(Carregando Índice)... (Carregando Índice)... |
Última revisão: 14/01/2015
Comentários de assinantes: 0
Quando há diminuição da sobrevida eritroide, que normalmente é de 110 a 120 dias, diz-se haver hemólise. A hemólise será compensada enquanto a hiper-regeneração medular reacional, que consegue multiplicar a eritropoese até 6 a 8 vezes, mantiver as cifras hematimétricas dentro do normal. Se a sobrevida diminuir aquém da capacidade máxima de reposição, haverá anemia hemolítica.
Nas anemias hemolíticas, além dos sinais e sintomas gerais de anemia, costuma haver icterícia e esplenomegalia. A icterícia não se acompanha de colúria e acolia; deve-se a aumento da bilirrubina indireta, por exagero do catabolismo hemoglobínico, superando a capacidade hepática de conjugação. Como circula ligada à albumina, a bilirrubina não-conjugada não é filtrada nos glomérulos renais; o turnover pigmentar exagerado favorece a formação de cálculos biliares. A esplenomegalia é persistente nas anemias hemolíticas crônicas e passageira quando houver apenas surtos de hemólise; pode desaparecer se a doença causar atrofia do baço. Na hemólise intravascular, há hemoglobinúria. Há outros exames também utilizados para caracterizar hemólise: dosagem da haptoglobina, que diminui, e da desidrogenase láctica, que se eleva muito.
Nas anemias hemolíticas congênitas, a constante hiperplasia eritroide, com expansão das áreas ósseas com medula vermelha durante a época do crescimento, causa deformidades ósseas como alongamento (em torre) do crânio, alargamento do díploe, que mostra aos raios X estrias semelhantes a cerdas de uma escova, proeminência dos malares e maxilares, ocasionando o aspecto descrito como fascies de roedor.
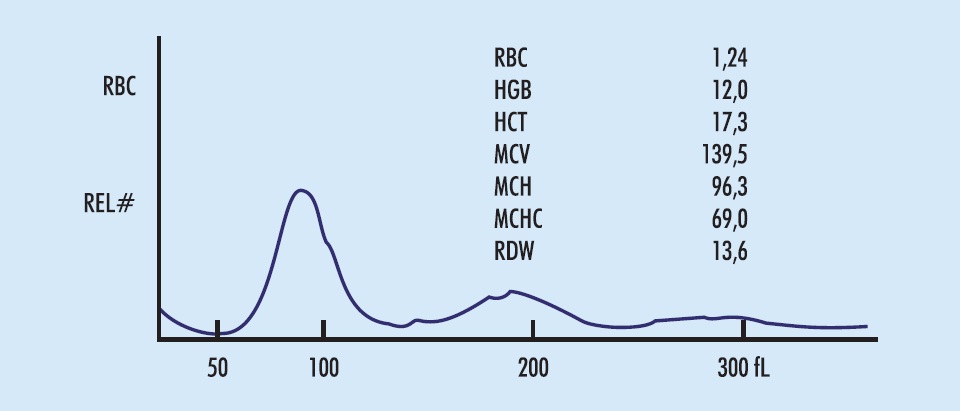
A Figura 5.1 mostra os hemogramas de dois casos de anemia hemolítica. Ambas são normocíticas (considerar o VCM infantil, na da esquerda) e têm os sinais de hiper-regeneração, constantes e persistentes nas anemias hemolíticas: policromatocitose, reticulocitose e IRF elevada. Originam-se da hiperplasia eritroide medular sempre presente, que torna inútil o exame da medula óssea. A contagem de reticulócitos e a IRF em 16 casos de anemias hemolíticas podem ser vistas nas Figuras 3.2 e 3.3, p. 114 e 115).
O diagnóstico diferencial entre as diversas anemias hemolíticas exige cuidadoso exame microscópico no hemograma. Nos exemplos da Figura 5.1, grande número de esferócitos no da esquerda e de drepanócitos no da direita permitem diagnóstico conclusivo em ambos. Nem sempre é assim: exames complementares são usualmente necessários.
Uma diminuição súbita da eritropoese, em paciente com anemia hemolítica, pela curta sobrevida eritroide, causa grave intensificação da anemia; essas crises aplásticas costumam decorrer da aplasia eritroide fugaz que acompanha a maioria das viroses; é constante e extrema na parvovirose e significativa na dengue. O esgotamento das reservas de folatos, cujas necessidades são muito aumentadas pela eritropoese exagerada, pode levar a uma anemia megaloblástica de rápida instalação; isso ocorre em épocas de consumo excessivo de folatos, como o crescimento (entre um e três anos e na puberdade) e a gestação.

Figura 5.1
Eritrogramas em anemias hemolíticas: esferocitose em paciente de 4 anos (E) e drepanocitose em paciente de 12 anos (D).
anemias hemolíticas decorrem de defeitos intrínsecos aos eritrócitos –anemias hemolíticas intracorpusculares – ou a fatores hemolíticos extrínsecos – anemias hemolíticas extracorpusculares. Mesmo tomadas em conjunto, excetuando-se a malária nas zonas endêmicas e a drepanocitose nas populações negras, as anemias hemolíticas são relativamente raras e justificam a consulta com um hematologista.
As anemias hemolíticas intracorpusculares, quase todas genéticas, são subclassificadas pela natureza do defeito causal (Tabela 5.1).
ias hemolíticas intracorpusculares
A membrana, como parte superficial do citoesqueleto, confere ao eritrócito flexibilidade e resistência, que lhe permite manter a integridade em traumas circulatórios e deformar-se de modo reversível na microcirculação. É composta de lipídios, continuamente renovados a partir do plasma, e de proteínas com múltiplas funções. Defeitos genéticos, qualitativos ou quantitativos, na síntese das principais proteínas, levam à instabilidade estrutural, à perda de vesículas lipoproteicas, à diminuição da superfície da membrana em relação ao volume do glóbulo e à deformação esferoide ou eliptoide. A Tabela 5.2 mostra os principais defeitos genéticos e suas consequências. A análise das proteínas ou dos genes, por biologia molecular, não é utilizada na clínica; o diagnóstico de esferocitose e ovalocitose costuma ser puramente hematológico.
É um defeito poligênico das proteínas da membrana, de acordo com a Tabela 5.2. Em 75% dos casos, é autossômico dominante; recessivo em aproximadamente 10%; de novo nos demais. A prevalência é elevada nas populações do norte da Europa (aproximadamente 50/100.000), menor nas populações brancas latinas (20 a 30/100.000) e ainda menor em negros e orientais. Os esferócitos têm sobrevida reduzida; são retidos e destruí dos precocemente no baço.
