(Carregando Índice)... (Carregando Índice)... |
Autores:
Murillo de Oliveira Antunes
Especialista em Cardiologia pelo Instituto do Coração do Hospital das Clínicas da Faculdade de Medicina da USP
Edmundo Arteaga
Médico Assistente, Doutor da Unidade Clínica de Miocardiopatias do InCor-HC-FMUSP.
Última revisão: 18/10/2010
Comentários de assinantes: 1
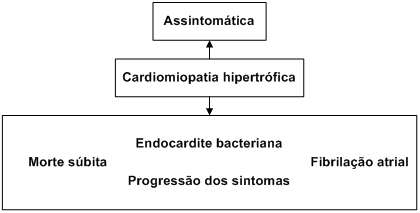
A cardiomiopatia hipertrófica (CMH) é uma doença cardíaca primária caracterizada por hipertrofia do ventrículo esquerdo (HVE), sem dilatação, na ausência de doença cardíaca ou sistêmica que justifique a magnitude dessa hipertrofia (como a hipertensão arterial ou valvulopatias).
A hipertrofia é assimétrica na maioria dos casos, acometendo principalmente o septo ventricular, e que pode gerar uma obstrução mecânica ao fluxo sanguíneo na via de saída do ventrículo esquerdo (VSVE) em mais de 25% dos casos. A função sistólica em repouso na cardiomiopatia hipertrófica é normal ou se observa um estado hiperdinâmico.
Sua prevalência é de 0,2% (2:1.000 habitantes) da população geral e de 0,5% dos pacientes selecionados encaminhados ao exame ecocardiográfico.
A morte súbita cardíaca (MSC) é a manifestação mais temível da doença, sendo que é a cardiomiopatia hipertrófica constitui a principal causa de MSC entre jovens e atletas.
A cardiomiopatia hipertrófica é uma doença genética, de caráter familiar e de transmissão autossômica dominante, em 50 a 60% dos casos (nos casos restantes, são de etiologia desconhecida, podendo ser decorrentes de mutações espontâneas). É a doença cardíaca de origem genética mais comum.
A cardiomiopatia hipertrófica decorre de mutações em genes codificadores das proteínas do sarcômero cardíaco. Foram identificados 13 genes diferentes relacionados aos componentes dos filamentos fino ou grosso da fibra muscular cardíaca com funções regulatórias, estruturais e contráteis. Mais de 400 mutações são descritas, sendo que os genes da cadeia pesada da betamiosina cardíaca, gene da troponina T cardíaca e da proteína C de ligação à miosina respondem por aproximadamente 70% dos casos (Tabela 1).
Uma característica própria da cardiomiopatia hipertrófica é a ampla diversidade na expressão fenotípica da doença. Conforme a mutação acometida, observa-se variabilidade na transmissão familiar, no grau de hipertrofia, na evolução e no prognóstico da doença.
Tabela 1: Genes causadores de cardiomiopatia hipertrófica
|
Cadeia pesada da betamiosina |
MYH7 |
|
Proteína C de ligação à miosina |
MYBPC3 |
|
Troponina T |
TNNT2 |
|
Alfatropomiosina |
TPM1 |
|
Cadeias leves da miosina essencial |
MYL3 |
|
Cadeias leves da miosina reguladora |
MYL2 |
|
Troponina I |
TNNI3 |
|
Alfa-actina |
ACTC |
|
Titanina |
TTN |
|
Troponina C |
TNTC1 |
|
Cadeia pesada da alfa miosina |
MYH6 |
|
Proteína muscular LIM |
CRP3 |
|
Teletonina |
TCAP |
Observa-se aumento da massa miocárdica com cavidades ventriculares normais ou pequenas. O ventrículo esquerdo (VE) é mais acometido que o ventrículo direito (VD), porém, em 30% dos casos, há o acometimento concomitante do VD. A hipertrofia miocárdica é assimétrica em 2/3 dos casos, apresentando o septo interventricular e a parede anterolateral do VE como segmentos mais envolvidos. O átrio esquerdo geralmente encontra-se dilatado em consequência da alta resistência ao enchimento ventricular causado pela disfunção diastólica e efeito do refluxo através da valva mitral. Há uma relação inversa entre grau de hipertrofia e a idade dos pacientes, com incerteza sobre a causa, se é devida à morte prematura dos pacientes com hipertrofia mais acentuada ou à redução progressiva do espessamento miocárdico em razão de processo de remodelação.
Os achados microscópicos incluem hipertrofia dos cardiomiócitos, desorganização dos feixes musculares e perda do alinhamento paralelo resultando em um padrão espiralado característico em 5% ou mais do miocárdio em geral.
Os elementos constituintes do tecido conectivo intersticial estão aumentados e a presença de fibrose é variável. Pode ocorrer de forma difusa ou localizada e quase sempre tais elementos são suficientes para produzir cicatrizes visíveis macroscopicamente.
As artérias coronárias intramurais apresentam lúmen reduzido, paredes espessadas e aumento de células musculares lisas e de colágeno na camada média e íntima.
As principais alterações fisiopatológicas são descritas a seguir.
A função diastólica encontra-se alterada na maioria dos pacientes, em diferentes graus, sendo a principal responsável pelos sinais e sintomas de insuficiência cardíaca.
A anormalidade no enchimento diastólico independe da extensão e da espessura da hipertrofia e os principais mecanismos responsáveis são alterações do relaxamento miocárdio e diminuição da complacência ventricular.
As alterações do relaxamento miocárdio se devem a desarranjo dos miócitos, anormalidades da cinética do cálcio intracelular, distorção da geometria ventricular, isquemia miocárdica e alterações da velocidade e sincronia da repolarização.
A redução da complacência do VE se deve a fibrose e desarranjo celular, prolongamento e assincronismo do relaxamento e alterações na geometria ventricular.
Desde as descrições iniciais da cardiomiopatia hipertrófica, a característica que chama atenção é o gradiente pressórico na via de saída do ventrículo esquerdo.
A forma obstrutiva – definida como gradiente em repouso na VSVE > 30 mmHg – é encontrada em 30% dos pacientes portadores de cardiomiopatia hipertrófica e possui implicações prognósticas que aumentam o risco de morte súbita cardíaca e de progressão para dilatação ventricular, quando comparado com os pacientes sem obstrução.
Várias hipóteses foram sugeridas sobre sua verdadeira origem. Admite-se que seja decorrente do estreitamento na via de saída do ventrículo esquerdo (em virtude da acentuada hipertrofia septal) associada a anormalidades da válvula mitral (principalmente movimento anterior sistólico da válvula mitral).
A origem do movimento anterior sistólico da válvula mitral permanece controverso, acreditando-se que seja decorrente do efeito Venturi associado a alterações anatômicas da válvula mitral.
