Esclerose múltipla e distúrbios relacionados
J. William Lindsey, MD*
Artigo original: Lindsey JW, MD. Multiple sclerosis and related disorders. ACP Medicine.
[The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2015 Decker Intellectual Properties Inc. All Rights Reserved.]
Tradução: Paulo Henrique Machado.
Revisão técnica: Dr. Lucas Santos Zambon.
A bainha de mielina é essencial para a condução rápida de sinais nervosos ao longo de axônios com grande diâmetro [ver a Figura 1]. Os oligodendrócitos produzem e mantêm mielina no sistema nervoso central (SNC). As células de Schwann produzem e mantêm mielina nos nervos periféricos. Inúmeras doenças afetam preferencialmente a mielina, sendo que os neurônios, axônios e outros tipos de células são relativamente preservados. Essas doenças desmielinizantes variam amplamente em termos de etiologia e patogênese e incluem infecções imunomediadas, infecções de origem genética e doenças nutricionais. Neste capítulo apresentaremos algumas discussões sobre as doenças desmielinizantes do SNC [ver a Tabela 1].
|
Tabela 1: Doenças Desmielinizantes no Sistema Nervoso Central | |
|
Tipo |
Doença |
|
Imunomediadas |
Recorrentes Esclerose múltipla Neuromielite óptica
Monofásicas Neurite óptica Mielite transversa Encefalomielite aguda disseminada
|
|
Hereditárias |
Adrenoleucodistrofia Leucodistrofia metacromática
|
|
Metabólicas |
Deficiência de vitamina B12 Mielinólise pontina central
|
|
Infecciosas |
Leucoencefalopatia multifocal progressiva Panencefalite esclerosante subaguda |
Esclerose múltipla
As doenças imunomediadas formam a categoria mais comum de doenças desmielinizantes, sendo que a esclerose múltipla (EM) é a mais prevalente entre essas doenças. A esclerose múltipla se caracteriza pela presença de sintomas recorrentes ou progressivos, desmielinização no SNC e disseminação de lesões no espaço e no tempo.
Epidemiologia
A esclerose múltipla é mais comum em mulheres do que em homens. Estudos recentes sugerem que a proporção entre mulheres e homens está crescendo e atualmente é de 3 por 1.1 A esclerose múltipla é mais comum na América do Norte e na Europa e menos frequente, embora ainda esteja presente, na África e na Ásia. Nos Estados Unidos, onde há uma grande mistura de grupos étnicos, a esclerose múltipla é mais comum na população branca, menos comum entre afro-americanos e menos comum ainda na população de origem asiática. Usualmente, o início da doença ocorre na fase inicial da vida adulta, com incidência máxima na quarta década de vida. Há um gradiente geográfico, sendo que a prevalência de EM aumenta com a distância em relação ao equador, tanto no hemisfério norte como no hemisfério sul. Essas diferenças geográficas e étnicas que foram observadas possivelmente tenham origem em fatores genéticos, ambientais ou culturais.2 Embora não haja estudos recentes rigorosos sobre a prevalência de EM nos Estados Unidos, as últimas estimativas eram de aproximadamente 350.000 casos.3 A prevalência de EM no Canadá varia de 55 a quase 300 casos por 100.000 pessoas, dependendo da localização geográfica. 4
Etiologia/Genética
A etiologia exata da esclerose múltipla é desconhecida, apesar de evidências substanciais que sugerem ser uma doença autoimune. A patologia demonstra que há infiltração de leucócitos em áreas desmielinizadas; há evidências de produção de imunoglobulinas específicas no SNC estimuladas por antígenos; e todos os tratamentos eficazes disponíveis de EM produzem efeitos imunossupressivos ou imunomoduladores. A infecção causada pelo vírus de Epstein-Barr está fortemente associada ao risco de esclerose múltipla, embora o papel do vírus na incidência da doença seja obscuro.5 A deficiência de vitamina D e o tabagismo podem agravar a doença.6
|
*O autor e os editores expressam seus sinceros agradecimentos às contribuições do autor da versão anterior, Jerry Wolinsky, MD, no desenvolvimento e na redação deste capítulo. |
As informações financeiras estão no final deste capítulo, antes das referências.