Na esferocitose, há todos os sinais clínicos e laboratoriais de hemólise, mas a severidade é variável, desde hemólise compensada, sem anemia, até anemia severa, com Hgb < 8 g/dL. O hemograma (Figura 5.1, [E]) mostra policromatocitose/reticulocitose constante. Nos casos com anemia severa, os esferócitos são numerosos e fáceis de notar à microscopia, pela falta do centro claro por perda da biconcavidade e pelo diâmetro menor; o VCM, entretanto, não costuma estar diminuído, sendo imprópria a denominação microesferócitos. Nos casos leves, os esferócitos são pouco numerosos, geralmente passam despercebidos na rotina do hemograma, e o diagnóstico não é feito. Para identificá-los ao microscópio, há necessidade de lâminas bem distendidas e coradas, uso das objetivas de imersão recomendadas e, principalmente, conhecimento e suspeita do defeito por parte do técnico. Devem ser procurados com atenção em todos os casos com policromatocitose/reticulocitose. Nas famílias com defeito da ß-espectrina, vários esferócitos são espiculados (esferoacantócitos); no defeito da proteína banda-3, há esferócitos em forma de ampulheta, como se tivessem sido pinçados no centro.
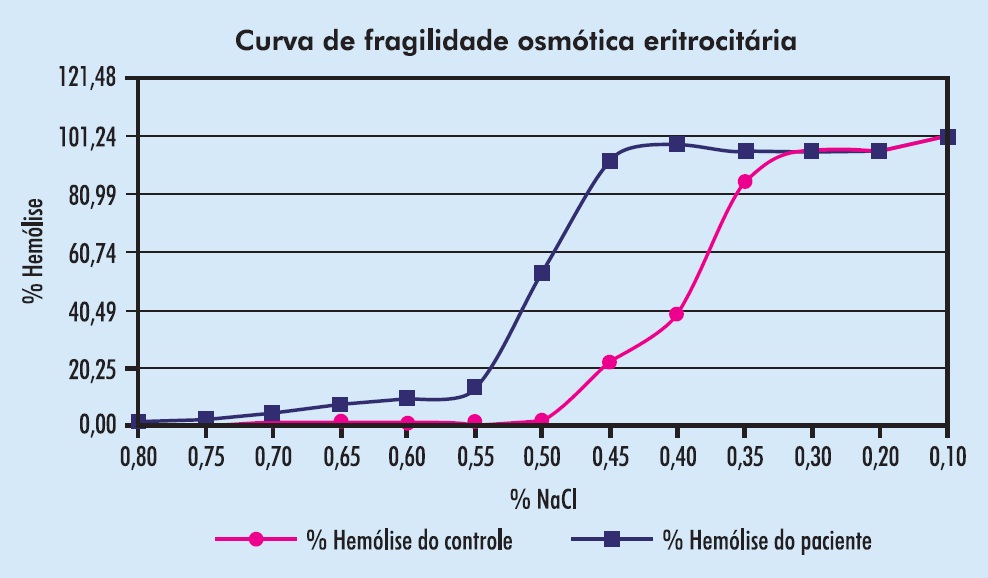
O teste diagnóstico para os casos duvidosos de esferocitose, a determinação da resistência globular à hipotonia osmótica, é manual, trabalhoso, poucas vezes solicitado e geralmente malfeito. Exige cuidadosa pesagem de NaCl não-hidratado, que é difícil de se obter e de se manter. O resultado pode ser expresso em tabela ou gráfico, correlacionando a porcentagem de hemólise a concentrações decrescentes de NaCl, de 0,9 a 0 g/dL. Exemplo de curva de resistência globular cuidadosamente feita é visto na Figura 5.2.
Figura 5.2
Resultado de teste de resistência globular em paciente com esferocitose.
(Cortesia do Laboratório da Santa Casa de Misericórdia de Porto Alegre.)
O pediatra, por sua vez, deve considerar a hipótese de esferocitose em todos os pacientes com anemia de longa data, com icterícia leve (bilirrubina indireta entre 1 e 4 mg/dL) e baço palpável. Às vezes, há hemólise perinatal, com anemia e icterícia, a ser distinguida da doença hemolítica do recém-nascido; na incompatibilidade ABO, os esferócitos também são numerosos. Dada a usual herança dominante, o exame dos pais, muitas vezes, é esclarecedor.
A anemia da esferocitose cura-se com a esplenectomia, embora persistam os esferócitos. Deve ser feita entre os cinco e os sete anos de idade.
O hemograma pós-esplenectomia costuma mostrar grande número de esferoacantócitos, além dos usuais corpos de Howell-Jolly. A contagem de reticulócitos no Cell-Dyn 4000, em seis casos do autor, mostrou um resultado discordante difícil de se interpretar, com contagens de reticulócitos em média 330.000/µL (extremos 263.000 e 574.000/ µL), mas exame microscópico sem policromatocitose nem reticulocitose à coloração própria e com IRF notavelmente diminuída, em média 0,084 (extremos 0,064 e 0,113). Ou o Cell-Dyn nota e lê como reticulocitose uma fluorescência inespecífica (?), ou os reticulócitos, que amadurecem no baço, na falta deste mantêm um mínimo de RNA, notado pela máquina, mas não à microscopia. O aumento espúrio do número total faz cair a fração imatura a um número impossível; os reticulócitos incluídos na IRF, entretanto, são os que estão mais de 30 canais acima do limiar de identificação, de modo que seu número absoluto independe dessa suposta situação. Se tomarmos as médias de Retics e IRF desses casos e calcularmos o número absoluto de reticulócitos imaturos, teremos: 330.000 X 0,084 = 27.720/µL. Ao experimentar o Cell-Dyn 4000, o autor encontrou 24% como valor de referência para os reticulócitos imaturos (da IRF) dentre os totais.Considerando-se o número acima como correspondente a essa porcentagem, teremos um número total de reticulócitos = 115.500/µL, um número coerente, dando verossimilhança à interpretação sugerida.
A designação eliptocitose está predominando na literatura sobre ovalocitose, mas a escolha é irrelevante. O defeito poligênico é sempre dominante. Como a maioria dos portadores é assintomática, a prevalência não pode ser definida com precisão, provavelmente da ordem de 50/100.000. Como os contadores eletrônicos não notam a eliptocitose e, não havendo anemia, não se faz mais microscopia, o defeito só ocasionalmente é notado nos hemogramas de rotina.
A sobrevida dos eliptócitos é próxima à normal; a hemólise mínima é facilmente compensada. Os pacientes anêmicos são minoria; geralmente são homozigotos ou duplamente heterozigotos para genes eliptocíticos. Os defeitos da a-espectrina e da proteína 4.1 causam eliptocitose assintomática nos heterozigotos e anemia hemolítica significativa, com icterícia e esplenomegalia, nos homozigotos; nos defeitos da glicoforina C, os heterozigotos têm eliptocitose assintomática, e os homozigotos, hemólise, geralmente compensada ou com mínima anemia.
