(Carregando Índice)... (Carregando Índice)... |
Autores:
Patrícia Martin
Especialista em Reumatologia pelo Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo (HC-FMUSP).
Cláudia Tereza Lobato Borges
Especialista em Reumatologia pelo Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo (HC-FMUSP).
Última revisão: 25/11/2010
Comentários de assinantes: 0
Constituem um grupo heterogêneo de doenças caracterizadas por fraqueza muscular proximal e inflamação dos músculos esqueléticos.
São doenças raras e estima-se que a incidência varie de
Para o diagnóstico de polimiosite e dermatomiosite são utilizados os seguintes critérios: fraqueza muscular proximal, aumento de enzimas musculares, alterações miopáticas na eletroneuromiografia e evidência histológica de inflamação muscular. A presença de lesões cutâneas características permite o diagnóstico de dermatomiosite (Tabela 1). Classicamente as poli/dermatomiosites são subdivididas em 5 grupos: dermatomiosite; polimiosite; dermatomiosite e polimiosite juvenil; miosites associadas a neoplasias e miosites associadas a doenças do colágeno como lúpus eritematoso sistêmico e esclerose sistêmica.
A miosite por corpúsculo de inclusão foi considerada como doença inflamatória, porém muitos investigadores atualmente a consideram primariamente degenerativa, apresentando secundariamente alterações inflamatórias.
Tabela 1: Critérios diagnósticos para dermatomiosite e polimiosite
|
Critério |
Definição |
|
Fraqueza muscular proximal |
|
|
Aumento de enzimas musculares |
CPK, podendo ocorrer elevação de aldolase, TGO, TGP e DLH |
|
Evidência na eletroneuromiografia |
Tríade característica: 1) aumento da atividade insercional, fibrilações e ondas pontiagudas positivas; 2) descargas espontâneas e bizarras de alta frequência; 3) unidades motoras polifásicas de baixa amplitude e curta duração |
|
Evidência histológica |
Necrose das miofibrilas, fagocitose, núcleo grande com nucléolo proeminente, atrofia perifascicular, variação no tamanho das fibras e infiltrado inflamatório |
|
Achados dermatológicos |
Heliótropo (pálpebras violáceas), edema periorbital, sinal e pápulas de Gotron (eritema nas superfícies extensoras dos cotovelos e joelhos; pápulas eritematosas nas articulações interfalângicas proximais e metacarpofalângicas) |
|
Diagnóstico definitivo: 3 de 4 critérios + quadro cutâneo característico (dermatomiosite) ou presença dos 4 critérios (polimiosite); sensibilidade de 75% Diagnóstico provável: 2 de 3 critérios + quadro cutâneo (dermatomiosite) ou presença de 3 critérios (polimiosite); sensibilidade de 20% | |
A etiologia é desconhecida, mas não há dúvidas de que, em indivíduos geneticamente predispostos, a doença seja imunologicamente mediada. A presença de auto-anticorpos séricos, bem como de infiltrado de células T nos tecidos afetados, mostra que há alteração tanto na imunidade humoral quanto na imunidade celular, resultando em lesão muscular e de outros órgãos.
Nas Tabelas 2, 3 e 4 estão resumidos os principais sintomas e sinais da dermato/polimiosite.