A prevalência de esclerose múltipla aumenta em parentes próximos de pacientes com EM, sugerindo a presença de um papel importante desempenhado por fatores familiares.7-9
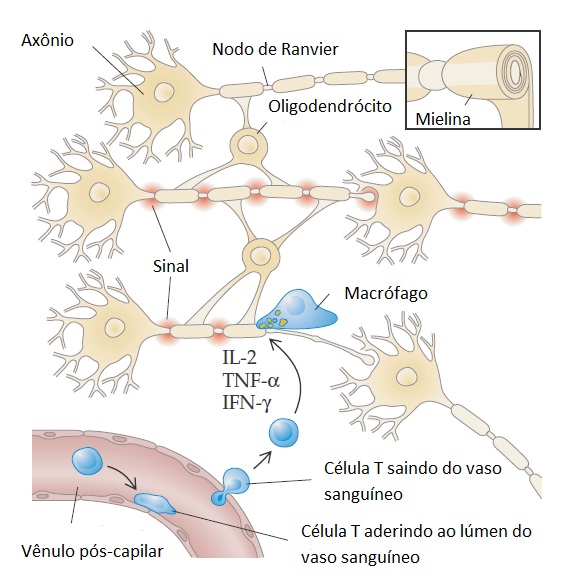
Figura 1 - Um único oligodendrócito pode servir de suporte para os segmentos intermodais de mielina para 60 ou mais axônios vizinhos. Uma das principais consequências da mielinização é a organização do microambiente dos axônios de grandes diâmetros. Os canais de sódio são agrupados em segmentos do axônio, entre os segmentos de mielina (nodos de Ranvier), e os canais de potássio são distribuídos de forma difusa sob os segmentos investidos de mielina dos axônios. Esse tipo de arranjo permite que os axônios mielinizados propaguem rapidamente os potenciais de ação a partir do corpo celular axonal, que se localiza em uma posição distal em relação a sua sinapse por meio de condução saltatória. A despolarização sequencial dos nodos de Ranvier resulta na propagação rápida dos impulsos neurais nas fibras nervosas com diâmetros maiores em velocidades superiores a 100 m/s. A parte superior da figura mostra um neurônio com axônio mielinizado. No canto direito, a imagem do neurônio é ampliada e cortada em uma secção transversal demonstrando a estrutura lamelar concêntrica da mielina. A parte central da figura mostra um neurônio com axônio mielinizado formando uma sinapse com outro neurônio. A figura mostra dois oligodendrócitos; cada célula faz a mielinização de múltiplos segmentos em mais de um axônio. O neurônio na parte inferior da figura foi parcialmente desmielinizado e é circundado por células T, que são responsáveis pela secreção das citocinas inflamatórias (interleucina 2 [IL-2], interferon gama [IFN-y] e fator de necrose tumoral alfa [TNF-alfa]) e pelos macrófagos, que são tiras mielínicas decapadas do axônio. Os macrófagos contêm resíduos de mielina em vacúolos fagocíticos. Há um bloqueio na condução no axônio desmielinizado. Um vaso sanguíneo em secção cruzada na parte inferior da figura mostra células T aderindo ao lúmen do vaso no interior do cérebro.
Estudos epidemiológicos simples realizados no Canadá demonstraram que o risco familiar é principalmente genético, sem nenhuma influência do compartilhamento de ambientes ou de estilos de vida.10,11 Uma análise cuidadosa dos padrões de recorrência familiar sugere o envolvimento de múltiplos genes, sendo que cada um deles é responsável por um pequeno efeito.12 Sabe-se já há algum tempo que a esclerose múltipla está associada ao haplótipo DR2 do complexo do antígeno leucocitário humano (HLA, do inglês human leukocyte antigen) com uma razão de chances de aproximadamente 2.13 Estudos recentes estabeleceram que muito provavelmente o gene que cria algum tipo de risco nesse complexo intimamente ligado é o DRB1*1501.14 Ultimamente, estudos cada vez mais ambiciosos e sofisticados de associação genômica ampla identificaram um grande número de outros genes, sendo que a maioria deles representa apenas um pequeno aumento no risco.15 A maioria desses genes foi identificada recentemente desempenha algum papel provável no sistema imune.
Patogênese
Acredita-se que a esclerose múltipla seja uma doença autoimune, ou pelo menos imunomediada, com fundamento em várias linhas diferentes de evidências. Assim como em muitas doenças autoimunes, o alvo antigênico da resposta autoimune é desconhecido, bem como os fatores predisponentes subjacentes e os estímulos desencadeadores imediatos da atividade da doença. O papel desempenhado pelas células T vem sendo foco de muita atenção, sendo que foi proposto um papel patogênico para as células auxiliares tipo 1 (Th1) ou, mais recentemente, para as células Th17. As células B também têm uma participação importante e cada vez mais valorizada.16 Possivelmente haja também um componente degenerativo na esclerose múltipla, principalmente em uma fase mais avançada da doença, quando a inflamação pode ser mínima, enquanto o processo de incapacitação continua a progredir.17,18
O exame macroscópico mostra que, nos casos de EM, o cérebro apresenta inúmeras placas endurecidas na substância branca, que deu origem ao termo “esclerose múltipla”. Usualmente essas placas são polivenulares. Elas podem ocorrer em qualquer local onde houver mielina, porém as localizações típicas são ao redor dos ventrículos, na substância branca subcortical imediata, nos nervos ópticos, no tronco cefálico e na medula espinhal. Um trabalho recente mostrou a importância e a abundância de lesões na substância cinza.19 Os exames microscópicos mostram que as placas ativas contêm infiltrados celulares de células T, células B e fagócitos com perda de mielina e com preservação dos axônios. As lesões crônicas apresentam escassez de infiltrado celular imune, com astrocitose e gliose. A transecção de axônios é comum, mesmo que eles sejam relativamente preservados em comparação com a mielina.20 Estudos patológicos cuidadosos mostraram que há padrões patológicos distintos nas lesões causadas por EM,21 sendo que alguns indivíduos têm uma deposição mais extensiva de imunoglobulina e de complemento ativado, sugerindo que há uma heterogeneidade na imunopatogênese subjacente.
