(Carregando Índice)... (Carregando Índice)... |
Autor:
Karina Gatz Capobianco
Médica reumatologista do Hospital Moinhos de Vento de Porto Alegre. Mestre em Clínica Médica pela PUCRS. Doutora em Clínica
Médica pela UFRGS.
Última revisão: 02/04/2014
Comentários de assinantes: 0
Uma paciente do sexo feminino, 40 anos, branca, comparece à consulta relatando que, há cerca de dois anos, nota alteração na cor das mãos, nos meses de inverno – inicialmente as mãos ficam brancas quando em contato com o frio e, à medida que o tempo de exposição aumenta, a cor se altera para roxa. Além disso, nota vermelhidão nas mãos quando aquecidas e sudorese excessiva nas palmas das mãos e plantas dos pés, independente da estação do ano. Relata também artralgias em mãos, punhos e pés, sem sinais inflamatórios locais, além de edema difuso dos dedos das mãos há seis meses. A paciente afirma apresentar dificuldade para deglutir alimentos sólidos, como carne, e/ou secos, como pão, progressivamente, neste mesmo período. Não relata história familiar de fenômeno de Raynaud e/ou de doença reumática. Não é tabagista e também não utiliza qualquer medicação de forma contínua.
Ao realizar exame, verifica-se que a paciente está em bom estado geral. Observa-se teleangiectasias em face, língua e palmas das mãos. Há fenômeno de Raynaud, fase cianótica, em mãos e pés; edema difuso dos dedos das mãos com esclerodactilia limitada às articulações metacarpofalangianas bilateral e simetricamente. Não há artrite e nem evidência de miopatia. Sua pressão arterial é de 110/70 mmHg, sua frequência cardíaca, de 70 bpm, e sua frequência respiratória, de 16 rpm. Não são verificadas alterações na ausculta cardíaca e pulmonar. Observa-se abdome indolor à palpação, sem visceromegalias. Os exames laboratoriais estão normais, exceto por velocidade sedimentação globular (VSG) de 60 mm/h, proteína C-reativa ultrassensível de 4,5 mg/dL, fator antinuclear (FAN) reagente, título superior a 1:640, padrão centromérico à imunofluorescência indireta.
A esclerose sistêmica (ES) é uma doença autoimune, multissistêmica, caracterizada por vasculopatia fibroproliferativa proeminente, inflamação e progressiva fibrose da pele (esclerodermia) e/ou de órgãos internos, como trato gastrintestinal, pulmão, coração e rim.1
Nos Estados Unidos (EUA), a incidência da ES varia entre 9 a 19/1.000.000 habitantes por ano, e a prevalência é de 286/1.000.000 habitantes.
A doença é predominante em indivíduos do sexo feminino (3 a 4:1), geralmente apresentando início entre 30 e 50 anos, sendo muito rara em crianças e em idosos com mais de 70 anos.1,2 Na infância, formas localizadas de ES são mais comuns, como a forma linear e a morfeia.2
Cerca de 1,6% dos pacientes com ES têm um familiar de primeiro grau com a mesma doença, o que evidencia claramente a influência do fator genético na patogenia dadoença.1-3 Fatores ambientais também podem estar envolvidos na etiologia da ES, assim como agentes infecciosos (p. ex., retrovírus, citomegalovírus); agentes químicos, como solventes orgânicos (p. ex., tolueno, benzeno e tricloroetano), cloreto de vinil, pesticidas, L-triptofano,resinas epóxi, fármacos (bleomicina, melfalan, docetaxel, pentazocina, cocaína, anorexígenos derivados da flenfuramina).3,4
Os principais fatores envolvidos na patogênese da ES estão bem definidos: disfunção vascular, ativação imunológica/ inflamação e fibrose.2,3,5,6
Ao contrário de outras doenças que se desenvolvem com fibrose, autoimunidade e vasculopatia precedem a ocorrência de fibrose tecidual em casos de ES.2,7 A liberação de mediadores solúveis a partir do dano das células do endotélio vascular desencadeia diversos eventos que resultam em diferenciação dos fibroblastos em células contráteis que secretam colágeno, os miofibroblastos. Ocorre, então, a substituição de células da musculatura lisa e estruturas epiteliais por matriz extracelular rica em colágeno, havendo consequente disfunção dos órgãos afetados.2,5-7
O evento mais precoce na patogênese da ES é a microangiopatia, embora ainda não se saiba qual é o mecanismo exato responsável por gerar lesão vascular. Sabe-se que atividade imunológica ocorre juntamente com a vasculopatia. Os anticorpos específicos, como anticorpos anticentrômero (ACA) e antitopoisomerase 1 (Anti-Scl70), estão associados à ES, e recentes estudos observaram que receptores específicos para autoanticorpos, como o fator de crescimento derivado de plaquetas (FCDP) nos fibroblastos e nas células endoteliais, contribuem para a ocorrência de dano tecidual.3
Os danos à microcirculação geram remodelação vascular, causando uma progressiva redução do diâmetro e obliteração da luz vascular. Em casos de ES, a patogenia da lesão vascular é mediada pela liberação de substâncias vasodilatadoras, como as prostaciclinas (PCs) e o óxido nítrico (ON), e vasoconstritoras, como as endotelinas 1 e 2 (ETs), a partir do dano às células endoteliais. A redução da produção de PCs e ON, junto com o aumento da liberação das ETs, afeta o tônus vascular e causa remodelação celular, hipertrofia vascular e inflamação. As ETs também parecem ter um papel na ativação dos fibroblastos, o que contribui para a ocorrência de fibrose tecidual por meio de estímulo à síntese de colágeno e inibição das colagenases.3,6
A patogênese da ES, portanto, consiste em microangiopatia obliterativa sistêmica, causando comprometimento da função de vários órgãos, que se traduz clinicamente como ulcerações digitais isquêmicas, hipertensão arterial pulmonar, crise renal esclerodérmica, miocardiopatia, entre outras manifestações.2,3,6,7
O dano vascular inicial que ocorre em indivíduos geneticamente suscetíveis desencadeia alterações funcionais e estruturais vasculares, inflamação e geração de autoimunidade.