A isquemia miocárdica na cardiomiopatia hipertrófica é comum e de origem multicausal. Está relacionada a vários mecanismos fisiopatológicos que atuam de forma isolada ou sinérgica e são responsáveis pelo sintoma de angina encontrado em 50% da população portadora da doença. São eles:
aumento excessivo da massa muscular, com consequente aumento da demanda de oxigênio pelo miocárdio;
estrutura microvascular coronariana insuficiente em relação ao aumento da massa ventricular;
elevação das pressões diastólicas ventriculares e compressão microvascular, diminuindo o fluxo sanguíneo principalmente na região subendocárdica;
espessamento das paredes das artérias coronárias com diminuição do lúmem arteriolar.
A maioria dos pacientes com cardiomiopatia hipertrófica são assintomáticos ou oligossintomáticos e tem seu diagnóstico suspeitado pela detecção de sopros cardíacos, alteração eletrocardiografica ou, ainda, durante a investigação de famílias acometidas.
Estudo realizado nos EUA com 227 pacientes, onde 90% eram assintomáticos no início da randomização, mostrou que, após o seguimento de 8 anos, apenas 25% dos pacientes evoluíram para sintomas incapacitantes ou morreram e 69% dos pacientes mantiveram-se assintomáticos ou apresentavam apenas sintomas leves (Tabela 2).
A morte súbita cardíaca infelizmente pode ser a primeira manifestação clínica da doença e acomete sobretudo adolescentes e adultos antes dos 35 anos, embora possa ocorrer em qualquer faixa etária.
Tabela 2: Manifestações clínicas na cardiomiopatia hipertrófica
|
Dispneia |
|
Dor torácica |
|
Sincope e pré-síncope |
|
Palpitações |
|
Morte súbita cardíaca |
É a principal manifestação clínica da doença e ocorre em 90% dos pacientes. É decorrente de vários mecanismos, porém a disfunção diastólica do VE juntamente com a obstrução do fluxo sanguíneo na via de saída do ventrículo esquerdo e a regurgitação mitral são os principais responsáveis.
Em aproximadamente 10% dos casos, a doença pode evoluir para forma dilatada, com diminuição da função sistólica do VE e dispneia.
Encontrada em
A associação de vários mecanismos fisiopatológicos colabora para que ocorra um desequilíbrio entre a oferta e o consumo do oxigênio do miocárdio, levando à isquemia miocárdia (Tabela 3).
Admite-se que cerca de 20% dos pacientes idosos com cardiomiopatia hipertrófica possam ter doença arterial coronariana obstrutiva concomitante.
Tabela 3: Principais mecanismos envolvidos na origem da dor torácica
|
Aumento da demanda O2 |
Redução da oferta O2 |
|
Hipertrofia miocárdica |
Pontes miocárdicas |
|
Disfunção diastólica |
Redução do lúmen arteriolar |
|
Estresss na parede VE |
Fibrose miocárdica |
|
Desarranjo dos miócitos |
Disfunção microvascular |
|
Arritmias |
|
Cerca de
A ocorrência de episódio de síncope inexplicada na cardiomiopatia hipertrófica (quando afastadas causas neurocardiogênicas) identifica pacientes com risco aumentado de morte súbita cardíaca, principalmente quando acontece em crianças e adolescentes.
É uma queixa muito comum nos portadores de cardiomiopatia hipertrófica. Pode estar associada a arritmias ventriculares e supraventriculares que são frequentemente observadas na eletrocardiografia dinâmica.
Em pacientes assintomáticos e sem obstrução da via de saída do ventrículo esquerdo, o exame físico costuma ser normal. O ictus é amplo, forte e desviado para a esquerda e pode-se palpar frêmito sistólico ao nível da ponta ou na borda esternal baixa.
O pulso venoso jugular pode apresentar onda “a” elevada pela contração atrial acentuada resultante da diminuição da distensibiladade do ventrículo.
O pulso carotídio tem um aumento rápido e depois diminui na mesossístole à medida que ocorre a formação do gradiente seguida por um aumento secundário, dando um aspecto de bisferiens.
À ausculta cardíaca, o achado mais comum é a presença de uma B4, que é explicada por uma vigorosa contração atrial. A B2 é desdobrada, sendo que, nos pacientes com obstrução acentuada da via de saída do ventrículo esquerdo, podem apresentar um desdobramento paradoxal. Na maioria dos pacientes com a forma obstrutiva, ausculta-se um sopro sistólico rude em “crescendo-descrescendo”, que se inicia logo após a B1, audível ao longo da borda esternal esquerda baixa resultante do fluxo turbulento na via de saída estreitada do VE. Este sopro é muito semelhante ao encontrado na estenose aórtica, porém não se irradia para o pescoço. Quando a regurgitação mitral está presente, pode-se ouvir um sopro holossistólico no ápice, suave e que se irradia para axila.
Algumas manobras modificam a intensidade do sopro e auxiliam para o diagnóstico clínico da doença (Tabela 4).
Tabela 4: Efeitos de intervenções sobre o gradiente da via de saída do ventrículo esquerdo e o sopro em pacientes com cardiomiopatia hipertrófica
|
Intervenção |
Gradiente e sopro |
|
Manobra de Valsalva |
Aumento |
|
Posição ortostática |
Aumento |
|
Pós-extrassístole |
Aumento |
|
Posição de cócoras |
Redução |
|
Manobra handgrip |
Redução |
Encontra-se alterado em até 95% dos casos e não há um padrão característico da doença. Altera-se precocemente, mesmo antes das alterações ecocardiográficas ocorrerem.