Diagnóstico
O diagnóstico de esclerose múltipla deve ser feito por médicos especialistas e exige a confirmação de lesões múltiplas no SNC, com disseminação no espaço e no tempo. A presença de lesões deve ser evidente em mais de um local no SNC e devem ocorrer mais de uma vez. Inúmeros critérios diagnósticos foram propostos ao longo dos anos. A Tabela 2 apresenta uma lista dos critérios mais recentes.22 Os critérios mais antigos consideravam somente os sintomas e as descobertas clínicas, enquanto que os critérios atuais incorporam descobertas de IRM e, em alguns casos, permitem fazer o diagnóstico no momento do primeiro episódio clínico. Levando-se em consideração que não há nenhuma descoberta clínica patognomônica ou teste laboratorial, é imprescindível avaliar as evidências com muita cautela e, com frequência, o diagnóstico é feito alguns meses ou alguns anos após os sintomas iniciais. Outras doenças com apresentação semelhante devem ser excluídas por testes laboratoriais apropriados.
Manifestações clínicas
Os sintomas de esclerose múltipla podem surgir em padrões temporais distintos, que definem os subtipos clínicos da doença. A forma mais comum da doença é a esclerose múltipla remitente-recorrente (EMRR) que se caracteriza por recidivas ou exacerbações. As recidivas típicas se caracterizam pelo surgimento de novos sintomas neurológicos ou pelo agravamento de algum problema existente. Tipicamente, os sintomas evoluem em um período de um dia ou de alguns dias, seguido por um período de estabilidade e então pela resolução parcial ou total em algumas semanas. Embora a frequência das recidivas varie amplamente, a média é de uma recidiva em intervalos de 2 a 3 anos.
|
Tabela 2: Critérios Diagnósticos para Esclerose Múltipla | |
|
Apresentação Clínica |
Dados Adicionais Necessários para o Diagnóstico de EM |
|
Dois ou mais ataques
|
Nenhum |
|
Evidências clínicas objetivas de duas ou mais lesões, ou
|
|
|
Evidências clínicas de uma lesão com evidências históricas razoáveis e um ataque anterior
|
|
|
Dois ou mais ataques
|
Disseminação espacial demonstrada pela presença de lesões em T2 em pelo menos 2 de 4 regiões típicas de esclerose múltipla, ou
Ataque clínico posterior em um sítio diferente.
|
|
Evidências clínicas objetivas de uma lesão | |
|
Um ataque |
Disseminação no tempo demonstrada pela presença simultânea de lesões com intensificação e sem intensificação na IRM, ou
Nova T2 ou lesão com intensificação na IRM de acompanhamento, ou
Segundo ataque clínico
|
|
Evidências clínicas objetivas de duas ou mais lesões
| |
|
Um ataque |
Disseminação no espaço e no tempo, demonstrada por: No espaço: Lesões em T2 em pelo menos 2 de 4 regiões típicas de EM, ou
Segundo ataque clínico em um sítio diferente do SNC
No tempo: Presença simultânea de lesões com realce e sem realce na IRM, ou
Nova T2 ou lesão com realce na IRM,ou
Segundo ataque clínico
|
|
Evidências clínicas objetivas de uma lesão
| |
|
Progressão insidiosa sugerindo a presença de esclerose múltipla |
Progressão da doença por 1 ano e 2 entre os 3 fatores abaixo IRM do cérebro com 1 ou mais lesões em T2 em regiões típicas de EM IRM da medula espinhal com 2 ou mais lesões em T2 LCE com bandas oligoclonais ou |
LCE = líquido cerebroespinhal; IRM = imagens por ressonância magnética; EM = esclerose múltipla.
O agravamento da esclerose múltipla também pode ocorrer de uma forma progressiva, sem recidivas discretas. A EM progressiva se divide em esclerose múltipla progressiva primária (EMPP), na qual a doença evolui a partir do início, e esclerose múltipla progressiva secundária (EMPS), em que a progressão da doença começa após um curso recorrente inicial.
Há diversos sintomas de esclerose múltipla com algumas variações entre os pacientes, porém muitos deles são comuns e alguns são característicos [ver a Tabela 3].23,24 A perda visual unilateral causada por neurite óptica ocorre em algum momento em quase metade dos pacientes, sendo que em 29% dos pacientes é o sintoma de apresentação da doença. A diplopia na fixação ocular lateral ocorre com oftalmoplegia internuclear. A presença de fraqueza é comum e pode ocorrer em qualquer distribuição, incluindo hemiplegia, paraplegia ou monoplegia, porém, de maneira geral, afeta mais intensamente as extremidades inferiores que as extremidades superiores. Os sintomas sensoriais, incluindo perda de sensibilidade e parestesias, também são muito frequentes. Podem ocorrer dificuldades de coordenação, sendo que alterações na marcha são bastante frequentes. O sinal de Lhermite se caracteriza pela presença de parestesias que irradiam no sentido descendente das costas ou nos membros com flexão do pescoço, sendo que usualmente se correlaciona com alguma lesão na coluna espinhal cervical. A sensibilidade ao calor, com agravamento transitório ou surgimento de sintomas neurológicos com elevação da temperatura corporal causada por exercícios, febre ou exposição a ambientes quentes, sugere a presença de lesões desmielinizantes. A presença de fadiga, que pode limitar gravemente as atividades, é uma ocorrência comum, assim como a depressão. A função cognitiva, principalmente a memória de curto prazo, e a função executiva possivelmente sejam comprometidas. A função dos intestinos e da bexiga pode ser afetada, sendo que as queixas mais comuns são frequência urinária, urgência urinária e constipação.
