(Carregando Índice)... (Carregando Índice)... |
Autores:
Fabio Pires de Souza Santos
Especialista em Hematologia pelo Hospital das Clínicas da Faculdade de Medicina da USP
Gustavo dos Santos Fernandes
Especialista em Hematologia pelo Hospital das Clínicas da Faculdade de Medicina da USP
Última revisão: 17/07/2009
Comentários de assinantes: 0
Os linfomas são proliferações neoplásicas malignas de linfócitos, que residem predominantemente em órgãos linfóides. Os linfomas são doenças extremamente heterogêneas, sendo alguns subtipos extremamente agressivos e outros muito indolentes. Os linfomas classificam-se em dois grandes grupos:
linfoma não-Hodgkin.
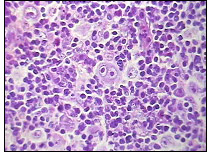
O linfoma de Hodgkin (LH) é um subtipo de linfoma que se caracteriza histologicamente pela presença no linfonodo de células malignas binucleadas, as chamadas células de Reed-Sternberg, associada à presença de infiltrado celular de composição variada, com células mononucleadas (células de Hodgkin) e linfócitos T. É responsável por cerca de 30% dos linfomas. Uma característica dos LH é que eles tendem a disseminar por contigüidade, seguindo para áreas contíguas de linfonodos, disseminando difusamente apenas em estádios mais avançados. A maioria destes linfomas é de células ß.
A patogênese do LH ainda é assunto de muito estudo, e discute-se o papel do vírus EBV na patogênese desta doença. Um grande avanço, porém, foi a descoberta de que a célula de Reed-Sternberg, a célula maligna do LH, descende de linfócitos B do centro germinativo do linfonodo.
O LH é uma doença rara, com uma incidência de 2 a 4 casos por 100.000 pessoas/ano. Sabe-se que apresenta uma distribuição por idade bimodal, apresentando dois picos de incidência: um ao redor dos 20 a 30 anos de idade, e o outro após os 50 anos de idade. A incidência dos LH vem se mantendo estável nas últimas décadas.
Vários sistemas de classificação foram propostos. Jackson e Parker propuseram dividir estes linfomas em paragranuloma, granuloma e sarcoma; posteriormente, Luke e Butler propuseram dividir os pacientes na categoria granulomatosa em esclerose nodular e celularidade mista; os mesmos autores propuseram a existência de uma variante com predomínio linfocítico ou histiocítico, que acabou substituindo na classificação o termo paragranuloma. Mais recentemente, o Linfoma Study Group propôs um novo sistema de classificação de nominado de REAL (Revised European-American Lymphoma); estes conceitos foram incorporados na classificação da OMS.
Segundo a classificação da OMS, os LH dividem-se em dois grandes grupos: LH com predominância linfocítica nodular e LH clássico. O segundo grupo corresponde a 95% dos casos e apresenta 4 subdivisões, conforme Tabela 1.
Tabela 1: Classificação dos linfomas de Hodgkin pela OMS
|
linfoma de Hodgkin com predominância linfocítica nodular |
|
linfoma de Hodgkin clássico (subtipos): |
|
Esclerose nodular |
|
Celularidade mista |
|
Rico em linfócitos |
|
Depleção linfocítica |
Os doentes com LH podem apresentar diversas manifestações clínicas, sendo as mais comuns a presença de massas linfonodais indolores e a presença de sintomas sistêmicos.
Cerca de 70% dos casos têm massa linfonodal indolor, de consistência fibroelástica. Os sítios mais comumente envolvidos são:
pescoço (60 a 80%);
mediastino (60%);
retroperitoneal (20%);
axilar (10 a 20%);
inguinal (6 a 12%).
Outro sítio comum de acometimento é o baço, presente em cerca de 30% dos casos. Além da linfadenomegalia assintomática, a segunda forma mais comum de apresentação é como massa mediastinal detectada em exames de imagem. Eventualmente, essas massas podem ser grandes o bastante para produzir sintomas como dor retroesternal, dispnéia ou tosse.
Como já mencionado, uma característica importante do LH, que tende a diferenciá-lo do LNH, é a tendência do linfoma surgir em um único linfonodo e se disseminar por contigüidade para linfonodos adjacentes, de maneira organizada. Mais tardiamente, tem-se uma disseminação hematogênica e presença infiltração ampla da doença.
