(Carregando Índice)... (Carregando Índice)... |
Autor:
Daniel Soares Freire
Endocrinologista pelo Hospital das Clínicas da Faculdade de Medicina da USP;
Médico Colaborador do Serviço de Endocrinologia do Hospital das Clínicas da Faculdade de Medicina da USP
Última revisão: 18/05/2009
Comentários de assinantes: 0
Lesões de diversas naturezas podem crescer e ocupar a região selar. Embora as síndromes endócrinas determinadas pelos tumores hipofisários sejam relativamente raras, na maioria das vezes, é o médico generalista quem recebe o paciente inicialmente; assim, faz-se necessário que o clínico saiba reconhecer e diagnosticar estas situações.
Nesta seção, serão abordados os aspectos clínicos e a investigação laboratorial inicial dos tumores hipofisários. Atenção especial será dada aos incidentalomas hipofisários, os tumores selares descobertos em exames de imagem realizados por outra causa que não doença pituitária.
Os tumores hipofisários podem ser funcionantes ou clinicamente não-secretores. Os tumores funcionantes determinam as síndromes de hipersecreção hipofisária, que compreendem a acromegalia, a hiperprolactinemia, a doença de Cushing e os raros casos de tumores secretores de TSH e de gonadotrofinas.
Na Tabela 1 estão listadas as principais lesões hipofisárias.
Tabela 1: Principais lesões hipofisárias
|
Adenomas funcionantes |
Prolactinoma (PRL) |
|
Somatotropinoma (GH) | |
|
Corticotropinoma (ACTH) | |
|
Tireotropinoma (TSH) | |
|
Gonadotropinoma (LH, FSH) | |
|
Adenomas clinicamente não-secretores |
|
|
Tumores malignos na hipófise |
Carcinoma de hipófise |
|
Tumores metastáticos para a hipófise | |
|
Cistos hipotálamo-hipofisários |
Craniofaringeomas |
|
Cistos da bolsa de Rathke | |
|
Mucoceles | |
|
Cistos aracnoides |
Os adenomas funcionantes mais comuns são os prolactinomas (secretores de prolactina), os somatotropinomas (secretores de GH) e os corticotropinomas (secretores de ACTH). Os tumores secretores de TSH e de gonadotrofinas são particularmente raros.
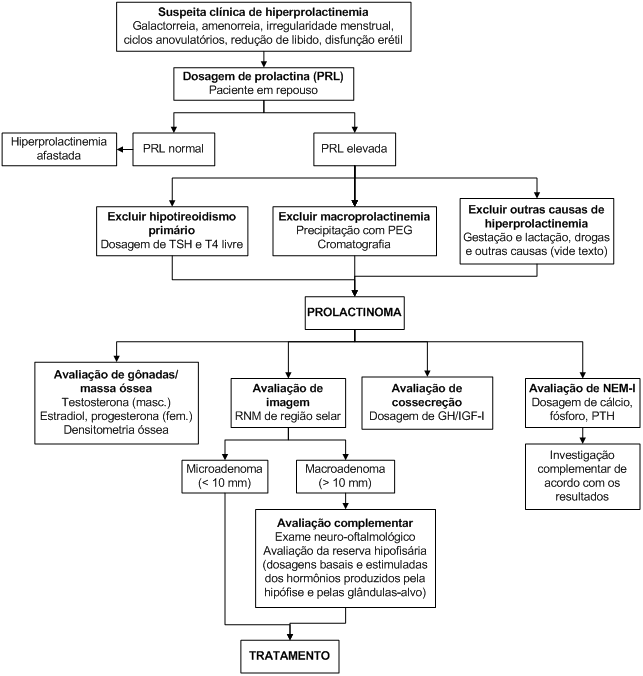
As causas de hiperprolactinemia podem ser agrupadas em três grupos: fisiológicas, farmacológicas e patológicas; neste último grupo, enquadram-se os prolactinomas (Tabela 2).
Tabela 2: Causas de hiperprolactinemia
|
Fisiológicas |
Farmacológicas |
Patológicas |
|
Gestação |
Psicotrópicos (antidepressivos tricíclicos, inibidores seletivos de recaptação de serotonina, haloperidol) |
Prolactinomas |
|
Lactação |
Anti-hipertensivos (reserpina, metildopa, bloqueadores de canais de Ca+2, inibidores de ECA) |
Lesões que comprimam a haste hipofisária |
|
Exercício físico extenuante |
Antagonistas H2 (cimetidina) |
Hipotireoidismo primário |
|
Estresse, dor, alimentação, sucção mamilar |
Estrogênio em dose alta |
Síndrome de ovários policísticos |
|
|
Opiáceos |
Causas neurogênicas (lesão de nervo intercostal, herpes zóster) |
|
|
Cocaína |
Insuficiência renal crônica (IRC) |
|
|
|
Cirrose |
Os prolactinomas são os tumores hipofisários funcionantes mais comuns. Os microadenomas são mais comuns em mulheres (proporção F:M de aproximadamente 20:1); já os macroadenomas apresentam uma distribuição quase equivalente entre homens e mulheres, embora costumem ser maiores nos homens que nas mulheres.
O quadro clínico dos prolactinomas está descrito minuciosamente na Tabela 3.
Tabela 3: Quadro clínico de hiperprolactinemia
|
Órgão ou sistema |
Achados clínicos |
|
Gônadas |
Tanto os pacientes com micro como com macroprolactinomas manifestam sintomas de hiperprolactinemia. O efeito negativo da prolactina sobre o eixo hipotálamo-hipófise-gônada se dá tanto por redução da pulsatilidade do GnRH quanto por um bloqueio direto sobre os ovários e testículos. |
|
Mulheres: distúrbios menstruais, galactorreia e redução de libido são os sintomas mais importantes. Os distúrbios menstruais incluem amenorreia (primária ou secundária), oligomenorreia, menorragia ou redução da duração da fase lútea (que pode ser causa de infertilidade). | |
|
Homens: os pacientes manifestam redução de libido, disfunção erétil, oligospermia ou azospermia. | |
|
Metabolismo ósseo |
Osteopenia ou osteoporose advêm do hipogonadismo induzido pelo excesso de prolactina. |
|
Neurológico |
Microadenomas podem ser assintomáticos do ponto de vista neurológico, embora alguns possam ser invasivos e a incidência de cefaleia seja maior nos pacientes do que na população geral. |
|
Macroadenomas podem invadir ou comprimir estruturas vizinhas à sela, determinando quadro de perda de campo visual por comprometimento do quiasma óptico. Outros sintomas, como epilepsia por comprometimento do lobo temporal ou paralisia de nervos cranianos por invasão do seio cavernoso, são relatados, embora pouco frequentes. |
Na avaliação de um paciente com hiperprolactinemia, especialmente se o quadro clínico for pouco importante frente aos níveis de prolactina, deve-se afastar a macroprolactinemia, que consiste na presença de complexos de moléculas de prolactina com elevado peso molecular, que apresentam imunoatividade (são dosáveis no soro), mas não têm bioatividade. A prevalência de macroprolactina na população geral é estimada em 0,2%, e esta pode ser a forma predominante da prolactina em até 25% dos soros hiperprolactinêmicos. O rastreamento da macroprolactinemia deve ser feito por meio de precipitação com polietilenoglicol (PEG), e os casos suspeitos, confirmados por cromatografia.
