterial de longa data, podendo haver alguns infartos lacunares, porém sem evidência clínica de lesão vascular na maioria das vezes. Raramente ocorre secundariamente a doenças tromboembólicas. O estado lacunar e a encefalopatia de Binswanger são os tipos de demência isquêmica subcortical.
A demência por infarto estratégico é caracterizada pela presença de lesão vascular única em região cerebral específica, resultando em demência. Não estão contemplados nessa nomenclatura lesões extensas em hemisfério dominante.
demência por múltiplos infartos: o quadro clínico depende do número e da quantidade de lesões, podendo haver características clínicas corticais e subcorticais, geralmente com predomínio dessas últimas. A evolução característica é dita em degraus, ocorrendo platôs entre os eventos clínicos. No entanto, essa característica nem sempre está presente. Alterações de marcha e incontinência urinária podem ocorrer em fases iniciais da doença. Sinais e sintomas de paralisia pseudobulbar (disfagia, disartria e labilidade emocional) também são característicos. Flutuações cognitivas e piora noturna são comuns. Os pacientes também podem apresentar labilidade emocional, apatia e acentuação da personalidade pré-mórbida.
demência isquêmica subcortical: o quadro clínico caracteriza-se por instalação insidiosa dos sintomas cognitivos, associados a sinais e sintomas motores, com características piramidais e extrapiramidais. O sinal mais comum ao exame é a apraxia de marcha. Os pacientes apresentam alterações em funções executivas, e lentificação da velocidade de pensamento, atribuídas ao maior comprometimento de circuitos frontais cortico-subcorticais. Alterações de humor e outros sintomas neuropsiquiátricos são comuns.
demência por infarto estratégico:
ü infarto talâmico medial: confusão mental ou coma de instalação aguda, e subseqüente amnésia. Pode haver apatia. Em alguns casos pode haver paralisia do olhar vertical e apraxia de pálpebra;
ü infarto de cápsula interna-tálamo lateral: flutuação do alerta, inatenção, apatia e alentecimento psicomotor na fase aguda, e posterior déficit de memória na fase crônica
ü infarto em globo pálido e núcleo caudado: infartos bilaterais dos núcleos da base podem resultar em abulia, apatia e depressão, ou ainda em quadro de hiperatividade, desinibição e inatenção;
ü infarto em artéria cerebral posterior: a oclusão da artéria cerebral posterior pode decorrer em 1) infarto do lobo occipital medial, levando à hemianopsia homônima; 2) da hipocampal, levando a déficit de memória, que pode ser grave se infarto bilateral; 3) da porção posterior do corpo caloso, podendo levar à alexia sem agrafia, em casos de infartos do lado esquerdo; e 4) infartos do lobo temporal inferior, podendo levar à agnosia para objetos, cores ou faces, a depender da extensão e lado da lesão;
ü síndrome de Gerstmann: o infarto do giro angular esquerdo por oclusão do ramo posterior da artéria cerebral média esquerda pode levar ao quadro clínico denominado de síndrome de Gerstmann, com acalculia, agrafia, desorientação esquerda-direita e agnosia digital. O mais comum é a ocorrência de alguns elementos da síndrome associados a afasia ou déficit de atenção.
ü infarto do mesencéfalo basal: pode resultar de complicação cirúrgica para correção de aneurisma da artéria comunicante anterior, com lesão dos vasos perfurantes da região. O infarto da região interrompe as vias colinérgicas que conectam a região ao hipocampo, levando a um quadro amnéstico. Infarto bilateral da artéria cerebral anterior pode levar a quadro de abulia intensa.
A investigação do quadro demencial deve ser realizada da mesma forma que na DA. Nos quadros vasculares, a RM de encéfalo fornece informação melhor em relação à localização e à extensão das lesões. A etiologia das lesões vasculares deve ser pesquisada, da mesma forma que em outros casos de acidente vascular cerebral.
O diagnóstico diferencial da DV envolve as outras causas de síndrome demencial apontadas na tabela 2.
A heterogeneidade clínica dos casos de DV limita a generalização dos achados dos estudos clínicos. Esses estudos mostram que tanto os inibidores da acetilcolinesterase quanto a memantina pouco têm benefício para a cognição em pacientes com DV. Os casos devem ser estudados individualmente para a escolha terapêutica. A prevenção secundária de novos eventos vasculares deve ser realizada, como nos casos de acidente vascular cerebral.
A DCL é a segunda causa de demência degenerativa em idosos.