Os sintomas sistêmicos clássicos do LH são os chamados sintomas B, que indicam um pior prognóstico:
febre – ocorre em 25 a 50% dos doentes, sendo mais comum com estádios mais avançados. A febre no LH é mais noturna, persistente, raramente pode ter um padrão intermitente-recidivante. Outro padrão característico é a chamada febre de Pel-Ebstein, quando pacientes apresentam temperaturas elevadas por vários dias, alternada com temperatura normal ou abaixo do normal;
perda de peso > 10% do peso basal em 6 meses;
sudorese noturna.
Outros sintomas que merecem ser citados incluem:
prurido (10 a 15% dos doentes no diagnóstico);
dor induzida pelo álcool (em locais de invasão óssea e massas linfonodais);
dor lombar e desconforto abdominal por linfonodomegalia retroperitoneal;
lesões de pele (ictiose, acroqueratose, urticária, eritema multiforme, eritema nodoso);
síndrome nefrótica (padrão lesões mínimas);
síndromes neurológicas paraneoplásicas.
Os exames complementares do LH visam confirmar o diagnóstico e realizar o estadiamento da doença para que se possa iniciar o tratamento. Os exames de maior importância são descritos a seguir.
Hemograma: a presença de leucocitose (> 15.000/mm3) e linfopenia (< 600/mm3) são marcadores de pior prognóstico;
velocidade de hemossedimentação (VHS): tende a estar aumentada;
cálcio: pode haver hipercalcemia no LH por aumento de produção de calcitriol, a exemplo das doenças granulomatosas;
função hepática;
função renal;
desidrogenase lática.
Biópsia linfonodal: essencial para fazer o diagnóstico; o ideal é ter um linfonodo inteiro.
Figura 1: Célula de Reed-Sternberg, que é a característica patológica do LH.

Radiografia de tórax (PA e perfil);
tomografia computadorizada de tórax, pescoço, abdome e pelve;
cintilografia com gálio ou PET-SCAN com FDG;
ultra-sonografia de abdome: melhor exame para avaliar acometimento esplênico;
biópsia de medula óssea bilateral e outros procedimentos invasivos: apenas em situações específicas;
laparotomia: raramente é feita hoje em dia.
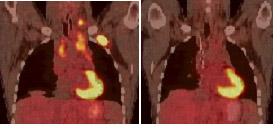
Figura 2: PET-CT de paciente com LH. Inicial à esquerda com lesão hipercaptante em mediastino, supraclavicular e cervical. Após 2 ciclos de tratamento, verifica-se o desaparecimento da atividade metabólica nas lesões, denotando um bom prognóstico.
Após a realização de todos os exames, o LH deve ser estadiado conforme a classificação de Ann Arbor.
O diagnóstico diferencial do LH deve ser feito, de modo semelhante ao LNH, com outras patologias que cursam com linfonodomegalia, esplenomegalia e com o próprio LNH. Quanto ao diagnóstico diferencial entre LNH e LH, vale citar as seguintes diferenças, descritas na Tabela 2.
Tabela 2: Diferenças entre linfoma de Hodgkin e linfoma não Hodgkin
|
Característica |
linfoma de Hodgkin |
Linfoma não-Hodgkin |
|
Apresentação clínica |
Variada, porém segue estereótipo de massa linfonodal indolor e sintomas sistêmicos. Não costuma dar sintomas compressivos. |
Bastante heterogênea, desde doenças muito indolentes e assintomáticas até grandes massas causando sintomas compressivos. |
|
Sítio de origem |
Nodal. Em geral, não apresenta acometimento extranodal isolado. |
Predomínio nodal, mas pode ser extranodal em até 35% dos casos. |
|
Disseminação linfonodal |
Contigüidade |
Sem contigüidade |
De uma maneira geral, olinfoma de Hodgkin apresenta um prognóstico favorável; no entanto, determinadas características são capazes de predizer um comportamento mais agressivo da doença tanto em doença precoce (estádios I e II) quanto em doença avançada (estádios III e IV).
Quando tratamos de pacientes com doença precoce, os fatores que impactam negativamente a sobrevida são:
doença mediastinal volumosa (> 10 cm), ou utilizando as definições clássicas de bulky disease, quando o maior diâmetro transversal é maior que 1/3 do diâmetro torácico;
envolvimento neoplásico de 4 ou mais cadeias ganglionares;
idade superior a 50 anos ao diagnóstico;
combinação entre sintomas B e VHS > 30 mm/h ou VHS > 50 mm/h, independentemente da presença de sintomas B.