Algoritmo 1: Investigação de hiperprolactinemia.

Existem duas armadilhas diagnósticas que merecem atenção especial do clínico na investigação de hiperprolactinemias: os pseudoprolactinomas e o efeito-gancho.
Os pseudoprolactinomas são, na verdade, tumores clinicamente não-secretores que provocam aumento discreto da secreção de prolactina pela hipófise normal residual pela compressão da haste hipofisária e interferência no aporte de dopamina à hipófise. Nestes casos, os níveis de prolactina são pouco elevados (em geral, menores que 100 ng/mL) e reduzem drasticamente após doses baixas de agonista dopaminérgico (p.ex., 1,25 a 2,5 mg de bromocriptina).
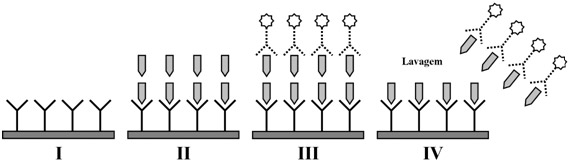
O efeito-gancho ocorre quando se utilizam métodos que empregam duplo anticorpo para a dosagem de prolactina. Se os níveis do hormônio forem extremamente elevados, pode haver perda do segundo anticorpo que se liga ao excesso de antígeno, fornecendo resultados falsamente baixos (< 100 ng/mL). O problema é resolvido com pré-diluição do soro ou lavagem após incubação com o primeiro anticorpo.
Figura 1: O efeito-gancho.
I – Anticorpo específico para prolactina na parede do tubo. II – Quando a concentração de prolactina é extremamente elevada, existe uma grande quantidade de antígeno livre que não se liga a nenhum anticorpo na parede do tubo. III – O segundo anticorpo pode se ligar a moléculas de prolactina não-ligadas ao anticorpo do tubo. IV – Após a lavagem final antes da leitura, o segundo anticorpo é perdido, pois se ligou ao excesso de antígeno disponível, fornecendo um resultado falsamente baixo.
Os objetivos do tratamento são controle da secreção de prolactina, normalização da função gonadal e regressão do tumor. O tratamento pode ser clínico, cirúrgico ou radioterápico.
Desde o seu advento, os agonistas dopaminérgicos representam a primeira linha de tratamento dos prolactinomas. No Brasil, dispomos de duas drogas, a bromocriptina e a cabergolina. Dada a maior eficácia, maior comodidade posológica e menor incidência de efeitos adversos, a cabergolina se estabeleceu como droga de escolha no tratamento dos prolactinomas. As principais características dos agentes dopaminérgicos usados no tratamento da hiperprolactinemia estão sumarizadas na Tabela 4.
Tabela 4: Agonistas dopaminérgicos
|
Droga |
Nome comercial |
Disponível no Brasil |
Dose inicial |
Comentários |
|
Bromocriptina |
Parlodel® Bagren® |
Sim |
1,25 mg/dia |
Náusea, vômito, hipotensão postural são muito comuns. É a droga de escolha no tratamento da gestante com prolactinoma. |
|
Cabergolina |
Dostinex® |
Sim |
0,25 mg/semana |
Maior duração de ação e maior afinidade pelo receptor de dopamina D2. Consequentemente, promove melhor controle da secreção de prolactina e redução tumoral. Além disso, é mais bem tolerado que outras drogas da classe. |
|
Pergolida |
Permax® |
Não |
0,025 mg/dia |
Retirado do mercado devido a risco de lesão cardíaca valvar. |
|
Quinagolida |
Norprolac® |
Não |
0,025 mg/dia |
Agonista dopaminérgico não-ergot. |
Recentemente, duas publicações apontaram para o risco de lesão cardíaca valvar relacionada aos agonistas dopaminérgicos cabergolina e pergolida. Contudo, os estudos avaliaram pacientes com doença de Parkinson, que normalmente utilizam doses bem mais altas dos agonistas dopaminérgicos. O risco era aumentado para os pacientes que receberam doses superiores a 3 mg/dia (cabergolina ou pergolida) por mais de 6 meses.
O insucesso do tratamento clínico pode decorrer de intolerância ou resistência aos agonistas dopaminérgicos. A intolerância é mais comum com a bromocriptina; se os níveis de prolactina ou a imagem tumoral indicarem necessidade de doses maiores do agente, uma possibilidade é a substituição por cabergolina, que cursa com menos efeitos adversos. A definição de resistência a agonistas dopaminérgicos não é uniforme entre os autores; ela pode ser definida como ausência de normalização da prolactina ou redução tumoral após 3 meses de tratamento com bromocriptina em dose igual ou superior a 15 mg/dia. O mecanismo de resistência ainda não é bem conhecido, mas já se demonstrou redução da expressão do receptor D2 nos prolactinomas resistentes em comparação com os responsivos a agonistas dopaminérgicos.
A cirurgia transfenoidal está indicada quando há risco iminente de perda visual, na apoplexia hipofisária e nos casos de resistência ou intolerância aos agonistas dopaminérgicos, após tentativa de troca por um agente que curse com menos efeitos adversos (cabergolina).
A radioterapia é reservada somente aos casos que não responderam ao tratamento clínico e cirúrgico. O tempo para se atingir o controle pode ser longo e esta modalidade terapêutica se associa a efeitos adversos como hipopituitarismo (até 60% dos pacientes em 10 anos), lesões de vias ópticas, radionecrose de lobo temporal e aumento do risco de neoplasias extra-hipofisárias.
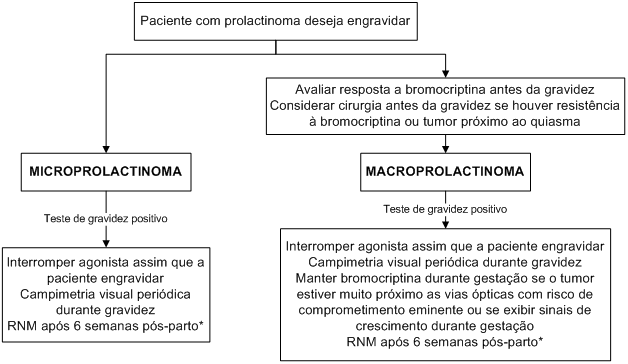
Durante a gestação, pode haver aumento dos prolactinomas. Estima-se que o crescimento tumoral ocorra em 1,4% das pacientes com microprolactinomas e 16 a 36% no caso dos macroprolactinomas.