A DCL tem etiologia desconhecida. Sua característica anatomopatológica é a presença de corpos de Lewy com distribuição cortical predominante, podendo haver concomitância de alterações patológicas da DA. Os corpos de Lewy também são encontrados na substância negra, porém em menor intensidade do que na doença de Parkinson.
A DCL é caracterizada por quadro demencial progressivo, com déficits atencionais, visoespaciais, e de funções executivas, e a presença de duas das seguintes características: 1) flutuação dos sintomas cognitivos com variação intensa na atenção e alerta; 2) alucinações visuais recorrentes, geralmente detalhadas e vívidas; 3) parkinsonismo. Outras características clínicas comuns são quedas repetidas, síncope, delírios, alucinações não visuais, depressão, distúrbio do sono REM, e sensibilidade ao neuroléptico.
A investigação deve ser feita como nos casos de DA. O exame de imagem estrutural pode ser normal no início do quadro. A atrofia hipocampal é menos importante do que na DA. O SPECT mostra redução do fluxo sangüíneo nos lobos occipitais.
O diagnóstico diferencial da DCL envolve as outras causas de síndrome demencial apontadas na tabela 2 e, principalmente, outras causas de demência e parkinsonismo.
Não há tratamento específico para a DCL. Os neurolépticos devem ser evitados, pela alta chance de impregnação. Os inibidores da acetilcolinesterase são utilizados e podem melhorar os sintomas cognitivos, além de reduzir os sintomas psicóticos.
A demência frontotemporal (DFT) é uma das síndromes clínicas que compõem os quadros de degeneração lobar frontotemporal, juntamente com a afasia progressiva primária e a demência semântica. Discutiremos os aspectos clínicos da DFT apenas, por ser a mais freqüente entre as três entidades.
É uma doença degenerativa, geneticamente determinada na minoria dos casos. Não existe um fator etiológico determinado nos casos esporádicos da doença. Os achados anatomopatológicos são divididos em três grupos principais: inclusões ubiquitinas positivas, inclusões tau positivas e ausência de alterações específicas.
O quadro clínico da DFT caracteriza-se por curso insidioso de alteração do comportamento ou da personalidade, por exemplo, desinibição, impulsividade, apatia, perda do insight, desinteresse, afastamento social. Há dificuldade em planejamento e na capacidade de julgamento. As alterações do comportamento podem ser divididas em desinibidas, apáticas ou estereotípicas. Alterações de humor, ansiedade e depressão são freqüentes. O paciente pode apresentar comportamento psicopático. São comuns sintomas de hiperoralidade, hipersexualidade e comportamentos estereotipados. Distúrbios de linguagem podem acompanhar o quadro clínico. A idade de início é em geral na quinta ou sexta década, e a duração da doença de aproximadamente oito anos.
O exame neurológico é geralmente normal nas fases iniciais da doença. Alguns casos apresentam sinais extrapiramidais (DFT associada a parkinsonismo – cromossomo 17) e podem estar associados a doença do neurônio motor. Sinais de frontalização, como reflexo de sucção, preensão palmar, e snouting, podem estar presentes.
A investigação deve ser feita como nos casos de DA. O exame de imagem estrutural pode ser normal no início do quadro, porém a presença de atrofia dos lobos frontais e temporais, com preservação relativa dos hipocampos, sugere o diagnóstico de DFT. O SPECT mostra redução do fluxo sangüíneo frontal e temporal, com relativa preservação das regiões posteriores.
O diagnóstico diferencial da DFT envolve as outras causas de síndrome demencial apontadas na tabela 2 e também doenças psiquiátricas que cursam com distúrbios de humor, distúrbios de ansiedade e psicoses tardias.
Não há tratamento específico para a DFT. Os sintomáticos mais utilizados são os antidepressivos e os neurolépticos.
As Doenças priônicas humanas são doença de Creutzfeldt-Jakob (DCJ), doença de Gerstmann-Sträussler-Scheinker (GSS), Insônia fatal (IF) e Kuru. A DCJ é a mais comum das Doenças priônicas e sua incidência aproximada é de
O agente causador é chamado príon, termo que deriva da expressão “proteinaceous infectious particle”, cunhada por Prusiner em 1982. Príon é o nome dado à isoforma patogênica da proteína prion celular, que está presente nos neurônios e na glia em situações normais. A transformação para a isoforma patogência (ou scrapie) ocorre de forma espontânea (nas doenças esporádicas), em decorrência de mutação genética no gene da proteína príon celular (nas formas genéticas), ou após contato com proteína patogênica exógena (na formas adquiridas).
