(Carregando Índice)... (Carregando Índice)... |
Autores:
Patrícia Andrade de Macedo
Especialista em Reumatologia pelo Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo (HC-FMUSP).
Samuel Katsuyuki Shinjo
Médico Assistente do Serviço de Reumatologia do Hospital das Clínicas da Faculdade de Medicina da USP
Clínico geral e Reumatologista pelo Hospital das Clínicas da Faculdade de Medicina da USP
Mestre e Doutor em Ciências pela Universidade Federal de São Paulo
Última revisão: 08/09/2010
Comentários de assinantes: 0
A síndrome de Sjögren (SS) é uma doença auto-imune sistêmica caracterizada por acometimento de glândulas exócrinas, além de presença de sinais/sintomas de doença sistêmica. As glândulas lacrimais e salivares são frequentemente afetadas, causando a síndrome sicca, caracterizada por ressecamento dos olhos (xeroftalmia) e da boca (xerostomia). É também conhecida como síndrome de Gougerot ou Mikulicz, exocrinopatia ou epitelite auto-imune.
Afeta principalmente mulheres na 4ª ou 5ª décadas de vida, sendo a proporção de 9 mulheres para cada homem afetado. Estima-se uma prevalência entre 3 e 4% na população adulta (
Apesar de numerosos estudos acerca da fisiopatologia e características clínicas predisponentes para SS primária, sua causa ainda não está bem estabelecida. Em linhas gerais, acredita-se que a patogênese é multifatorial, tendo importância fatores genéticos e ambientais (triggers de doenças auto-imunes).
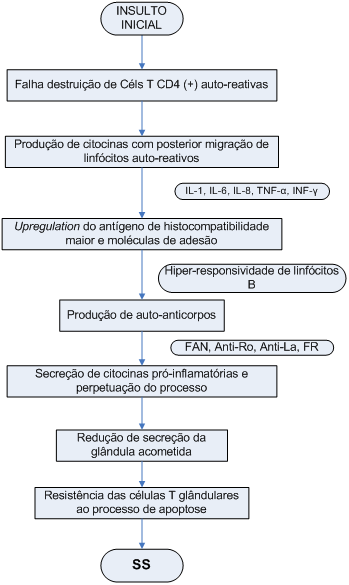
A proposta de fisiopatologia mais recente para SS considera o início da doença com um estímulo (infeccioso ou não) das glândulas salivares que leva a um processo inflamatório, necrose e apoptose celular. Esse estímulo inicial levaria à estimulação de células T auto-reativas que não sofreram processo de destruição na área tímica. Segue-se então a produção de citocinas pró-inflamatórias que estimulariam a migração linfocitária e de células dendríticas para as zonas glandulares afetadas, além do upregulation do HLA-DR(+) com subsequente re-estimulação de linfócitos. Essa cascata inflamatória levaria à hiperprodução de auto-anticorpos e à secreção de interleucinas (principalmente interferons tipo I) que proporcionariam uma perpetuação do processo inflamatório. Atividade glandular é então bastante reduzida tanto pela destruição acinar como pela desnervação glandular crônica nesses pacientes (Esquema 1).
Entre os possíveis agentes que atuam como desencadeantes da doença estão os vírus Epstein-Barr (EBV), hepatite C (HCV), HTLV-1 e outras retroviroses. Fatores genéticos predisponentes também estão sendo estudados e acredita-se que os principais envolvidos na patogênese da SS primária são o HLA-DQA1*0501 e HLA-DQB1*0201. Relação da doença com baixos níveis androgênicos em mulheres foi relatada e tem sido estudada.
Um erro comum é acreditar que os sintomas sicca resultam de total destruição imune do epitélio. O grau de destruição glandular e os sintomas de xerose não parecem estar relacionados ao grau de infiltrado inflamatório glandular. Postula-se que parte desses sintomas sejam devidos à denervação crônica epitelial e, em biópsias, o restante de ácinos viáveis, sem infiltrado inflamatório, se mostram disfuncionais.
Esquema 1: Fisiopatologia da síndrome de Sjögren.

Geralmente, a síndrome de Sjögren apresenta um curso benigno e lento de evolução. Suas manifestações clínicas baseiam-se em dois pilares básicos: sintomas glandulares e extraglandulares.
A SS pode acometer qualquer glândula exócrina do corpo humano, sendo as principais representantes as parótidas e as oculares, com redução das secreções oculares.