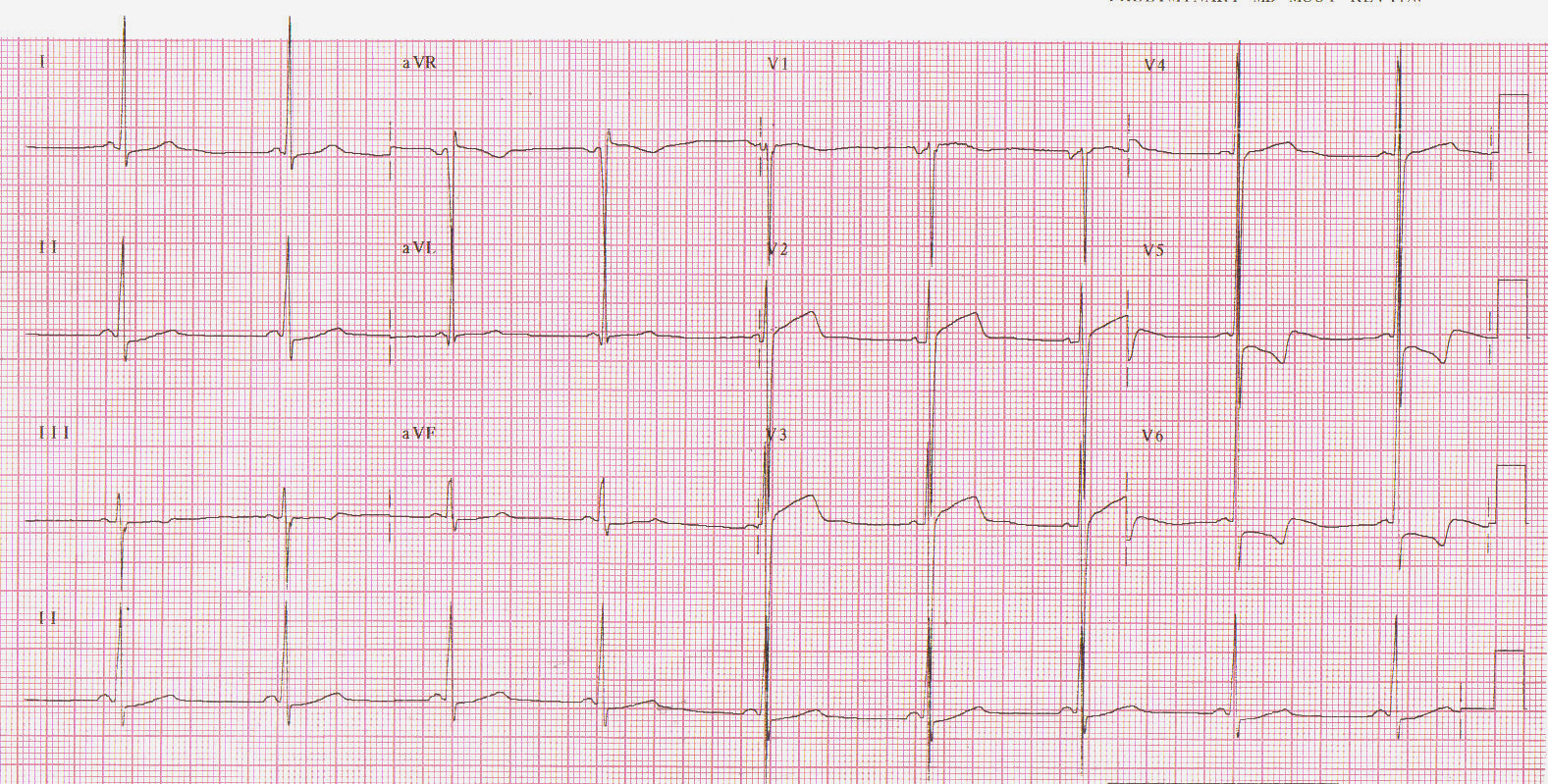
As anormalidades mais comuns são as alterações do segmento ST e ondas T, seguidas pela evidência de hipertrofia ventricular esquerda. Presença de ondas S profundas em V1 e V2 e ondas R amplas em V5 e V6, alterações de repolarização tipo “strain” são achados comuns (Figura 1).
Observam-se anormalidades da onda P relacionadas com sobrecarga atrial esquerda (duração > 120 mS, onda P entalha – P mitrale, índice de Morris). A fibrilação atrial é observada em 5 a 8% dos pacientes e 1 a 2% têm síndrome de Wolff-Parkinson-White associada.
Ondas Q proeminentes envolvendo as derivações inferiores (DII, DIII e AVF) ocorrem em
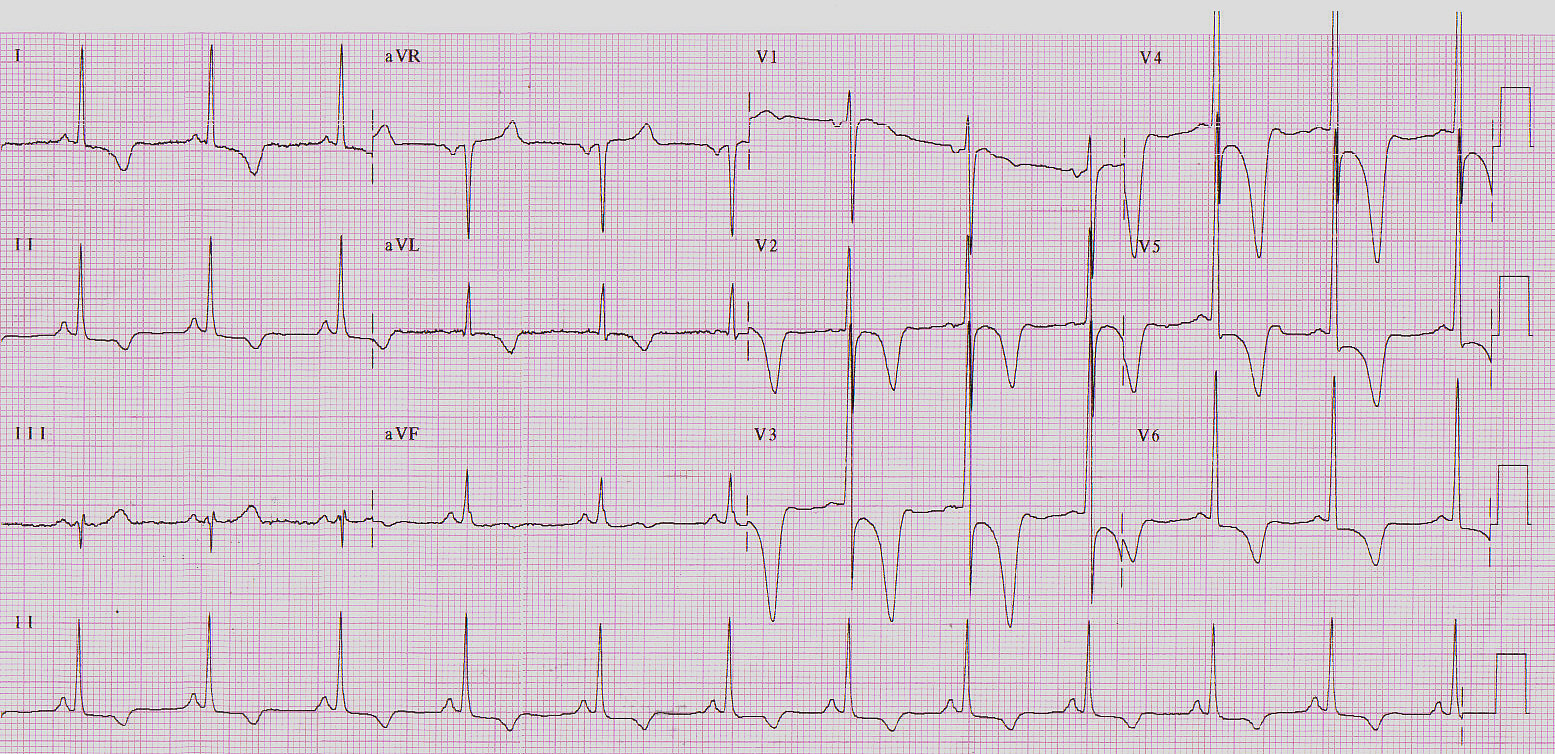
A presença de ondas T invertidas e gigantes nas derivações precordiais são características de uma forma especial de cardiomiopatia hipertrófica, descrita inicialmente no Japão e denominda Yamagushi, onde a presença da hipertrofia é localizada na ponta do VE – forma apical (Figura 2).