No recém-nascido, a eliptocitose não é notada; os eliptócitos surgem após o quarto mês. Paradoxalmente, as combinações genéticas que incluem defeitos da a-espectrina, às vezes, causam severa anemia hemolítica, com presença no hemograma de extrema pecilocitose, com eliptócitos, esferócitos, eritrócitos fragmentados e numerosos macrócitos policromáticos; o quadro denomina-se piropecilocitose por assemelhar-se ao da hemólise ocasionada por queimaduras extensas ou por aquecimento acidental do sangue no laboratório. A tendência é a melhora em alguns meses, com lenta transformação para um quadro usual de eliptocitose. Há descrição de casos com aspectos morfológicos semelhantes e anemia hemolítica duradoura, em que a piropecilocitose decorre de combinação de genes eliptocíticos com raros genes que retardam a síntese de a-espectrina; são prevalentes apenas em populações africanas. Os eritrócitos desses pacientes são particularmente frágeis ao calor, fragmentando-se in vitro a 46°C, temperatura inferior à necessária para fragmentar eritrócitos normais; a designação piropecilocitose familiar é particularmente apropriada.
Há um gene, que altera a proteína banda-3, com prevalência de 20 a 30% nos indígenas da Melanésia, ilhas do Pacífico a leste da Austrália e sul da Malásia. O defeito é chamado ovalocitose do sudeste da Ásia; os eritrócitos mostram uma combinação única de ovalocitose (são mesmo ovoides, não eliptoides) com estomatocitose. Os heterozigotos são assintomáticos; crê-se que a homozigose cause morte fetal.
É uma rara doença dependente de um defeito clonal adquiridodas células primitivas da hematopoese. O gene responsável pela síntese das moléculas de fosfatidil-inositol-glican (gene PIG-A), no braço curto do cromossomo X, sofre mutação; há inúmeras variantes. Esse glicolipídio serve de âncora para várias proteínas da membrana do eritrócito, necessárias à regulação das moléculas do complemento (CD59, CD55 e outras). A falta ocasiona uma suscetibilidade à lise mediada pelo complemento, principalmente em meio acidulado. A mutação pode estender-se também à granulocitopoese e à trombocitopoese, daí haver uma curiosa combinação de anemia hemolítica crônica com insuficiente regeneração, neutrocitopenia e trombocitopenia. Pacientes em recuperação parcial ou total de anemia aplástica muitas vezes apresentam uma população clonal do tipo HPN.
A hemólise é intravascular, acentua-se nas horas de sono, e o paciente tem hemoglobinúria ao despertar. Na experiência do autor, a maioria dos pacientes não refere espontaneamente a passagem de urina escura, só quando especificamente questionado. Outra tendência que dificulta o diagnóstico dessa rara doença é a dos laboratoristas não se darem conta, no exame de urina, de que a positividade do teste para hemoglobina não se acompanha de eritrócitos no sedimento; dão a hemoglobinúria como hematúria.
A hemólise, na HPN, não se acompanha de uma hiper-regeneração no nível usual das demais anemias hemolíticas. Nas contagens de reticulócitos mostradas na Figura 3.2, p. 114, no grupo das anemias hemolíticas, os dois casos quase idênticos, de contagem mais baixa (reticulócitos aproximadamente 180.000/µL), são de HPN. A filtração renal continuada de hemoglobina causa depósito de hemossiderina nas células tubulares e hemossiderinúria; pode ser notada pela coloração de Perls do sedimento. Alguns pacientes desenvolvem anemia ferropênica, com a microcitose usual, pela perda crônica de ferro; o tratamento com ferro melhora esse componente da anemia, mas intensifica a hemólise pelo defeito basal.
A comprovação do diagnóstico se faz pelo teste de Ham: pesquisa de hemólise dos eritrócitos do paciente em soro fresco (para conter complemento) compatível, acidificado com HCl. Há um teste de gel-centrifugação para HPN, simples e sensível. O diagnóstico, com quantificação da população acometida pode ser feito por citometria em fluxo, com anticorpos monoclonais anti-CD59 ou anti-CD55.
A HPN não tem tratamento curativo, mas a evolução costuma ser lenta e geralmente benigna. O tratamento com anticorpo anticomplemento C5, eculizumab, reduz significativamente a hemólise.
São defeitos genéticos em que há trocas de aminoácidos na sequência das cadeias globínicas da hemoglobina. A identificação das hemoglobinas anormais por eletroforese está sendo substituída pela técnica de cromatografia líquida de alta resolução (HPLC = high performance liquid chromatography). Pela elevada prevalência e significativa morbidade de suas combinações genéticas, as síndromes falcêmicas merecem discussão detalhada. As demais hemoglobinopatias são mais raras no Brasil.
Incluem-se, sob essa denominação, as eventualidades em que há teste de afoiçamento positivo, pela presença de hemoglobina S (de sickle = foicinha), pura ou associada a outras hemoglobinas anormais. A Hgb S tem prevalência entre 5 e 10% nas populações negras da África Equatorial; é comum, também, na Arábia Saudita, na Grécia e no sul da Itália e da Ásia. Crê-se que a considerável prevalência africana deva-se a séculos de seleção natural, pela maior resistência dos eritrócitos com Hgb S à infecção pelo Plasmodium falciparum. A Hgb S veio para o Brasil com a escravatura; a prevalência africana original diluiuse pela miscigenação racial; por outro lado, passou a ser encontrada mesmo em pessoas aparentemente brancas. As síndromes falcêmicas mais comuns estão resumidas na Tabela 5.3.
Traço drepanocítico (ou falciforme): os portadores heterozigóticos de Hgb S (com Hgb A e S), embora tenham teste de afoiçamento positivo, são sadios. A Hgb S, em concentrações inferiores a 50% nos eritrócitos, não cristaliza nas tensões de oxigênio existentes in vivo, salvo em raras eventualidades de extrema anoxemia, tais como subida a grandes altitudes, falta de O2 durante anestesia, sobrevivência a afogamentos, etc. As variações osmóticas no córtex renal podem, entretanto, causar afoiçamento local: os drepanócitos obliteram glomérulos, causando glomerulite focal e hematúria; o defeito, embora persistente, não é progressivo. Anemia, ou alterações do hemograma, salvo a presença de raras células falciformes e target cells na cauda da distensão, nunca devem ser atribuídas ao traço falciforme.
A anemia ferropênica, em crianças com traço drepanocítico, tem causado um erro embaraçante; o pediatra recebe o hemograma com anemia (ferropênica) e o teste de afoiçamento positivo (pedido por tratar-se de paciente negro) e interpreta o conjunto como drepanocitose (anemia de células falciformes), apesar da diferença óbvia entre os hemogramas das duas anemias. Essa confusão do traço heterozigótico com a doença homozigótica tem sido feita por médicos até em pacientes negros não-anêmicos. Para evitá-la, o autor recomenda que o laboratório, quando tiver teste de afoiçamento positivo e hemograma simultâneo que não mostre anemia drepanocítica, anote no resultado:teste positivo (traço drepanocítico). O teste em si já mostra uma diferença: é rapidamente positivo em todas as células no homozigoto e tardiamente positivo (> 24 horas), e só numa fração de células, no heterozigoto. O melhor é substituir definitivamente o teste de afoiçamento pela HPLC (Figura 5.3, [E]); o teste do pezinho atualmente inclui essa última técnica, de modo que hemoglobinas patológicas são identificadas já no recém-nascido (ver Capítulo 19).
