(Carregando Índice)... (Carregando Índice)... |
Autor:
Carla Bastos Valeri
Especialista em Pneumologia pelo Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo
Pneumologista do Hospital Sírio Libanês
Médica da Unidade de Terapia Intensiva do Hospital Universitário da Universidade de São Paulo
Última revisão: 29/08/2009
Comentários de assinantes: 0
O pulmão é sustentado por uma rede de fibras de tecido conectivo chamada interstício pulmonar, o qual se distribui ao longo do pulmão em três sistemas de fibras: peribroncovascular, subpleural e intralobular. Eles cumprem basicamente duas funções: manter as vias aéreas e os vasos pérvios e permitir a troca gasosa, pois faz a interface entre o alvéolo e o capilar. Portanto, qualquer estímulo ou agente que altere o interstício causará uma doença pulmonar intersticial. Sendo assim, definimos essas doenças como um grupo heterogêneo de situações que podem causar inflamação e/ou cicatrização do tecido pulmonar, afetando diretamente a troca gasosa e a sustentação vascular, alveolar e de vias aéreas.
Habitualmente, a doença intersticial pulmonar tem seu processo fisiopatológico desencadeado por uma injúria ao epitélio alveolar ou ao endotélio vascular. Em seguida, ocorre inflamação alveolar (alveolite), composta de macrófagos alveolares e um número menor de linfócitos, neutrófilos, eosinófilos e mastócitos, todos presentes na luz alveolar e no interstício. As alterações seguintes são: danos às paredes alveolares, com mudança no epitélio e espessamento fibrótico das paredes, e colapso alveolar.
Essa inflamação intensa do tecido pode, com tratamento e afastamento do agente etiológico, regredir ou causar uma fibrose, ou seja, cicatrização do tecido pulmonar, por meio da proliferação de tecido de reparação nas áreas acometidas. A estrutura (interstício) que antes era delgada, agora se encontra espessada, dificultando a troca gasosa e retraindo o parênquima pulmonar. Observa-se neste instante uma das características marcantes de algumas doenças intersticiais: um desarranjo das estruturas alveolares e a perda de unidades alveolocapilares funcionantes.
Essa é a sequência fisiopatológica dos eventos que causam redução dos volumes pulmonares, infiltrado intersticial ao exame de imagem e piora a troca gasosa.
Como já foi citado na definição das doenças intersticiais, este é um grupo grande e heterogêneo de doenças, tanto em sua apresentação clínica, como em sua etiologia. As causas podem ser divididas em grupos quanto ao tipo de exposição e agente desencadeante.
Poeira inorgânica: sílica, asbesto, metais pesados, berílio.
Poeira orgânica: bactérias termofílicas/Actinomices, fungos (Aspergillus, Penicillin spp), poeiras animais/de aves (doença do criador de pombos), produtos bacterianos (bissinose) – pneumonite de hipersensibilidade.
Gases, fumaça, fontes químicas, aerossol, vapores (isocianatos, paraquat, dióxido sulfúrico).
Radiação ionizante.
Talco (usuários de drogas endovenosas).
Agentes quimioterápicos: bleomicina, metotrexato, ciclofosfamida, nitrosureias.
Antibióticos: nitrofurantoína, sulfonamidas.
Antiarrítmicos: amiodarona.
Agentes antirreumáticos: sais de ouro, penicilamina.
Outros: fenitoína.
Infecção fúngica disseminada: coccidioidomicose, blastomicose, histoplasmose.
Micobacteriose disseminada.
Pneumocistose.
Viroses.
Lúpus eritematoso sistêmico.
Doença reumatoide.
Esclerose sistêmica progressiva.
Síndrome de Sjögren.
Polimiosite e dermatomiosite.
Doença mista do colágeno.
Espondilite anquilosante.
Fibrose pulmonar idiopática (FIP).
Pneumonia em organização criptogênica (COP) ou mais conhecida como bronquiolite obliterante com pneumonia em organização (BOOP).
Pneumonia intersticial não específica (NSIP).
Pneumonia intersticial aguda (AIP).
Pneumonia intersticial associada à bronquiolite respiratória (RB-ILD).
Pneumonia intersticial descamativa (DIP).
Pneumonia intersticial linfocitária (LIP).
Vasculites: granulomatose de Wegener, síndrome de Churg Strauss.