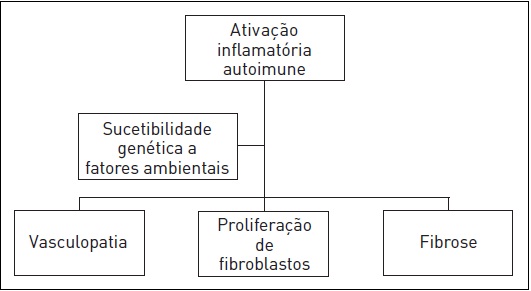
As respostas inflamatória e imune iniciam e causam ativação sustentada dos fibroblastos e diferenciação, resultando em fibrogênese patológica e dano tecidual irreversível (Fig. 120.1). 2,3,5-8
As manifestações clínicas da ES são o resultado de três processos fisiopatogênicos principais: vasculopatia, autoimunidade e fibrose.5,6
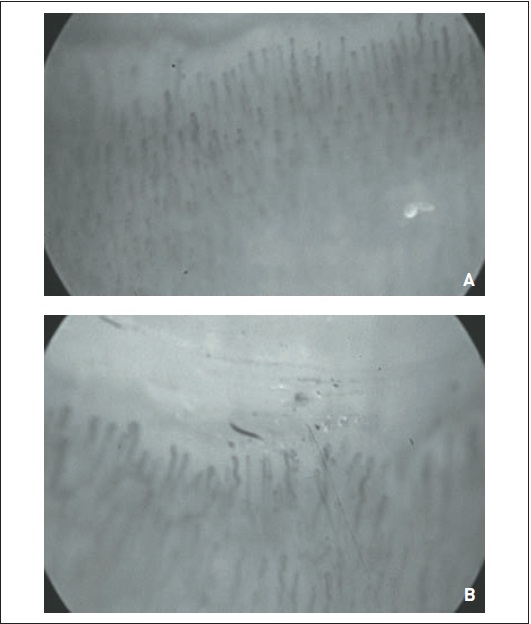
A vasculopatia de pequenos vasos é um evento precoce na patogenia da doença, podendo preceder em anos as demais manifestações clínicas decorrentes de fibrose.3,4 A microangiopatia esclerodérmica pode ser identificada precocemente por meio de capilaroscopia periungueal (CPU). Essa técnica possibilita o acesso à microcirculação in vivo pela observação dos capilares da fileira distal periungueal (Fig. 120.2) com lupa estereoscópica, fonte de luz externa fria (halógena) e meio diafanizador sobre a superfície periungueal.8,9
A identificação precoce de alterações funcionais e morfológicas da microcirculação permite também avaliar a evolução e o prognóstico da doença.8,10
A CPU é o melhor preditor da vasculopatia esclerodérmica que se manifesta clinicamente por meio do fenômeno de Raynaud (FRy) secundário. Nesses casos, há alteração na disposição normal de capilares na fileira distal da região periungueal, atipias capilares morfológicas patológicas, como ectasias, e megacapilares (capilares aneurismáticos, cerca de 10 vezes o diâmetro normal). Ocorrem perda de capilares, formando áreas de desvascularização ou deleção, micro-hemorragias e alteração do fluxo sanguíneo capilar, que se torna lento e com áreas de estase total. Essas alterações caracterizam o padrão SD ou padrão esclerodérmico (Figs. 120. 3, 120.4, 120.5 e 120.6).8,9,12.
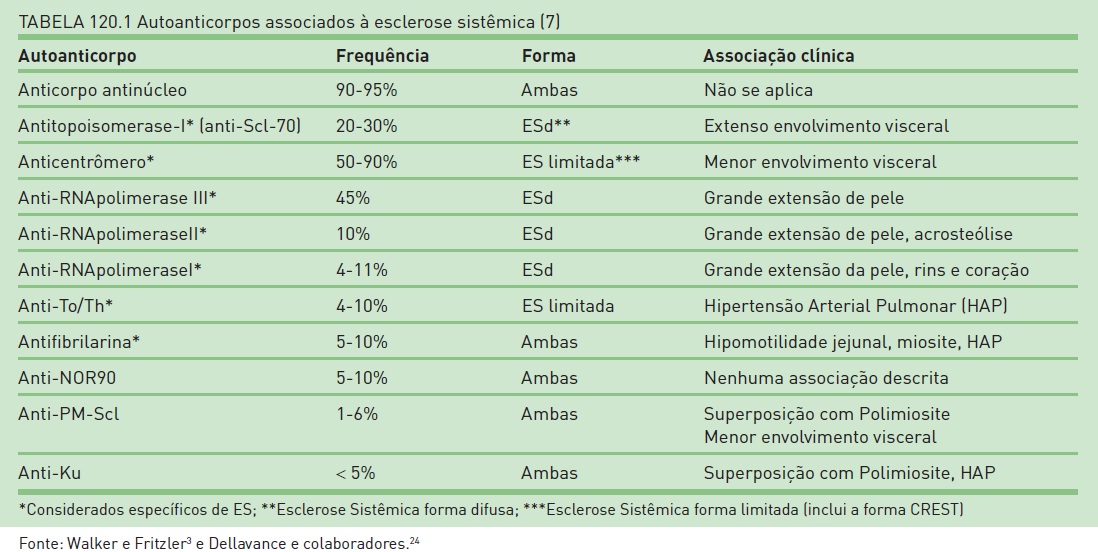
Na Tabela 120.1 são apresentados os autoanticorpos específicos e sua frequência nas diferentes formas clínicas da ES (ESd ou CREST).