Drepanocitose (ou anemia de células falciformes): a hemoglobi nopatia S homozigótica, pela dupla herança (pai e mãe heterozigóti cos), é uma grave anemia hemolítica. O eritrograma está na Figura 5.1 (D). A presença dos drepanócitos patognomônicos é usual, mas não constante; costuma faltar nos pacientes em tratamento com hidroxicarbamida. Eritroblastemia é usual; há neutrofilia, às vezes acentuada, e trombocitose. A reticulocitose é algo inferior a esperada para a intensidade da hemólise e gravidade da anemia; a Hgb S tem baixa afinidade ao O2 de modo que a oxigenação periférica não estimula a síntese apropriada de eritropoetina para o grau de anemia. Há icterícia com bilirrubina indireta entre 2 e 6 mg/dL, grande aumento da DHL e baixa da haptoglobina. À esplenomegalia dos primeiros dois anos de vida, sucede-se atrofia do baço pela sequência de pequenos infartos; a asplenia funcional faz surgirem no hemograma corpos deHowell-Jolly, leptocitose, raros acantócitos, aumento do número de eritroblastos e linfocitose.
Os pacientes com drepanocitose, além da anemia, têm outras complicações: crises de sequestração esplênica (nos primeiros meses de vida), crises dolorosas por microinfartos múltiplos, decorrentes do afoiçamento in vivo, suscetibilidade aumentada a infecções e significativa diminuição da sobrevida. Só há tratamento paliativo. A hidroxicarbamida (Hydrea ) aumenta a síntese de Hgb F e diminui a frequência das crises dolorosas; causa macrocitose e deforma ainda mais o histograma eritroide (ver Figura 2.12 [D], p. 93). Nos casos mais graves, se houver doador compatível, o transplante de célulastronco deve ser considerado.
Figura 5.3
Cromatografia líquida de alta resolução (HPLC Bio-Rad Variant) em traço drepanocítico (E) e portador heterozigótico de hemoglobina C (D).
Microdrepanocitose: a herança concomitante de um gene S com um gene ß-talassêmico causa anemia hemolítica semelhante à drepanocitose, porém menos severa. Um gene faz sintetizar Hgb S, o outro causa déficit na síntese de Hgb A; a predominância de Hgb S causa afoiçamento in vivo, com suas consequências. Nesses pacientes, não há atrofia precoce do baço; mantém-se a esplenomegalia. A microdrepanocitose é comum no Rio Grande do Sul e nas demais áreas geográficas onde coexistem populações negra e italiana. O hemograma difere do da drepanocitose por haver anemia menos intensa, microcitose e número chamativo de leptócitos (target cells). O exame da hemoglobina mostra os valores da Tabela 5.3.; se a combinação genética for S ß0-Thal, haverá virtual ausência de Hgb A, só S, F e A2; se for S ß+Thal, haverá 5 a 20% de Hgb A, e a anemia será menos severa.
Hemoglobinopatia SC: a troca de ácido glutâmico por lisina na posição 6 da cadeia ß dá origem à Hgb C. O defeito acomete os mesmos grupos raciais que a Hgb S, mas com prevalência 10 vezes inferior. A dupla heterozigoticidade SC não chega a ser rara. A anemia hemolítica é moderada; o hemograma mostra numerosos leptócitos, raros drepanócitos e alguns eritrócitos com uma forma peculiar, ditos pecilócitos SC, decorrentes da dupla cristalização das hemoglobinas anormais; a CHCM costuma estar diminuída (hipocromia sem microcitose). Crises dolorosas são raras; os pacientes têm suscetibilidade aumentada a fenômenos tromboembólicos.
Hemoglobinopatia C: a heterozigótica (AC) é assintomática; o resultado de HPLC está na Figura 5.3 (D). A homozigótica (CC) causa hemólise moderada que deveria ser facilmente compensada por hiperplasia eritroide; contudo há anemia, com Hgb entre 9 e 12 g/dL. Essa aparente incongruência deve-se à baixa afinidade da Hgb C pelo oxigênio, pois, mesmo com anemia, há satisfatória oxigenação teci dual, daí não haver estímulo à síntese de eritropoetina e correção da anemia. Não há crises dolorosas; há esplenomegalia. O hemograma mostra considerável leptocitose, ou seja, mais de 80% dos eritrócitos têm aspecto de target cells; a procura cuidadosa pode evidenciar a presença de cristais de Hgb C em alguns eritrócitos. A herança da Hgb C com um gene ß-talassêmico causa um quadro clínico semelhante ao da talassemia intermédia.
Hemoglobinopatia D: a heterozigótica (AD) é assintomática; alguns casos de DPunjab têm sido notados no Brasil pelo teste do pezinho. A homozigótica é raríssima no Brasil. O autor só viu um caso; o eritrograma mostrava anemia com curiosa combinação de hipocromia, esferocitose e policromatocitose. A eletroforese é enganadora; a Hgb D migra com a S em pH alcalino e com a Hgb A em pH ácido; a HPLC distingue-a com facilidade.
Hemoglobinopatia E: só é prevalente na Tailândia e Laos. A síntese de cadeias ß da Hgb E é inapropriadamente lenta, como ocorre em mutações talassêmicas; a herança simultânea de um ß-gene talassêmico causa quadro de talassemia maior ou intermédia. A Hgb E, em HPLC, elui com a Hgb A2.
Hemoglobinas instáveis: há dezenas de variantes genéticas das cadeias globínicas, todas muito raras, originando hemoglobinas instáveis, que desnaturam e precipitam nos eritrócitos, causando anemia hemolítica crônica ou crises hemolíticas quando há exposição a drogas oxidantes. Pesquisam-se pelo teste da desnaturação da hemoglobina pelo calor (50oC) ou pela solução de isopropanol a 17%. A coloração apropriada pode mostrar corpos de Heinz.
Os eritrócitos obtêm a energia necessária para a manutenção do gradiente catiônico em relação ao plasma e para manter o glutatião em estado reduzido (para a defesa da hemoglobina e das enzimas contra agentes oxidantes) por meio da glicólise anaeróbica. As deficiências enzimáticas genéticas na via Embden-Meyerhoff causam esgotamento energético prematuro e hemólise; as deficiências na via hexose-fosfato, suscetibilidade a hemólise por agentes oxidantes. A carência de enzimas necessárias ao metabolismo de nucleotídios também pode causar anemia hemolítica.