Sarcoidose.
Síndrome do desconforto respiratório agudo (SDRA).
Síndromes hemorrágicas: hemossiderose pulmonar idiopática, síndrome de Goodpasture.
Histiocitose de células de Langerhans ou histiocitose X ou granuloma eosinofílico.
Linfangioleiomiomatose.
Aspiração gástrica crônica/doença do refluxo gastroesofágico (DRGE).
Edema pulmonar crônico.
Proteinose alveolar.
Amiloidose.
Pneumonia eosinofílica crônica.
Linfangite carcinomatosa.
Doenças inflamatórias intestinais.
Doença venoclusiva/hipertensão venosa pulmonar crônica.
A história clínica é de fundamental importância nas doenças intersticiais pulmonares, pois normalmente é por ela que serão feitas hipóteses quanto ao agente etiológico envolvido. Inicialmente, deve-se atentar para idade, sexo e raça, por exemplo: a linfangioleiomiomatose e algumas doenças do colágeno ocorrem quase que exclusivamente em mulheres em idade fértil. Já a fibrose pulmonar idiopática acomete mais os homens, e acima de 60 anos de idade. A idade também é importante para as doenças do colágeno e sarcoidose pulmonar, pois tendem a aparecer dos 20 aos 40 anos de idade. A sarcoidose está mais associada à raça negra, e a panbronquiolite à raça amarela.
A história ocupacional deve ser detalhada, buscando uma ordem cronológica das ocupações profissionais e as respectivas exposições a poeiras, gases e substâncias químicas. É imprescindível determinar a duração e a intensidade da exposição, bem como o uso ou não de máscaras ou outros materiais de proteção.
A história ambiental deve conter informações sobre exposição a animais de estimação (principalmente pássaros), ar condicionado, umidificadores, aquecedores e ambientes úmidos ou com mofo. A pneumonia de hipersensibilidade está muito relacionada com criadores de aves, contato com feno (pulmão do fazendeiro, pulmão do criador de ave).
Algumas doenças intersticiais estão intimamente relacionadas ao tabagismo: histiocitose X, pneumonite intersticial descamativa, bronquiolite respiratória e fibrose pulmonar idiopática. Portanto, não se pode esquecer de questionar sobre o hábito tabágico, seu início e término, além da quantidade consumida.
O uso prévio de fármacos também é relevante, pois várias medicações podem causar lesão pulmonar intersticial. A injúria pode acontecer semanas ou anos após o fim da utilização do remédio.
A história familiar pode ser útil em algumas doenças com associação genética, como fibrose pulmonar idiopática, sarcoidose e neurofibromatose (Tabela 1).
Dispneia: queixa mais comum. Geralmente o inicio é insidioso, com piora progressiva. Não é raro encontrar pacientes que demoram a procurar ajuda, pois acham que a dispneia advém do descondicionamento físico, envelhecimento ou aumento de peso recente. Quanto a este sintoma, o principal diagnóstico diferencial é a doença cardiológica.
Tosse: tosse seca é o segundo sintoma mais comum. Na maioria dos casos, a tosse é perturbadora e impede atividades habituais. Quanto maior o envolvimento das vias aéreas, mais intensa será a tosse, por exemplo, bronquiolite respiratória, sarcoidose, BOOP.
Hemoptise: não é comum, mas quando presente (sangue vivo ou escarro com raias de sangue), fala a favor de doenças específicas como hemorragia alveolar por vasculites granulomatosas, lúpus eritematoso sistêmico, valvopatia mitral de longa data, doença veno-oclusiva, linfangioleiomiomatose. Se, apesar do tratamento, a hemoptise persistir, fazer diagnóstico diferencial com neoplasias pulmonares.
Dor torácica: pode ocorrer dor pleurítica em algumas patologias, como lúpus eritematoso sistêmico, doença reumatoide e doenças induzidas por fármacos. Dor torácica e desconforto retroesternal são comuns na sarcoidose.
Sibilo e roncos: os sibilos são manifestações raras e podem acontecer na linfangite carcinomatosa, pneumonite eosinofílica crônica, bronquiolite respiratória e síndrome de Churg-Strauss. Os roncos inspiratórios podem decorrer de acometimento de vias aéreas, como na bronquiolite.