Por outro lado, a exposição do feto a agonistas dopaminérgicos deve ser evitada na maior parte dos casos. Das drogas disponíveis, a bromocriptina é a que acumula maior quantidade de estudos demonstrando sua segurança. Estudos retrospectivos que avaliaram pacientes que engravidaram durante o uso de bromocriptina não demonstraram aumento do risco de abortamentos, prematuridade, multiparidade ou malformações fetais.
Durante o aleitamento materno, é recomendada a suspensão dos agonistas dopaminérgicos, exceto se houver sinais de crescimento tumoral. Nesta situação, o aleitamento deve ser interrompido e o tratamento com agonista dopaminérgico, iniciado.
O Algoritmo 2 ilustra as recomendações para as pacientes com prolactinomas durante a gestação.
Algoritmo 2: Manejo dos prolactinomas durante a gestação.
* considerar a realização da RNM durante a gestação se houver necessidade absoluta.
Os tumores secretores de GH podem causar gigantismo ou acromegalia, dependendo de o excesso de GH ter se instalado antes ou após o fechamento das epífises dos ossos longos. Trata-se de uma doença rara, com incidência anual de 3 a 4 casos por 1.000.000 de pessoas. Afeta homens e mulheres de forma igual, e é mais comum entre a 5ª e a 7ª décadas de vida.
Em mais de 98% dos casos, a acromegalia decorre de um tumor hipofisário benigno (adenoma secretor de GH). Casos anedóticos de secreção ectópica de GHRH já foram descritos. Aproximadamente 5% dos casos podem estar associados a tumores pancreáticos endócrinos (gastrinomas, insulinomas, tumores não-secretores) e a hiperparatireoidismo, constituindo a síndrome de neoplasia endócrina múltipla do tipo 1.
Além do crescimento de extremidades e partes moles e do ganho de estatura, o hipersomatotropismo também cursa com artralgias, cefaleia, sudorese, visceromegalias, crescimento de nódulos tireoidianos e pólipos intestinais, hipertensão arterial sistêmica, miocardiopatia acromegálica (podendo evoluir até insuficiência cardíaca congestiva grave) e distúrbios do metabolismo glicêmico (intolerância à glicose e diabetes mellitus).
Mais de 80% dos somatotropinomas são macroadenomas hipofisários, isto é, lesões com mais de 10 mm no maior eixo. As lesões podem apresentar crescimento suprasselar, comprimindo o quiasma óptico e determinando prejuízo de campo visual (como a hemianopsia bitemporal), ou crescimento parasselar, podendo levar a oftalmoplegia.
O quadro clínico da acromegalia é de instalação insidiosa, podendo evoluir durante anos. Na maioria dos casos, pode-se identificar um intervalo de mais de uma década entre o início dos sintomas e o diagnóstico da doença. As principais características clínicas estão sumarizadas na Tabela 5.
Tabela 5: Quadro clínico de acromegalia
|
Órgão ou sistema |
Achados clínicos |
|
Neurológico |
Cefaleia é presente em até 65% dos pacientes, e pode ser desproporcional ao tamanho tumoral. Quando o tumor cresce para além da região selar, pode determinar perda de campo visual, hidrocefalia, paralisias de nervos cranianos e epilepsia temporal. |
|
Cardiovascular |
Doença cardiovascular é a principal causa de morte de pacientes acromegálicos, contabilizando até 60% dos óbitos. |
|
Hipertensão arterial sistêmica decorre de retenção de sódio e água, expansão volêmica e hiperatividade simpática. | |
|
Já foi descrita a cardiomiopatia acromegálica, resultado da exposição do miocárdio a concentrações elevadas de GH por longos períodos, que cursa com hipertrofia concêntrica de ventrículos esquerdo e direito, aumento do potencial arritmogênico e disfunção ventricular (predominantemente diastólica). | |
|
A função endotelial também está prejudicada na acromegalia, o que pode contribuir para o processo aterosclerótico. | |
|
Respiratório |
A principal alteração é a apneia obstrutiva do sono, relacionada principalmente com a macroglossia e o aumento das partes moles na orofaringe, mas também associada a um componente central de hipoventilação. |
|
Metabolismo de carboidratos e lipídios |
Diabetes mellitus e intolerância à glicose decorrem da resistência insulínica determinada pelo GH, que é um hormônio contrarregulador. Até 70% dos pacientes apresentam hiperinsulinemia, denotando a resistência à ação da insulina. É responsável, em conjunto com a hipertensão e a cardiomiopatia acromegálica, pelo excesso de mortalidade cardiovascular. |
|
Hipertrigliceridemia pode ocorrer e decorre da menor atividade da lipoproteína-lipase, devido à resistência insulínica. Geralmente se acompanha de baixos níveis de HDL-colesterol, o que agrava o potencial aterogênico. | |
|
Gônadas |
Hipogonadismo hipogonadotrófico pode decorrer de disfunção dos gonadotrofos por compressão ou por hiperprolactinemia concomitante (tumor secretor de GH e prolactina ou compressão da haste hipofisária pelo tumor). Aproximadamente metade das pacientes tem amenorreia, enquanto que a mesma proporção dos homens tem disfunção erétil. |
|
Músculo-esquelético |
Quando o hipersomatotropismo se instala antes da puberdade, o não fechamento das epífises ósseas (pelo hipogonadismo) associado ao excesso de GH causa um importante ganho em estatura, caracterizando o gigantismo. |
|
Na vida adulta, o excesso de GH provoca marcadas alterações músculo-esqueléticas. Crescimento de extremidades (mãos, pés, nariz, orelhas, lábios) ocorre em todos pacientes, em menor ou maior intensidade. | |
|
Até 30% exibem importante poliartralgia, que frequente melhora com a redução dos níveis de GH. | |
|
Síndrome de túnel do carpo está presente em mais de 2/3 dos pacientes. | |
|
Dermatológico |
Sudorese excessiva, acantose nigricante, acne, cistos sebáceos, papilomas e skin tags são frequentemente observados em pacientes acromegálicos. |
|
Neoplasias |
Pacientes acromegálicos são mais propensos a nódulos tireoidianos e pólipos intestinais, porém não há evidência clara de aumento no número de casos de nódulos malignos. Há controvérsias quanto à maior incidência de tumores prostáticos e de mama. |
|
Visceromegalias |
Diversos órgãos apresentam aumento do seu tamanho: rins, fígado, próstata, glândulas salivares, baço, tireoide e língua. |
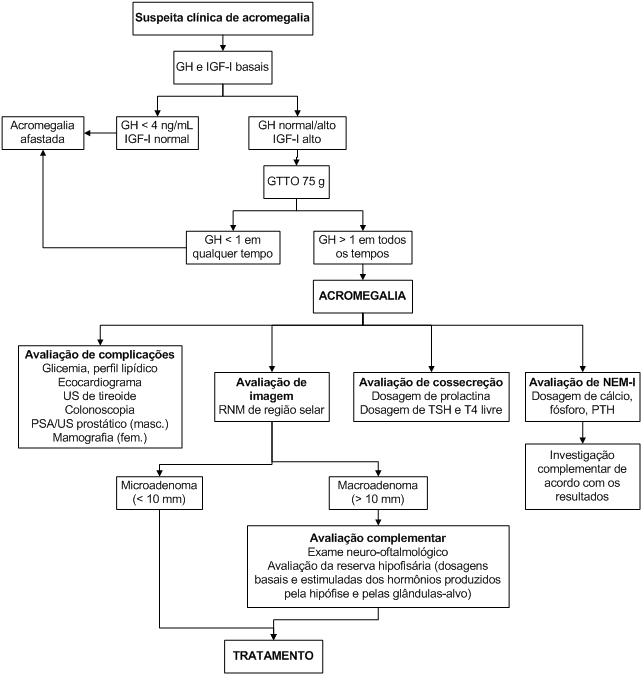
O diagnóstico clínico deve ser confirmado laboratorialmente, por meio das dosagens de GH e IGF-I.