Figura 1: Sobrecarga ventricular esquerda.

Figura 2: Forma apical.
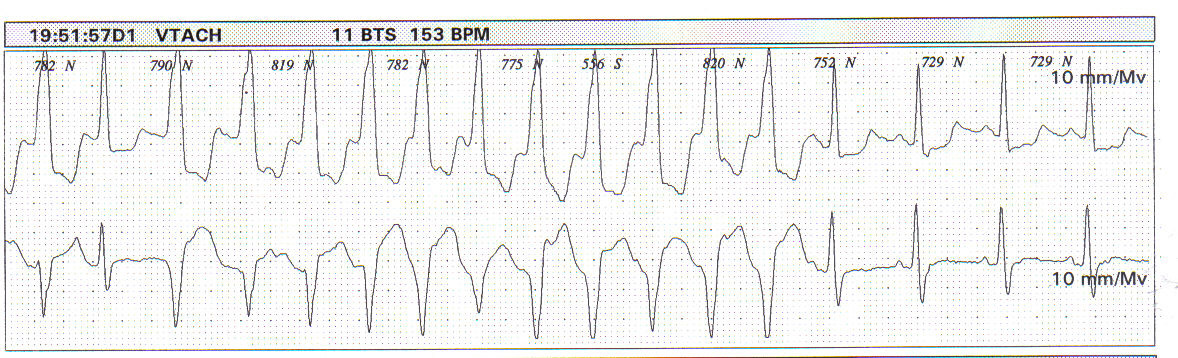
Deve ser solicitado de rotina para os pacientes com cardiomiopatia hipertrófica, já que a presença de taquicardia ventricular não sustentada (TVNS) tem importância prognóstica, revelando um grupo com alto risco para morte súbita cardíaca (Figura 3).
Figura 3: TVNS registrada no Holter em paciente assintomático.
O ecocardiograma constitui o principal método de diagnóstico da cardiomiopatia hipertrófica. Permite estabelecer o diagnóstico, a localização e o grau de hipertrofia, podendo avaliar, ainda, a presença e o grau de obstrução, assim como o acompanhamento evolutivo da doença. Também é utilizado na avaliação e no acompanhamento de familiares dos pacientes com cardiomiopatia hipertrófica, possibilitando o diagnóstico de formas mais discretas, que poderiam passar despercebidas sem a associação com uma história familiar bem definida. O diagnóstico de hipertrofia ventricular é definido por espessura do septo anterior ou parede livre do VE =
Outros achados ecocardiográficos encontrados são:
hipertrofia assimétrica do septo interventricular, com relação entre a espessura do septo e da parede posterior do VE > 1,5;
gradiente obstrutivo na via da saída de VE, resultado da hipertrofia do septo e do deslocamento do aparelho valvar mitral (Figura 5);
insuficiência mitral e movimentação anterior sistólica da válvula mitral, resultado do efeito Venturi;
cavidade ventricular normal ou diminuída;
aumento de átrio esquerdo;
disfunção diastólica com padrão de onda E < A, no fluxo mitral;
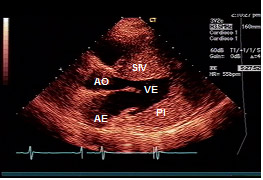
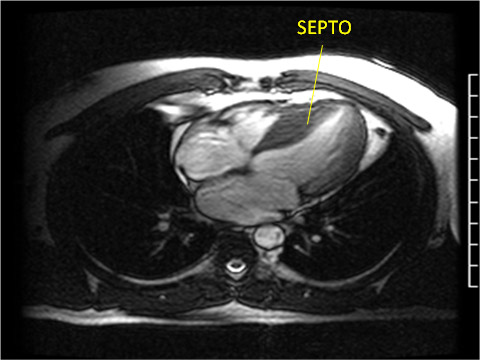
Figura 4: Ecocardiograma bidimensional, corte longitudinal, destacando hipertrofia do septo interventricular (SIV =
SIV PI VE AO AE
AO = aorta; AE = átrio esquerdo; VE = ventrículo esquerdo.
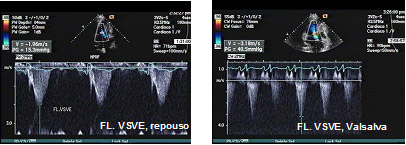
Figura 5: Fluxo sistólico na via de saída do VE, estimando gradiente em 15 mmHg, em repouso. No pico da manobra de Valsalva, gradiente sistólico estimado em 40 mmHg.
Nos últimos anos, a ressonância magnética cardiovascular (RMC) emergiu como um instrumento bastante preciso no diagnóstico de cardiomiopatia hipertrófica, especialmente nos casos em que as áreas de hipertrofia se tornam tecnicamente difíceis de serem avaliadas pela ecocardiografia convencional (como nas formas apicais).
A grande precisão da RMC na análise anatomofuncional dos ventrículos esquerdo e direito na quantificação do volume e da massa ventricular aumentou a sensibilidade e a especificidade do diagnóstico de cardiomiopatia hipertrófica, permitindo identificar precisamente diversas formas de hipertrofia; auxilia no diagnóstico diferencial (hipertrofia vs. infiltração) e na identificação da presença de fibrose miocárdica (Figuras 6 e 7).
Alguns estudos iniciais demonstraram que a presença de fibrose, assim como a extensão e o padrão de distribuição desta, está relacionada com maior risco para as arritmias ventriculares, morte súbita cardíaca e dilatação do ventrículo esquerdo.
Figura 6: Paciente de 23 anos com septo de
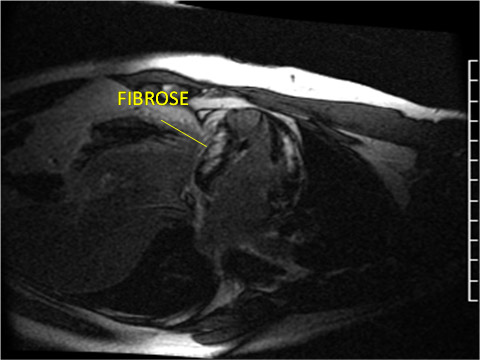
Figura 7: Realce tardio evidenciando fibrose multifocal.