Tabela 1: Exemplos de pneumopatias intersticiais e seus dados de história característicos
|
Patologias |
Idade |
Sexo |
Raça |
Tabagismo |
Exposição |
História familiar |
|
Fibrose pulmonar idiopática |
> 60 anos |
Comum em homens |
Ind. |
+ |
- |
+ |
|
Pneumopatias do colágeno |
20 a 40 anos |
Comum em mulheres, exceto a DRe |
Ind. |
Ind. |
- |
++ |
|
Sarcoidose |
20 a 40 anos |
Ind. |
Negra |
- |
- |
+ |
|
Linfangioleiomiomatose |
Idade fértil |
Apenas mulheres |
Ind. |
- |
- |
+ |
|
Histiocitose X |
20 a 40 anos |
Ind. |
Ind. |
+++ |
- |
? |
|
Panbronquiolite |
|
Comum em homens |
Amarela |
+ |
- |
+ |
|
Bronquiolite respiratória |
> 30 anos |
Ind. |
Ind. |
+++ |
- |
- |
|
Pneumoconioses |
Ind. |
Comum em homens |
Ind. |
Ind. |
+++ |
- |
|
Pneumonite de hipersensibilidade |
Ind. |
Ind. |
Ind. |
“Tabagismo protege” |
+++ |
- |
DRe: doença reumatoide; Ind.: indiferente; +: presente; ++; fortemente presente; +++: condição indispensável; -: ausente; ?: não se sabe.
O exame físico pode ser normal ou apresentar alterações pouco específicas para a doença. O achado mais comum é a ausculta pulmonar com estertores crepitantes grosseiros ou “em velcro”, teleinspiratórios e localizados predominantemente nos terços inferiores dos pulmões. Isso pode ocorrer em qualquer pneumopatia intersticial, mas tende a estar presente sobretudo quando há fibrose do parênquima. Em alguns casos, pode-se observar taquipneia e diminuição da expansibilidade pulmonar.
Hipocratismo digital é comum em fibrose pulmonar idiopática e asbestose, e raro em sarcoidose, histiocitose X e pneumonite de hipersensibilidade. Geralmente esta é uma manifestação tardia de doença avançada.
Hipertensão pulmonar e sinais de cor pulmonale são vistos em fases mais avançadas de algumas doenças, como fibrose pulmonar idiopática, pneumonite de hipersensibilidade e histiocitose X, mas, na esclerose sistêmica progressiva, essas alterações são mais precoces.
As alterações extrapulmonares de exame físico podem ajudar a desvendar a etiologia da doença intersticial pulmonar. É o caso das doenças do colágeno, vasculites e sarcoidose, que cursam com artrites, anemia, alterações em pele, rins e olhos, e vários outros achados (Tabela 2).
Tabela 2: Alterações extrapulmonares de exame físico e a etiologia associada
|
Achados físicos |
Patologias relacionadas |
|
Hipertensão arterial sistêmica |
Doenças do colágeno e neurofibromatose |
|
Eritema nodoso |
Doenças do colágeno, sarcoidose, histoplasmose |
|
Erupção maculopapular |
Fármacos, amiloidose, doença de Gaucher, doenças do colágeno |
|
Gotron e heliotropo |
Dermatomiosite |
|
Fenômeno de Raynaud |
Doenças do colágeno, fibrose pulmonar idiopática |
|
Nódulos subcutâneos |
Doença reumatoide |
|
Vasculite cutânea |
Vasculites sistêmicas, doenças do colágeno |
|
Calcinose |
Dermatomiosite, escleroderma |
|
Uveíte |
Sarcoidose, doença de Behçet, espondilite anquilosante |
|
Esclerite |
Vasculites sistêmicas, sarcoidose, escleroderma, lúpus |
|
Ceratoconjuntivite seca |
Síndrome de Sjögren, pneumonia intersticial linfocitária |
|
Linfadenopatia e hipertrofia da parótida |
Pneumonia intersticial linfocitária, sarcoidose |
|
Miosite e fraqueza muscular |
Doenças do colágeno, fármacos |
|
Hepatoesplenomegalia |
Doenças do colágeno, sarcoidose, amiloidose, pneumonia intersticial linfocitária, histiocitose X |
Exames laboratoriais: nenhum exame laboratorial é específico para doença pulmonar, e sim para outras etiologias, como as doenças do colágeno e vasculites. Hemograma para ver a presença de anemia, leuco ou linfopenia, comuns em doenças do colágeno, vasculites e sarcoidose. Ureia e creatinina e exame de urina, para descartar acometimento renal. Provas de atividade inflamatória (VHS). Dosagem de autoanticorpos como o fator antinúcleo (FAN), fator reumatoide (FR), anticorpo anticitoplasma de neutrófilo (ANCA). Dosagem de enzima de conversão da angiotensina (ECA); um aumento desta enzima é muito sugestivo de sarcoidose, mas não é específico.