A secreção de GH é pulsátil durante as 24 horas, de maneira que uma dosagem pouco elevada pode representar um pico, e não ser patológica. Por outro lado, a dosagem randômica de GH ajuda a afastar o diagnóstico, uma vez que valores inferiores a 0,4 ng/mL (associado a IGF-I normal) tornam o diagnóstico muito improvável.
A hipófise normal suprime a secreção de GH frente ao estímulo glicêmico. Assim, dosagens de GH após sobrecarga de glicose (75 g por VO) devem ser realizadas no diagnóstico da acromegalia. Indivíduos normais devem suprimir o GH para níveis abaixo de 1 ng/mL após a sobrecarga de glicose.
A dosagem de IGF-I é bastante útil no diagnóstico e no seguimento de pacientes acromegálicos. Trata-se de um fator de crescimento de produção hepática e tecidual, responsivo ao GH. Como os níveis de IGF-I não oscilam durante o dia, sua dosagem fornece ideia da secreção integrada de GH nas 24 horas. Os resultados devem ser interpretados levando-se em consideração o sexo e a idade do paciente. Virtualmente, todos os pacientes acromegálicos apresentam IGF-I aumentado para sexo e idade.
Após o diagnóstico da acromegalia, o paciente deverá ser investigado quando às seguintes complicações:
Avaliação do metabolismo lipídico e de carboidratos: glicemia de jejum, teste de tolerância oral à glicose, hemoglobina glicada (se diabético) e perfil lipídico com dosagem de triglicérides, HDL e LDL colesterol.
Avaliação cardiovascular: medida de PA (monitoração ambulatorial de PA durante 24 horas se indicado), eletrocardiograma, ecocardiograma, radiografia de tórax para avaliação da área cardíaca.
Avaliação respiratória: polissonografia.
Avaliação da função hipofisária: 1/3 dos pacientes pode apresentar hiperprolactinemia concomitante, e os tumores maiores podem cursar com pan-hipopituitarismo.
Avaliação da integridade das vias ópticas: por meio de exame neuro-oftalmológico que inclui acuidade visual, campo visual, avaliação da papila ao exame de fundo de olho e medida da pressão intraocular.
Avaliação da presença de neoplasias: ultrassonografia de tireoide, avaliação prostática (PSA, ultrassonografia e biópsia, se indicado), mamografia e colonoscopia.
Avaliação quanto ao diagnóstico de NEM-1: dosagem de cálcio, fósforo, PTH, insulina e gastrina.
Algoritmo 3: Investigação de acromegalia.
Os objetivos do tratamento são redução de massa tumoral, supressão dos níveis de GH e IGF-I, tratamento das sequelas crônicas do hipersomatotropismo e manutenção da função hipofisária.
Há três modalidades terapêuticas para a acromegalia: tratamento cirúrgico, tratamento clínico e tratamento radioterápico.
A cirurgia transfenoidal é o tratamento de escolha para a maioria dos casos. A via transcraniana cursa com maior morbidade, mas pode ser adequada para tumores grandes com componente suprasselar importante. As complicações mais graves (meningites, lesão de vias ópticas e morte) ocorrem em menos de 2%. Remissão (definida como GH < 1 ng/mL durante GTTO e IGF-I normal) é observada em aproximadamente 70% dos microadenomas, mas em somente 50% dos macroadenomas.
Pode ser utilizado nos pacientes que não responderam à cirurgia ou no período pré-operatório, para tratar as complicações relacionadas à acromegalia e minimizar o risco cirúrgico.
1. Análogos de somatostatina
Dos 5 subtipos de receptores de somatostatina, SSTR2 e SSTR5 são os mais expressos pelos somatotropinomas. Diversos ligantes destes receptores foram desenvolvidos e são utilizados com sucesso no controle dos adenomas hipofisários secretores de GH.
O primeiro análogo de somatostatina utilizado foi a octreotida (Sandostatin®), dada em múltiplas injeções diárias. Há atualmente uma formulação de depósito para aplicações mensais (Sandostatin®LAR). Em média, ocorre normalização do GH e IGF-I em 65% e redução tumoral em 30% dos pacientes em uso da formulação de depósito (resultados um pouco mais modestos com a formulação de ação curta).
Os principais efeitos adversos dos análogos de somatostatina são gastrintestinais (diarreia, náusea, flatulência, litíase biliar). Contudo, a tolerabilidade desta classe de hormônios é muito boa.
O outro análogo de somatotatina (lanreotida) não está disponível no Brasil, mas tem eficácia e segurança semelhantes à octreotida.