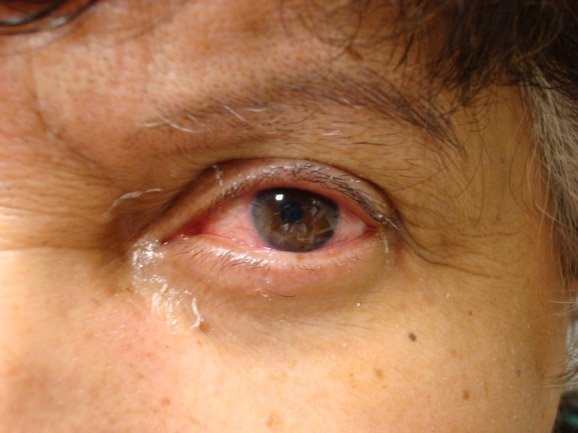
A xeroftalmia é o envolvimento glandular mais frequente nos casos de SS. A redução da produção lacrimal ocorre em 95% dos casos e, assim como a xerostomia, é evento precoce na doença. Pode haver destruição dos epitélios corneano e conjuntival levando ao aparecimento de sintomas e sinais clínicos de ceratoconjuntive seca. O paciente usualmente apresenta sintomas como sensação de areia, prurido ocular, vermelhidão e fotossensibilidade. Ao exame oftalmológico, podemos encontras sinais como hiperemia ocular, irregularidade corneana, hipertrofia das glândulas lacrimais e dilatação dos vasos conjuntivais.
Figura 1: Xeroftalmia em paciente portador de SS secundária à artrite reumatóide.
A xerostomia encontra-se presente em 90% dos casos ao diagnóstico, é expressa clinicamente pela sensação de boca seca e decorre da redução da produção de saliva pelas glândulas salivares. Os pacientes podem apresentar incapacidade para mastigar e deglutir alimentos secos e/ou falar continuamente, alterações de paladar, aumento da incidência de cáries, halitose, além de dificuldade para uso de próteses dentárias. Pode estar associada à hipertrofia de parótidas (unilateral ou bilateral mais frequentemente), é perceptível ao exame clínico e ocorre em até 60% dos pacientes portadores de SS primária. Ao exame físico, podem ser encontrados sinais como mucosa oral seca e eritematosa, cáries dentárias e atrofia das papilas filiformes encontradas no dorso da língua. Redução da secreção de faringe e hipofaringe também ocorrem, levando à disfagia.
Outros envolvimentos glandulares podem estar presentes, embora sejam clinicamente menos significativos: redução da secreção de orofaringe e trato respiratório superior pode levar a sintomas como rouquidão e desenvolvimento de bronquite e/ou pneumonite recorrentes; redução da secreção vaginal pode gerar dispareunia; e redução da secreção pancreática causa sintomas de insuficiência pancreática exócrina (síndromes desabsortivas) e hipocloridia.
Manifestações sistêmicas da doença podem estar presentes em até 50% dos casos e geralmente são precedidas de sintomas constitucionais como fadiga, febre baixa, mialgia e artralgias. Os acometimentos extraglandulares podem ser divididos em acometimentos periepiteliais (decorrente da infiltração linfocítica adjacente a glândulas epiteliais) e não-periepiteliais (decorrentes da hiper-reatividade de linfócitos B, formação e deposição de imunocomplexos) que acometem cerca 1/3 dos casos.
Artrite pode estar presente em até 50% dos casos e pode preceder sintomas sicca. Geralmente apresenta-se como poliartrite simétrica e aditiva de grandes e pequenas articulações (lupus-like), não erosiva na radiografia convencional.
Pele seca é a manifestação cutânea mais frequente (55% dos casos). O fenômeno de Raynaud acomete até 35% dos pacientes, podendo preceder em anos os sintomas principais da doença, e é mais frequente em pacientes com acometimento articular. Eritema anular ou eritema pérnio são outras manifestações cutâneas da SS primária, assim como vasculite cutânea manifestada por púrpura palpável.
Envolvimento pulmonar é comum, encontrado em até 20% dos casos, mas raramente tem importância clínica. Além de xerotraquéia (manifestação glandular) manifestada por tosse seca, dispnéia e hipoxemia moderada secundária à obstrução aérea de pequenas vias podem acontecer.