Radiografia de tórax: essencial para diagnosticar pneumonias intersticiais, já que em muitas delas os sintomas demoram a surgir e tendem a ser pouco específicos. O exame pode ser normal em até 10% dos casos. O padrão mais comum e menos específico é o reticular ou reticulonodular, com diminuição dos volumes pulmonares. Não é raro o padrão de preenchimento alveolar associado ao padrão intersticial. Os achados menos frequentes podem ajudar na sugestão das hipóteses diagnósticas, como adenomegalias sugerindo malignidade ou sarcoidose, cardiomegalia e congestão, pneumotórax e histiocitose X. A maioria das doenças intersticiais afeta predominantemente as porções inferiores dos pulmões.
Tomografia de tórax: o ideal é realizar tomografia de alta resolução (TCAR), porque mostra o parênquima pulmonar com muito mais detalhes do que a tomografia convencional. Este exame apresenta vantagens em relação à radiografia simples, pois possibilita a visualização do parênquima pulmonar sem sobreposição de imagens, como na radiografia. Portanto, tornou-se um exame imprescindível para quem tem intersticiopatia. Após o consenso de pneumonias intersticiais idiopáticas, ficou estabelecido que não há necessidade de biópsia para firmar o diagnóstico de FIP, se o paciente apresentar história clínica e alterações tomográficas compatíveis com a doença. Estas são algumas vantagens da TCAR: detecção e confirmação precoce da suspeita de doença, melhor avaliação da distribuição e da extensão do acometimento, pode mostrar alterações em outros locais do corpo, capacidade para selecionar o melhor local para biópsia. Em algumas doenças, existe uma boa correlação entre os achados tomográficos e o resultado da biópsia. Dentre as alterações mais comuns, destacamos o vidro fosco e faveolamento.
Tabela 3: Padrão da tomografia nas doenças intersticiais
|
Doença |
Localização |
Padrão |
|
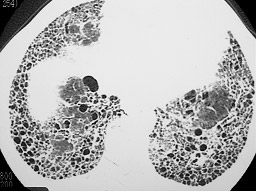
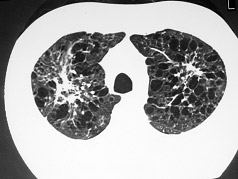
Fibrose pulmonar idiopática – FIP (Figura 1) |
Periferia e regiões inferiores |
Faveolamento |
|
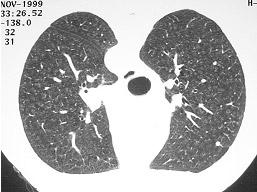
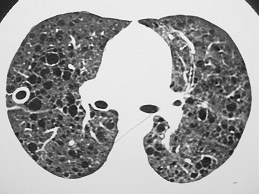
Pneumonite de hipersensibilidade – PH (Figura 2) |
Regiões superiores |
Reticular e vidro fosco |
|
BOOP |
Periferia |
Consolidação migratória |
|
Colagenoses |
Periferia |
Vidro fosco, nódulos |
|
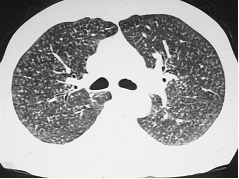
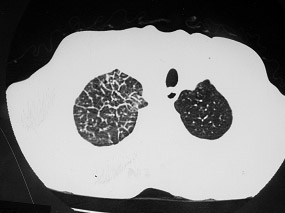
Pneumoconioses (Figura 3) |
Regiões superiores |
Nódulos e massas |
|
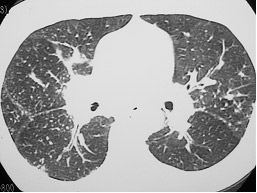
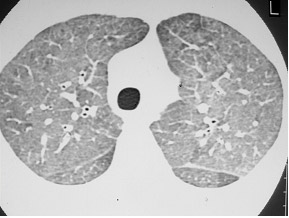
Sarcoidose (Figura 4) |
Regiões superiores, central/axial |
Nódulos, vidro fosco |
|
Histiocitose X (Figura 5) |
Regiões inferiores, periferia |
Cistos, vidro fosco |
|
Linfangioleiomiomatose (Figura 6) |
Regiões superiores |
Cistos, nódulos, derrame pleural |
|
Bronquiolite respiratória |
Regiões superiores |
Micronódulos centrolobulares |
|
Pneumonite intersticial linfoide (LIP) |
Periferia |
Consolidação com broncograma aéreo |
|
Linfangite carcinomatosa (Figura 7) |
Qualquer região |
Espessamento do septo interlobular |
Figura 1: TC de tórax de alta resolução evidenciando o padrão de faveolamento alveolar, característico de fibrose pulmonar idiopática (FIP).