Tabela 6: Análogos de somatostatina
|
Droga |
Nome comercial |
Disponível no Brasil |
Dose usual |
|
Octreotida |
Sandostatin® |
Sim |
50 a 100 mcg SC 3 vezes/dia (máximo: 1.500 mcg/dia) |
|
Octreotida LAR |
Sandostatin®LAR |
Sim |
20 mg IM a cada 28 dias |
|
Lanreotida |
Somatuline® |
Não |
30 mg IM a cada 10 a 14 dias |
|
Lanreotida Autogel |
Somatuline®Autogel |
Não |
60 a 120 mg SC a cada 28 dias |
2. Agonistas dopaminérgicos
Bromocriptina (Parlodel®) e Cabergolina (Dostinex®) promovem controle da secreção de GH em uma minoria de pacientes (especialmente naqueles com tumores cossecretores de prolactina e GH). As doses necessárias no tratamento da acromegalia geralmente são superiores àquelas usadas no tratamento de prolactinomas.
3. Antagonista de receptor de GH
Recentemente, foi introduzido um antagonista do GH, o pegvisomant (Somavert®), que atua nos tecidos impedindo a ação do GH e reduzindo os níveis de IGF-I. Contudo, a secreção de GH pode até aumentar, de forma que a concentração sérica do hormônio não pode mais ser usada como parâmetro de tratamento. A eficácia em normalizar os níveis de IGF-I se aproxima de 90%. Como a medicação não interfere no tumor, apenas nos níveis de IGF-I, a redução da retroalimentação sobre o hipotálamo e a hipófise pode, em teoria, facilitar crescimento tumoral. Por medidas de segurança, o tamanho tumoral deve ser monitorado durante o uso do pegvisomant. A dose usual é de 10 a 40 mg/dia SC. A associação do pegvisomant com um análogo de somatostatina (octreotida LAR) é possível e há evidências de que permite um controle da secreção de GH e IGF-I superior a cada uma das drogas isoladamente.
A radioterapia convencional fracionada (dose total de 4.500 a 5.000 cGy) possibilita controle da secreção de GH em 65 a 80% dos casos, mas o tempo para atingir este controle pode ser de até 20 anos, o que impõe a necessidade de outra modalidade terapêutica (p.ex., análogos de somatostatina) durante o intervalo.
Todas as causas de síndrome de Cushing, incluindo os corticotropinomas (doença de Cushing), são abordadas no capítulo específico sobre Síndrome de Cushing.
São tumores muito raros, compreendendo menos de 1% dos tumores de hipófise. Geralmente são macroadenomas que podem apresentar invasão de estruturas vizinhas e que determinam quadro de hipertireoidismo com bócio e TSH elevado. Duas situações devem ser diferenciadas dos tireotropinomas: a síndrome de resistência aos hormônios tireoidianos e a hiperplasia hipofisária relacionada ao hipotireoidismo primário.
Causada por uma mutação no receptor de hormônio tireoidiano (TR-beta), também cursa com TSH inapropriadamente elevado para os níveis de T4 livre, mas não há tumor hipofisário. O quadro clínico é peculiar, pois os pacientes apresentam sintomas de hipo e hipertireoidismo ao mesmo tempo (uma vez que o receptor TR-alfa não está mutado e a quantidade de cada um dos tipos de receptores varia nos diversos órgãos-alvo).
Pacientes com hipotireoidismo primário grave de longa data podem apresentar hipertrofia do setor tireotrófico da hipófise devido à redução da retroalimentação negativa pelos hormônios tireoidianos. A hipófise aparece globosa, mas sem tumor evidente, e o paciente apresenta hipotireoidismo, e não hipertireoidismo. Com a reposição do hormônio tireoidiano, há normalização do tamanho da hipófise.
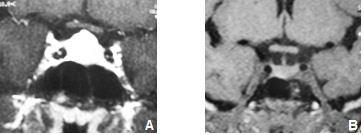
Figura 2: Hiperplasia hipofisária associada a hipotireoidismo primário.
A B
Paciente do sexo feminino, com 17 anos de idade, apresenta-se com amenorreia secundária e galactorreia. Os níveis de prolactina eram de 91 ng/mL e o TSH de 660 mU/L, com T4 livre < 0,3 ng/dL. A RNM inicial (A) mostra aumento difuso da hipófise, que quase toca o quiasma óptico. Após reposição com levotiroxina, houve reestabelecimento dos ciclos menstruais, remissão do quadro de galactorreia e normalização dos níveis de hormônios tireoidianos e da prolactina. A imagem hipofisária realizada 6 meses após normalização do TSH mostra hipófise de aspecto normal (B).
O tratamento é essencialmente cirúrgico, mas, em até 70% dos casos, pode haver controle da secreção de TSH com análogos de somatostatina. Há relatos de casos em que a ablação definitiva da função tireoidiana (com 131I ou tireoidectomia) provocou aumento significativo do tumor hipofisário, à semelhança da síndrome de Nelson. Radioterapia pode ser usada como tratamento adjuvante quando a cirurgia e os análogos de somatostina não permitem controle da doença.
Os tumores secretores de gonadotrofinas são muito raros, embora seja frequente a demonstração destes hormônios (especialmente FSH) em imuno-histoquímica de tumores clinicamente não-secretores.
Em geral, são macroadenomas agressivos e laboratorialmente caracterizam-se por aumento do FSH e subunidade alfa. Paradoxalmente, os pacientes podem apresentar hipogonadismo por down-regulation do receptor de gonadotrofinas na gônada. Algumas mulheres têm quadro de dor pélvica associada à hiperestimulação ovariana. Tumores secretores de LH são excepcionalmente raros e, nos homens, causam aumento dos níveis de testosterona com LH elevado.
Utiliza-se cirurgia, radioterapia ou ambos para o tratamento dos tumores secretores de gonadotrofinas. Há relatos anedóticos de resposta tumoral ao tratamento com agonistas dopaminérgicos, agonistas de GnRH ou análogos de somatostatina, mas os resultados são modestos demais para que o seu emprego seja recomendado.
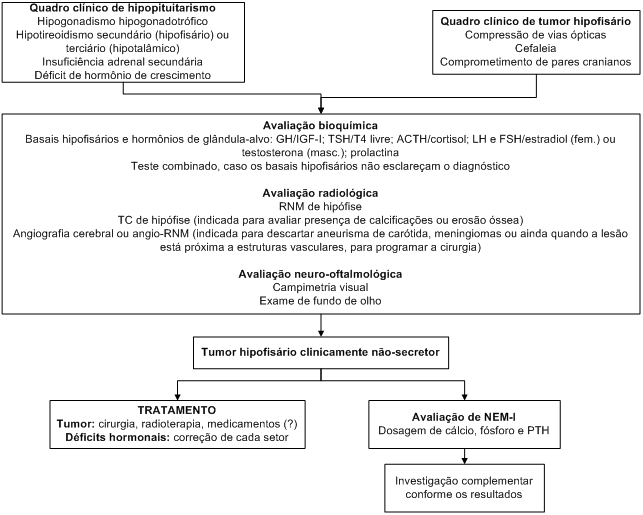
Aproximadamente 25 a 35% dos tumores hipofisários são lesões clinicamente não-secretoras. Mais de 90% das lesões são adenomas hipofisários. A denominação “adenoma não-funcionante” não deve ser usada, pois, na realidade, estes tumores podem ser capazes de sintetizar diversos peptídios (LH, FSH, TSH e subunidade alfa, principalmente), o que é observado em imuno-histoquímica, hibridização in situ e em cultura de células. Porém, esta produção não é grande o suficiente para alterar os níveis séricos dos hormônios.