Tabela 2: História clínica
|
Início insidioso ( |
|
Sintomas sistêmicos: fadiga, emagrecimento, febre (mais comum em crianças) |
|
Artralgia simétrica, de pequenas articulações |
|
Disfagia |
|
Dispnéia |
|
Tosse |
Tabela 3: Exame físico na polimiosite
|
Dificuldade para levantar da cadeira na sala de espera: apoio na cadeira ou ajuda de familiares |
|
Mãos de mecânico: fissuras e hiperqueratose na pele da região lateral das mãos e dedos |
|
Poliatrite simétrica, em geral leve; pode cursar com deformidades e instabilidade do polegar na síndrome anti-sintetase |
|
Fenômeno de Raynaud: presente na síndrome anti-sintetase |
|
Fraqueza muscular: em geral proximal e simétrica Incapacidade de levantar a cabeça do traveseiro Pode ser assimétrica e distal na miosite por corpúsculo de inclusão Não atinge músculos oculares ou da face |
Tabela 4: Exame físico na dermatomiosite
|
Achados da polimiosite |
|
Edema de pálpebras ou na face (Figura 1); mais comum em crianças |
|
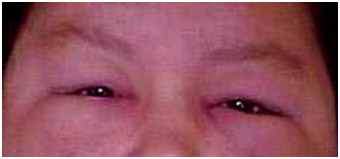
Heliótropo: manchas violáceas nas pálpebras (Figura 2) |
|
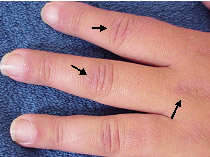
Pápulas de Gottron: pápulas rosadas, eritematosas ou violáceas nas proeminências ósseas: articulações metacarpofalângicas e interfalângicas proximais e distais das mãos ( |
|
Sinal de Gottron: pápulas e/ou placas rosadas, eritematosas ou violáceas na superfície extensora dos cotovelos, joelhos e tornozelos (50% ou menos dos pacientes) |
|
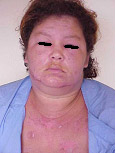
Eritema em face, acometendo região malar, área nasolabial e testa (em pacientes com lúpus, estas áreas são poupadas) (Figura 1) |
|
Fotossensibilidade |
|
Sinal do V: região eritematosa na área do decote |
|
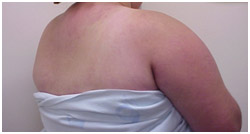
Sinal do xale: eritema no colo, braços e dorso, lembrando um xale (Figura 4) |
|
Hipertrofia de cutículas e eritema periungueal |
|
Calcinose: calcificações subcutâneas, na fáscia ou intramusculares, em áreas sujeitas a traumas |
Figura 1: Edema e eritema na face; manchas em região anterior do tórax. Observe que o sulco nasolabial é poupado.

Figura 2: Heliótropo: coloração lilás nas pálpebras.
Figura 3: Pápulas de Gottron. Pápulas violáceas nas superfícies extensoras das interfalângicas proximais e metacarpofalâgicas.
Figura 4: Sinal do xale. Manchas e placas róseas distribuídas em dorso e braços. Observe também que as lesões se localizam em áreas foto expostas.
Alguns aspectos de história e exame clínico merecem ser ressaltados, uma vez que permitem o diagnóstico diferencial com outras enfermidades, bem como a presença de doenças associadas. Além disso, têm importância prognóstica e terapêutica, conforme descrito a seguir.
A idade do paciente é um dado fundamental, uma vez que doença de início após 50 anos de idade sugere miosite por corpúsculo de inclusão, associação com neoplasia, além de ser um fator de mau prognóstico. Deve-se questionar sobre uso de drogas lícitas e ilícitas (Tabela 5) que podem causar miopatias. Finalmente devem ser pesquisados os fatores de mau prognóstico, pois estes definirão a conduta terapêutica inicial (Tabela 6). Quando há história familiar positiva associada a curso muito lento, mialgia após esforços e hipertrofia de panturrilhas, deve-se pensar em distrofia muscular. Na revisão de sistemas, o paciente deve ser interrogado sobre sintomas sugestivos de neoplasias associadas.
No exame físico, devem ser pesquisados sinais de acometimento cutâneo, articular, cardíaco (taquicardia) e pulmonar. Quando houver áreas de vasculite cutânea, estas devem ser cuidadosamente inspecionadas a fim de excluir infecção secundária. Deve ser realizado exame neurológico completo, uma vez que diminuição de força muscular distal sugere miopatia por corpúsculos de inclusão e acometimento de musculatura ocular e facial excluem o diagnóstico de dermato/polimiosite.