Figura 120.1
Representação esquemática do mecanismo fisiopatológico da esclerose sistêmica.
Fonte: Clements e Furst.2
989
Figura 120.2
Alterações capilares na região periungueal em um paciente com esclerodermia limitada.
Fonte: Longo e colaboradores.11
O fenômeno de Raynaud (FRy) é o resultado de uma resposta vascular exagerada a estímulos como o frio e o estresse.4,8,9
A vasoconstrição das artérias digitais, arteríolas pré-capilares e shunts arteriovenosos cutâneos resulta em uma isquemia digital transitória, manifestando-se clinicamente como palidez ou cianose das mãos e dos pés. Essa fase isquêmica, uma vez cessados os estímulos para a vasoconstrição, é seguida por vasodilatação desses pequenos vasos e retorno do fluxo sanguíneo capilar ao normal. Nessa segunda fase, mãos e pés apresentam eritema secundário à rápida reperfusão dos dígitos. O FRy pode manifestar-se também na face, no nariz e no pavilhão auricular externo.4,7-9
O FRy é primário quando não está associado a nenhum fator desencadeante identificável ou doença sistêmica subjacente e secundário quando acompanha doença crônica ou é decorrente do uso de medicamentos, por exemplo.2,4 O primeiro ocorre em cerca de 20 a 30% da população do sexo feminino entre 15 e 30 anos de idade, entretanto, a prevalência do FRy primário pode chegar até 2 a 3% da população considerando os diferentes grupos étnicos e variações climáticas de cada região do país.9 O primeiro caracteriza-se por crises intermitentes de palidez e cianose dos dígitos quando expostos ao frio ou ao estresse, sem evidência de dano estrutural vascular associado, ulcerações digitais (UD) ou gangrenas.7 A CPU de pacientes é normal, não há autoanticorpos específicos, e a velocidade de sedimentação globular (VSG) também é normal.3,7,9,12
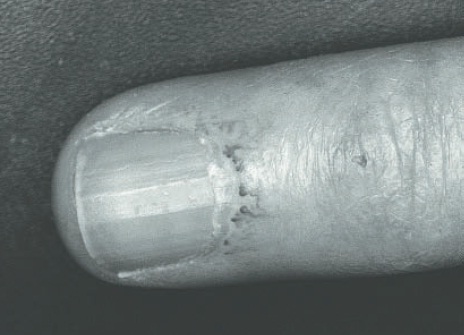
Em casos de FRy secundário, as crises estão associadas a dano estrutural dos vasos sanguíneos, dor decorrente da isquemia digital sustentada e UD (Fig. 120.7). Pode haver assimetria dos dígitos comprometidos, além de sinais e sintomas da doença reumática sistêmica associada (como a ES, doença mista do tecido conectivo [DMTC], lúpus eritematoso sistêmico [LES], artrite reumatoide [AR], dermatomiosite [DM], síndrome de Sjögren [SSj]), das doenças endócrinas (como o hipotireoidismo e o feocromocitoma) e hematológicas (como as crioglobulinemias e paraproteinemias). Os testes laboratoriais dos pacientes com FRy secundário à ES evidenciam alterações, como a presença de autoanticorpos específicos e VSG elevada. A CPU invariavelmente está alterada mostrando o padrão SD.8,9,13 Outras causas que devem ser pesquisadas em pacientes com FRy secundário são o uso de fármacos simpaticomiméticos (p. ex., betabloqueadores, ergotamina, cocaína, nicotina, cafeína, narcóticos, anfetaminas), quimioterápicos (p. ex., bleomicina, vincristina, cisplatina, carboxiplatina) além de antibióticos macrolídeos ou inibidores de proteases. Devem ser realizados diagnósticos diferenciais entre FRy secundário e acrocianose, síndrome do túnel do carpo, aterosclerose, embolia, oclusão vascular de artérias de médio e grande calibre, como a que ocorre na granulomatose de Wegener (GW), poliarterite nodosa (PAN) e na doença de Buerger.1,2,4-6,10,15
Figura 120.3
A e B - Padrões capilaroscópios normais.
Figura 120.4
Padrão “SD” precoce.
Figura 120.5
Padrão esclerodérmico ativo.
Figura 120.6
Padrão esclerodérmico tardio.
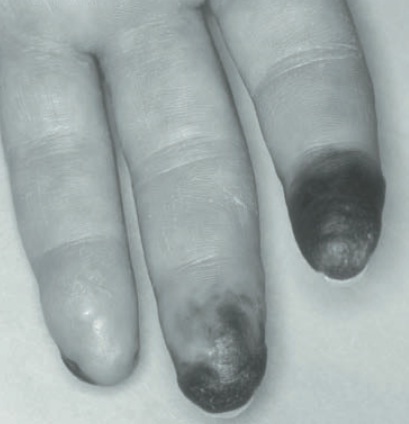
Figura 120.7
Necrose digital em um paciente com esclerose sistêmica associada ao FRy grave.