É infrequente, mas tem sido vista no Brasil, inclusive pelo autor. Há mais de 130 variantes genéticas com atividade defeituosa em lócus poligênico; por isso, os pacientes afetados costumam ser duplamente heterozigotos para o defeito, daí não depender de consanguinidade nos pais, usual nas doenças recessivas raras.
É uma anemia hemolítica de severidade dependente da combinação de mutantes herdadas, sem características clínicas e hemato lógicas próprias; deve-se pensar em deficiência de PK diante de qualquer caso de anemia hemolítica congênita não-esferocítica.
Não há tratamento curativo. A esplenectomia é indicada nos casos severos; causa melhora marginal, elevando a hemoglobina em 1 ou 2 g/dL. Após a esplenectomia, há presença no sangue de numerosos acantócitos, e a contagem eletrônica mostra absurda reticulocitose entre 30 e 40% (dado da literatura); o fenômeno parece o mesmo que o autor observou em casos esplenectomizados de esferocitose, e sobre o qual emitiu hipótese esclarecedora (ver p. 126).
O gene que codifica a sequência de aminoácidos da enzima localiza-se no cromossomo X, de modo que só há defeito clinicamente relevante nos homens (hemizigotos). As mulheres (portadoras heterozigóticas) são geralmente indenes, salvo pequena porcentagem negativamente afetada pela lyonização, e as raras homozigotas, filhas de mãe portadora e pai afetado. É um lócus poligênico; há mais de 250 variantes conhecidas de G6FD, algumas instáveis, outras de atividade deficiente, acometendo, em conjunto, mais de 400 milhões de pessoas.Duas têm relevância clínica e alta prevalência:
Variante africana (ou A-): tem prevalência superior a 10% nos negros da África Equatorial; no Brasil, diluiu-se como a Hgb S. Crê-se que os portadores têm resistência aumentada ao Plasmodium falciparum, com aumento da prevalência por séculos de seleção natural. É rara em populações brancas. A enzima mutante tem atividade entre 5 e 15% da normal. Os portadores são assintomáticos, a menos que haja agressão aos eritrócitos por agentes oxidantes: naftalina, anilinas, antimaláricos, sulfonamidas, nitrofurantoína, ácido nalidíxico e, em doses muito elevadas (intoxicações), AAS, acetaminofen e vitamina K sintética. Nesse caso, há crise hemolítica grave, com extrema anemização.
Variante mediterrânea: muito comum no sul da Itália, na Grécia (há áreas com prevalência >30%), no norte da África, na Ásia menor e no sul da Ásia. A deficiência é mais grave que a variante africana; a enzima mutante tem atividade virtualmente zero. Há crises hemolíticas pelos mesmos agentes supracitados, mas também por substâncias oxidantes existentes na fava, alimento tradicional naquelas áreas geográficas. O favismo foi importado para o Rio Grande do Sul com a colonização italiana; o autor acompanhou várias crises hemolíticas dessa etiologia.
As crises hemolíticas por deficiência de G6FD são fáceis de diagnosticar: anemia súbita, grave, sem sinais de perda sanguínea, geralmente com hemoglobinúria, em paciente das etnias conhecidas, com história de ingestão ou uso do agente oxidante 24 a 48 horas antes. A icterícia surge em 24 horas. O hemograma mostra eritrócitos irregularmente contraídos e fragmentados e eritrócitos com a hemoglobina concentrada numa calota da célula, e o estroma vazio no restante (hemiestromas). A reticulocitose começa entre o 3º e o 4º dias; a IRF eleva-se já no 2º dia. Durante a crise, a coloração própria pode mostrar corpos de Heinz.
Há testes bioquímicos qualitativos fáceis para detectar a falta da enzima. São falsamente negativos na convalescença das crises, porque os eritrócitos novos, da regeneração, são mais ricos em enzima, que decai progressivamente durante a sobrevida na circulação. Pelo mesmo motivo, nesse período, há resistência à repetição da crise no caso de nova exposição ao agente oxidante.
Os recém-nascidos com deficiência de G6FD frequentemente têm icterícia neonatal. Não há sinais óbvios de hemólise, nem agente oxidante causal; a patogênese é imprecisa. Há casos descritos de anemia neonatal severa e mesmo de hidropisia fetal, decorrentes da ingestão de drogas oxidantes pela gestante. O teste do pezinho pode inclur dosagem de G6FD (ver Capítulo 19).
Há variantes genéticas de G6FD, muito raras, que causam anemia hemolítica congênita não-esferocítica crônica.
A deficiência genética, autossômica recessiva, é rara. No único caso visto pelo autor, havia consanguinidade nos pais. O diagnóstico é fácil; a falta da enzima causa um bloqueio no catabolismo do RNA, cujos metabólitos acumulam-se nos ribossomos e são vistos como fino pontilhado basófilo na maioria dos eritrócitos. É necessário que as lâminas sejam feitas a partir de sangue nativo, pois a conservação do sangue in vitro, com EDTA, inibe a precipitação dos ribossomos pelo corante. A anemia hemolítica é moderada (Hgb 8-9 g/dL) e melhora marginalmente com a esplenectomia.
A intoxicação pelo chumbo (saturnismo), tanto aguda como crônica, causa inibição da atividade da pirimidina-5-nucleotidase e anemia hemolítica, além do quadro de doença sistêmica. É uma anemiaextracorpuscular; descrita aqui por sua similitude com a deficiência genética da enzima. O saturnismo crônico costuma ser doença profissional, acometendo operários que trabalham com baterias, tintas contendo chumbo, linotipia (com a tecnologia antiga). A intoxicação, ocorrida há décadas, pode manifestar-se na velhice pela liberação, causada pela osteoporose, do chumbo armazenado nos ossos; nesses casos, a dosagem de chumbo no sangue pode não estar aumentada. A ingestão de tinta de parede, por crianças com pica por anemia ferropênica, é uma causa pediátrica. O saturnismo agudo, causado pela ingestão de aves de caça conservadas em vinagre, substância que dissolve os projéteis de chumbo, não era raro no Rio Grande do Sul: o autor viu vários casos, mas nenhum nas últimas duas décadas.
A anemia hemolítica do saturnismo não é severa, tem Hgb entre 8 e 11 g/dL, reticulócitos aproximadamente 200.000/µL. O leucograma não é afetado; o autor acompanhou um caso com trombocitopenia irresponsiva aos corticoides. O diagnóstico é feito pelo pontilhado basófilo grosseiro, visto em muitos eritrócitos, examinando-se lâminas com os cuidados mencionados. O tratamento prolongado com EDTA cálcico intravenoso elimina lentamente o excesso de chumbo.