Exame físico
As descobertas dos exames neurológicos de pacientes com esclerose múltipla refletem a perda funcional em tratos mielinizados. As lesões no nervo óptico podem produzir palidez no disco óptico nos exames fundoscópicos, defeitos pupilares aferentes, déficits na acuidade visual que não podem ser corrigidos com refração e problemas na visão colorida. As lesões no tronco cefálico podem causar anormalidades nos movimentos dos olhos, com a presença típica de oftalmoplegia internuclear. O examinador deve observar se a adução da fixação ocular lateral é ausente ou reduzida, porém sem convergência. As lesões nos tratos corticoespinhais causam fraqueza com espasticidade e hiperreflexia. De maneira geral, a perda sensorial é mais acentuada nas extremidades inferiores e usualmente afeta as grandes fibras mielinizadas que fazem a mediação da sensação de vibração e de posição articular, em comparação com as fibras menores que são mediadoras da dor e da temperatura. Ataxia é uma presença comum, sendo que a marcha anormal é frequente e poderá resultar em fraqueza, espasticidade, má coordenação, perda sensorial, ou uma combinação desses fatores.
Testes laboratoriais
Exames de sangue. Não há nenhum exame de sangue com valor preditivo positivo para esclerose múltipla, porém, usualmente, são realizados vários exames de sangue para excluir outras doenças no diagnóstico diferencial. Esses exames incluem anticorpos de Lyme, sorologia para sífilis, anticorpos para o vírus linfotrópico de células T humanas tipos 1 e 2 (HTLV-1/2, do inglês human T-cell lymphotropic vírus), anticorpos antinucleares, anticorpos de Sjögren, anticorpos anticitoplasmáticos de neutrófilos, enzima conversora da angiotensina, vitamina B12 e antiaquaporina-4 IgG. Costuma-se fazer também o teste dos níveis de vitamina D.
Exames de outros líquidos corporais. Com frequência, o exame do líquido cerebroespinhal (LCE) é bastante útil para diagnósticos precoces ou em casos duvidosos.25 A imunoeletroforese do LCE mostra a presença de bandas oligoclonais (BOCs) de imunoglobulina no LCE – porém não no soro – em até 95% de pacientes com esclerose múltipla.26
|
Tabela 3: Distribuição Aproximada de Deficiências Neurológicas no Início da Esclerose Múltipla e depois de 5 a 10 Anos de Doença
| ||
|
Deficiências |
No Início (%) |
5 a 10 Anos após o Início (%)
|
|
Deficiências cognitivas |
<5 |
30
|
|
Deficiências visuais |
20 a 30 |
50
|
|
Diplopia |
10 a 20 |
35
|
|
Fraqueza |
40 |
80
|
|
Ataxia |
5 a 20 |
65
|
|
Déficits sensoriais |
40 |
80
|
|
Sintomas intestinais ou na bexiga |
5 a 10 |
55
|
Embora sejam relativamente específicas para esclerose múltipla, as BOCs podem também estar presentes em infecções crônicas, tais como neurossífilis e panencefalite esclerosante subaguda (PEES) e, com menos frequência, em outras doenças inflamatórias do sistema nervoso central. De maneira geral, o índice IgG, calculado a partir das concentrações da IgG e da albumina no soro e no líquido cerebroespinhal (LCE), também é elevado nos casos de esclerose múltipla, indicando que há produção intratecal de anticorpos. Usualmente o nível proteico do LCE é normal ou ligeiramente elevado. O número de eritrócitos também é normal ou modestamente elevado, com predominância de linfócitos.
Estudos de Imagens. As imagens por ressonância magnética (IRM) do cérebro e da medula espinhal são extremamente úteis nos casos de esclerose múltipla (EM). A IRM desempenha um papel importante no diagnóstico de EM, além de ser extremamente útil para avaliar o prognóstico na fase inicial da doença. A maior parte dos pacientes já teve lesões múltiplas na substância branca do cérebro no momento dos sintomas iniciais, sendo que as varreduras seriais frequentes por IRM demonstram que as novas lesões cerebrais são 10 vezes mais frequentes que os novos sintomas. Embora a IRM seja bastante sensível, a aparência das lesões não é específica. As características consistentes com esclerose múltipla e que têm muita utilidade no diagnóstico [ver a Figura 2] incluem lesões periventriculares ovoides com os respectivos eixos longos perpendiculares aos ventrículos laterais, lesões subcorticais, presença de múltiplas lesões, lesões infratentoriais e lesões na medula espinhal.27,28 Usualmente, as lesões medulares são menos que um segmento vertebral na extensão longitudinal e envolvem apenas uma parte da secção transversa da medula. Uma proporção significativa de pacientes, particularmente na fase inicial da doença ou na doença ativa, possivelmente tenha algumas lesões que realçam com agentes de contraste à base de gadolínio. Se eventualmente estiver presente, o realce talvez seja pontilhado, difuso envolvendo toda a lesão ou ao redor da borda da lesão. Tipicamente o realce dura entre 4 a 6 semanas, indicando a presença de inflamação ativa com rompimento da barreira hematoencefálica. As IRMs seriais obtidas em intervalos de vários meses devem demonstrar a evolução das lesões ao longo do tempo com resolução do realce, redução no edema e surgimento de novas lesões.