A análise do DNA constitui o método mais definitivo para a identificação da cardiomiopatia hipertrófica, permitindo a realização do diagnóstico na fase pré-clínica da doença e mesmo antes de ocorrerem as alterações estruturais cardíacas. O substrato molecular heterogêneo, representado por centenas de mutações em múltiplos genes, confere complexidade ao diagnóstico genético e limita sua aplicação clínica de rotina.
Atualmente, o custo elevado e a demora na obtenção de resultados têm restringido o diagnóstico genético-molecular apenas a centros de pesquisa.
Não está indicado de rotina para o diagnóstico de cardiomiopatia hipertrófica e só deve ser solicitado na suspeita de doença arterial coronariana ou nos casos em que se pretende realizar o tratamento invasivo da doença (cardiomiectomia ou alcoolização septal).
A contribuição do estudo eletrofisiológico para avaliação do substrato arritmogênico da cardiomiopatia hipertrófica ainda não se encontra definida. Embora alguma relação entre indutibilidade e prognóstico tenha sido demonstrada, a acurácia preditiva é discutível, com valor limitado e, deste modo, não deve ser rotineiramente indicado.
A cardiomiopatia hipertrófica possui uma história natural variável, podendo ocorrer em qualquer fase da vida, desde a infância até a terceira idade (Figura 8). Para a maioria dos pacientes, o curso é relativamente benigno, mantendo-se assintomáticos ou com sintomas discretos. Cerca de 5 a 10% dos casos têm progressão dos sintomas com dilatação e disfunção ventricular e uma minoria evolui para apresentação clínica restritiva grave.
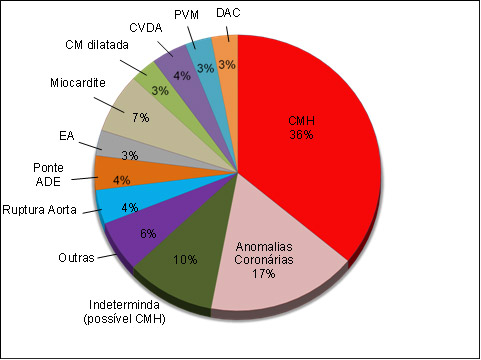
Figura 8: Causas de morte súbita em atletas competitivos jovens.
ADE: artéria coronária descendente esquerda; EA: estenose aórtica; CM: cardiomiopatia; CVDA: cardiomiopatia arritmogênica do ventrículo direito; PVM: prolapso de válvula mitral; DAC: doença arterial coronariana.
Adaptado de: Maron et al., 1996 94:850-56.
A taxa de mortalidade anual gira em torno de 3% em adultos selecionados em centros de referências, mas provavelmente gire em torno de 1% quando todos pacientes com cardiomiopatia hipertrófica são incluídos
A complicação mais temida da doença e que deve ser avaliada em cada consulta é a morte súbita cardíaca, que pode correr em qualquer época da vida, sendo, muitas vezes, a primeira manifestação da doença. Acomete com maior frequência as crianças entre 8 e 16 anos de idade e atletas com mortalidade anual em cerca de 6%. Está relacionado com diversos fatores, como: mutação responsável pela doença, história familiar de morte súbita em parente de 1º grau com menos de 45 anos de idade, espessura miocárdica do septo = 30, TVNS no Holter, história de síncope e resposta anormal da pressão arterial sistólica ao esforço.
A endocardite infecciosa pode ocorrer em 5% dos pacientes portadores da forma obstrutiva da doença (gradiente > 30 mmHg na via de saída do ventrículo esquerdo); deste modo, a profilaxia com antibioticoterapia nos procedimentos cirúrgicos ou odontológicos é mandatória. A infecção pode ocorrer nas válvulas aórtica, mitral ou no endocárdio, no local do contato da válvula mitral com a parede septal.
A fibrilação atrial tem sido documentada em 20 a 25% dos doentes e sua ocorrência está associada à piora da dispneia. Também tem sido relacionada com aumento do risco para morte súbita cardíaca, já que funcionaria como um “gatilho” para a ocorrência de arritmias ventriculares.
Figura 9: História natural da cardiomiopatia hipertrófica.
O diagnóstico diferencial da cardiomiopatia hipertrófica deve ser feito com todas as patologias que podem gerar uma hipertrofia ventricular (HVE), sendo que as mais frequentes são: hipertensão arterial sistêmica (HAS), estenose da válvula aórtica e coração do atleta.
Cursa geralmente com hipertrofia ventricular concêntrica do tipo simétrica, onde a espessura da parede ventricular não atinge grandes tamanhos (em média 15 a 18 mm). Diferentemente do que se vê na cardiomiopatia hipertrófica, esta hipertrofia regride quando instituído um controle adequado dos níveis pressóricos. Como a prevalência de HAS na população em geral é alta, não é raro encontrar a associação das duas patologias, tornando-se difícil a diferenciação de hipertrofia ventricular por HAS ou CMH (ou vice-versa).
Também acarreta grandes hipertrofias do tipo simétricas, de modo que um exame físico minucioso, com realização de algumas manobras durante a ausculta, pode auxiliar para o diagnóstico diferencial (Tabela 5).
A hipertrofia ventricular neste caso é decorrente de treinamento físico intenso (atletas de competição), que pode ocorrer tanto nos exercícios estáticos ou de resistência (aeróbicos).
O diagnóstico diferencial com cardiomiopatia hipertrófica nem sempre é fácil de fazer, porém é de grande importância, já que determina a possibilidade do paciente realizar ou não atividades físicas (Tabela 5).
Tabela 5: Diagnóstico diferencial entre cardiomiopatia hipertrófica e coração do atleta
|
Parâmetros |
Miocardiopatia hipertrófica |
Coração do atleta |
|
Espessura da parede VE e morfologia |
Maior > |
Geralmente < |
|
Ventrículo esquerdo |
< 45 mm |
> 55 mm |
|
Átrio esquerdo |
Aumentado |
Normal |
|
Função diastólica |
Déficit de relaxamento (E < A) |
Normal |
|
Resposta ao descondicionamento |
Não |
Diminuição da hipertrofia |
|
História familiar |
Presente |
Ausente |
Adaptado de: Maron, Pellicia e Spirito, 1995.
A morte súbita cardíaca é a principal e mais temida complicação da cardiomiopatia hipertrófica. Acomete sobretudo os indivíduos adultos jovens, sendo a maior responsável pelas mortes súbitas em atletas (ver Figura 8). A taquicardia e a fibrilação ventricular são a causa de morte nessa doença.