Figura 2: TCAR apontando a presença de infiltrado reticular difuso e algumas áreas de vidro fosco, presentes na pneumonite de hipersensibilidade (PH).
Figura 3: TCAR mostrando a presença de diversos micronódulos pulmonares difusos. O diagnóstico diferencial é extenso, mas inclui diversas pneumoconioses, como a silicose. Para o estabelecimento do diagnóstico de pneumoconiose, é obrigatória a comprovação da exposição ao agente causal. Neste caso específico, é uma pneumoconiose por exposição a grafite.
Figura 4: TCAR evidenciando a presença de micronódulos pulmonares e subpleurais, inclusive na cissura, aumento do hilo pulmonar bilateral compatível com linfonodomegalia e espessamento do interstício peribroncovascular. A soma destes achados é compatível com o diagnóstico de sarcoidose pulmonar.
Figura 5: TCAR apresentando cistos, alguns mal delimitados, opacidades lineares e nódulos pulmonares de predomínio nos lobos superiores, compatível com histiocitose X. Para estabelecer o diagnóstico, é fundamental a presença de tabagismo na história clínica.
Figura 6: TCAR mostrando múltiplas imagens císticas, difusas, compatíveis com linfangioleiomiomatose. À direita, evidencia-se um dreno colocado para drenagem de pneumotórax espontâneo.
Figura 7: TCAR evidenciando espessamento dos septos interlobulares e nódulos intralobulares no pulmão direito. Compatível com linfangite carcinomatosa.
Prova de função pulmonar: o padrão mais comum é o restritivo com redução da capacidade pulmonar total, volume residual e capacidade residual funcional. Os fluxos estão reduzidos (CVF e VEF1), mas com a relação VEF1/CF normal a aumentada. A capacidade de difusão do monóxido de carbono (DLCO) encontra-se diminuída, já que muitas doenças têm perda de unidades alveolares e espessamento da membrana alveolocapilar. O grau de queda da difusão não se correlaciona com gravidade da doença. O padrão obstrutivo não descarta doença intersticial, pois algumas cursam associadas com DPOC ou são consequentes do tabaco (histiocitose X e bronquiolite).
Biópsia transbrônquica e lavado broncoalveolar: em algumas situações pode ajudar, evitando a biópsia pulmonar a céu aberto. A maioria das doenças intersticiais acomete predominantemente a periferia pulmonar, e, por esta razão, a biópsia transbrônquica raramente elucida o diagnóstico. A sarcoidose é uma exceção, já que muitos casos são diagnosticados por meio da broncoscopia. Na Tabela 4, destacam-se alguns achados do lavado broncoalveolar que afastam e/ou sugerem algum diagnóstico.