Testes fisiológicos. As respostas auditivas evocadas do tronco encefálico, visuais e somatossensoriais, possivelmente mostrem que há um retardo na condução indicando a presença de desmielinização. Esses testes não são utilizados com muita frequência, mas, mesmo assim, eles são ocasionalmente úteis para demonstrar a presença de lesões subclínicas. A tomografia de coerência óptica é uma técnica sensível e não invasiva para medir a espessura retinal e atualmente é utilizada em pesquisas.
Diagnóstico diferencial
O diagnóstico diferencial de esclerose múltipla pode ser bastante extenso e varia de acordo com a apresentação clínica da doença.29,30 Embora o número de doenças que causam recidivas agudas com melhora espontânea em adultos jovens seja limitado, há inúmeras considerações em relação à mielopatia progressiva em adultos mais velhos. A Tabela 4 apresenta uma lista das considerações mais comuns que incluem doenças autoimunes ou inflamatórias, infecções e doenças genéticas.
Manejo
O manejo médico da esclerose múltipla se divide em tratamento para recidivas agudas; tratamentos modificadores da doença cuja finalidade é evitar a ocorrência de recidivas e a progressão da incapacidade; e tratamentos sintomáticos, com o objetivo de melhorar a qualidade de vida através do alívio de sintomas específicos.
Recidivas agudas
O tratamento padrão para recidivas agudas é um curso curto com altas doses de corticosteroides, como, por exemplo, 1.000 mg por dia de metilprednisolona por um período de 3 ou 5 dias. Nas situações em que forem administrados durante inflamações ativas, os corticosteroides possivelmente agilizem a recuperação de sintomas recentes, permitindo que os pacientes retornem mais cedo às atividades plenas. A melhora clínica pode iniciar ao final de um curso de cinco dias ou nas semanas seguintes. Nenhum benefício de longo prazo chegou a ser comprovado em termos de uma melhor recuperação ou de ampliação no intervalo da próxima exacerbação. Recidivas menores que não afetam as atividades cotidianas, como sintomas sensoriais leves, não precisam, necessariamente, ser tratadas. Os pacientes que já estiverem passando por um processo de melhora, ou que permaneceram estáveis por várias semanas, provavelmente não tenham nenhum benefício. De maneira geral, essas altas doses de corticosteroides são administradas por infusão intravenosa. As doses orais equivalentes (p.ex., 600 mg de prednisona duas vezes ao dia durante cinco dias) produzem benefícios e efeitos colaterais semelhantes,31,32 porém a falta de uma formulação oral com dosagem adequada e as ligações telefônicas de farmacêuticos apavorados complicam essa abordagem. Uma das opções é fazer uma breve redução gradual na dosagem oral de prednisona, iniciando com 60 mg por dia. É preferível interromper a administração de corticosteroides o mais rapidamente possível e raramente utilizar uma redução gradual na dosagem oral. A administração de doses mais baixas de corticosteroides, como, por exemplo, 1 mg/kg/dia de prednisona não produz nenhum benefício nos casos de recidiva de esclerose múltipla.
Terapia modificadora da doença para EMRR
Está comprovado que um número cada vez maior de tratamentos diminui a frequência ou o acúmulo de incapacidade em casos de esclerose múltipla remitente-recorrente (EMRR) (ver a Tabela 5). A maior parte dos pacientes com EMRR deve iniciar um desses tratamentos no momento do diagnóstico e permanecer na modalidade escolhida em longo prazo.33 A escolha do agente deve ser feita por um neurologista com experiência no tratamento de esclerose múltipla. As considerações mais relevantes incluem a gravidade anterior e a previsão da gravidade futura da doença, os benefícios e os riscos conhecidos do medicamento e as preferências dos pacientes.
Antes de iniciar a discussão sobre tratamentos individuais, é importante mencionar algumas dificuldades relevantes no processo de definição de tratamentos efetivos para esclerose múltipla. O curso clínico da esclerose múltipla varia entre os pacientes e é imprevisível em pacientes individuais, de modo que experiências anedóticas e testes não controlados não têm nenhuma utilidade. A demonstração da eficácia exige um número muito grande de pacientes e de seu acompanhamento em um período de tempo relativamente longo, fazendo-se a comparação entre pacientes tratados e pacientes de grupos de controle. Testes clínicos recentes incluíram várias centenas de pacientes por braço de tratamento, que foram acompanhados por um período de 2 a 3 anos. As medições usuais dos resultados clínicos incluem o número de recidivas, geralmente expresso como a taxa anual de recidivas, alteração na incapacidade medida pelo Kurtke Expanded Disability Status Score, e a pontuação no Multiple Sclerosis Functional Composite. A maioria dos testes inclui também várias medições por IRM da atividade da doença, incluindo a alteração no volume total da lesão, número e volume de novas lesões, número e volume de lesões com realce, e alteração no volume do cérebro.