Nos pacientes com doença avançada, que intrinsecamnete apresentam prognóstico pior, é possível ainda uma subdivisão prognóstica importante. De forma similar ao que foi feito com LNH agressivos, um índice prognóstico foi concebido incluindo os fatores mais importantes na fase avançada da doença. O International Prognostic Score (IPS) inclui 7 fatores prognósticos desfavoráveis a serem considerados: albumina < 4 g/dL, hemoglobina menor do que 10,5 g/dL, sexo masculino, idade superior a 45 anos, estádio IV, leucócitos = 15,000/microL e linfócitos < 600/microL ou representando menos de 8 % das células totais. A contagem destes fatores é determinante no prognóstico dos pacientes, conforme segue:
nenhum fator: 84% de sobrevida em 5 anos;
1 fator: 77% de sobrevida em 5 anos;
2 fatores: 67% de sobrevida em 5 anos;
3 fatores: 60% de sobrevida em 5 anos;
4 fatores: 51% de sobrevida em 5 anos;
5 ou mais fatores: 42% de sobrevida em 5 anos.
Novos fatores prognósticos vêm aparecendo. Neste contexto, é de especial destaque a avaliação da atividade metabólica da doença por meio do PET após o segundo ciclo de quimioterapia, sendo que os pacientes que apresentam negativação da atividade metabólica na lesão apresentam um melhor prognóstico (Figura 2).
O tratamento dos LH baseia-se em uso de quimioterapia e radioterapia. De modo geral, o esquema mais utilizado hoje em dia é o ABVD (adriamicina, bleomicina, vimblastina e dacarbazina). O número de ciclos a serem realizados varia de 4 a 8, de acordo principalmente com a extensão da doença e com os fatores prognósticos. Procura-se fazer radioterapia após a quimioterapia nos pacientes com doença precoce (pois recebem menos ciclos de quimioterapia) e naqueles com doença avançada que apresentam de lesões bulky ou lesão residual importante, que dificilmente são eliminadas com quimioterapia isolada. De modo geral, tem-se obtidos bons resultados, com taxas de remissão completa de até 80% em 5 anos para doença em estádio inicial e 55 a 60% para doença em estádio avançado.
Em pacientes com doença mais avançada e prognóstico ruim, o esquema BEACOPP (bleomicina, etoposídeo, adriamicina, ciclofosfamida, vincristina – cujo nome comercial é Oncovin® –, procarbazina e prednisona) é opção. Outro esquema quimioterápico recentemente estudado é o Stanford V, que utiliza vimblastina, vincristina, adriamicina, prednisona, bleomicina, etoposídeo e mecloretamina. Pacientes com recidiva precoce (menos de 12 meses após o tratamento), segunda recidiva após tratamento convencional para primeira recidiva ou recidiva sistêmica indicando comportamento biológico agressivo do tumor fazem considerar a possibilidade da realização de transplante de medula óssea.
No subtipo histológico de predominância linfocítica, se for encontrado apenas cadeias cervicais isoladas (estádio I-IIA), pode-se optar por radioterapia apenas de campo envolvido.
Os linfomas de Hodgkin representam cerca de 30% dos linfomas.
Caracteristicamente apresentam disseminação por contiguidade, com disseminação difusa apenas em estágios avançados.
O linfoma de Hodgkin é um subgrupo de linfomas que costuma acometer principalmente jovens e idosos; caracteriza-se pela presença da célula maligna de Reed-Sternberg na biópsia linfonodal.
A classificação da OMS divide os pacientes em linfoma clássico (divididos em subtipos como esclerose nodular, celularidade mista, rico em linfócitos e depleção linfocítica) e pacientes com predominância linfocítica nodular.
Linfonodomegalia assintomática é a apresentação em cerca de 70% dos pacientes.
A segunda forma mais comum de apresentação é na forma de massa mediatinal.
Os sítios mais comuns de linfonodomegalia são o pescoço e o mediastino. Podem ter sintomas sistêmicos associados. O LH costuma surgir em um único linfonodo e disseminar-se de modo contíguo para linfonodos adjacentes.
Presença de linfocitose e leuccocitose implicam pior prognóstico.
Hipercalcemia pode ocorrer devido à hiperprodução de calcitriol.
O diagnóstico é feito pela biópsia linfonodal, e o estadiamento por meio de exames de imagem (tomografia, cintilografia). Deve-se, então, estadiar o LH segundo o estadiamento de Ann Arbor.