Tabela 4: Achados do lavado broncoalveolar (LBA) nas pneumonites intersticiais
|
Doença |
Achado no LBA |
|
Linfangite neoplásica linfoma |
Células malignas |
Hemorragia alveolar |
Macrófagos com hemossiderina, eritrócitos |
|
Proteinose alveolar |
Material lipoproteináceo nos alvéolos corado com ácido Schiff-PAS |
|
Histiocitose X |
Histiócitos positivos para o anticorpo T6 e macrófagos vistos na microscopia eletrônica com grânulos de Birbeck em seu interior |
|
Silicose |
Partículas de pó vistas através da luz polarizada |
|
Pneumonia lipídica |
Grânulos de gordura dentro dos macrófagos |
Biópsia pulmonar: método que oferece o diagnóstico com maior precisão, além de definir o estágio da doença. A biópsia pulmonar a céu aberto via toracotomia ou videotoracoscopia é um procedimento relativamente seguro, com baixa morbimortalidade (< 1%). As vantagens deste tipo de procedimento são o fornecimento de um diagnóstico preciso, excluindo patologias graves e que necessitam de tratamento imediato como as neoplasias e infecções, além de fornecer uma avaliação precisa da atividade da doença, possibilitando a escolha do melhor tratamento e com menos efeitos colaterais. Outra vantagem é acabar com a ansiedade do paciente e seus familiares quanto ao diagnóstico de certeza. Geralmente não realizamos biópsia pulmonar a céu aberto se pudermos chegar ao diagnóstico por meio de outros exames complementares, , como na fibrose pulmonar idiopática, sarcoidose e proteinose alveolar. Existem algumas contraindicações relativas ao procedimento: doença cardiovascular grave, estágio terminal da doença, disfunção pulmonar grave e outras doenças que aumentem muito o risco cirúrgico.
Como a quantidade de doenças que se encaixam no grupo de pneumopatias intersticiais é muito grande, quando nos deparamos com alguma doença intersticial, automaticamente todas as outras causas tornam-se os principais diagnósticos diferenciais: pneumonites intersticiais idiopáticas, doenças do colágeno, doenças ocupacionais e de exposição, doenças vasculares, neoplasias, doenças cardiogênicas, infecções, pneumopatias por fármacos e várias outras. Muitas vezes, a insuficiência cardíaca congestiva pode simular doenças intersticiais pulmonares, sendo fundamental a sua exclusão (Figura 8).
Figura 8: Presença de vidro fosco difuso, engurgitamento vascular e espessamento septal devem alertar para a possibilidade de insuficiência cardíaca, sendo a história clínica e os exames complementares (ecocardiograma, BNP) fundamentais nestes casos.
Devido ao grande número de causas de doenças intersticiais, citaremos o tratamento das mais prevalentes (Tabela 5).
Tabela 5: Tratamentos mais prevalentes
|
Doenças |
Tratamento |
|
Fibrose pulmonar idiopática |
Prednisona 0,5 a 1 mg/kg/dia por 3 meses, associado ou não a ciclofosfamida ou azatioprina. Considerar transplante pulmonar para os refratários. |
|
BOOP |
Poucos melhoram sozinhos. Prednisona 0,5 a 1 mg/kg/dia por 6 meses. Cursos mais curtos podem causar maior índice de recorrência. |
|
Lúpus eritematoso sistêmico |
Corticoide é terapia de escolha. Resgate com azatioprina e ciclofosfamida. |
|
Esclerodermia |
Ciclofosfamida via oral associado à baixa dose de corticoide. D-penicilamina pode ser usada no início. |
|
Sarcoidose |
Tratamento apenas de formas com doença pulmonar e extrapulmonar progressivas. Dose não padronizada de prednisona: 40 mg a 1 mg/kg/dia por 6 a 18 meses. |
|
Granulomatose de Wegener |
Prednisona 1 mg/kg/dia por 2 a 3 meses + ciclofosfamida 2 mg/kg/dia por 6 a 12 meses. |
|
Histiocitose X |
Cessar o tabagismo é imprescindível. Prednisona 0,5 a 1 mg/kg/dia por 6 a 12 meses. |
|
Linfangioleiomiomatose |
Progesterona 10 a 20 mg/dia VO ou 400 a 800 mg/mês injetável. Ooforectomia. Agonista do GnRH. Considerar transplante pulmonar para os refratários. |
|
Proteinose alveolar |
Lavagem pulmonar. Administração de GMCSF. |
As doenças intersticiais pulmonares constituem um grupo heterogêneo de patologias que cursam com inflamação e, por vezes, cicatrização do parênquima pulmonar.
São inúmeras as causas das intersticiopatias. Dentre elas, destacam-se as idiopáticas, as relacionadas as doenças do colágeno, as secundárias a fármacos, as ocupacionais, as ambientais e aquelas provenientes de doenças sistêmicas como vasculites, sarcoidose, cardiopatias e outras.