O interferon beta foi o primeiro tratamento modificador da doença aprovado para esclerose múltipla. Diversos testes clínicos diferentes mostraram que o interferon beta diminui a taxa de recidivas em aproximadamente 30% em comparação com placebo e reduz significativamente a atividade da doença na IRM, medida pelo número de novas lesões ou de lesões com realce.34-36 Atualmente há diversas formas de interferon beta no mercado para aplicação específica no tratamento de esclerose múltipla, incluindo os medicamentos Betaseron, Rebif, Avonex e Extavia. Todos esses medicamentos são injetáveis, sendo que a via, a frequência e a dosagem variam entre as preparações.
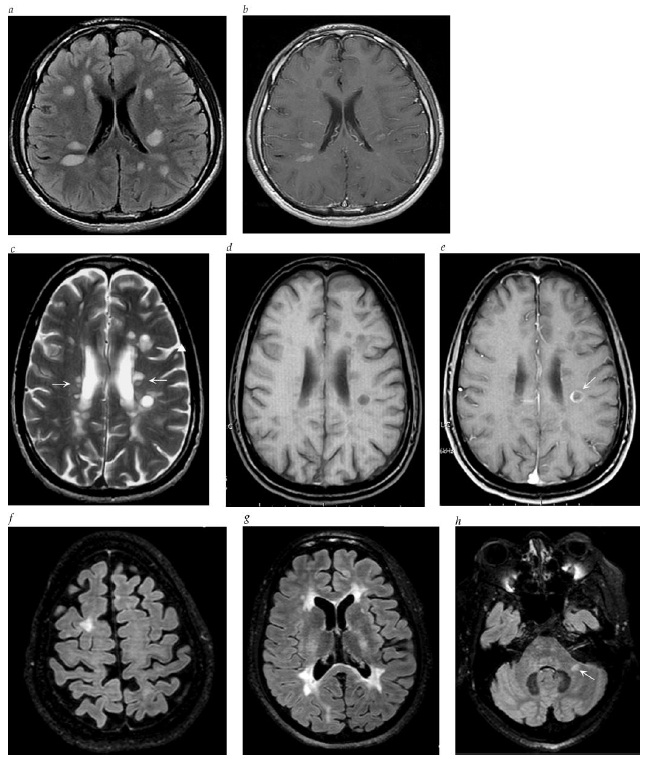
Figura 2 - Descobertas típicas de imagens por ressonância magnética em esclerose múltipla (EM) e neuromielite óptica (NMO). (a, b) Recuperação de inversão com atenuação do líquido (FLAIR, do inglês fluid-attenuated inversion recovery) e imagens pós-gadolínio em T1 de um paciente com EM e doença extensiva e ativa. Muitas das lesões evitadas são subagudas ou crônicas; (c) T2; (d) e (e) Imagens pós-gadolínio em T1 de um paciente diferente com EM. Este paciente tem também lesões periventriculares extensivas (setas) e uma lesão subcortical (ponta de seta). Algumas dessas lesões são escuras em T1. Uma lesão está com realce anular (seta). (f, g) Imagens FLAIR mostrando lesões subcorticais de EM. (f) Uma única lesão subcortical na substância branca frontal direita alta. (g) Lesões subcorticais extensivas que se localizam em sítios confluentes com as lesões periventriculares. (h) Imagens FLAIR mostrando uma lesão infratentorial no pedúnculo cerebelar médio esquerdo (seta).
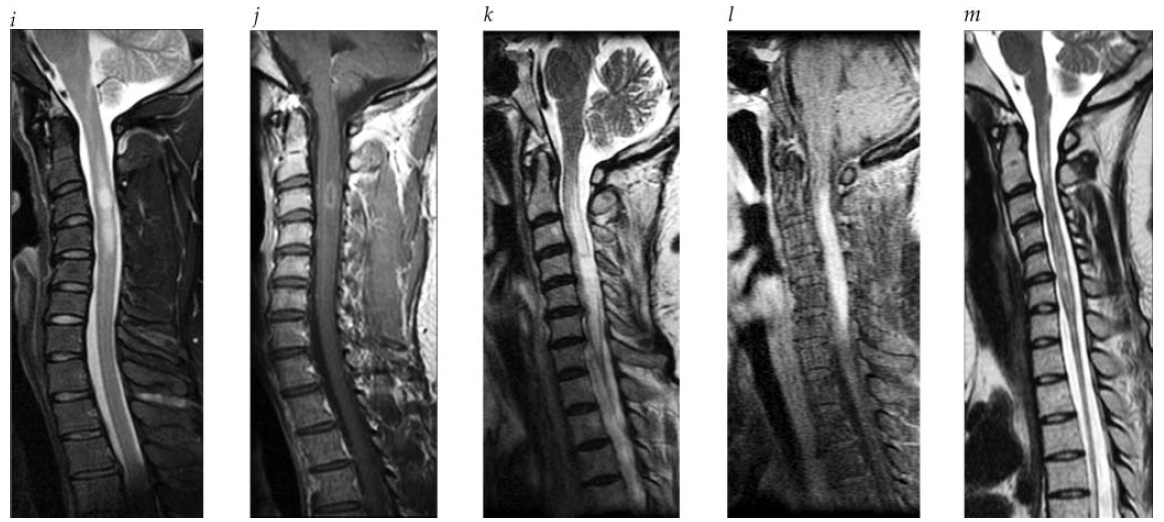
Figura 2 - Continuação. (i) T2; e (j) Imagens pós-gadolínio em T1 da coluna cervical em EM. Há duas lesões medulares, uma das quais está com realce. (k) Sagital em T2 e (l) Imagens pós-gadolínio em T1 da coluna cervical de um paciente com NMO em recidiva ativa. A lesão medular envolve quase toda a área transversal da medula de C2 a C6, com realce intenso do contraste de C3 a C6, ao contrário das lesões de EM demonstradas em “i”. (m) Imagens em T2 de um paciente diferente com NMO crônica. Há lesões difusas na medula cervical com atrofia grave na medula torácica.