Tabela 5: Drogas que podem causar miopatia
|
Drogas hipolipemiantes: estatinas, fibratos, ácido nicotínico |
|
Corticóides |
|
Zidovudina (AZT) |
|
Cloroquina |
|
Colchicina |
|
D-penicilamina |
|
Interferon-alfa |
|
Antipsicóticos (na ausência de síndrome neuroléptica maligna): clozapina, risperidona, olanzapina, haloperidol |
|
Drogas em que há relatos de casos: procainamida, amiodarona, valproato, fenitoína, ciclosporina (especialmente se associada a colchicina), labetalol, derivados da vitamina A (etretinato, isotretinoína), quinolonas, esomeprazol, micofenolato mofetil |
|
Ecstasy, cocaína ou heroína podem provocar rabdomiólise |
|
Álcool |
Tabela 6: Fatores de mau prognóstico
|
Idade superior a 60 anos |
|
Intervalo entre início dos sintomas e início do tratamento maior que 8 meses |
|
Queixa pulmonar |
|
Disfagia grave |
|
História pessoal ou familiar de neoplasia |
|
Paciente na cadeira de rodas ou acamado |
|
Fraqueza proximal e distal |
O hemograma pode mostrar anemia de doença crônica. Entretanto, sobretudo na dermatomiosite, as anemias devem ser cuidadosamente investigadas, pois podem ser uma pista de neoplasia oculta.
A velocidade de hemossedimentação (VHS) e a proteína C reativa não são bons indicadores de atividade de doença, embora possam estar levemente elevados.
A CK sérica é a enzima muscular mais sensível e específica, útil tanto no diagnóstico quanto na monitoração do tratamento, uma vez que diminui antes da melhora clínica e, na reativação da doença, seu aumento precede a fraqueza muscular. Em quadros muito avançados, quando já houve consumo da massa muscular, a CK pode ser normal, o que indica mau prognóstico.
Em geral, atinge até 50 vezes o limite superior; incrementos superiores a 100 vezes colocam o diagnóstico em questão. Na miosite por corpúsculo de inclusão, o aumento de CK é mais discreto e, nas distrofias musculares e miopatias metabólicas, esta enzima também pode estar elevada.
Em ordem decrescente de especificidade, TGO, aldolase, TGP e desidrogenase lática também indicam lesão muscular.
Autoanticorpos (anti Jo-1, anti-Mi2, anti-SRP, anti-PL7, PL12, EJ, OJ, KS) estão presentes em
Devem ser solicitados para diagnóstico diferencial com miopatias infecciosas (sorologia para toxoplasmose, HIV) e metabólicas (TSH, PTH intacto quando cálcio sérico anormal).
Particularmente útil, mostrando padrão miopático característico (ver Tabela 1). Também permite o diagnóstico diferencial com doenças neurológicas como síndrome de Guillain-Barré, esclerose lateral amiotrófica e miastenia grave. Deve-se poupar um membro para a realização da biópsia muscular.
O papel da ressonância magnética ainda não está definido.
Considerada o padrão-ouro no diagnóstico da polimiosite, deve ser realizada precocemente em todos os pacientes com diagnóstico sugestivo. Na dermatomiosite não é necessária, pois as lesões cutâneas são características.
A alteração histológica mais comum é a degeneração e regeneração de miofibrilas refletindo-se por variação em seu tamanho, porém não é específica. Infiltrado inflamatório crônico perivascular e ao redor das miofibrilas, invasão linfocítica de miofibrilas não-necróticas, fibras fantasmas e núcleos periféricos também podem ser observados. Na doença crônica, tecido conectivo fibroso ou gordura substituem as fibras.
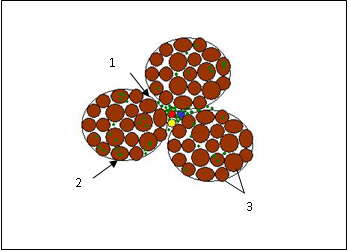
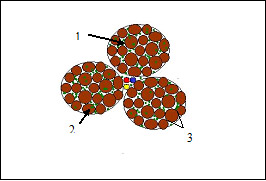
São observadas importantes diferenças histológicas entre a dermatomiosite (infiltrado inflamatório perivascular) e a polimiosite (infiltrado inflamatório perifascicular) (Figuras 5 e 6). Na miosite por corpúsculo de inclusão, são encontradas inclusões circulares avermelhadas.