Dismotilidade esofágica secundária à redução da secreção glandular por infiltrado inflamatório linfocítico pode ocorrer, levando a disfagia ou sintomatologia compatível com refluxo gastroesofágico. Epigastralgia e náuseas são secundárias ao acometimento gástrico. Acometimento da parede gástrica leva a gastrite crônica atrófica, hipopepsinogenemia, elevação da gastrina sérica e anemia perniciosa.
A associação entre SS e cirrose biliar primária é bem estabelecida. Pacientes portadores de SS primária podem desenvolver hepatomegalia (25% dos casos) associada à elevação de enzimas colestáticas e presença de anticorpo antimitocondrial (5% dos casos). Já em pacientes portadores de cirrose biliar primária, o desenvolvimento de sintomas sicca é encontrado em até 50% dos pacientes, sendo 10% dos casos de sintomas severos.
Envolvimento renal é observado em 5% dos pacientes, sendo as manifestações mais comuns a nefrite intersticial, considerada um envolvimento periglandular levando à acidose tubular renal (principalmente acidose tubular renal distal) e glomerulonefrite membranosa ou membranoproliferativa secundária a deposição de imunocomplexos.
Cistite intersticial levando a polaciúria, noctúria e disúria é um achado casual e a biópsia demonstra o intenso infiltrado linfocítico característico da doença.
Vasculite sistêmica de pequenos e médios vasos é encontrada em 5% dos pacientes e usualmente se manifesta por púrpura palpável (mais comum), urticária recorrente, úlceras cutâneas ou mononeurite múltipla. Lesões em órgãos como fígado, rim, baço e trato gastrintestinal já foram relatadas.
O principal acometimento neurológico encontrado na SS é a mononeurite múltipla secundária à vasculite de pequenos vasos. O achado de acometimento de pares cranianos, principalmente nervo óptico e trigêmeo, é comum em pacientes com envolvimento neurológico. Envolvimento de SNC é raro, mas casos de hemiparesias, convulsões, mielite transversa, meningite asséptica, demência e encefalopatia já foram associados a SS.
Pacientes portadores de SS têm um risco 44 vezes maior de desenvolver linfoma, sendo o principal tipo o linfoma de células B de zona marginal. Fatores de risco para o desenvolvimento desse processo neoplásico são hipertrofia de parótidas, esplenomegalia, linfadenopatia, baixos níveis de C4 e de presença de crioglobulinemia mista. Geralmente é de localização extranodal e o envolvimento neoplásico de glândulas salivares está presente em 55% dos casos. A sobrevida desses pacientes está relacionada ao prognóstico do linfoma, ou seja, depende do baixo ou alto grau de malignidade.
Tabela 1: Achados clínicos na síndrome de Sjögren
|
Sintomas glandulares | |
|
Xeroftalmia |
Sintomas: sensação de areia, prurido ocular, vermelhidão e fotossensibilidade; Exame físico: hiperemia ocular, irregularidade corneana, hipertrofia das glândulas lacrimais e dilatação dos vasos conjuntivais |
|
Xerostomia |
Sintomas: sensação de boca seca, incapacidade para mastigar e deglutir alimentos secos e/ou falar continuamente, alterações de paladar, aumento da incidência de cáries, halitose, dificuldade para uso de próteses dentárias Exame físico: mucosa oral seca e eritematosa, cáries dentárias e atrofia das papilas filiformes encontradas no dorso da língua |
|
Sintomas extraglandulares | |
|
Articular |
Artrite presente em até 50% dos casos Característica: poliartrite simétrica e aditiva de grandes e pequenas articulações (lupus-like), não erosiva |
|
Cutâneo |
Pele seca em 55% dos casos Fenômeno de Raynaud em 35% dos casos Outros: eritema anular e vasculite cutânea (púrpura palpável) |
|
Pulmonar |
Em até 20% dos casos Xerotraquéia (manifestação glandular): tosse seca, dispnéia e hipoxemia por obstrução aérea de pequenas vias |
|
Trato geniturinário |
Acometimento renal em 5% dos pacientes: nefrite intersticial, acidose tubular renal, glomerulonefrite membranosa ou membranoproliferativa, cistite intersticial |
|
Trato gastrintestinal |
Dismotilidade esofágica: disfagia ou refluxo gastroesofágico Estômago: epigastralgia e náuseas, gastrite crônica atrófica Cirrose biliar primária |
|
Neurológico |
Principal achado: mononeurite múltipla Pares cranianos, principalmente nervo óptico e trigêmeo |
Pacientes com diagnóstico de SS podem apresentar alterações laboratoriais sugestivas de processo inflamatório sistêmico. Os achados mais frequentes em pacientes com doença ativa são anemia de doença crônica (25% dos pacientes), leucopenia (10%), elevação da velocidade de hemossedimentação (VHS) em 90% dos pacientes com proteína C reativa (PCR) normal e hipergamaglobulinemia refletindo a ativação policlonal de linfócitos B.