DCJ: classificada em esporádica (eDCJ), genética (gDCJ), iatrogênica (iDCJ), e nova variante (vDCJ). A eDCJ corresponde a cerca de 85% dos casos de Doenças priônicas. A idade média de início é de 60 anos e a sobrevida é de oito meses. Demência de evolução rápida e mioclonias, presentes em 80% dos pacientes, são os sinais mais característicos. Também podem estar presentes sinais piramidais e extrapiramidais, cerebelares e cegueira cortical. Na fase final da doença, os pacientes encontram-se em mutismo acinético. A gDCJ é herdada segundo padrão autossômico dominante com alta penetrância, e são descritas mais de 50 mutações patogênicas. Quadros típicos e atípicos, com evolução mais lenta e idade de início mais jovem, ocorrem a depender da mutação. A forma iatrogênica está associada à exposição ao príon por meio de procedimentos neurocirúrgicos, transplantes de córnea ou ingestão de hormônio de crescimento extraído de cadáveres humanos. A vDCJ ocorre em indivíduos mais novos, com idade média de início aos 28 anos e sobrevida mais longa, com média de 15 meses. O quadro clínico da vDCJ inicia-se com sintomas de natureza psiquiátrica, como ansiedade, depressão ou insônia, que duram aproximadamente seis meses. Algumas vezes, os pacientes desenvolvem distúrbios de marcha, dores e/ou parestesias difusas, além de déficits cognitivos. Em seguida, as alterações neurológicas se desenvolvem: ataxia cerebelar, movimentos involuntários (mioclonias, coréia ou distonia), disfasia, sinais piramidais e demência franca ocorrem em rápida sucessão.
GSS: doença genética caracterizada por ataxia axial e de membros progressiva, sinais piramidais, disartria, alteração da personalidade e declínio cognitivo. Inicia-se na quinta e sexta década de vida, com sobrevida mais longa, em torno de
IF: doença geneticamente determinada na maioria das vezes, podendo ocorrer de forma esporádica. Os sintomas iniciais compreendem insônia, ataxia, distúrbios autonômicos e disartria. Pode haver déficits atencionais, que evoluem para demência. Com a progressão da doença, ocorrem alucinações complexas, mioclonias, sinais piramidais e sonhos vívidos. O paciente evolui para o estado de coma, com intensificação dos distúrbios autonômicos e das mioclonias. A idade média de início da doença é de 50 anos, com sobrevida média entre 13 e 15 meses. Na maioria das vezes, a doença está associada à mutação no códon 178 e à presença de metionina no códon 129 do alelo mutado.
Kuru: os principais sintomas são ataxia de marcha, tremor e fala escandida, que podem ser precedidos por longos períodos de incubação. A ocorrência de demência é rara. Tem sobrevida média de
Os exames mais importantes para o diagnóstico das Doenças priônicas são o EEG, a pesquisa da proteína 14-3-3 no LCR e a RM de crânio, além da pesquisa de mutações no gene da proteína príon celular. As alterações anatomopatológicas são importantes para o diagnóstico definitivo.
O EEG característico da DCJ demonstra atividade periódica, em torno de 1-2 hertz, associada ou não a mioclonias, além de alentecimento da atividade de base. O LCR geralmente é normal. A proteína 14-3-3 encontra-se elevada na DCJ, mas não é específica para esta situação. Na vDCJ não ocorre atividade periódica, apenas o alentecimento da atividade de base. Na IF, o exame eletrográfico do sono mostra alterações no sono REM e nos fusos do sono não REM, que podem estar reduzidos ou ausentes.
A TC de crânio é normal ou demonstra redução do volume encefálico. A RM pode identificar hipersinal cortical e/ou nos gânglios da base, particularmente visibilizados com a técnica de difusão na eDCJ. Na vDCJ, o achado característico é o hipersinal simétrico no pulvinar do tálamo. Na GSS, a RM pode mostrar atrofia cerebelar.
A biópsia de tonsila palatina é um recurso diagnóstico nos casos da vDCJ e pode revelar a presença da isoforma scrapie, confirmando o diagnóstico clínico.
As principais doenças que podem cursar com demência rapidamente progressiva e que devem ser pesquisadas são encefalopatias metabólicas, principalmente hipercalcemia, encefalites virais, neurossífilis, abscessos cerebrais múltiplos, tumores cerebrais, encefalopatias autoimunes como a encefalite límbica paraneoplásica e a encefalopatia de Hashimoto, encefalopatias associadas a medicamentos (amitriptilina, mianserina, lítio e baclofeno), DA de evolução rápida, DV, DFT, degenerações cerebelares e demência associada a AIDS. Dessa forma, a investigação habitual para pacientes com demência também deve ser realizada quando há suspeita de DCJ.