Apesar dos achados característicos, alguns pacientes podem apresentar biópsia muscular normal, por várias razões: o infiltrado inflamatório é focal, tratamento prévio, escolha do músculo errado (para evitar, deve-se biopsiar um músculo fraco do lado oposto àquele em que houve alterações na eletroneuromiografia).
Figura 5: Dermatomiosite. Observar: 1– infiltrado linfocítico perivascular; 2 – invasão linfocítica das miofibrilas; 3 – variação no tamanho das fibras.
Figura 6: Polimiosite. Observar: 1 – infiltrado linfocítico perifascicular; 2 – invasão linfocítica das miofibrilas; 3 – variação no tamanho das fibras.
Uma vez que pode ocorrer envolvimento pulmonar, inicialmente assintomático, e que o diagnóstico precoce altera o curso da doença, tomografia de tórax de cortes finos e prova de função pulmonar completa devem ser realizadas anualmente. Também sugere-se realização de eletrocardiograma e ecocardiograma uma vez por ano.
A distribuição das neoplasias associadas a dermato/polimiosite é similar ao esperado para idade na população geral. Além disso, há maior incidência de câncer de ovário, pulmões e trato gastrintestinal na dermatomiosite. Já em pacientes com polimiosite, linfoma de Hodking, câncer de pulmão e bexiga são mais frequentes.
Sugerimos a seguinte rotina:
1. Investigação de acordo com sinais e sintomas de neoplasia.
2. Pacientes sem achados clínicos sugestivos: investigação para neoplasia oculta de acordo com o esperado para idade.
3. Pacientes com alto risco de neoplasia associada (Tabela 7): ultra-sonografia transvaginal (preferenciamente com Doppler), mamografia, pesquisa de sangue oculto nas fezes, CA-125 e tomografia computadorizada de tórax e abdome.
4. Uma vez que o surgimento de neoplasias pode suceder o diagnóstico de dermatomiosite/polimiosite em até 60 meses, recomenda-se rastreamento anual nos primeiros 5 anos da doença.
Tabela 7: Pacientes de alto risco para malignidade
|
Dermatomiosite em mulheres > 60 anos de idade |
|
Emagrecimento importante e/ou albumina baixa |
|
História pessoal ou familiar de neoplasia |
|
Anti Jo-1 negativo |
Os principais diagnósticos diferenciais podem ser observados na Tabela 8. Em alguns casos, a história e o exame físico direcionam para a possível causa, confirmada por exames complementares simples. Muitas vezes, entretanto, apenas a biópsia com técnicas e colorações especiais define o diagnóstico.
Tabela 8: Diagnóstico diferencial
|
Toxoplasmose |
|
Endocrinopatias: síndrome de Cushing, hipo e hipertireoidismo, hiperparatireoidismo |
|
Miosite por corpúsculo de inclusão |
|
Distrofias musculares |
|
Miopatias metabólicas: doenças de depósitos de lipídios ou glicogênio, miopatias mitocondriais |
|
Miopatias induzidas por drogas |
|
Doenças de neurônio motor |
|
Miastenia grave |
Uma vez que dermatomiosite e polimiosite são doenças incomuns, existem poucos estudos controlados e randomizados, a maioria deles com poucos pacientes avaliados, de forma que ainda não está definida qual a melhor terapia.
Propomos tratamento descrito a seguir.
Prednisona 1 mg/kg/dia durante
após 6 semanas, se CK normal: diminuir 10 mg/mês até 20 mg;
a partir de 20 mg, diminuir 2,5 mg/mês;
observar que o tempo de corticoterapia é longo.
Pacientes inicialmente graves (disfagia com risco de broncoaspiração, doença pulmonar, insuficiência respiratória e acamados) devem receber pulsoterapia com metilprednisolona 1 g/dia durante 3 dias.
A dermatomiosite pode cursar com vasculite difusa associada. Se ocorrer infecção secundária em um doente inicialmente grave, pode ser administrada gamaglobulina endovenosa, na dose de 1 g/kg/dia durante 3 dias, para evitar a imunossupressão causada por pulsoterapia com corticóide.