Os tumores clinicamente não-secretores só provocam sintomas se causam compressão do quiasma e vias ópticas ou se interferem com a função da hipófise normal adjacente. Logo, a maioria dos tumores não-secretores sintomáticos é composta por macroadenomas (> 10 mm), alguns dos quais apresentam importante crescimento suprasselar e invasão de estruturas vizinhas.
Os setores hormonais da hipófise anterior obedecem a uma ordem de acometimento pelos macroadenomas clinicamente não-secretores, sendo o setor somatotrófico o mais acometido (100%), seguido pelos setores gonadotrófico (96%), tireotrófico (81%) e corticotrófico (62%). O hipopituitarismo pode decorrer de compressão da hipófise pelo tumor ou ainda de compressão da haste hipofisária, impedindo o aporte de hormônios tróficos hipotalâmicos (GHRH, GnRH, TRH e CRH) à hipófise.
A compressão da haste também diminui o tônus dopaminérgico sobre os lactotrofos normais, determinando elevação discreta da prolactina (em geral, níveis inferiores a 100 ng/mL). É importante que o clínico reconheça esta situação e saiba diferenciá-la de um macroprolactinoma, que geralmente cursa com níveis muito mais elevados de prolactina (acima de 200 ng/mL).
Diabetes insipidus é raro, e sua ocorrência deve fazer pensar em outra etiologia para a massa selar (craniofaringioma, hipofisite, metástase para a hipófise, histiocitose X, sarcoidose e outras).
Os sintomas neurológicos incluem cefaleia, hidrocefalia, compressão quiasmática (determinando perdas de campo visual) e paralisias de pares cranianos (por extensão ao seio cavernoso).
Algoritmo 4: Diagnóstico e conduta dos tumores hipofisários clinicamente não-secretores.
O tratamento dos tumores clinicamente não-secretores é cirúrgico. Melhora visual parcial ou completa é possível desde que o efeito compressivo não tenha sido muito prolongado, o que pode determinar atrofia de papila e déficit visual permanente. Se as deficiências hormonais da hipófise anterior decorrem principalmente de compressão da haste, pode haver melhora com a cirurgia; todavia, se forem causados por efeito compressivo sobre a hipófise, tendem a ser definitivos.
Quando a cirurgia não é curativa e há significativa massa de tumor residual, e para os casos de recidiva, está indicado tratamento radioterápico, com objetivo de impedir crescimento adicional do tumor. O seguimento deve ser feito com exames de imagem, uma vez que não há marcador sérico da doença. Os inconvenientes da radioterapia (hipopituitarismo e predisposição para neoplasias intracranianas, acidentes vasculares cerebrais e demência) devem ser levados em consideração no momento da indicação do tratamento.
Há relatos de redução de adenomas clinicamente não-secretores com análogos de somatostatina e agonistas dopaminérgicos, mas não existe ainda uma recomendação formal para tratamento medicamentoso destes tumores.
Compreendem os raros carcinomas de hipófise e os tumores metastáticos para a hipófise.
São tumores extremamente raros, constituindo 0,1 a 0,2% dos tumores hipofisários. O diagnóstico de carcinoma é clínico, com demonstração de metástases a distância.
A maior parte dos carcinomas de hipófise secreta algum hormônio (principalmente ACTH e prolactina). Há casos de carcinomas secretores de ACTH que surgiram após adrenalectomia (no contexto da síndrome de Nelson).
Os sítios de metástase dos carcinomas de hipófise são cérebro, medula espinal, meninges, ossos, fígado, linfonodos, ovários, coração e pulmões.
Opções terapêuticas incluem cirurgia, agonistas dopaminérgicos (para carcinomas secretores de prolactina), análogos de somatostatina (para carcinomas secretores de GH), radioterapia e quimioterapia.
Os tratamentos, na maioria absoluta dos casos, é paliativo, com sobrevida média dos pacientes de 2 anos. Contudo, há diversos relatos de sobrevida mais longa.
Grandes séries de necrópsias apontam para ocorrência de metástases para a hipófise em 1 a 3,6% dos pacientes com câncer.
O principal sítio primário que origina metástases para a hipófise é a mama, seguido por pulmão, próstata, rins, trato gastrintestinal, linfoma, leucemia, tireoide e plasmocitoma.
Quase 2/3 das metástases acometem exclusivamente a neuro-hipófise, enquanto pouco mais de 13% localizam-se na adeno-hipófise, 12% nos dois lobos e o restante na haste e na cápsula. O acometimento preferencial da neuro-hipófise decorre provavelmente do suprimento sanguíneo deste órgão, que é diretamente arterial (em oposição à adeno-hipófise, que recebe sangue do sistema porta-hipofisário).
Estudos de autópsia sugerem que mais de 90% das metástases para hipófise são assintomáticas. Dentre os sintomas mais comumente reportados, destacam-se diabetes insipidus, cefaleia, oftalmoplegia, perda de campo visual e hipopituitarismo. Algumas características de imagem podem auxiliar no diagnóstico diferencial entre um adenoma hipofisário e uma metástase para a hipófise. Contudo, nenhum destes sinais é específico. São eles:
espessamento da haste;
perda do hipersinal da neuro-hipófise;
isossinal em imagens ponderadas em T1 e T2;
invasão do seio cavernoso;
alterações escleróticas ao redor da sela túrcica.
Não há estudos comparativos entre as diversas modalidades terapêuticas devido à raridade do tumor e ao contexto em que se enquadra (paciente com neoplasia disseminada). A ressecção cirúrgica por via transesfenoidal foi amplamente realizada e cursa com melhora de sintomas locais (dor, oftalmoplegia, acuidade visual). Esta modalidade de tratamento pode ser complementada com radioterapia, que, por sua vez, pode ser direcionada para a região selar ou aplicada em todo encéfalo. A quimioterapia também pode ser usada, embora a eficácia no controle de metástases para a hipófise nunca tenha sido publicada na literatura.
São tumores parasselares que correspondem a 3% de todos os tumores intracranianos e até 10% dos tumores no SNC na faixa pediátrica.