Não há, no momento, nenhum tratamento de comprovada eficácia para as Doenças priônicas. Existem fármacos que exibem efeito in vitro e que foram testados em alguns estudos. O emprego de flupirtina demonstrou discreto efeito sobre a deterioração mental, mas não interferiu na sobrevida, em estudo contra placebo. Estudos controlados estão em andamento para avaliar a eficácia da quinacrina e da clorpromazina, mas os dados disponíveis até o momento não são animadores.
O comprometimento cognitivo leve (CCL) é definido com uma entidade clínica em que há algum declínio cognitivo, porém de intensidade insuficiente para a caracterização de quadro demencial. Portanto, trata-se de um estágio intermediário entre o estado cognitivo normal e a demência, principalmente a DA. O diagnóstico do CCL permite a identificação de indivíduos com alto risco de desenvolver demência, possibilitando intervenção clínica precoce, principalmente no controle de fatores de risco.
Existem três tipos de CCL: CCL amnéstico, CCL de múltiplos domínios e CCL domínio único não-amnéstico. O CCL amnéstico evolui, na maioria das vezes, para DA.
As principais etiologias do CCL são degenerativas, vasculares, metabólicas e traumáticas. Dentre as degenerativas, a DA é a mais comum, entretanto todos os casos demenciais degenerativos podem ser precedidos por fase de sintomatologia mínima, classificado como CCL. Os quadros de DV também podem ser precedidos pelo CCL vascular. Outra etiologia importante é a depressão. Quadros de hipotiroidismo e de deficiência de vitamina B12 são exemplos de CCL de etiologia metabólica.
O quadro clínico do CCL depende da etiologia e das funções cognitivas comprometidas. O tipo mais comum é o CCL amnéstico. Nesse caso, o quadro clínico caracteriza-se pelo déficit de memória, que pode ser progressivo e estar associado a outros déficits cognitivos. A característica clínica que diferencia o CCL das demências é o fato de que o déficit cognitivo é leve, e de intensidade incapaz de interferir de forma significativa nas atividades do paciente. A maioria dos quadros de CCL evolui para demência, com taxa de conversão anual de 10% a 15%, em média. O CCL pode ainda manter-se estável por alguns anos, ou melhorar, como no caso do CCL secundário à depressão.
Os casos de CCL devem ser investigados da mesma forma que os de demência. Fatores de risco para demência devem ser investigados, por exemplo, hipertensão arterial e diabetes, para que haja intervenção terapêutica precoce, diminuindo a chance de progressão para demência.
O diagnóstico diferencial dos quadros de CCL engloba todas as etiologias de demência, uma vez que é um diagnóstico de uma condição clínica intermediária entre o normal e o quadro demencial.
O tratamento do CCL é dividido em tratamento dos fatores de risco e tratamento sintomático das alterações cognitivas. O tratamento dos fatores de risco deve ser priorizado, principalmente o tratamento de fatores de risco vasculares.
Em relação ao tratamento sintomático, alguns estudos com antidepressivos e com inibidores da acetilcolinesterase para os quadros de CCL amnéstico não mostraram eficácia satisfatória. Alguns estudiosos especialistas no assunto preconizam o uso de inibidores da acetilcolinesterase nos casos de CCL amnéstico, porém não há evidência em literatura médica disponível até o momento que aponte para uma eficácia inequívoca no tratamento medicamentoso do CCL.
A síndrome demencial é caracterizada por um declínio cognitivo adquirido que interfira nas atividades profissionais e sociais do indivíduo. O déficit cognitivo deve compreender alteração de memória associado à alteração de outro domínio cognitivo.
O diagnóstico de demência não deve ser feito na vigência de delirium.
As causas mais comuns de demência são a DA e a DV, seguidas pela DCL.
Devemos suspeitar de quadro demencial quando o paciente apresentar alterações cognitivas (principalmente perda de memória), sintomas psiquiátricos, alterações de personalidade, mudanças no comportamento ou diminuição na capacidade de realizar atividades da vida diária.
É fundamental pesquisar sintomas depressivos precedendo o quadro, pois muitas vezes a depressão simula quadros demenciais
O Miniexame do Estado Mental (Mini-mental) é um bom exame de rastreamento e acompanhamento de um quadro demencial, embora tenha limitações por ser influenciado por idade e escolaridade
Deve ser realizada avaliação clínica e neurológica completa nos pacientes com suspeita de demência, não apenas para se procurar por causas tratáveis de declínio cognitivo, mas também para realizar suporte adequado ao paciente e seus familiares e para se iniciar tratamento quando indicado.