O interferon beta tem um grande número de efeitos diferentes sobre o sistema imune, com vários mecanismos possíveis de ação. Os efeitos colaterais principais são sintomas semelhantes à influenza que ocorrem logo após a injeção do medicamento. Usualmente, esses efeitos colaterais são mais incômodos na fase inicial da terapia.
|
Tabela 4: Diagnóstico Diferencial de Esclerose Múltipla
|
|
Doenças inflamatórias ou autoimunes Neuromielite óptica Neurossarcoidose Vasculite cerebral Doença de Behçet Lúpus eritematoso sistêmico Síndrome de Sjögren Síndrome de Susac
|
|
Doenças infecciosas Doença de Lyme Neurossífilis HTLV-1/2 LMP
|
|
Doenças metabólicas Deficiência de vitamina B12 Deficiência de cobre
|
|
Doenças hereditárias Adrenoleucodistrofia Leucodistrofia metacromática CADASIL Doenças mitocondriais
|
CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts) = arteriopatia cerebral autossômica dominante com infartos subcorticais; HTVL-1/2 (human T cell lymphotropic vírus types I and II) = vírus linfotrópico de células T humanas tipos I e II; LMP = leucoencefalopatia multifocal progressiva.
O acetato de glatirâmer (AG) foi o segundo tratamento modificador da doença aprovado para esclerose múltipla.37 Trata-se de um copolímero randômico de ácido glutâmico, lisina, alanina e tirosina com uma faixa bem definida de pesos moleculares. O AG deve ser administrado uma vez ao dia por injeção subcutânea. O AG reduz a taxa de recidiva em 28% e diminui a atividade IRM por um valor semelhante.38 O acetato de glatirâmer possui vários mecanismos de ação, incluindo indução de células reguladoras e deslocamento de respostas imunes para uma via menos inflamatória. Os efeitos colaterais principais são reações nos sítios de injeção, que em geral são leves; e uma rara reação sistêmica pós-injeção.
O natalizumabe é um anticorpo monoclonal que se liga ao antígeno-4 de ativação muito tardia (VLA-4, do inglês very late activation antigen-4), um antígeno importante para a passagem de leucócitos da corrente sanguínea para o interior dos tecidos. O natalizumabe reduz a taxa de recidiva em 68%, lentifica o acúmulo de incapacidades e diminui a atividade da doença nas imagens por ressonância magnética.39 A administração é feita por infusão intravenosa em intervalos de quatro semanas. O efeito colateral mais problemático é a rara ocorrência de leucoencefalopatia multifocal progressiva (LMP) causada por infecção cerebral [ver abaixo]. O risco de complicações varia de acordo com tempo de duração do tratamento, sendo que o risco atinge o pico máximo no terceiro ano de tratamento e provavelmente permaneça elevado a partir de então. A estimativa atual de incidência de LMP no terceiro ano de tratamento é de 1,99 casos em cada 1.000 pacientes tratados. O risco é mais elevado em pacientes que foram previamente tratados com agentes citotóxicos. Aproximadamente 50% de adultos foram infectados pelo vírus JC, que é a causa de LMP, sendo que não existe atualmente no mercado nenhum teste sanguíneo para anticorpos do vírus JC.40 Esse tipo de teste ajuda a identificar pacientes com risco mais alto ou mais baixo de LMP.41 Nos Estados Unidos, a administração de natalizumabe é feita apenas através de programas rigorosos de vigilância.