Os craniofaringiomas originam-se de remanescentes embrionários da bolsa de Rathke. Em 60% dos casos, a lesão origina-se na sela, enquanto que, no restante, advém de remanescentes parasselares.
Os craniofaringiomas podem ser grandes (até 10 cm), e causar sintomas por compressão de estruturas vizinhas. As vias de drenagem de líquido cefalorraquidiano podem ser comprimidas, determinando quadro de hipertensão intracraniana (cefaleia, vômitos, papiledema e redução de nível de consciência). O crescimento suprasselar pode levar a acometimento das vias ópticas, causando defeitos do campo visual. A invasão dos seios cavernosos causa paralisias de pares cranianos. Meningite asséptica pode ocorrer devido a vazamento do conteúdo da parte cística do craniofaringioma para o espaço subaracnoide. Ao contrário dos adenomas hipofisários, que raramente causam deficiência da neuro-hipófise, os craniofaringiomas frequentemente cursam com diabetes insipidus, que pode inclusive ser o primeiro sintoma da doença. Deficiência completa ou parcial da adeno-hipófise também pode ser observada. A compressão da haste hipofisária pode reduzir o aporte dopaminérgico aos lactotrofos hipofisários levando à hiperprolactinemia discreta por desconexão.
O aspecto clássico do craniofaringioma na tomografia computadorizada, presente na maioria das crianças e em 50% dos adultos, é de uma lesão cística com uma parte sólida contendo calcificações.
O tratamento dos craniofaringiomas é cirúrgico e pode ser complementado com radioterapia. A taxa de recorrência é de aproximadamente 20%.
A adeno-hipófise e a porção intermédia originam-se embriologicamente da bolsa de Rathke; alterações na obliteração desta bolsa durante o desenvolvimento podem levar à formação de cistos na interface entre a adeno-hipófise e a neuro-hipófise, encontrados em até 20% em séries de autópsias.
A maioria dos pacientes é assintomática. Contudo, cistos grandes podem causar déficits visuais ou hipopituitarismo. Ao contrário dos craniofaringiomas, o diabetes insipidus não é comumente observado.
À tomografia computadorizada, os cistos de bolsa de Rathke aparecem como lesões homogêneas com hipoatenuação, sem calcificações. A RNM pode evidenciar hipo ou hipersinal em T1 ou T2.
Pacientes com cistos assintomáticos devem ser observados com imagens periódicas. Em caso de déficit visual ou comprometimento da função hipofisária, a cirurgia é a melhor opção terapêutica.
São acúmulos de material mucoso nos seios paranasais, ocorrendo quando a drenagem do seio é obstruída. As mucoceles do seio esfenoidal são raras (< 1% dos casos) e elas podem causar compressão de estruturas parasselares.
Os pacientes apresentam cefaleia, déficits visuais (geralmente unilaterais), exoftalmo unilateral, anosmia e, raramente, hipopituitarismo. A RNM mostra uma massa homogênea no osso esfenoide.
O tratamento é cirúrgico, por meio de marsupialização da parede do cisto por via transesfenoidal.
São lesões císticas delimitadas por uma parede de pia-máter e preenchidas por líquido cefalorraquidiano. A origem dos cistos aracnoides é embrionária; uma teoria explica o seu surgimento pela herniação da aracnoide na fossa hipofisária na presença de um diafragma incompetente, e posterior perda da comunicação com o espaço subaracnoide.
Os pacientes podem ser assintomáticos ou apresentar cefaleia, defeitos de campo visual ou hipopituitarismo. Na RNM, o cisto apresenta o mesmo sinal em T1 e T2 que o líquido cefalorraquidiano.
O tratamento é a cirurgia transesfenoidal.
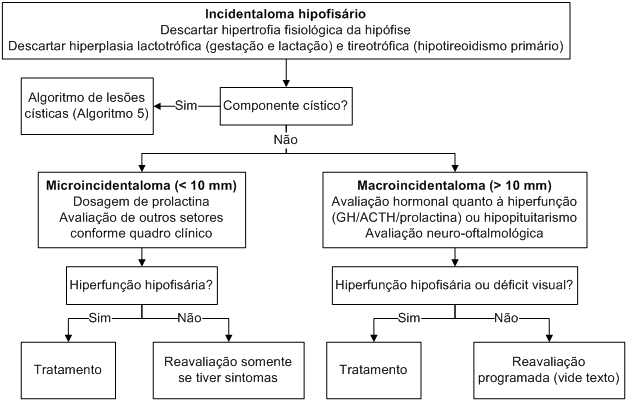
Com a melhoria das técnicas e popularização dos métodos de imagem, cada vez mais alterações hipofisárias são diagnosticadas em pacientes assintomáticos (os chamados incidentalomas hipofisários).
Estudos de autópsia mostram grande prevalência destas lesões (principalmente microadenomas e cistos da fenda de Rathke), que acometem até 10% da população geral. Estes dados são confirmados por estudos de radiologia com RNM realizada em indivíduos assintomáticos. Contudo, apenas 0,2% destes incidentalomas são macroadenomas.
É fundamental, portanto, que o médico generalista avalie inicialmente o paciente que alberga um tumor hipofisário incidental. Frente a uma possível lesão hipofisária em um paciente assintomático, algumas questões se impõem.
A hipertrofia fisiológica da hipófise consiste no aumento da altura da hipófise, que, em alguns casos, pode se aproximar do quiasma óptico. É um achado relativamente frequente em mulheres jovens e adolescentes (até 10% podem apresentar hipófise com altura maior que 7 mm), mas também pode ser evidenciada em mulheres após a menopausa.
Gestantes e lactantes podem apresentar hiperplasia hipofisária, relacionada ao setor lactotrófico. Como citado anteriormente, uma forma de hiperplasia hipofisária não-fisiológica se apresenta em casos de hipotireoidismo primário grave (hiperplasia tireotrofocítica).
Raramente, tumores ectópicos secretores de CRH ou GHRH podem levar à hiperplasia dos setores corticotrópicos e somatotrópicos, respectivamente.
Analisando as prevalências na população de microincidentalomas hipofisários (10%) e tumores secretores de prolactina (0,03 a 0,15%), GH (0,003 a 0,009%) e ACTH (0,0006 a 0,003%), fica evidente que apenas uma minoria dos incidentalomas é clinicamente funcionante e, nestes, a maior parte é composta por prolactinomas.
Frente à baixa probabilidade de diagnóstico de um corticotropinoma ou de um somatotropinoma assintomáticos, e ao elevado custo da investigação hormonal completa, recomenda-se somente a dosagem de prolactina para os microincidentalomas, a não ser que a história clínica levante suspeita de disfunção hipofisária.