A presença de massa volumosa, sintomas sistêmicos e a idade são fatores que influenciam o prognóstico.
O IP é um novo sistema prognóstico que consegue correlacionar-se com a sobrevida dos pacientes.
O tratamento de LH envolve basicamente o uso de quimioterapia (p. ex., esquema ABVD), com radioterapia adjuvante em casos selecionados. Nos pacientes com doença avançada e com fatores prognósticos desfavoráveis, uma abordagem mais agressiva pode ser considerada. Os resultados obtidos costumam ser muito bons, especialmente nos estádios iniciais da doença.
Quando há recorrência após a primeira linha de tratamento, o LH persiste sendo curável por meio de modalidades de quimioterapia de resgate seguida de transplante autólogo de medula.
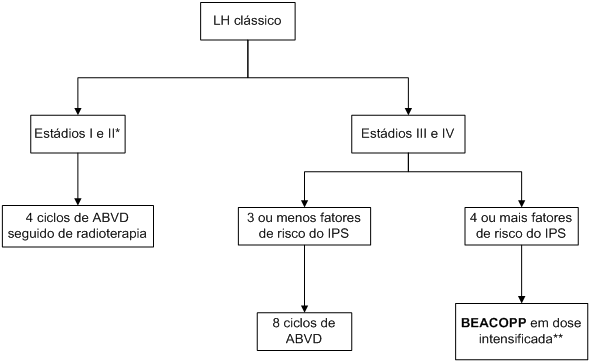
Algoritmo 1: Manejo inicial de linfoma de Hodgkin clássico.
* Pacientes com estádio IIb em geral são tratados com 6 ciclos de ABVD. ** O esquema BEACOPP é mais efetivo nos pacientes de alto risco, no entanto é muito tóxico e só deve ser tentado em pacientes jovens (<60 anos) e com saúde geral perfeita; nos outros pacientes, o ABVD em 8 ciclos está bem indicado.
1. Aleman BM, Raemaekers JM, Tirelli U, et al. Involved-field radiotherapy for advanced Hodgkin’s lymphoma. N Engl J Med 2003; 348:2396.
2. Bierman PJ, Anderson JR, Freeman MB, et al. High-dose chemotherapy followed by autologous hematopoietic rescue for Hodgkin’s disease patients following first relapse after chemotherapy. Ann Oncol 1996; 7:151.
3. Boleti E, Mead GM. ABVD for Hodgkin’s lymphoma: full-dose chemotherapy without dose reductions or growth factors. Ann Oncol 2007; 18:376.
4. Carella AM, Carlier P, Congiu A, et al. Autologous bone marrow transplantation as adjuvant treatment for high- risk Hodgkin's disease in first complete remission after MOPP/ABVD protocol. Bone Marrow Transplant 1991; 8:99.
5. DeVita VJ, Hubbard SM. Hodgkin’s disease. N Engl J Med 1993; 328:560.
6. Diehl V, Thomas RK, Re D. Part II: Hodgkin’s lymphoma -- diagnosis and treatment. Lancet Oncol 2004; 5:19.
7. Engert A, Franklin J, Eich HT, et al. Two cycles of doxorubicin, bleomycin, vinblastine, and dacarbazine plus extended-field radiotherapy is superior to radiotherapy alone in early favorable Hodgkin's lymphoma: final results of the GHSG HD7 trial. J Clin Oncol 2007; 25:3495.
8. Jaffe ES, Harris NL, Stein H, Vardiman JW, eds. World Health Organization Classification of Tumors. Pathology and genetics of tumors of haematopoietic and lymphoid tissues. Lyon:IARC Press , 2001.
9. Mauch PM, Kalish LA, Kadin M, et al. Patterns of presentation of Hodgkin disease. Implications for etiology and pathogenesis [see comments]. Cancer 1993; 71:2062.
10. Press OW, LeBlanc M, Lichter AS, Grogan TM. Phase III randomized intergroup trial of subtotal lymphoid irradiation versus doxorubicin, vinblastine, and subtotal lymphoid irradiation for stage IA to IIA Hodgkin’s disease. J Clin Oncol 2001; 19:4238.
11. Yahalom J, Ryu J, Straus DJ, et al. Impact of adjuvant radiation on the patterns and rate of relapse in advanced-stage Hodgkin’s disease treated with alternating chemotherapy combinations. J Clin Oncol 1991; 9:2193.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.