Fonte: Longo e colaboradores.11
Telangiectasias. As telangiectasias são capilares dilatados observados mais frequentemente na região periungueal, na face, na superfície palmar das mãos e mucosas. Ocorrem de forma predominante em pacientes com a forma CREST (Fig. 120.8).2,3
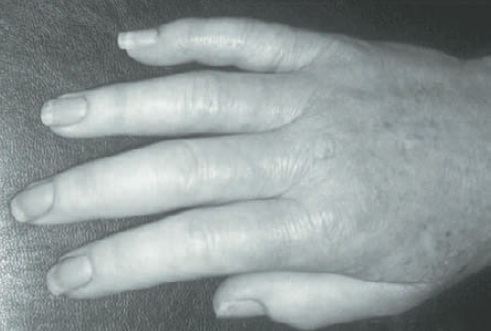
Esclerodermia ou espessamento da pele. Essa manifestação é a mais facilmente reconhecida nas formas difusa (ESd) e CREST da ES; entretanto não é proeminente em todos os casos, pois, em alguns pacientes, a vasculopatia se estabelece sem haver envolvimento cutâneo que possa ser identificado clinicamente.2,6,13,16 A avaliação clínica da extensão e da gravidade do comprometimento cutâneo em casos de ES pode ser por meio de escore modificado de Rodnan, ultrassonografia de alta resolução e ressonância nuclear magnética.6,16
Nas fases mais precoces da ESd, a pele fica edemaciada e inflamada, apresentando prurido, alterações de pigmentação (áreas hiperpigmentadas intercaladas com áreas vitiligo-like) (Fig. 120.9).1,2,4,16
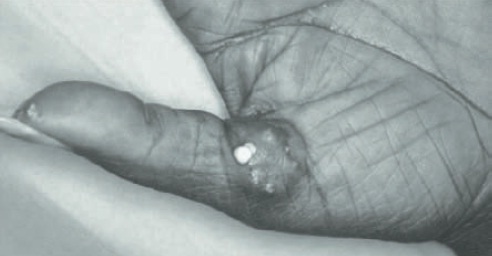
Calcinose. Em pacientes com calcinose, há depósitos de cálcio no tecido subcutâneo que podem ser identificados nos dedos das mãos, dos pés e nas superfícies extensoras dos antebraços. A radiografia das áreas afetadas evidencia as calcificações e/ou deformidades ósseas (Fig. 120.10).1,2,15
Outras manifestações cutâneas que ocorrem nas fases mais tardias da doença são pele seca, atrófica e descamativa (devido ao dano às glândulas sebáceas) e liquenificação.1,4,15
A manifestação muscular mais frequente é a miosite, ocorrendo geralmente em casos de síndromes de sobreposição de ES e polimiosite (PM). A relação entre a miopatia e o envolvimento cardíaco tem sido estudada.15
A queixa de artralgias é comum, entretanto, artrite evidente não é frequente, ocorrendo mais nos casos de sobreposição de ES e AR.1,2,4,5 As deformidades ósseas ocorrem com frequência em pacientes com ESd, sendo que, na maioria dos casos, são observadas nas mãos e podem ser identificadas radiologicamente como erosões, redução dos espaços articulares, desmineralização, acrosteólise, deformidades em flexão dos dedos e calcinose. As calcificações distróficas podem afetar tendões, articulações, cartilagem articular e discos intervertebrais.1,2,4,6
Figura 120.8
Telangiectasias na face.
Fonte: Longo e colaboradores.11
Figura 120.9
Espessamento da pele em paciente com esclerose sistêmica limitada.
Fonte: Longo e colaboradores.11
Figura 120.10
Calcinose em paciente com esclerose sistêmica limitada.
Fonte: Longo e colaboradores.11
As manifestações de comprometimento do trato gastrintestinal (TGI) são as que ocorrem com maior frequência na ES, afetando 75 a 90% dos pacientes.1,5 Qualquer porção do TGI pode ser comprometida. Clinicamente, os pacientes podem se apresentar desde assintomáticos, disfagia, odinofagia, epigastralgia, náuseas, diarreia, constipação, refluxo gastro-esofágico, pseudo-obstrução intestinal, má absorção e incontinência fecal por doença anorretal.1,2,4,6,17
A cardiopatia sintomática pode ser verificada em 20 a 25% dos casos, mas a maioria dos pacientes é assintomática. Caracteristicamente, as artérias coronárias não são afetadas, mas sim a microvasculatura do miocárdio e o sistema de condução cardíaco. Numa fase precoce da doença pode ocorrer disfunção sistólica e diastólica ventricular esquerda, e evoluir de forma silenciosa até insuficiência cardíaca congestiva clinicamente evidente. A arritmia pode ser a primeira manifestação clínica de pacientes com fibrose do sistema de condução cardíaco. A ocorrência de pericardite com tamponamento cardíaco é rara. 2,4-7,15,17,18
Mesmo não havendo sintomas, preconiza-se o acompanhamento dos pacientes com ES objetivando realizar o diagnóstico precoce por meio de métodos não invasivos: eletrocardiograma, ecocardiograma transtorácico com Doppler, ressonância magnética nuclear. 2,4-6,15
O NT-pro-brain peptídeo natriurético (NT-proBNP) é um biomarcador de doença cardíaca em pacientes com ES e pode auxiliar a identificar cardiopatia de evolução precoce ou subclínica.5,6,19,20
Cerca de metade dos pacientes com ES apresenta alguma evidência de nefropatia, como proteinúria, discretas elevações na creatinina sérica e/ou hipertensão arterial sistêmica (HAS).1,5
O comprometimento renal pode ser também o resultado de doença pré-renal associada à insuficiência cardíaca, HAS, estenose de artéria renal, uso de anti-inflamatórios não esteroides, diuréticos, hipovolemia decorrente do comprometimento do TGI e/ou má absorção. Outras causas de disfunção renal são a ação tóxica direta de fármacos como a D-penicilamina, efeito de HAS crônica e, raramente, glomerulonefrite.15
A CRE afeta precocemente 10 a 20% dos pacientes com ESd que apresentam menos de quatro anos de evolução da doença. Em raros casos, pode ser a primeira manifestação dessa forma de ES.5,6,15