O uso de imunossupressores está indicado desde o início do tratamento em pacientes com fatores de mau prognóstico (ver Tabela 6), quando não há resposta à corticoterapia e quando ocorre recidiva na retirada da prednisona.
1. Metotrexato: pode ser usado nas polimiosites e na presença de anti-Jo-1. Inicia-se na dose de 10 mg/semana; podendo-se aumentar até 50 mg/semana (doses acima de 25 mg/semana são administradas via intramuscular). Deve-se associar ácido fólico 1 mg/dia e monitorar enzimas hepáticas e hemograma.
2. Azatioprina: pode ser utilizada na dermatomiosite, na dose de 3 mg/kg. Também devem ser monitoradas enzimas hepáticas e hemograma.
3. Associação de azatioprina com metotrexato está indicada na falha ou recidiva quando usada apenas uma das drogas.
4. Ciclosporina: em casos resistentes a metotrexato e azatioprina, a ciclosporina pode substituir ou ser associada (dose máxima 2,5 mg/kg/dia). A pressão arterial e creatinina devem ser cuidadosamente monitoradas.
5. Ciclofosfamida: quando ocorre fibrose pulmonar grave sem resposta ao metotrexato.
Ainda há relatos de uso de rituximabe em casos refratários a outras formas de tratamento.
Devido ao tratamento prolongado e com altas doses de corticóides, todos os pacientes devem receber cálcio e vitamina D, mesmo aqueles com calcinose.
Em relação à calcinose, também não existem estudos controlados indicando o melhor tratamento, mas o controle rigoroso da doença previne o surgimento desta complicação. Na Tabela 9 estão listadas algumas opções terapêuticas, porém nenhuma delas evita ou elimina a calcinose.
Tabela 9: Opções de tratamento para calcinose
|
Varfarina 1 mg/dia |
|
Colchicina 1 mg/dia |
|
Probenicida 250 a 2.000 mg/dia |
|
Alendronato de sódio 10 mg/dia |
|
Dilitazem 240 mg/dia |
|
Excisão cirúrgica |
|
Hidróxido de alumínio (em média 2 g/dia) |
|
Minociclina 50 a 100 mg/dia |
|
Salicilatos 80 mg/kg/dia |
|
Esteróides intralesionais |
|
Anti-TNF |
As miopatias inflamatórias são doenças raras, mas de grande morbidade.
Fraqueza muscular proximal simétrica e lesões cutâneas em áreas fotoexpostas sugerem o diagnóstico.
Os principais critérios diagnósticos da polimiosite e dermatomiosite são fraqueza muscular proximal, aumento de enzimas musculares, alterações miopáticas na eletroneuromiografia, evidência histológica de inflamação muscular e, no caso da dermatomiosite, a presença de lesões cutâneas características.
Pápulas de Gottron e heliótropo são os achados mais típicos da dermatomiosite.
Doença de início após 50 anos sugere miosite por corpúsculo de inclusão ou associada à neoplasia, além de ser um fator de mau prognóstico.
Muitas drogas lícitas e ilícitas podem causar miopatia.
Ao exame neurológico, diminuição de força muscular distal sugere miopatia por corpúsculos de inclusão, e acometimento de musculatura ocular e facial exclui o diagnóstico de dermato/polimiosite.
A CK sérica é a enzima muscular mais sensível e específica, útil tanto no diagnóstico quanto na monitoração do tratamento. Diminui antes da melhora clínica e, na reativação da doença, seu aumento precede a fraqueza muscular.
A eletroneuromiografia e a biópsia muscular são exames particularmente úteis para o diagnóstico da polimiosite.
Na ausência de lesões cutâneas características a biópsia muscular é obrigatória.
Eletroneuromiografia com padrão miopático característico sugere o diagnóstico, mas não é específica.
Tomografia de tórax, prova de função pulmonar, ecocardiograma e eletrocardiograma devem ser realizados anualmente para rastrear acometimento precoce pulmonar e cardíaco.
Neoplasia oculta deve ser investigada nos doentes com dermato/polimiosite de acordo com os sinais e sintomas apresentados e conforme a rotina de investigação de neoplasia habitual para a idade.