Há outras deficiências enzimáticas genéticas no mecanismo glicolítico, todas muito raras, tendo sido descritas apenas algumas dezenas de casos de cada. Na maioria, a anemia hemolítica acompanha-se de malformações, de neuropatias com retardo mental e de miopatias. Estão listadas, em ordem alfabética, na Tabela 5.4.
ela
eficiências enzimáticas raras que causam anemia hemolítica
Dentre as anemias hemolíticas extracorpusculares, isto é, causadas por agentes hemolíticos alheios ao eritrócito (Tabela 5.5), a única de grande prevalência é a malária, estimada pela OMS em 300 milhões de casos novos por ano. Todas exigem a especial atenção diagnóstica de um hematologista.
ela 5.5
Nos residentes, ou viajantes vindos de zona endêmica, a malária deve ser a primeira suspeita a ser considerada no diagnóstico de doença febril. O paroxismo, com febre, calafrio e extrema prostração, repetido com periodicidade dependente da espécie de Plasmodium, é característico, mas inconstante.
Em mais de 50% dos casos, há sinais precoces de hemólise; na infecção por P. falciparum, pode haver até hemoglobinúria. Eritrogramas, repetidamente normais, sem anemização nem policromatocitose/reticulocitose, e bilirrubina indireta normal são contrários à suposição de malária como causa de febre obscura. A reticulocitose na malária é inferior à das demais crises hemolíticas, pela ativação imunológica e resposta anemizante às citoquinas (mecanismo de anemia de doença crônica).
A pesquisa de hematozoários deve ser feita várias vezes, porque o número no sangue varia com o estágio do ciclo: é máximo nas horas que precedem as crises; há que prevê-las pelo retrospecto. O técnico precisa ter enorme experiência e paciência para encontrá-los, quando poucos. Para a descrição e a diferenciação das espécies, o leitor deve procurar um atlas de hematologia*.
A parasitemia interfere na contagem automatizada de reticulócitos do Cell-Dyn 4000: o hematozoário fluoresce. A máquina pode, também, identificar os eritrócitos parasitados como eritroblastos.
O leucograma geralmente mostra desvio à esquerda com neutro citopenia ou número normal de neutrófilos; neutrofilia é incomum. Há eosinofilia na convalescença dos casos tratados. A contagem eletrônica de eosinófilos pode ser afetada pela polarização da luz do pigmento malárico. Na infecção por P. falciparum, costuma haver trombocitopenia.
* Obra recomendada: BAIN, Barbara J. Células sangüíneas: um guia prático. 4. ed. Porto Alegre: Artmed, 2007.
Na infecção pelos hematozoários do gênero Babesia, transmitidos por carrapatos, os eritrócitos são parasitados como na malária, mas as áreas geográficas endêmicas não correspondem; a babesiose tem sido descrita algumas vezes nos EUA e não foi descrita no Brasil.
Na bartonelose (febre de Oroya), prevalente nos países andinos do norte e nas Guianas, a Bartonella bacilliformis infecta os eritrócitos, causando esferocitose e anemia hemolítica.
As infecções por Clostridium, Streptococcus e Staphilococcus (raramente) podem acompanhar-se de anemia hemolítica.
São causadas por anticorpos antieritrocitários.
Nas isoimunes (ou aloimunes), os anticorpos formam-se em outra pessoa e têm acesso à circulação do(a) paciente por via transpla centária ou transfusional. A anemia hemolítica da incompatibilidade materno-fetal será discutida no capítulo sobre hemograma do recém-nascido (Capítulo 19). A hemólise por incompatibilidade transfusional é dos glóbulos transfundidos, não dos glóbulos do receptor, daí não haver, propriamente, anemia hemolítica, salvo no caso incomum de infusão de grande volume de plasma incompatível.
Nas autoimunes, os anticorpos são formados no organismo do(a) paciente. Distinguem-se e são classificadas pela natureza do autoanticorpo:
São anticorpos IgM, fixadores de complemento, com ótimo térmico entre 5 e 25ºC. As crioaglutininas são notadas no laboratório aomanipular-se o sangue conservado à temperatura ambiente em dias frios ou em refrigerador. A aglutinação pode ser vista a olho nu ou ao microscópio: há conglomerados de eritrócitos, distintos do rouleau. As crioaglutininas interferem na contagem de eritrócitos e na medida do VCM, gerando resultados incoerentes (Figura 5.4), com aumento impossível da CHCM. O aquecimento do sangue a 37°C, com passagem imediata no contador, costuma gerar resultados corretos.
Figura 5.4
Hemograma (Coulter STKS) em paciente com alto título de crioaglutininas.
A presença de crioaglutininas deve ser anotada no resultado do hemograma, porque:
1. Embora ocorram em pessoas idosas sadias (até título 1/4), são muito mais comuns e em títulos mais altos em pessoas com doenças do colágeno, neoplasias e infecções crônicas.
2. Aparecem transitoriamente no decurso da pneumonia por Mycoplasma. Crioaglutinação em paciente jovem, com febre e tosse, deve sugerir esse diagnóstico. A dosagem de crioaglutininas, feita por titulação do soro, anotando-se a diluição máxima ainda aglutinante, mostra títulos 1/4 a 1/256. Podem surgir crioaglutininas também na mononucleose infecciosa. Em nenhuma das duas infecções a amplitude térmica das crioaglutininas é suficiente para causar hemólise significativa in vivo.
3. Há uma doença de crioaglutininas, rara, em que um título acima de 1/256 (às vezes > 1/2.000) e uma atividade até acima de 30°C provocam distúrbios da circulação periférica, com púrpura, síndrome de Raynaud e anemia hemolítica. O eritrograma, às vezes, é impossível de ser feito nos contadores eletrônicos (Figura 5.4), mesmo com o sangue reaquecido; deve ser coletado e mantido a 37°C até o exame; se nem assim for possível, é preciso basear-se na dosagem da hemoglobina e no micro-hematócrito por centrifugação. A sintomatologia aparece com a exposição ao frio eameniza-se com o calor. Aceita-se que a doença seja decorrente de uma proliferação clonal de linfócitos B, isto é, de um linfoma indolente, sem organomegalias; a IgM é monoclonal.
Autoanticorpos IgG ligam-se aos eritrócitos e causam seques tração no tecido macrofágico do baço, do fígado e da medula óssea. A autoimunidade causal pode ser iatrogênica, parte do quadro de doenças imunológicas mais amplas, como o lúpus eritematoso sistêmico (LES), a leucemia linfocítica crônica, os linfomas e a aids, mas, na maioria das vezes, é idiopática.