A CRE é caracterizada por início abrupto de HAS maligna em pacientes com pressão arterial normal prévia, rápida perda de função renal (rápida elevação da creatinina, proteinúria e hematúria macroscópica), retinopatia hipertensiva (hemorragias e exsudatos no exame de fundo de olho), encefalopatia hipertensiva, acidente vascular cerebral e insuficiência cardíaca. Pode haver anemia e/ou trombocitopenia, que melhoram com a normalização da função renal.5-7,15 Essa condição deve ser tratada precoce e agressivamente com o uso de inibidores da enzima conversora da angiotensina (IECAs) associados ou não ao de outros anti-hipertensivos. A função renal tende à normalizar poucos dias após o início do tratamento.5,6,15
Na fisiopatogenia da CRE, ocorre redução significativa do fluxo sanguíneo renal e ativação do sistema renina-angiotensina.7 Anteriormente ao uso dos fármacos IECAs (captopril, enalapril, ramipril), a CRE era a causa mais comum de morte dos pacientes com ES.2,4,5,15 Em indivíduos com CRE, há microangiopatia trombótica semelhante ao que ocorre na nefroesclerose maligna, púrpura trombocitopênica, nefrite por radiação, rejeição crônica à transplante e síndrome antifosfolipídeos.6 A biópsia renal não possibilita o estabelecimento do diagnóstico definitivo de CRE porque os achados histológicos são semelhantes aos das demais situações clínicas citadas.5,6,17
São descritos como fatores preditivos de CRE: ESd, doença com menos de quatro anos de evolução, presença dos anticorpos anti-RNA polimerase III, anemia, eventos cardíacos, uso prévio de altas doses de corticosteroides.2,5,6 Os fatores que determinam pior prognóstico da CRE são creatinina sérica maior do que 3 mg/dL (no momento do diagnóstico), duração de tratamento para normalização da pressão arterial de mais de três dias, sexo masculino, idade avançada, insuficiência cardíaca, mulheres jovens com CRE, mas com pouco aumento da pressão arterial além do basal.1,5,6,15,18
Pneumopatia esclerodérmica é atualmente a principal causa de morte dos pacientes com ES de ambas as formas. Pode ocorrer em até 75% dos pacientes, e em cerca de 25% dos casos é a causa do óbito.6
Pode haver duas formas principais de comprometimento pulmonar em casos de ES: alveolite fibrosante (AF) com evolução para fibrose intersticial pulmonar (FIP) e hipertensão arterial pulmonar (HAP). Esses dois processos podem ocorrer de forma independente ou concomitante.15,17 Raramente a HAP pode apresentar-se como manifestação isolada da ES, indicando pior prognóstico da doença e sobrevida de 10% em cinco anos.5,6,10,15
A maioria dos pacientes com AF e FIP é assintomática até que a doença pulmonar esteja avançada. A radiografia de tórax não é um método sensível para o diagnóstico precoce de FIP e não possibilita a identificação de doença ativa.6,10 Os estertores crepitantes em bases à ausculta pulmonar não são identificados precocemente; quando existentes, indicam FIP estabelecida.1,4,10
A tomografia computadorizada pulmonar de alta resolução (TCPAR) proporciona a identificação da doença inicial e fornece indicativos de atividade da doença.5,6,10,15,17 As alterações típicas da FIP incluem espessamento dos septos interlobulares, cistos subpleurais, opacidades em bases pulmonares com aspecto de favo de mel (inflamação ativa, alveolite). Uma vez identificada a alveolite na TCPAR, indica-se a realização do lavado broncoalveolar (LBA), que pode evidenciar predomínio de neutrófilos e eosinófilos. A constatação de neutrofilia no LBA está associada à mortalidade precoce, mas os achados desse procedimento não se correlacionam com a velocidade de progressão da doença.6,10,15 Micronódulos, bronquiectasias, bronquioloectasias e opacidades em vidro fosco verificados na TCPAR indicam FIP estabelecida.2,4,5,10,15
Os testes de função pulmonar nos pacientes com ES podem evidenciar alterações restritivas: redução da capacidade vital forçada (CVF), de volumes pulmonares e da difusão de monóxido de carbono (DMCO) por meio da membrana alveolocapilar. Esses testes de função pulmonar devem ser realizados periodicamente, mesmo em pacientes assintomáticos.5,6,10,15
A CPU, em fases muito iniciais da ESd (menos de cinco anos de evolução), pode apresentar alterações como altos graus de deleção capilar (desvascularização), que podem ser preditores de comprometimento pulmonar.10
A HAP ocorre quando há comprometimento vascular pulmonar com vasoconstrição, proliferação da camada íntima dos vasos e obliteração da luz por trombose ou fibrose irreversível. O aumento da resistência vascular pulmonar dificulta o fluxo sanguíneo na microcirculação, semelhante ao que ocorre no FRy e, consequentemente, há sobrecarga do ventrículo direito.1,5,15,18,19,21 Também na evolução da HAP, evidenciam-se níveis séricos elevados de ETs, potentes vasoconstritores, liberados em resposta ao dano das células do endotélio vascular.5,6,19,20,22