Auto-anticorpos séricos sempre estão presentes na doença, sendo os mais comuns o fator reumatóide (FR) e o fator antinúcleo (FAN), os quais são considerados não-específicos para o diagnóstico da doença. Anticorpos clássicos da SS são encontrados em até 70% dos casos, porém são pouco específicos para definição diagnóstica. São eles o anti-Ro (SS-A) e anti-La (SS-B), sendo este último o menos comum (
A avaliação da síndrome sicca é feita pelo teste específico para cada envolvimento glandular.
Tabela 2: Exames na xeroftalmia.
|
Teste de Schimmer |
Usado para avaliar a secreção das glândulas lacrimais. O teste é realizado com a colocação de um filtro de papel na pálpebra inferior por 5 minutos. É feita uma medida da área molhada: área menor que |
|
Teste de Rosa Bengala |
Avalia-se a sequela da baixa produção lacrimal no epitélio corneano. O corante Rosa Bengala é colocado no olho e cora uma área de lesão epitelial vista ao exame pela lâmina de fenda, considerando, então, o teste positivo. |
Tabela 3: Exames na xerostomia.
|
Sialometria |
Usada para avaliar o fluxo das glândulas parótidas, submandibulares e sublinguais. Tem baixa especificidade, visto que alterações como idade, sexo ou uso de medicações podem estar presentes e nenhum valor específico é diagnóstico de SS. |
|
Sialografia |
Método radiológico utilizado para avaliar alterações anatômicas do sistema de glândulas salivares. É tão sensível e específico quanto a biópsia de glândulas salivares e o achado de sialectasias é o característico da SS. |
|
Cintilografia de parótidas |
Cintilografia com 99Tc: excelente método funcional de avaliação das glândulas salivares. Redução de captação do contraste após 60 minutos da injeção tem boa correlação com alterações de sialometria e sialografia, além de correlacionar-se com a intensidade do infiltrado inflamatório nas glândulas salivares. Tem alta sensibilidade, mas baixa especificidade para diagnóstico de SS primária. |
|
Outros métodos de imagem |
Ultra-sonografia, RNM de parótidas, sialografia por RNM e RNM de parótidas dinâmicas (pré e pós-estímulo de glândulas salivares) são métodos recentes de avaliação do comprometimento de parótidas. São técnicas não-invasivas e bastante sensíveis, porém necessitam de validação adequada para indicação de seu uso rotineiro. |
Considerada o padrão-ouro para a confirmação da síndrome de Sjögren. A característica histopatológica consiste em agregados de 50 linfócitos, células plasmáticas e macrófagos adjacentes ao ácino glandular, caracterizando uma sialoadenite linfocítica crônica. No entanto, uma biópsia de glândula menor pode ser considerada específica para SS se apresentar
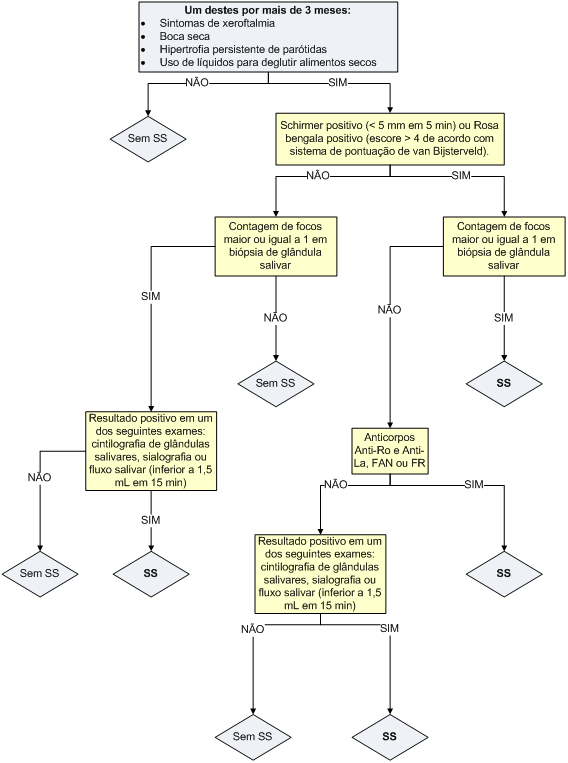
O diagnóstico da SS é baseado em critérios clínicos e na comprovação dos 2 pilares básicos da doença – evidência de déficit glandular e comprovação de sinais de auto-imunidade. Reconhecimento e diagnóstico precoce dessa doença são importantes para adequada intervenção terapêutica e prevenção de complicações, visto que a doença tratada tem curso lento e evolução benigna.