Os exames necessários para investigação de síndrome demencial em faixa etária senil são hemograma, função renal (dosagem de eletrólitos, uréia e creatinina), hepática e tiroidiana, dosagem sérica de vitamina B12 e ácido fólico, sorologia para sífilis, glicemia de jejum e exame de neuroimagem.
Exames adicionais conforme o quadro clínico são análise do líquor, sorologia para HIV e EEG.
A principal e mais precoce característica clínica da DA é o déficit de memória. O paciente tem dificuldade em memorizar novas informações, experiências e eventos recentes. Com a progressão do quadro, o déficit de memória acentua-se e se associam déficits em outros domínios cognitivos como linguagem, praxia, atenção, funções executivas e orientação espacial. O paciente passa a ter dificuldades progressivas nas atividades do dia-a-dia.
O diagnóstico é feito mediante história clínica e exames neurológico sugestivos, devendo ser excluídas outras causas de síndrome demencial por meio de exames complementares
A DV ocorre secundariamente a insultos vasculares ao sistema nervoso central. A evolução característica é em degraus, ocorrendo platôs entre os eventos clínicos. O paciente pode apresentar sintomas neurológicos focais, caracteristicamente tem fatores de risco para doença cerebrovascular (hipertensão, diabetes, tabagismo, dislipidemia e idade avançada) e os exames de imagem podem revelar os eventos isquêmicos prévios.
O quadro clínico típico da DCL é o de um déficit cognitivo flutuante com variação intensa na atenção e alerta associado a alucinações visuais recorrentes e parkinsonismo.
Os neurolépticos devem ser evitados na DCL, pela alta chance de impregnação.
O CCL é uma entidade clínica em que há algum declínio cognitivo, freqüentemente déficit de memória, porém de intensidade insuficiente para a caracterização de quadro demencial.
O diagnóstico do CCL permite a identificação de indivíduos com alto risco de desenvolver demência, possibilitando intervenção clínica precoce.
Algoritmo 1: Avaliação diagnóstica dos quadros demenciais

1. Bottino CMC, Laks J, Blay SL. Demência e transtornos cognitivos em idosos. Rio de Janeiro: Guanabara-Koogan; 2006.
2. Caramelli M, Ru G, Acutis P, Forloni G. Príon diseases: current understanding of epidemiology and pathogenesis, and therapeutic advances. CNS Drugs 2006;20:15-28.
3. Collinge J. Molecular neurology of prion disease. J Neurol Neurosurg Psychiatry 2005;76:906-19.
4. Cummings JL, Mega MS. Neuropsychiatry and behavioral neuroscience. New York: Oxford University Press; 2003.
5. Farlow MR, Cummings JL. Effective pharmacologic management of Alzheimer’s disease. Am J Med 2007; 120:388-97.
6. Feldman HH, Ferris S, Winblad B et al. Effect of rivastigmine on delay to diagnosis of Alzheimer’s disease fro mild cognitive impairment: the InDDEx study. Lancet Neurol 2007;6:501-12.
7. Glatzel M, Stoeck K, Seeger H, Lührs T, Aguzzi A. Human prion diseases: molecular and clinical aspects. Arch Neurol 2005;62:545-52.
8. Hodges JR. Early-onset dementia: a multidisciplinary approach. New York: Oxford University Press; 2001.
9. Kavirajan H, Schneider LS. Efficacy and adverse effects of cholinesterase inhibitors and memantine in vascular dementia: a meta-analysis of randomised controlled trials. Lancet Neurol 2007;6:782-92.
10. Mesulam MM. Principles of behavioral and cognitive neurology. 2. ed. New York: Oxford University Press; 2000.
11. Petersen RC, Morris JC. Mild cognitive cmpairment as a clinical entity and treatment target. Arch Neurol 2005;62:1160-3.
12. Petersen RC, Thomas RG, Grundman M et al. Vitamin E and donepezil for the treatment of mild cognitive impairment. N Engl J Med 2005;352(23):2379-88.
13. Pickering-Brown SM. The complex aetiology of frontotemporal lobar degeneration. Experim Neurol 2007;206:1-10.
14. Waldemar G, Dubois B, Emre M et al. Recommendations for the diagnosis and management of Alzheimer’s disease and other disorders associated with dementia: EFNS guideline. J Neurol 2007; 14:e1-e26.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.