Além dos testes de função pulmonar realizados para identificar comprometimento pulmonar restritivo precoce em pacientes com ES, com ou sem FIP, são necessárias outras formas de investigação a fim de avaliar o grau de limitação funcional, estabelecer diagnóstico definitivo e realizar planejamento terapêutico. O teste da caminhada de 6 minutos (TC6M) é obrigatório para todos os pacientes em investigação de hipertensão pulmonar (HP) e deve ser realizado tanto na etapa diagnóstica quanto no seguimento.20
Na Tabela 120.2, verifica-se a interpretação do TC6M aplicado nos pacientes com ES e HP.19,20
O grau de limitação funcional também precisa ser avaliado; nesse caso, o desempenho funcional é classificado de acordo com os critérios propostos pela New York Heart Association, adaptados para HAP pela Organização Mundial da Saúde (OMS) (Tab. 120.3).20
Deve-se realizar o ecocardiograma com Doppler regularmente no acompanhamento dos pacientes com ES a fim de determinar a pressão estimada da artéria pulmonar (PAP) e o gradiente tricúspide (velocidade de regurgitação tricúspide). Quando a PAP estimada por meio do ecocardiograma é maior ou igual a 40 mmHg, indica-se a realização de cateterismo cardíaco do ventrículo direito (CAT) para o diagnóstico definitivo de HAP. Durante esse procedimento, pode-se realizar testes com vasodilatadores para determinar se há redução da PAP, o que auxilia a elaboração do plano terapêutico do paciente.5,6,15,19,20,22
O diagnóstico de ES é estabelecido de acordo com a classificação apresentada no Quadro 120.1
Os valores de VHS e de proteína C-reativa elevados são frequentes, de forma inespecífica, em pacientes com doenças inflamatórias crônicas que apresentam envolvimento sistêmico.
Diagnóstico diferencial1-5,16
•Hipotireoidismo com escleromixedema.
•Escleredema de Buschke.
•Fasciíte eosinofílica.
•Escleredema associado a diabetes .
•Fenilcetonúria (lesões esclerodermia-símiles).
•Esclerodermia associado à porfiria cutânea tarda.
•Esclerodermia induzido por fármacos (bleomicina, pentazocina, implante de silicone)
•Exposição a cloreto de vinil.
•Uso crônico de instrumentos vibratórios.
•Micose fungóide.
•Amiloidose primária.
•Síndrome carcinoide.
•Doença enxerto versus hospedeiro.
•Líquen escleroso.
•Variante esclerodermiforme da acrodermatite atrófica crônica.
•Atrofoderma de Pasini-Pierini.
•Hemiatrofia facial progressiva.
•Algoneurodistrofia
•Lipodistrofias localizadas, distrofia fascial congênita.
•Fibrodisplasia ossificante progressiva.
•Síndrome de Werner.
•Progéria neonatal esclerodermiforme.
•Dermopatia restritiva.
Atualmente, na prática diária, o tratamento da ES é feito de acordo com as manifestações clínicas que cada paciente apresenta. Até o momento, não existe comprovação de que um tratamento específico possa impedir a progressão da doença, pois um vez instalada a fibrose, este processo continua de forma autônoma. Por este motivo faz-se necessário um diagnóstico precoce, se possível numa fase onde somente a microangiopatia é identificada antes que a fibrose esteja estabelecida.6,15,17
O transplante de medula óssea parece ser um tratamento promissor para pacientes com ES, principalmente naqueles com doença recente, refratária e rapidamente progressiva.5,6
D-penicilamina. Esse medicamento foi utilizado durante muitos anos para tratamento específico do envolvimento cutâneo (esclerodermia), devido ao seu possível efeito antifibrótico. Entretanto, recentes estudos controlados não comprovaram a eficácia da droga. 2,4-6
Outros fármacos como EDTA, óleos vegetais, vitaminas, dextran e colchicina têm sido utilizadas na ESd mas os resultados são desanimadores.2,4-6
Metotrexato. Utilizado em baixas doses, por via oral, pode ser eficaz para o tratamento do comprometimento cutâneo, principalmente em pacientes com ESd rapidamente progressiva, mas não para o comprometimento visceral. É utilizado também em pacientes com síndrome de superposição de ES com AR e PM.5-6
Corticosteroides. Seu uso fica restrito à fase edematosa do comprometimento cutâneo.5-6
Nifedipina: pode ser utilizada doses entre 20 e 120mg/dia, mas se dá preferência às composições de liberação prolongada. Outros fármacos recomendados como anlodipina, captopril, losartana, prazosina parecem menoseficazes.5-6
Agentes como quetanserina, pentoxifilina e fluoxetina foram investigados, mas os resultados foram discretos ounegativos.5-6
O tratamento visa à analgesia, à melhora da circulação digital, à prevenção de infecções, à cicatrização, à prevenção de recidiva das UDs e de amputações. Para isso, é indicada a realização das seguintes medidas:
•Usar cremes e emulsões tópicas para manter a flexibilidade da pele.
•Evitar frio, nicotina e estresse emocional para reduzir a vasoconstrição.
•Utilizar hidrocoloides tópicos e curativos oclusivos.
•Administrar prostanoides via intravenosa (iloprosta intravenoso) para rápida cicatrização das UDs.