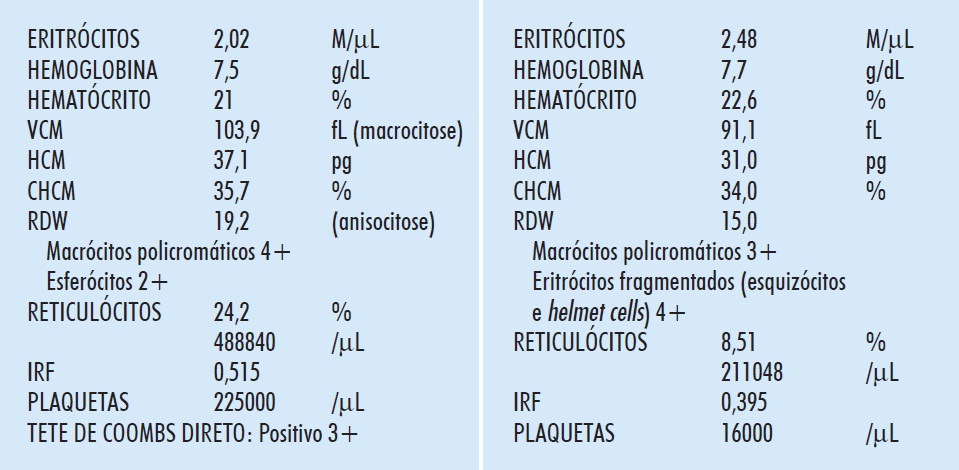
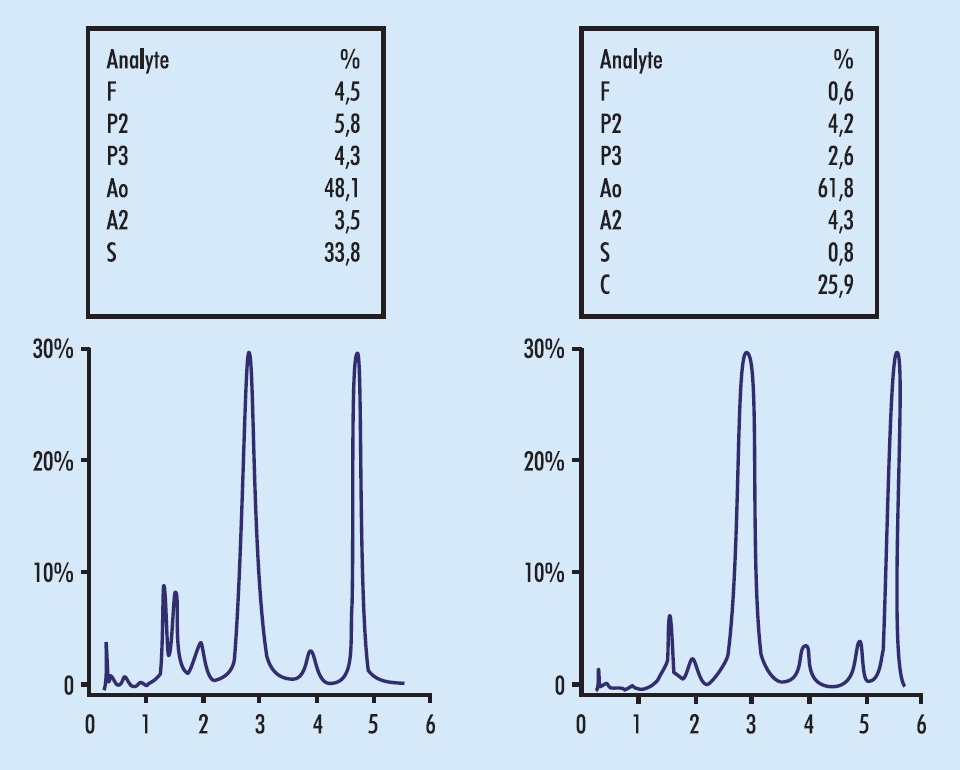
A Figura 5.5 (E) mostra um eritrograma de anemia hemolítica autoimune (AHAI). O grande número de macrócitos policromáticos, pela elevada atividade eritropoetínica, aumenta o VCM. A população macrocítica, às vezes, pode ser notada no histograma (ver Figura 2.11 [E], p. 90). A agressão imunológica causa perda de componentes da membrana e esferocitose pré-hemolítica. O aumento da bilirrubina indireta e da DHL e a baixa da haptoglobina são constantes. Em alguns pacientes com AHAI crônica de longa duração, a atividade esplênica desencadeia a formação de dacriócitos, e o hemograma pode lembrar mielofibrose, ou anemia perniciosa se a macrocitose for significativa.
O teste direto antiglobulina humana, mais conhecido no Brasil como teste de Coombs direto, que mostra a presença de anticorpos ligados aos eritrócitos, quando positivo, é patognomônico, o que ocorre em aproximadamente 90% dos casos; a positividade depende da distância entre as moléculas do anticorpo na membrana, de modo que pode ser transitoriamente negativo e positivar em exames ulteriores, ou vice-versa. O teste de Coombs indireto, que pesquisa anticorpos livres no plasma, não é indicado, pois se esgotam nos receptores dos eritrócitos.
O fármaco anti-hipertensivo a-metildopa (Aldomet ), atualmente menos usado, causa AHAI de modo previsível. Tomado por mais de três meses, em 20 a 30% dos pacientes, formam-se autoan ticorpos epositiva-se o teste de Coombs. Persistindo-se o uso do fármaco, 10% dos pacientes desenvolvem AHAI; parando-se o tratamento, há melhora lenta, com cessação da hemólise e negativação do teste de Coombs em seis meses. O tratamento com doses intravenosas elevadas de penicilinas e cefalosporinas também pode desencadear AHAI limitada.
A leucemia linfocítica crônica origina-se de linfócitos B CD5+, correlacionados com a autoimunidade; a proliferação neoplásica causa positividade do teste de Coombs em 10 a 15% dos pacientes e AHAI sintomática em uma fração desses; o tratamento com fludarabina torna essa complicação mais frequente. Os linfomas B indolentes e a doença de Hodgkin também se correlacionam com a AHAI; esta pode preceder (até por 2 a 3 anos), acompanhar ou suceder a doença linfoproliferativa.
A AHAI pode ser uma complicação do LES; é rara nas demais colagenoses.
A positividade do teste de Coombs na aids, com ou sem AHAI, notada desde o início da endemia, está se tornando mais frequente com o aumento da sobrevida média decorrente da eficácia do tratamento antiviral.
Exames pertinentes para as proliferações linfoides, colagenoses e aids são indispensáveis em todos os pacientes com AHAI.
A despeito desse amplo espectro etiológico, a AHAI idiopática é a de maior prevalência. Não porque tenha grande incidência: esta não passa de 1 caso/100.000/ano, mas porque é crônica e tratável, permitindo longa sobrevida. Os mesmos pacientes retornam periodicamente à consulta, durante anos. O tratamento com corticoides causa remissões rápidas, mas as recaídas são a regra ao suspendê-lo. A es plenectomia cura alguns pacientes e melhora outros. Tratamento com azatioprina, ciclofosfamida, anticorpos monoclonais e corticoides como manutenção são necessários se a esplenectomia não for curativa.
Figura 5.5
Eritrograma e exames pertinentes em anemia hemolítica autoimune (E) e púrpura trombocitopênica trombótica (D).
Os eritrócitos fragmentam-se quando sofrem trauma mecânico ou agressão física ou química; se a fragmentação for significativa, haverá anemia hemolítica.