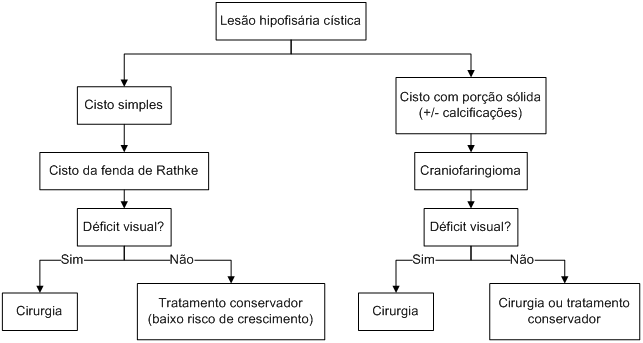
As principais possibilidades diagnósticas para os cistos na região hipotálamo-hipofisária são os cistos da fenda de Rathke e craniofaringiomas.
Os cistos da fenda de Rathke tendem a apresentar curso clínico indolente, e raramente levam a disfunção hipofisária, enquanto os craniofaringiomas podem crescer de forma espantosa, determinando hipopituitarismo e diabetes insipidus, além de comprometimento visual. Alguns dados clínicos, hormonais e radiológicos auxiliam no diagnóstico diferencial entre cisto da fenda de Rathke e craniofaringioma (Tabela 7).
Tabela 7: Diagnóstico diferencial entre cisto da fenda de Rathke e craniofaringioma
|
|
Cisto da fenda de Rathke |
Craniofaringioma |
|
Tamanho |
Geralmente < 20 mm |
Variável |
|
Porção sólida |
Ausente |
Presente |
|
Calcificações |
Ausentes |
Presentes (50%) |
|
Hipopituitarismo |
Raro |
Frequente |
|
Sinal em T1* |
Hipossinal ou hipersinal |
Isossinal ou Hipossinal |
|
Sinal em T2* |
Hipossinal, isossinal ou hipersinal |
Hipossinal |
* Achados mais comuns.
A conduta frente às lesões císticas depende da suspeita diagnóstica; sabe-se que os cistos da fenda de Rathke têm evolução benigna e raramente progridem; o mesmo não pode ser dito para os craniofaringiomas, mesmo aqueles de tamanho pequeno. Logo, quando se suspeita de craniofaringioma, a conduta tende a ser mais agressiva (cirurgia, podendo ser complementada com radioterapia para evitar recidiva tumoral). Se a conduta for expectante, recomenda-se avaliação radiológica anual com RNM.
Algoritmo 5: Tratamento das lesões hipofisárias císticas.
No seguimento, o clínico deve ter em mente que os microincidentalomas não apresentam crescimento significativo e não comprometem a função hipofisária. Assim, admite-se não fazer qualquer exame de acompanhamento (hormonal ou radiológico), exceto se o paciente vier a apresentar algum sintoma durante a evolução.
Os macroincidentalomas são bem mais raros (0,2% dos incidentalomas) e a sua ocorrência justifica uma avaliação hormonal mais extensa (à procura de hiperfunção de algum setor ou de hipopituitarismo). A avaliação neuro-oftalmológica é obrigatória para todo tumor com mais de 10 mm de diâmetro.
A conduta pode ser expectante ou terapêutica, a depender da funcionalidade do tumor e do comprometimento das vias ópticas. Sempre que há déficit de campo visual, o tratamento está indicado.
Os tumores secretores de ACTH e GH devem ser tratados cirurgicamente, assim como os adenomas clinicamente não-secretores que comprimem o quiasma óptico. Prolactinomas devem ser tratados primariamente com agonistas dopaminérgicos, sendo a cirurgia uma segunda opção terapêutica (ver seção “Hiperprolactinemia”).
Os casos sem indicação cirúrgica (macroincidentalomas clinicamente não-secretores que não afetaram o campo visual) devem ser seguidos com avaliação hormonal e radiológica periódica (semestral no primeiro ano, anual por 3 anos e bi- ou trianual em diante).
Algoritmo 6: Abordagem diagnóstica e terapêutica dos incidentalomas hipofisários.
O conhecimento das formas de investigação dos tumores de hipófise é essencial para todos os clínicos, principalmente nos últimos anos, com o aumento importante da sua incidência graças aos incidentalomas hipofisários.
1. Melmed S, Casanueva FF, Cavagnini F, Chanson P, Frohman L, Grossman A, et al. Guidelines for acromegaly management. J Clin Endocrinol Metab. 2002;87:4054-4058.
2. Merza Z. Modern treatment of acromegaly. Postgrad Med J. 2003;79:189-194.
3. Katznelson L. The diagnosis and treatment of acromegaly. Endocrinologist. 2003;13:428-434.
4. Carroll PV, Jenkins PJ. Acromegaly. 2007. In: Grossman A. (ed.). endotext.org.
5. Wand GS. Diagnosis and management of hyperprolactinemia. Endocrinologist. 2003;13:52-57.
6. Thorner MO, Bronstein MD. Hyperprolactinemia. 2007. In: Grossman A. (ed.). endotext.org.
7. Vallette-Kasic S, Morange-Ramos I, Selim A, Gunz G, Morange S, Enjalbert A, et al. Macroprolactinemia revisited: a study on 106 patients. J Clin Endocrinol Metab. 2002;87:581-588.
8. Schade R, Andersohn F, Suissa S, Haverkamp W, Garbe E. Dopamine agonists and the risk of cardiac-valve regurgitation. N Engl J Med. 2007;356:29-38.
9. Zanettini R, Antonini A, Gatto G, Gentile R, Tesei S, Pezzoli G. Valvular heart disease and the use of dopamine agonists for Parkinson’s disease. N Engl J Med. 2007;356:39-46.
10. Musolino NR, Passos VQ. Dopamine-agonist resistant prolactinomas: diagnosis and management. Arq Bras Endocrinol Metabol. 2005;49:641-650.
11. Katznelson L, Alexander JM, Klibanski A. Clinical review 45: clinically nonfunctioning pituitary adenomas. J Clin Endocrinol Metab. 1993;76:1089-1094.
12. Ragel BT, Couldwell WT. Pituitary carcinoma: a review of the literature. Neurosurg Focus. 2004;16:E7.
13. Fassett DR, Couldwell WT. Metastases to the pituitary gland. Neurosurg Focus. 2004;16:E8.
14. Shin JL, Asa SL, Woodhouse LJ, Smyth HS, Ezzat S. Cystic lesions of the pituitary: clinicopathological features distinguishing craniopharyngioma, Rathke’s cleft cyst, and arachnoid cyst. J Clin Endocrinol Metab. 1999;84:3972-3982.
15. Chanson P, Young J. Pituitary incidentalomas. Endocrinologist. 2003;13:124-135.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.