Não é raro haver dificuldades para se firmar o diagnóstico e a etiologia. Para começar a investigação, é imprescindível uma história clínica minuciosa a respeito dos sintomas e principalmente buscar agentes que possam ter desencadeado o problema.
Dentre as ferramentas diagnósticas, temos radiografia de tórax, exames laboratoriais, tomografia computadorizada de tórax de alta resolução, prova de função pulmonar, broncoscopia com lavado broncoalveolar, biópsia transbrônquica e, por fim, biópsia pulmonar a céu aberto.
O tratamento varia de acordo com a etiologia. Nos casos ocupacionais e de exposição, é importante o afastamento imediato do agente; já nos casos idiopáticos e autoimunes, são usados corticoides e outros imunomoduladores, como ciclofosfamida e azatioprina.
O prognóstico em algumas etiologias é benigno e observa-se a remissão parcial e até total dos sintomas, alterações funcionais e radiológicas. Porém, a maioria evolui com sequelas ou até progridem, a despeito de qualquer tratamento.
Nos casos refratários, considerar a possibilidade de transplante pulmonar.
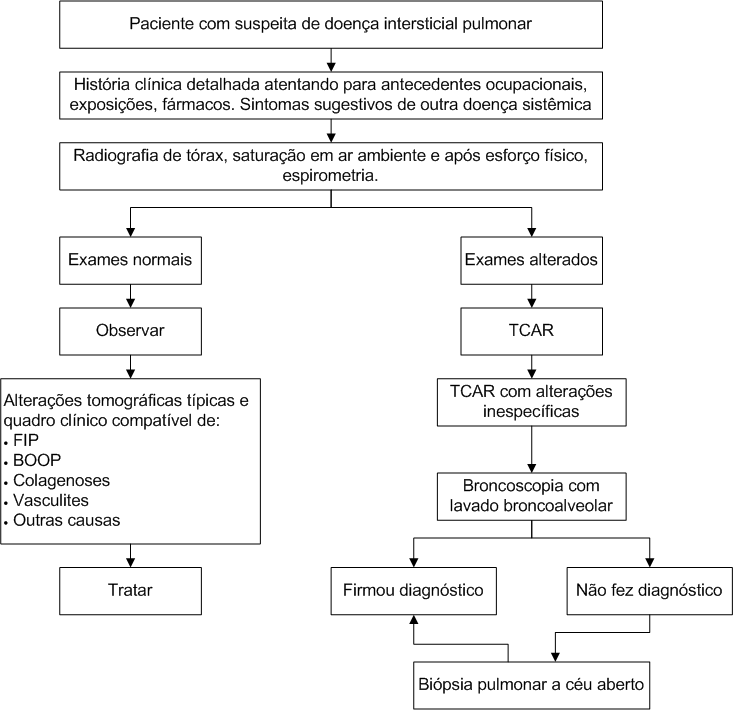
Abordagem da doença intersticial pulmonar.
1. American Thoracic Society. Idiopathic pulmonary fibrosis: diagnosis and treatment. International Consensus Statement. American Journal of Respiratory and Critical Care Medicine. 2000;161:646-664.
2. Raghu G. Interstitial lung disease: a diagnostic approach: are CT scan and lung biopsy indicated in every patient? American Journal of Respiratory and Critical Care Medicine. 1995;151:909-914.
3. Raghu G, et al. The accuracy of the clinical diagnosis of new-onset idiopathic pulmonary fibrosis and other interstitial lung disease. Chest. 1999;116:1168-70.
4. Fishman AP. Pulmonary diseases and disorders. 2.ed. New York: McGraw Hill.
5. American Thoracic Society and European Respiratory Society. ATS/ERS: International Consensus Classification of the Idiopathic Interstitial Pneumonia. American Journal of Respiratory and Critical Care Medicine. 2002;165(2):277-304.
6. Joint Statement of American Thoracic Society and European Respiratory Society and the World Association of Sarcoidosis and Other Granulomatous Disorders (WASOG). American Journal of Respiratory and Critical Care Medicine. 1999;160(2):736-55.
7. Webb WR, Müller NL, Naidich DP. High-resolution CT of the lung. 3.ed. Lippincott Williams & Wilkins; 2001.
8. King Jr. TE. Approach to the patient with interstitial lung disease. In: Kelley W. (ed.). Textbook of internal medicine. 3.ed. Philadelphia: J.B. Lippincott; 1996.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.