O trauma da colisão com próteses deslocadas, ou a passagem por pertuitos justavalvulares (leaking), causa fragmentação dos eritrócitos e anemia hemolítica. O diagnóstico pelo hemograma exige cuidadosa observação ao microscópio; há entre 0,1 e 10% de formas fragmentadas (queratócitos e esquizócitos). Raramente, a fragmentação é tão intensa que a hemólise é instantânea, e os fragmentos praticamente não circulam e não são vistos; nesses casos, a hemólise intravascular causa hemoglobinúria. A história da cirurgia, associada aos sinais de anemia hemolítica, torna fácil o diagnóstico; o tratamento é a reoperação.
O trauma aos eritrócitos na circulação plantar em marchas ou corridas de longa duração causa hemólise intravascular e hemoglobinúria. A anemia hemolítica é autolimitada. A generalização do uso de calçados para corrida com solas amortecedoras diminuiu a incidência. Tem sido descrita hemoglobinúria similar, por trauma palmar, em tocadores de bongô.
A exposição a altas temperaturas causa lesão irreversível dos eritrócitos. A hemólise pós-queimaduras extensas ocorre nas primeiras 48 horas; pode haver hemoglobinúria. Notam-se eritrócitos com protrusões citoplasmáticas (blebs), que se desprendem e circulam como esférulas, formas fragmentadas de todos os tipos e esferócitos. A anemia hemolítica não gera uma resposta eritropoetínica apropriada devido ao estado de doença crítica do paciente; reposição transfusional é indispensável.
A púrpura trombocitopênica trombótica (PTT), a síndrome urêmico-hemolítica (SUH), os raros casos de coagulação intravascular disseminada que chegam a causar fragmentação e a síndrome HELLP (descrita nas trombocitopenias da gestação, no Capítulo 18) são denominadas anemias microangiopáticas, porque em todas há anemia hemolítica com fragmentação eritrocitária e algum tipo de lesão na microvasculatura.
Púrpura trombocitopênica trombótica e síndrome urêmico-hemolítica: são doenças graves, potencialmente fatais. Caracterizam-se por anemia hemolítica com eritrócitos fragmentados (AHEF) e trombocitopenia. Na PTT, costumam acompanhar-se de distúrbios neurológicos flutuantes (>70%) e febre (30-40%), raramente insuficiência renal. Na SUH, a insuficiência renal é constante; a febre e os sintomas neurológicos, raros.
Na PTT, o processo decorre da agregação plaquetária, poten cialmente reversível, na microcirculação de múltiplos órgãos, principalmente no sistema nervoso central e nos rins. Microtrombos hialinos podem ser evidenciados em pequenas artérias em biópsias da gengiva ou da medula óssea, mas não há indicação para esse método invasivo. Os microtrombos contêm alta concentração de fator von Willebrand (vWF); no plasma, evidenciam -se multímeros exageradamente grandes do fator, em detrimento dos fragmentos de clivagem, de menor peso molecular, que predominam no plasma normal. Atribui-se o desencadear da síndrome, quando idiopática, à inativação por um autoanticorpo IgG da metaloprotease ADAMST13, responsável pela clivagem do fator vWF; há raríssimos casos de PTT recidivante por deficiência congênita de ADAMST13. A baixa atividade da protease do vWF não é um fator único na patogênese da PTT; o desencadeamento depende também de condições do microambiente endotelial, sujeito à ação de citoquinas e sensível a atrito circulatório excessivo em áreas localizadas.
A PTT é quatro vezes mais frequente em mulheres, predominantemente jovens. Em 10 a 20% dos casos, correlaciona-se com a gravidez; surge no primeiro semestre, enquanto a HELLP, síndrome semelhante, mas limitada, costuma ocorrer no terceiro trimestre. Quando a PTT surge em até seis semanas da concepção, costuma reaparecer em gestações ulteriores. Há pacientes com PTT recidivante, independente de gestação, algumas pela deficiência congênita da metaloprotease.
O hemograma é típico (Figura 5.5 [D]), com macrócitos policromáticos, reticulocitose (inferior à da AHAI), eritrócitos fragmentados e trombocitopenia. Esses achados podem faltar nas primeiras 48 horas do início dos sintomas; em casos suspeitos, o hemograma, com plaquetas e reticulócitos, deve ser repetido diariamente. A DHL é muito elevada. A PTT é uma urgência médica. Todo o hemograma com anemia e trombocitopenia deve ter microscopia cuidadosa, feita por técnico experiente, que inclua a pesquisa de eritrócitos fragmentados, além dos demais achados próprios às hemopatias. No caso de suspeita clínica, pela presença de sintomas neurológicos e púrpura, mais ainda se houver febre, o médico deve solicitar hemograma com pesquisa de eritrócitos fragmentados.
O tratamento, de início imediato, com plasmaferese intensiva, baixou a mortalidade de >90% para <10%.
Casos esporádicos de SUH só costumam ser vistos em crianças abaixo de dois anos de idade. A insuficiência renal predomina no quadro; é grave, mas geralmente reversível; sintomas neurológicos são fugazes ou ausentes. A doença, inclusive surgindo em pequenos surtos epidêmicos, também em crianças maiores e adultos, pode ser causada por uma toxina Shiga-like produzida pela Escherichia coli, especialmente a cepa O157:H7. O contágio faz-se por alimentos contaminados e, talvez, de paciente a paciente. A SUH surge alguns dias após a disenteria; o tratamento com antibióticos não a previne e pode ser prejudicial.
O hemograma da SUH é indistinguível do da PTT, mas a trombocitopenia é menos severa. A síntese de eritropoetina persiste algumas semanas apesar da insuficiência renal com elevação da creatinina, daí a manutenção da reticulocitose.
Síndrome semelhante à PTT (ou a própria) tem sido descrita em pacientes com aids em estágio avançado; é, também, uma rara complicação do transplante alogênico de medula óssea. O prognóstico desses casos é sombrio.
Certos fármacos oxidantes provocam desnaturação da hemoglobina mesmo em eritrócitos normais. A sulfassalazina e a dapsona provocam-na nas doses terapêuticas usuais; o piridium e o acetaminofen, só em doses muito elevadas. Formam-se corpos de Heinz, que são rapidamente removidos pelo baço, deixando os eritrócitos irregularmente contraídos e mordidos; o aspecto é patognomônico. A hemólise só excepcionalmente causa anemia com hemoglobina abaixo de 9 g/dL, mas a policromatocitose costuma ser óbvia, com reticulocitose proporcional à anemização. A bilirrubina não ultrapassa 3 mg/dL. A suspensão da droga causa rápida melhora do quadro; a retomada, quando indispensável, pode ser tentada com doses menores e cuidadoso controle do hemograma.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.

 s hemolíticas intracorpusculares
s hemolíticas intracorpusculares

 eficiências enzimáticas raras que causam anemia hemolítica
eficiências enzimáticas raras que causam anemia hemolítica