Mulheres maiores de 60 anos com dermatomiosite, história pessoal ou familiar de neoplasia, emagrecimento importante e/ou albumina baixa, anti-Jo-1 negativos devem ser investigadas extensamente no diagnóstico e a cada 5 anos, com ultra-sonografia transvaginal, mamografia, pesquisa de sangue oculto nas fezes, CA-125 e TC de tórax e abdome.
A base do tratamento da dermato/polimiosite é a prednisona em altas doses, que deve ser instituída por tempo prolongado.
Pacientes com fatores de mau prognóstico (Tabela 6), sem resposta à corticoterapia ou com recidiva na retirada da prednisona devem receber imunossupressores.
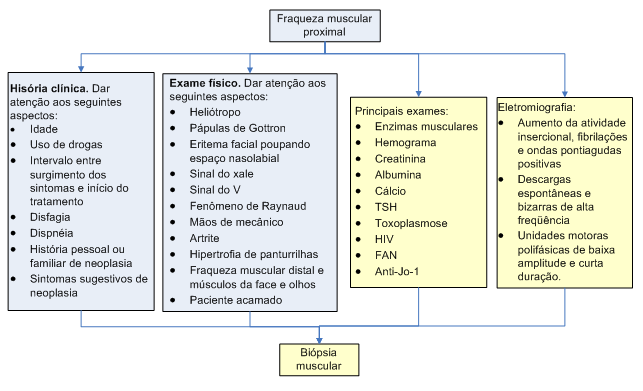
Algoritmo 1: Abordagem diagnóstica da dermato/polimiosite.
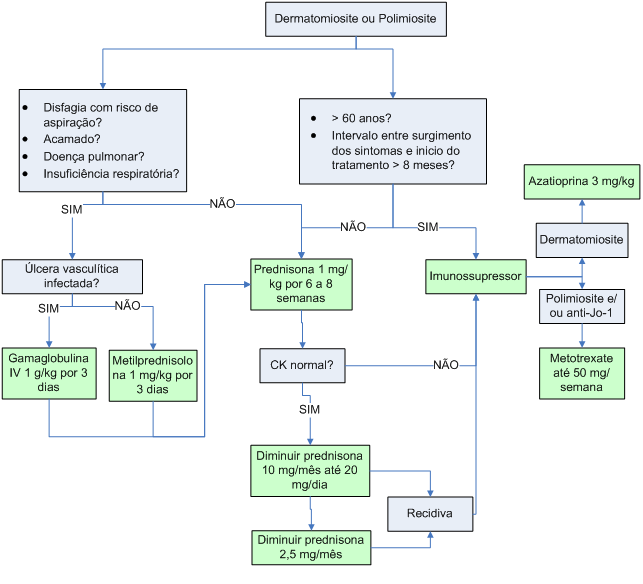
Algoritmo 2:
1. Amoura Z, Duhaut P, Huong Du LT, et al. Tumor antigen markers for the detection of solid cancers in inflamatory miopathies. Cancer Epidemiol Biomarkers Prev 2005; 14:1279-1282.
2. Askansas V, Engel WK. Proposed pathogenic cascade of inclusion body myositis: importance of amyloid-beta, misfolded proteins, predisposing genes and aging. Curr Opin Rheumatol 2003; 15:737-44.
3. Bohan A, Peter JB. Polimyositis and dermatomyositis: first of two parts. N Engl J Med 1995; 292:344-47.
4. Boulman NB, Slobodin G, Rozenbaum M, Rosner I. Calcinosis in rheumatic diseases. Sem Arthritis Rheuma 2005; 34:805-812.
5. Levine SM. Cancer and myositis: new insigths into an old association. Curr Opin Rheumatol 2006; 18:620-624.
6. Medsger TA, Oddis CV. Inflammatory muscle disease: clinical features. In: Rheumatology. 3. ed. Mosby, p.1537-1554.
7. Nirmalanthan N, Holton JL, Hanna Micahel G. Is it really myositis? A consideration of the differential diagnosis. Curr Opin Rheumatol 2004; 16:684-691.
8. Yazici Y, Kagen LJ. The association of malignancy with myositis. Curr Opin Rheumatol 2000; 12:498-500.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.