Os mais recentes critérios de classificação propostos pelo American-European Consensus Group datam de 2002 e ajudam no diagnóstico da doença (Tabela 4).
Tabela 4: Critérios de Classificação do American-European Consensus Group para síndrome de Sjögren
|
I. Sintomas oculares à resposta positiva a pelo menos 1 das perguntas abaixo: |
|
a. Você apresenta diariamente desconforto ocular persistente por mais de 3 meses? b. Você apresenta sensação recorrente de areia ou prurido nos olhos? c. Você usa lubrificantes oculares por mais de 3 vezes durante o dia? |
|
II. Sintomas orais à resposta positiva a pelo menos 1 das perguntas abaixo: |
|
a. Você apresenta uma sensação de boca seca diariamente por mais de 3 meses? b. Você apresenta hipertrofia de glândulas salivares recorrente ou persistente na idade adulta? c. Você necessita beber líquidos para ingerir alimentos secos? |
|
III. Sinais oculares à resultado positivo em pelo menos 1 dos seguintes testes: |
|
a. Teste de Schirmer sem anestesia (< 5 mm em 5 min) b. Escore de Rosa Bengala ou outro escore de olho seco (> 4 de acordo com o sistema de van Bijsterveld) |
|
IV. Histopatologia |
|
a. Biópsia de glândula salivar menor obtida de mucosa aparentemente normal mostrando sialoadenite focal linfocítica com um escore focal > 1, definido como um número de focos linfocíticos (que estão adjancentes ao ácino glândulas contendo ao menos 50 linfócitos) para cada 4 mm2 de tecido glândular |
|
V. Testes para envolvimento de glândulas salivares à resultado positivo em 1 dos seguintes testes: |
|
a. Baixo fluxo salivar sem estimulação prévia (< 1,5 mL em 15 min) b. Sialografia mostrando sialectasias difusas (padrão pontilhado, cavitário ou destrutivo), na ausência de obstrução dos ductos maiores c. Cintilografia de parótidas Tc99 demonstrando retardo de captação, concentração ou excreção do radiofármaco |
|
VI. Auto-anticorpos |
|
a. Anti-Ro (SSA), anti-La (SSB) ou ambos |
|
Regras para classificação: |
|
1. SS primária: a. 4 dos 6 critérios presentes, sendo obrigatório a presença dos itens IV ou VI b. Presença de 3 dos 4 critérios objetivos (III, IV, V ou VI) 2. SS secundária a. Doença associada a SS secundária + presença dos itens I ou II associados a 2 dos seguintes: III, IV ou V. 3. Critérios de exclusão a. Presença de radioterapia cervical prévia, infecção por hepatite C ou HIV, linfoma pré-existente, sarcoidose, doença enxerto versus hospedeiro, uso de drogas anticolinérgicas (pelo menos por tempo superior a 4 meias-vidas da droga) |
O diagnóstico diferencial de SS primária inclui todas as condições que podem levar à síndrome sicca (xerostomia e xeroftalmia) e hipertrofia de parótidas, além da identificação da síndrome de Sjögren secundária a doenças do tecido conectivo.
Entre as causas mais comuns de síndrome sicca, estão o uso de medicações como antidepressivos, diuréticos, medicações anticolinérgicas, neurolépticos, entre outras. Outras causas incluem radioterapia em região cervical e patologias que podem desencadear processos imunológicos e infiltrativos glandulares como sarcoidose, linfoma, lipoproteinemias, doença enxerto versus hospedeiro, amiloidose, infecções virais pelos vírus da hepatite C, HIV e HTLV I, além das próprias doenças do colágeno, sendo a artrite reumatóide e o lúpus eritematoso sistêmico as principais colagenoses associadas a SS (Tabela 5).