•Administrar inibidores dos receptores de ETs 1 e 2 (bosentana): eficácia na cicatrização das UDs e na prevenção de recorrência das UDs na ESd.
•Administrar inibidor de fosfodiesterases (sildenafil): terapia combinada com os inibidores de ET em pacientes que não respondem à monoterapia.5,6,17
•Analgésicos e anti-inflamatórios não esteroides: são utilizados para controle da dor e da inflamação articular.
•Corticosteroides: são administrados para tratamento da miosite, serosite, artrite e na fase edematosa do comprometimento cutâneo. Deve ser usado com cautela devido ao risco de desencadear CRE.6
•Fisioterapia, massoterapia, correção postural, terapia ocupacional: essas terapias podem ser realizadas para alívio da dor, inflamação, prevenção e correção de deformidades e reabilitação funcional.6
•Fármacos antirrefluxo que agem também na motilidade esofágica, como os inibidores da bomba de prótons (omeprazol, pantoprazol) são utilizados para controle do refluxo gastroesofágico, prevenção de ulcerações esofágicas e estenoses.
•Fármacos pró-cinéticos (domperidona): usadas para controlar os sintomas dos distúrbios da motilidade do TGI.
•Antibióticos: são utilizados de forma alternada (ciclos rotatórios de diferentes antibióticos) em infecçõesintestinais.1,2,4-6
•IECAs (enalapril, captopril, ramitril): utilizadas para controlar rapidamente a pressão arterial.
•Tratar as comorbidades, como insuficiência cardíaca, hipovolemia e efeito nefrotóxico de fármacos, como D-penicilamina e anti-inflamatórios não esteroides.
•Evitar o uso de corticosteroides.2,4-6
•Realizar o diagnóstico e o tratamento específico de arritmias, insuficiência cardíaca congestiva, miocardiopatia e pericardite sintomáticas.2,4-6
•Ciclofosfamida intravenosa (tratamento em pulsoterapia) é utilizada para controle de alveolite fibrosante (AV) e fibrose intersticial pulmonar (FIP), entretanto são necessários novos estudos para demonstrar sua real eficácia como droga modificadora de doença.5,6
•Na HAP, o objetivo principal do tratamento é a vasodilatação. São utilizados fármacos pertencentes a quatro principais classes terapêuticas:6,19,20,22
–Bloqueadores dos canais de cálcio (nifedipina e anlodipina).
–Antagonistas dos receptores de endotelina (bosentana, sitaxsentana e ambrisentana).
–Inibidores das fosfodiesterases (sildenafila).
–Prostanoides ou derivados da prostaciclina: epoprostenol (uso intravenoso; padrão-ouro para tratamento de pacientes de classe funcional IV da OMS).
•Treprostinil: uso subcutâneo em pacientes com HAP secundária a doenças do tecido conectivo, de classe funcional III e IV da OMS.
•Iloprosta inalatório ou intravenoso: pacientes com HAP de classe funcional III e IV da OMS.
•Beraprost: uso oral.
O diagnóstico de ES da paciente do caso clínico em questão fundamenta-se na história clínica e no exame físico, que caracteriza a forma limitada da ES, ou forma CREST (calcinose, Raynaud, esofagopatia, esclerodactilia e telangiectasias). Os valores da VSG e da proteína C-reativa estão elevados, achados estes que mesmo inespecíficos, são frequentes nestes pacientes com doenças inflamatórias crônicas que apresentam envolvimento sistêmico. À pesquisa de autoanticorpos específicos através do teste de imunofluorescência indireta, detectou-se títulos superiores a 1/640 em padrão centromérico de fluorescência. Este padrão refere-se a autoanticorpos anticentrômero, específicos para ES forma CREST, estando presentes em 50 a 90% destes pacientes. Outros exames laboratoriais solicitados, incluídos os para a avaliação da função renal, foram normais.
A cintilografia de esôfago – exame de alta sensibilidade para identificar estágios precoces da dismotilidade esofágica – solicitada para esta paciente evidenciou estase no terço inferior do esôfago tanto para alimentos sólidos quanto líquidos nas posições sentada e deitada.
Embora clinicamente a paciente não apresentasse sinais ou sintomas de comprometimento pulmonar, foram solicitados raio X de tórax e TCPAR (esta última mais sensível para identificar fases iniciais, ainda assintomáticas, de alveolite fibrosante e fibrose pulmonar). Os resultados destes exames, assim como do eletrocardiograma em repouso, foram normais, mas serão repetidos a cada ano para acompanhamento da evolução da doença. Ecocardiograma Doppler foi solicitado para se ter uma estimativa da pressão arterial pulmonar (PAP), uma vez que a prevalência de hipertensão arterial pulmonar (HAP) é maior nestes pacientes com a forma CREST da ES. Se a PAP estimada no ecocardiograma fosse maior ou igual a 40mmHg, estaria indicado o CAT de ventrículo direito para diagnóstico definitivo de HAP e indicar o tratamento mais adequado para esta paciente. Neste caso, será repetido também anualmente o ecocardiograma ou se houver suspeita clínica de HAP no acompanhamento da paciente.
O tratamento instituído para esta paciente foi o uso de vasodilatador como nifedipina (10 a 30mg/dia), medidas anti-refluxo (omeprazol 20mg/dia, em jejum pela manhã) e fármacos pró-cinéticos (domperidona 10mg, 2x/dia, antes das refeições principais). Corticosteroide (prednisona 20mg/dia) foi utilizado para a fase edematosa da ES (a paciente apresentava edema difuso dos dedos das mãos) e das artralgias, entretanto seu uso não foi contínuo nem por tempo prolongado.