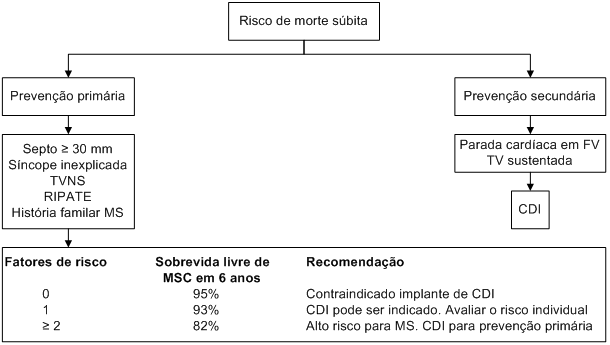
A identificação de indivíduos de risco elevado para morte súbita cardíaca, nos quais o implante de cardiodesfibrilador implantável (CDI) é recomendado, é um dos maiores desafios da cardiomiopatia hipertrófica. Em 2003, um consenso de especialistas definiu, com base em minucioso levantamento de dados publicados na literatura sobre o tema, uma série de fatores de risco que foram agrupados em duas categorias, de acordo com a sua importância (Tabela 6).
A presença de um ou mais fatores de risco maior identifica indivíduos com alto risco para morte súbita cardíaca. A categoria de fatores de risco possíveis contém os elementos coadjuvantes para a tomada de decisão terapêutica.
Uma importante observação a ser feita é que a Diretriz Brasileira de Dispositivo Eletrônico (Tabela 7), diferentemente das recomendações da ACC/AHA/ESC para Prevenção de Morte Súbita publicada em 2006, não considera como fator de risco maior para morte súbita cardíaca a resposta inadequada da pressão sistólica (PS) no teste ergométrico (definida como queda da PS ou elevação menor = 20 mmHg no pico do exercício).
Tabela 6: Fatores de risco de morte súbita cardíaca em portadores de cardiomiopatia hipertrófica
|
Fatores de risco maior |
Fatores de risco possíveis |
|
Prevenção secundária |
Fibrilação atrial |
|
Parada cardíaca (TV ou FV) |
Obstrução de via de saída |
|
Prevenção primária |
Mutação de alto risco |
|
TVS espontânea |
|
|
História familiar de MS (< 50 anos) |
|
|
Síncope inexplicada |
|
|
Espessura de parede = 30 mm |
|
|
TV não sustentada |
|
Tabela 7: Recomendações das Diretrizes Brasileiras de dispositivos cardíacos eletrônicos implantáveis para implante de cardiodesfibrilador implantável em pacientes com cardiomiopatia hipertrófica
|
Classe I |
Pacientes com cardiomiopatia hipertrófica que tenham apresentado TV/FV sustentada de causa não reversível e expectativa de vida de pelo menos 1 ano (NE B) |
|
Classe IIa |
Pacientes com cardiomiopatia hipertrófica que apresentem 1 ou mais fatores de risco maiores para morte súbita cardíaca (Tabela 6) e expectativa de vida de pelo menos 1 ano (NE C) |
|
Classe III |
Pacientes com cardiomiopatia hipertrófica sem fatores de risco (NE C) |
Classe I – Condições para as quais há evidências conclusivas ou, na sua falta, consenso geral de que o procedimento é seguro e útil/eficaz. Classe IIa – Condições para as quais há evidências conflitantes. Peso ou evidência/opinião a favor do procedimento. A maioria aprova. Classe III – Condições para as quais há evidências e/ou consenso de que o procedimento não é útil/eficaz e, em alguns casos, pode ser prejudicial. NE – nível de evidência: B – Dados obtidos a partir de meta-análise menos robusta, a partir de um único estudo randomizado ou de estudos não randomizados (observacionais). C – Dados obtidos de opiniões consensuais de especialistas.
Em julho de 2007, foram publicados os achados de um registro multicêntrico de 506 pacientes com cardiomiopatia hipertrófica submetidos a implante de cardiodesfibrilador implantável. Foi avaliada a importância dos seguintes fatores de risco para a ocorrência de terapias apropriadas:
história de morte súbita cardíaca prematura em 1 ou mais parentes de 1º grau com idade igual ou inferior a 50 anos;
hipertrofia ventricular expressiva (espessura de parede máxima >
pelo menos 1 episódio de TVNS com FC >120 bpm, ao Holter de 24 horas;
síncope inexplicada, afastada origem neurocardiogênica.
Observou-se incidência anual de 10,6% de intervenções apropriadas do cardiodesfibrilador implantável para prevenção secundária (probabilidade cumulativa em 5 anos de 39%) e 3,6% para prevenção primária (probabilidade cumulativa em 5 anos de 17%). A probabilidade de terapias apropriadas para prevenção primária foi similar nos pacientes com 1, 2, 3 ou mais fatores de risco, levando à conclusão de que a presença de um único marcador poderia justificar o implante de CDI (Figura 10).
Figura 10: Indicação do implante de CDI.
CDI: cardiodesfibrilador implantável; FV: fibrilação ventricular; TV: taquicardia ventricular; TVNS: taquicardia ventricular não sustentada; RIPATE: resposta da pressão arterial anormal no teste ergométrico; MS: morte súbita.
Adaptado de: Elliott et al., 2000.
Estudos prévios não conseguiram demonstrar o benefício da amiodarona na prevenção de morte súbita. A droga mostrou ser capaz de suprimir as arritmias ventriculares complexas, porém não protege contra a morte súbita cardíaca. Poderia ser indicada como alternativa ao cardiodesfibrilador implantável em pacientes com TV/FV prévia onde o implante de CDI não é factível (Classe IIa) ou em pacientes com algum dos fatores de risco maior e nos quais implante de CDI também não seria possível (Classe IIb).
Alguns outros achados na cardiomiopatia hipertrófica, ainda que não façam parte dos fatores de risco estabelecidos atualmente, são importantes na avaliação do paciente e devem ser considerados.
Vem ganhando espaço nos últimos anos à medida que avançam os conhecimentos genéticos da doença. Atualmente, descrições de mutações malignas fundamentam a aplicação da análise gênica na estratificação de risco de morte súbita. Se for construído o heredograma do paciente, observa-se a prevalência da doença dentro da família e o grau de evoluções fatais.
Assim como na cardiomiopatia isquêmica, as arritmias ventriculares observadas nos portadores de CMH provavelmente decorrem das múltiplas áreas de condução elétrica anormais causadas pela fibrose miocárdica, que podem causar morte súbita. Estudos de necrópsia em portadores de cardiomiopatia hipertrófica revelaram uma porcentagem mais elevada de fibrose nos indivíduos vitimados por morte súbita do que nos que morreram de causas não cardíacas. Em 2003, Moon et al. demonstraram a presença de realce tardio representando áreas de fibrose miocárdica pela RMC em 79% dos portadores de cardiomiopatia hipertrófica. A porcentagem de realce tardio era significativamente maior quando os pacientes apresentavam mais de 2 fatores de risco para morte súbita (15,7% vs. 8,6% p = 0,02). O mesmo ocorria em pacientes com doença progressiva, na forma de dilatação ventricular (28,5% vs. 8,7% p < 0,001). Dois padrões de fibrose são descritos: confluente (localizado e homogêneo) e difuso (focal e heterogêneo). Os portadores de cardiomiopatia hipertrófica com realce tardio difuso foram relacionados com porcentagem mais elevada de fibrose e pior prognóstico para morte súbita e dilatação do ventrículo esquerdo, ao contrário dos portadores com realce tardio confluente que apresentavam menor quantidade de fibrose.