Tabela 5: Diagnóstico diferencial da síndrome de Sjögren
|
Doenças auto-imunes (SS secundária) |
Drogas |
Doenças infiltrativas |
|
Artrite reumatóide Lúpus eritematoso sistêmico Esclerose sistêmica progressiva Doença mista do tecido conectivo Cirrose biliar primária Miopatias inflamatórias Vasculites sistêmicas Tireoidite Hepatite crônica ativa Crioglobulinemia mista |
Diuréticos Anticolinérgicos Antidepressivos Neurolépticos |
Sarcoidose Amiloidose |
|
Infecções |
Miscelânea | |
|
Hepatite C HIV Parotidite viral HTLV I |
Síndrome da fadiga crônica Fibromialgia Lipoproteinemias (II, IV e V) |
O primeiro estágio do tratamento da SS é a realização de um diagnóstico acurado (Algoritmo 1), baseado sobretudo nos critérios de classificação recentemente propostos. Por ter um diagnóstico diferencial amplo e por ter muitas características clínicas inespecíficas de doenças auto-imunes, os critérios de classificação dessa patologia em particular são muito importantes para ajudar na definição e no tratamento da doença.
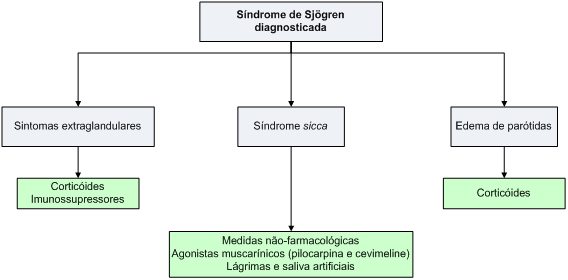
As manifestações clínicas da SS são variáveis;, por este motivo, o tratamento é dirigido para a forma específica de acometimento de cada paciente.
Citamos as medidas gerais e o tratamento farmacológico da síndrome sicca nas Tabelas 6 e 7.
Tabela 6: Tratamento para xeroftalmia
|
Medidas não-farmacológicas |
Evitar ambiente de baixa umidade e com ventilação abundante, leitura prolongada, uso de computadores. Umidificadores utilizados em acessórios como óculos Lentes de contato terapêuticas (indicação e acompanhamento oftalmológico) Evitar uso de drogas agravantes do quadro: diuréticos, antidepressivos tricíclicos, anti-histamínicos Oclusões temporárias de ductos de drenagem lacrimal |
|
Reposição de volume lacrimal |
Lágrimas artificiais Gotas oculares feitas a partir de soro autólogo |
|
Estimulação de secreção lacrimal |
Agonistas muscarínicos: pilocarpina e cevimeline Novos secretagogos: INS365, 15S-HETE, rebamipide, ecabet sodium, gefarnate (experimentais) |
|
Drogas imunomodulatórias |
Ciclosporina A 0,05% Tacrolimo e pimecrolimo (experimentais) Corticóides tópicos (curto período) |
|
Outros |
Testosterona tópica transdérmica (não aprovado pelo FDA) |
Tabela 7: Tratamento para xerostomia
|
Medidas não-farmacológicas |
Adequada hidratação, ingestão de líquidos sem açúcar Evitar fatores irritantes/predisponentes: café, álcool, nicotina etc. Cicletes e balas sem açúcar Substituição ou redução de drogas que podem levar a xerostomia Higiene oral meticulosa: aplicação de flúor regular, tratamento dentário adequado e frequente, tratamento de possíveis focos infecciosos, como monilíase oral |
|
Agentes lubrificantes |
Saliva artificial Gel e pastilhas lubrificantes, creme dentários adequados, soluções de higiene oral Implantes orais hidratantes de longa permanência Spray de mucina |
|
Secretagogos orais |
Agonistas muscarínicos: pilocarpina e cevimeline |
|
Drogas imunomodulatórias |
Corticóides orais, irrigação de parótidas com corticóides |
Hipertrofia de parótidas pode estar presente em até 60% dos casos de SS primária. Casos recorrentes respondem bem à dose moderada de corticóides (
Parotidite infecciosa, principalmente bacteriana, é difícil de distinguir da parotidite primária da SS. Sintomas constitucionais e febre são pouco frequentes, de modo que, em casos de parotidite refratária ou recorrente, antibioticoterapia deve ser considerada.