1.Hummers LK, Wigley FM. Scleroderma. In: Imboden J, Hellmann D, Stone J, editors. Current rheumatology: diagnosis and treatment. 2nd ed. New York: McGraw-Hill; 2006. p. 228-36.
2.Clements PJ, Furst DE, editors. Systemic sclerosis. 2nd ed. Philadelphia: Lippincott Williams and Wilkins, 2004.
3.Walker JG, Fritzler MJ. Systemic sclerosis. In: Shoenfeld Y, Cervera R, Gershwin ME, editors. Diagnostic criteria in autoimmune diseases. Totowa: Humana; 2008. p.31-6.
4.Mayes MD, Varga J, Buch MH, Seibold JR. Systemic sclerosis. In: Klippel JH, Stone JH, Crofford LeJ, White PJ, editors. Primer on the rheumatic disease.13th ed. New York: Springer; 2008. p. 343-62.
5.American College of Rheumatology. UpToDate in rheumatology [Internet]. Waltham: UpToDate; c2012 [capturado em 15 set. 2012].
Disponível em: http://www.uptodate.com/home/clinicians/specialties/ rheumatology.html.
6.Carol D, Guillevin L. Raising the profile of systemic sclerosis. In: 3rd International Systemic Sclerosis Forum; 2009 Feb 29- Mar 01; Basel, Switzerland; 2009.
7.Carpentier PH, Maricq HR. Microvasculature in systemic sclerosis. Rheum Dis Clin North Am. 1990;16(1):75-91.
8.Maricq HR, LeRoy EC, D’Angelo WA, Medsger TA Jr, Rodnan GP, Sharp GC, et al. Diagnostic potential of in vivo capillary microscopy in scleroderma and related disorders. Arthritis Rheum. 1980;23(2):183-9.
9.Capobianco KG, Xavier RM, Bredemeier M, Restelli VG, Brenol JC. Nailfold capillaroscopic findings in primary Sjögren’s syndrome: clinical and serological correlations. Exp Rheumatol. 2005;23(6):789-94.
10.Bredemeier M, Xavier RM, Capobianco KG, Restelli VG, Rohde LE, Pinotti AF, et al. Nailfold capillary microscopy can suggest pulmonary disease activity in systemic sclerosis. J Rheumatol. 2004;31(2):286-94.
11.Longo DL, Fauci AS, Kasper DL, Hauser SL, Jameson JL, Loscalzo J. Harrison’s principles of internal medicine. 18th ed. New York: McGraw- Hill; 2012. v. 2.
12.Cutolo M, Sulli A, Secchi ME, Olivieri M, Pizzorni C. The contribution of capillaroscopy to the differential diagnosis of connective autoimmune diseases. Best Pract Res Clin Rheumatol. 2007;21(6):1093-108.
13.Lonzetti LS, Joyal F, Raynauld JP, Roussin A, Goulet JR, Rich E, et al. Updating the American College of Rheumatology preliminary classification criteria for systemic sclerosis: addition of severe nailfold capillaroscopy abnormalities markedly increases the sensitivity for limited scleroderma. Arthritis Rheum. 2001;44(3):735-6.
14.Dellavance A, Gabriel Júnior A, Nuccitelli B, Taliberti BH, von Mühlen CA, Bichara CDA, et al. 3º Consenso Brasileiro para pesquisa de autoanticorpos em células HEp-2 (FAN). Recomendações para padronização do ensaio de pesquisa de autoanticorpos em células HEp-2, controle de qualidade e associações clínicas. Rev Bras Reumatol. 2009;49(2):89-109.
15.Steen VD, Conte C, Owens GR, Medsger TA Jr. Severe restrictive lung disease in systemic sclerosis.Arthritis Rheum. 1994;37(9):1283-9.
16.Brennan P, Silman A, Black C, Bernstein R, Coppock J, Maddison P, et al. Reliability of skin involvement measures in scleroderma. The UK Scleroderma Study Group. Br J Rheumatol. 1992;31(7):457-60.
17.Matucci-Cerinic M, Miniati I, Denton CP. Systemic sclerosis. In: Bijlsma JWJ, editor. EULAR Compendium on Rheumatic Diseases. London: BMJ; 2009.
18.Denton DP, Black C. Targeted theraphy comes of age in scleroderma. Trends Immunol. 2005;26(11):596-602.
19.Galiè N, Torbicki A, Barst R, Dartevelle P, Haworth S, Higenbottam T, et al. Guidelines on diagnosis and treatment of pulmonary arterial hypertension. The Task Force on Diagnosis and Treatment of Pulmonary Arterial Hypertension of the European Society of Cardiology. Eur Heart J.2004;25(24):2243-78.
20.Diretriz da Sociedade Brasileira de Cardiologia: diagnóstico, avaliação e terapêutica da hipertensão arterial pulmonar [Internet]. São Paulo: SBC; 2005 [capturado em 15 set. 2012]. Disponível em: http:// publicacoes.cardiol.br/consenso/2005/039.pdf.
21.Herrick A. Diagnosis and management of scleroderma peripheral vascular disease. Rheum Dis Clin North Am. 2008;34(1):89-114.
22.Denton CP, Humbert M, Rubin L, Black CM. Bosentan treatment for pulmonary arterial hypertension related to connective tissue disease: a subgroup analysis of the pivotal clinical trials and their open-labelextensions. Ann Rheum Dis. 2006;65(10):1336-40.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.