Em torno 30% dos pacientes portadores de cardiomiopatia hipertrófica possuem gradiente = 30 mmHg na via de saída do ventrículo esquerdo. Embora sua presença esteja relacionada com progressão dos sintomas e insuficiência cardíaca, sua associação com morte súbita cardíaca ainda é controversa. Em 2003, Maron et al. analisaram retrospectivamente 1.101 pacientes seguidos durante 18 anos, sendo que 25% destes eram portadores da forma obstrutiva. Concluíram que o risco relativo anual de morte súbita cardíaca foi de 2,1, sendo este risco proporcionalmente maior conforme a magnitude do gradiente na via de saída do ventrículo esquerdo. A mortalidade anual foi baixa quando comparada com os pacientes não obstrutivos (0,6% por ano). Deste modo, a presença isolada do gradiente na via de saída do ventrículo esquerdo não indicaria o implante do CDI para profilaxia primária para morte súbita cardíaca, porém é um fator que pode conferir um risco maior de morte súbita cardíaca aos portadores de cardiomiopatia hipertrófica.
Além de estar relacionada com aumento da morbimortalidade cardiovascular devido a fenômenos tromboembólicos e progressão de insuficiência cardíaca, poderia estar relacionada com morte súbita cardíaca, já que funcionaria como “gatilho” para episódios de taquicardia ventricular sustentada (TVS).
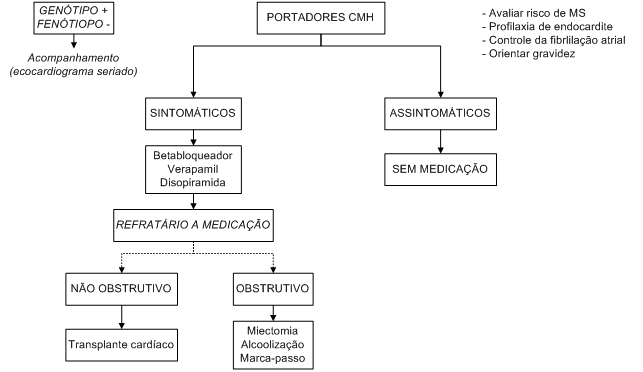
O tratamento da cardiomiopatia hipertrófica está reservado para pacientes sintomáticos e pode ser dividido em tratamento farmacológico e intervenções invasivas.
A maioria dos pacientes necessita apenas de terapia farmacológica, sendo que as intervenções invasivas são necessárias em apenas em
Os pacientes assintomáticos apresentam evolução clínica benigna e não devem receber nenhum tratamento (Tabela 8).
As principais drogas empregadas são os betabloqueadores e os bloqueadores dos canais de cálcio.
São as principais drogas no tratamento farmacológico da cardiomiopatia hipertrófica. Aliviam os sintomas em 2/3 dos pacientes e reduzem a obstrução da via de saída do ventrículo esquerdo durante o esforço físico, sendo, portanto, a droga de escolha nesses pacientes. Tem-se demonstrado uma boa droga para o alívio dos episódios sincopais decorrentes da disfunção autonômica.
Os efeitos benéficos são atribuídos à redução do inotropismo, do consumo de oxigênio miocárdico, à melhora do relaxamento e do enchimento ventricular cardíaco.
A dosagem deve ser aumentada gradualmente até que se consiga alívio dos sintomas ou uma frequência cardíaca em repouso entre 50 e 60 bpm. Os efeitos colaterais incluem fadiga, impotência, distúrbio do sono, predisposição a depressão e incompetência cronotrópica.
Constituem uma alternativa ao bloqueio beta-adrenérgico, sendo que não há consenso a respeito de qual droga deve ser primeiramente utilizada: betabloqueadores ou antagonistas do cálcio. Em pacientes que respondem inadequadamente a monoterapia, a associação das duas drogas pode ser tentada.
O verapamil é o antagonista do cálcio que tem sido mais amplamente utilizado e os efeitos mais importantes devem-se à melhora do enchimento diastólico, redução do assincronismo do relaxamento, melhora regional do fluxo coronariano e redução da contratilidade miocárdica. A experiência com outros agentes antagonistas do cálcio é limitada.
Efeitos colaterais incluem congestão pulmonar, edema, hipotensão e bloqueios atrioventriculares (quando associado com os betabloqueadores).
Indicado para pacientes com sintomas importantes de insuficiência cardíaca, refratários à medicação e que possuem gradiente de via de saída do ventrículo esquerdo. Esse importante subgrupo corresponde a aproximadamente 5% do total de pacientes com cardiomiopatia hipertrófica.
Para pacientes que não apresentam obstrução na via de saída do ventrículo esquerdo e em fases avançadas com disfunção sistólica, o transplante cardíaco seria a única opção.
Método terapêutico idealizado em 1982 por Sigwart, na Suíça, e realizado pela primeira vez em Londres 10 anos depois, em 3 pacientes com obstrução que não haviam melhorado com o tratamento clínico, nem com o implante de marca-passo.
Esse procedimento consiste na oclusão do ramo septal principal da artéria descendente anterior, pela injeção de até 5 mL de álcool absoluto por meio da técnica de cateterismo coronariano percutâneo, causando infarto da região septal. É realizado sob anestesia geral, com marca-passo temporário instalado. Antes da injeção do álcool, é feita a oclusão do ramo septal com cateter-balão e são realizadas medidas pressóricas e ecocardiografia para se ter segurança quanto à artéria a ser abordada. Após o procedimento, o paciente é encaminhado para a unidade de tratamento intensivo, como pós-infarto do miocárdio.
Os resultados demonstram redução da espessura do septo interventricular basal, aumento da área da via de saída do ventrículo esquerdo, diminuição do gradiente da via de saída do ventrículo esquerdo e diminuição do tamanho do átrio esquerdo.
As complicações são dor precordial, aumento de enzimas cardíacas, arritmias ventriculares complexas, bloqueio atrioventricular total precisando de marca-passo definitivo em 10%, e dissecção ou oclusão da artéria descendente anterior. A mortalidade em serviços com experiência é de 1 a 2%.
A nossa experiência é muito pequena, com pouquíssimos procedimentos realizados, porém seria uma alternativa à cirurgia, sobretudo em pacientes idosos com risco elevado para a cirurgia, pacientes previamente operados ou por livre escolha do paciente.