Linfoma sempre deve ser excluído em casos de parotidites recorrentes, de consistência nodular ou endurecida, associada à hipogamaglobulinemia e em pacientes portadores de fatores de risco, como doença de longa evolução e manifestações secundárias a depósito de imunocomplexos. Nesses casos, biópsia deve ser realizada e, em caso de confirmação diagnóstica, o paciente deve ser prontamente encaminhado para o especialista para tratamento do linfoma.
Em indivíduos jovens, as manifestações extraglandulares podem acarretar significativa morbidade. Até o presente momento, não existem tratamentos que definidamente reduzam a progressão da doença, e alternativas terapêuticas para alívio sintomático dos sintomas sicca, descritas anteriormente, devem ser instituídas. Envolvimento extraglandular exige terapêutica dirigida para o devido acometimento, com corticóides e imunossupressores, usualmente de maneira semelhante àquela aplicada para o lúpus eritematoso sistêmico.
O acometimento articular inflamatório tem características benignas e raramente, nos casos primários, está associado à erosão óssea radiológica. O quadro tende a se apresentar como períodos de artrite remitente, a qual é bem responsiva a hidroxicloroquina na dose de 400 mg/dia ou metotrexato em doses que variam de 10 a 25 mg/semana.
Acometimento pulmonar em pacientes com SS é frequente e pode variar desde quadros leves de xerotraquéia a pneumonia criptogênica em organização. Doença pulmonar inflamatória sintomática em SS respondem bem a corticóides associados a azatioprina e raramente progride durante seguimento.
Acometimentos severos na SS são raros, mas podem estar presentes. Mielite transversa já foi descrita como associada a esta patologia e seu tratamento consiste em ciclofosfamida e corticóides. Corticóides, em pulsoterapia, devem ser aplicados nas fases iniciais do quadro clínico e, em caso de má resposta a estes agentes, gamaglobulina endovenosa ou plasmaférese deve ser considerada. Pulsoterapia de ciclofosfamida mensal deve ser adicionada ao esquema terapêutico, em doses variando de
Crioglobulinemia associada à vasculite cutânea ou glomerulonefrite respondem à associação corticóides e ciclofosfamida. Aplicação de gamaglobulina pode ser útil em casos refratários.
Manifestação renal clínica é rara e o principal acometimento é nefrite intersticial, quadro associado a infiltrado linfocítico periglandular. Usualmente é uma condição benigna e o tratamento com corticóides não é necessário. Tratamento de suporte com reposição de K+ e bicarbonato de sódio pode ser usado.
Alterações hematológicas como citopenias, hipergamaglobulinemia e, em raros casos, anemia hemolítica auto-imune são tratadas com corticoterapia e imunossupressores, podendo-se usar azatioprina, metotrexato ou ciclofosfamida. Agranulocitose grave pode requerer uso de gamaglobulina. Danazol pode ser útil em plaquetopenia refratária.
Novas terapias estão sendo avaliadas para o tratamento da SS primária. Um tratamento promissor parece ser o uso de rituximabe, anticorpo anti-CD-20. O CD-20 é considerado um marcador específico de células B, altamente exposto na superfície de membrana de precursores de linfócitos B, assim como em células maduras. Trabalhos recentes mostraram benefício do uso desta droga nas manifestações sicca, articulares, em neuropatia periférica, glomerulonefrite, crioglobulinemia, esclerite, citopenias severas e linfoma associado a SS. Alternativas como o epratuzumabe, anti-CD22 específico para linfócitos B maduros, em estudos fase I/IIe mostraram benefícios na redução de atividade clínica da doença, porém mais estudos de segurança e eficácia são necessários até a validação dessa droga para o tratamento da SS.
síndrome de Sjögren: doença auto-imune sistêmica caracterizada por acometimento de glândulas exócrinas, além de presença de sinais/sintomas de doença sistêmica. Pode ser classificada em SS primária ou secundária.
Síndrome sicca: caracterizada por ressecamento dos olhos (xeroftalmia) e da boca (xerostomia). A atividade glandular é bastante reduzida tanto pela destruição acinar como pela denervação glandular crônica nesses pacientes.