Foi o primeiro tipo de tratamento proposto para essa doença, sendo descrita por Morrow e Brockenbrough. Consiste na retirada de uma fatia de músculo com aproximadamente
Método consagrado, com mais de 30 anos de acompanhamento, determina diminuição ou até abolição do gradiente da via de saída do ventrículo esquerdo, assim como a normalização das pressões e diminuição do refluxo mitral, levando à melhora dos sintomas e da tolerância ao exercício.
A mortalidade operatória é baixa, chegando a 0% em centros especializados, e tem a vantagem de poder ser combinado com outros procedimentos cirúrgicos, como revascularização do miocárdio. Os resultados cirúrgicos a longo prazo são bons e mostram melhora dos sintomas e capacidade funcional em 70 a 90% dos pacientes. É o principal método invasivo indicado pela nossa equipe.
A presença do eletrodo na ponta do ventrículo direito determina uma mudança de ativação contrátil do miocárdio, que passa a ser de baixo para cima e da direita para esquerda, ocorrendo movimentação paradoxal do interventricular, que se afasta da parede posterior do ventrículo esquerdo durante a sístole, aumentando a câmara ventricular e reduzindo o gradiente VSVE.
Estudos não controlados iniciais demonstraram redução do gradiente de pressão na via de saída do VE por meio da estimulação AV, acompanhada de melhora sintomática, na maioria dos casos. Entretanto, estudos randomizados (duplo-cegos) e cruzados foram realizados, e todos incluíram um grupo controle e período de
Assim, a indicação de marca-passo definitivo AV na cardiomiopatia hipertrófica seria reservada apenas aos pacientes muito sintomáticos, refratários ao tratamento farmacológico, não candidatos à miectomia septal cirúrgica ou à ablação septal percutânea – Classe IIb.
Tabela 8: Comparação entre as estratégias invasivas
|
Terapia |
Mortalidade |
Efetividade |
Seguimento em anos |
Complicações tipo |
% pacientes |
|
Miectomia |
< 2 a 3% |
> 90% |
> 30 |
BAVT Defeito de septo Insuficiência de válvula aórtica |
< 3 < 1 < 1 |
|
Alcoolização |
< 2 a 3% |
70 a 80% |
< 5 |
BAVT Defeito de septo IAM extenso |
10 a 40 Desconhecido Desconhecido |
|
Marca-passo |
< 1 |
10 a 40% |
10 |
Infecção ou perfuração |
< 2 |
Adaptado de: Nishimura et al., 2004.
Figura 11: Acompanhamento dos portadores de cardiomiopatia hipertrófica.
Todo indivíduo com história familiar para cardiomiopatia hipertrófica deve ser acompanhado de perto quanto ao risco do desenvolvimento da doença. Para parentes de 1º grau, o ecocardiograma deve ser realizado anualmente dos 12 aos 20 anos de idade; antes deste período, apenas em pacientes que cursam com sintomas ou que desejam participar de competições esportivas de alta intensidade. Após os 20 anos de idade, reavaliações trianuais com ecocardiograma seriam racionais.
O prognóstico adverso materno-fetal, com risco de prematuridade e mortalidade materna aumentada, de portadoras de cardiomiopatia hipertrófica e o alto potencial de transmissão genética da doença induzem a reflexão sobre a restrição da gravidez nesse grupo de pacientes.
A prática de atividades físicas competitivas de intensidade moderada e alta deve ser contraindicada para todos os indivíduos portadores de cardiomiopatia hipertrófica devido ao risco de morte súbita cardíaca durante tais atividades. As atividades de baixa intensidade, com consumo máximo de O2 < 40%, podem ser liberadas (pela 26ª Conferência de Bethesda: boliche, golfe, bilhar, tiro ao alvo).
1. Almeida DR, Viégas RFM, Carvalho A, et al.Tratamento clínico da cardiomiopatia hipertrófica. Socesp. 2000;10(4):470-9.
2. Arteaga E, Ianni BM, Fernandes F, Mady C. Benign outcome in a long-term follow-up of patients with hypertrophic cardiomyopathy in Brazil. Am Heart J. 2005;1949:1099-105.
3. Braunwald E, Zipes DP, Libby P, Bonow RO. Tratado de doenças cardiovasculares. 7.ed. Elsevier; 2006.
4. Elliott PM, et al. J Am Coll Cardiol. 2000;36:2212.
5. Maron BJ, et al. Circulation 1996; 94:850-6.
6. Maron BJ, McKenna WJ, Danielson G, Kappenberger LJ, Kuhn HJ, Seidman CE, et al. American College of Cardiology/European Society of Cardiology clinical expert consensus document on hypertrophic cardiomyopathy. Eur Heart J. 2003;24:1965-90.
7. Maron BJ, Pellicia A, Spirito P. Circulation. 1995;91:1596.
8. Maron BJ, Spirito P, Shen WK, et al. Implantable cardioverter-defibrillators and prevention of sudden cardiac death in hypertrophic cardiomyopathy. JAMA. 2007;298:405-12.
9. Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA. 2002;287:1308-20.
10. Martinelli Filho M, Zimerman LI, Lorga AM, Vasconcelos JTM, Rassi Jr. A. Guidelines for implantable electronic cardiac devices of the Brazilian Society of Cardiology. Arq Bras Cardiol. 2007;89(6):e210-e238.
11. Mattos e Piva B, Torres M, Freitas V. Avaliação diagnóstica da cardiomiopatia hipertrófica em fase clínica e pré-clínica. Arq Bras Cardiol. 2008;91(1):55-62.
12. Miller MA, Gomes JA, Fuster V. Risk stratification of sudden cardiac death in hypertrophic cardiomyopathy. Nature. 2007;4(12):667-76.
13. Nishimura RA, et al. N Engl M Med. 2004;350:1320.
14. Nishimura RA, Holmes Jr. DR. Hypertrophic obstructive cardiomyopathy. N Engl M Med. 2004;350:1320-7.
15. Nobre F, Serrano Jr. CV. Tratado de cardiologia da SOCESP. Barueri: Manole; 2005.
16. Shiozaki AA, Kim RJ, Parga JR, Tassi EM, Arteaga E, Rochitte CE. Ressonância magnética cardiovascular na cardiomiopatia hipertrófica. Arq Bras Cardiol. 2007;88:243-8.
17. Tirone A, Arteaga E, Pereira Barreto AC, Krieger JE, Buck PC, Ianni BM, et al. Pesquisa de marcadores para os genes da cadeia pesada da ß miosina cardíaca e da proteína C de ligação à miosina em familiares de pacientes com cardiomiopatia hipertrófica. Arq Bras Cardiol. 2005;84:467-72.
18. Zipes DP, et al. ACC/AHA/ESC Guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death — Executive summary. Circulation. 2006;114:1088-132.
"Excelente revisão!!!!"
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.