Afeta principalmente mulheres na 4ª ou 5ª décadas de vida.
A patogênese é multifatorial, tendo importância fatores genéticos e ambientais.
Possíveis agentes desencadeantes da doença: vírus Epstein-Barr (EBV), hepatite C (HCV), HTLV-1 e outras retroviroses.
Envolvimento glandular: a xeroftalmia está presente em 95% dos casos e a xerostomia em 90%, e são os eventos precoces da doença.
Manifestações extraglandualres da doença podem estar presentes em até 50% dos casos. As principais manifestações são: artrite lupus-like, pele seca e fenômeno de Raynaud.
Pacientes portadores de SS têm um risco 44 vezes maior de desenvolver linfoma. Os fatores de risco são hipertrofia de parótidas, esplenomegalia, linfadenopatia, baixos níveis de C4 e presença de crioglobulinemia mista.
Os achados laboratoriais mais frequentes em pacientes com doença ativa são anemia de doença crônica (25%), leucopenia (10%), elevação da VHS em 90% dos pacientes com proteína C reativa (PCR) normal e hipergamaglobulinemia.
Auto-anticorpos mais comuns: FR e FAN. Os anti-Ro (SS-A) e Anti-La (SS-B) são encontrados em até 70% dos casos, sendo o último o mais específico e menos frequente.
Para avaliação de xeroftalmia, usam-se os testes de Schimmer e de Rosa Bengala.
Para avaliação de xerostomia, usam-se a sialometria e a cintilografia de parótidas. Entretanto, o melhor teste é a sialografia (o achado de sialectasias é característico), sendo tão específico e sensível quanto a biópsia de glândulas salivares.
A biópsia de glândulas salivares é o padrão-ouro para a confirmação da síndrome de Sjögren.
O diagnóstico da SS é baseado em critérios clínicos e na comprovação dos 2 pilares básicos da doença: evidência de déficit glandular e comprovação de sinais de auto-imunidade.
O diagnóstico diferencial de SS primária inclui todas as condições que podem levar a síndrome sicca (xerostomia e xeroftalmia) e hipertrofia de parótidas, além da identificação da síndrome de Sjögren secundária a doenças do tecido conectivo.
O tratamento da xeroftalmia e da xerostomia inclui basicamente medidas não-farmacológicas e o uso de agonistas muscarínicos (pilocarpina e cevimeline).
Hipertrofia de parótidas pode estar presente em até 60% dos casos de SS primária. Casos recorrentes respondem bem a dose moderada de corticóides.
Os sintomas extragladulares são tratados basicamento com corticóides e imunossupressores.
Algoritmo 1: Guia para diagnóstico da síndrome de Sjögren.
Algoritmo 2: Guia para tratamento da síndrome de Sjögren.
1. Tzioufas AG, Moutsopoulos H. Sjögren’s syndrome. In: Hochberg MC, Silman AJ, Smolen JS, Weinblatt ME, Weisman MH, editores. Rheumatology. 3. ed. London: Mosby, 2003. p.1431-1443.
2. Tzioufas AG, Voulgarelis M. Update on Sjögren’s syndrome autoimmune epithelitis: from classification to increased neoplasias. Best Pract Res Clin Rheumatol 2007; 21:989-1010.
3. Ramos-Casals M, Brito-Zerón P. Emerging biological therapies in primary Sjögren’s syndrome. Rheumatology 2007; 46:1389-1396.
4. García-Carrasco M, Fuentes-Alexandro S, Escárcega RO, Salgado G, Riebeling C, Cervera R. Pathophysiology of Sjögren’s syndrome. Arch Med Res 2006; 37:921-932.
5. Venables PJ. Management of patients presenting with Sjogren’s syndrome. Best Pract Res Clin Rheumatol 2006; 20:791-807.
6. Seror R, Sordet C, Guillevin L, Hachulla E, Masson C, Ittah M, et al. Tolerance and efficacy of rituximab and changes in serum B cell biomarkers in patients with systemic complications of primary Sjögren’s syndrome. Ann Rheum Dis 2007; 66:351-357.
7. Thanou-Stavraki A, James JA. Primary Sjogren’s syndrome: current and prospective therapies. Semin Arthritis Rheum 2008; 37:273-292.
8. www.sjogrens.org.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.