O fingolimod é o medicamento mais recente que foi aprovado para tratamento de esclerose múltipla nos Estados Unidos. Trata-se de um agente oral administrado uma vez ao dia. O fingolimod bloqueia o receptor da esfingosina-1 fosfato, que é imprescindível para a saída de linfócitos do nodo linfático. O fingolimod reduz a taxa de recidiva em 54%, assim como o acúmulo de incapacidades, e diminui a atividade da doença nas imagens por ressonância magnética.42,43 Há inúmeros efeitos colaterais que precisam ser monitorados, incluindo a ocorrência de bradicardia com a primeira dose, edema macular em aproximadamente 0,3% e um risco ligeiramente elevado de determinados tipos de infecção. O fingolimod vem sendo comercializado desde outubro de 2010, sendo que sua segurança em longo prazo ainda não foi determinada. As complicações raras – porém graves – incluem eventos cardíacos ou infecciosos.
|
Tabela 5: Terapias Modificadoras da Doença para Esclerose Múltipla Recidivante-Remitente
| |||||
|
Medicação |
Via e Frequência |
Eficácia (%)* |
Efeitos Colaterais Graves |
Segurança† |
Conveniência† |
|
Interferon |
SC 3x/semana ou em dias alternados |
32 |
Semelhante a influenza, TFH, reações colaterais |
+++++ |
+ |
|
|
IM semanalmente |
18 |
|
|
|
|
Glatiramer |
SC diariamente |
29 |
Reações colaterais |
+++++ |
+
|
|
Natalizumabe |
IV a cada 4 semanas |
68 |
LMP |
++ |
++
|
|
Fingolimod |
Diariamente por via oral |
54 |
Edema macular, condução cardíaca, infecção.
|
+++ |
++++ |
|
Mitoxantrona |
IV a cada 3 meses |
63 |
Toxicidade cardíaca, LMA
|
+ |
++ |
LMA = leucemia mielocítica aguda; IM = intramuscular; TFH =teste da função hepática; LMP = leucoencefalopatia multifocal progressiva; SC = subcutânea;
*Redução na taxa de recidiva versus placebo.
†Classificação arbitrária: + pior segurança ou conveniência; +++++ melhor segurança ou conveniência.
A mitoxantrona, um agente citotóxico, foi aprovada para esclerose múltipla remitente-recorrente (EMRR) e esclerose múltipla progressiva secundária (EMPS). Usualmente, esse agente é administrado por via intravenosa, em intervalos de três meses, durante dois anos. A mitoxantrona diminui em 63% o número de recidivas e lentifica a progressão da doença.44 Esse medicamento produz inúmeros efeitos colaterais preocupantes, tais como imunossupressão, toxicidade cardíaca relacionada à dose e leucemia.45
Ao escolher um desses tratamentos para um paciente específico é importante saber quais produzem mais benefícios com menos efeitos colaterais, embora apenas alguns poucos estudos tenham feito a comparação direta entre esses agentes. Doses elevadas de alta frequência de interferon beta são modestamente mais eficazes do que a administração semanal do medicamento.46,47 Dois grandes estudos mostraram que não houve diferença significativa entre o interferon beta e o acetato de glatirâmer, seja em termos de eficácia ou de tolerabilidade.48,49 A comparação direta com o interferon mostrou que o fingolimod é significativamente mais eficaz.43 Embora nenhum estudo tenha feito a comparação entre o natalizumabe ou a mitoxantrona com outros agentes, muito provavelmente ambos sejam mais eficazes que o interferon ou o acetato de glatirâmer.
Geralmente na prática clínica utiliza-se o interferon beta ou o acetato de glatirâmer como tratamento de primeira linha na maior parte dos pacientes com esclerose múltipla remitente-recorrente (EMRR) [ver a Figura 3]. A grande maioria de pacientes se dá bem no primeiro tratamento e permanece nele por muito tempo. Nas situações em que o paciente não tolerar o tratamento inicial por causa dos efeitos colaterais, muda-se para o tratamento alternativo de primeira linha. Nos casos em que, a despeito do tratamento, o paciente apresentar atividade excessiva da doença, altera-se para o natalizumabe ou o fingolimod como tratamento de segunda linha. A escolha entre natalizumabe ou fingolimod está ligada ao fato de o paciente ter anticorpos do vírus JC ou qualquer uma das contraindicações relativas para o fingolimod e da tolerância do paciente para riscos. Raramente se utiliza a mitoxantrona ou qualquer outro agente citotóxico ou imunossupressivo, a não ser nos casos mais agressivos.
Provavelmente a estratégia de tratamento se altere no futuro próximo. Diversos agentes novos, incluindo alguns que aparentam ter uma excelente combinação de eficácia, segurança e conveniência, estão em processo de teste e aprovação.
Terapia modificadora da doença para EMPS e EMPP
Ao contrário da esclerose múltipla remitente-recorrente (EMRR), os tratamentos para doença progressiva são decisivamente inadequados. Somente um número limitado de estudos foi realizado para a doença progressiva. O interferon beta não lentifica a progressão da esclerose múltipla progressiva secundária (EMPS) e o acetato de glatirâmer não produz nenhum benefício nos casos de esclerose múltipla progressiva primária (EMPP). Possivelmente, a mitoxantrona tenha alguma utilidade na fase inicial da EMPS, porém devem-se levar em conta os efeitos colaterais. O rituximabe, um anticorpo monocloclonal que exaure os linfócitos B, produziu algum benefício somente no subgrupo de pacientes de EMPP com idade inferior a 50 anos e portadores de lesões com realce na IRM.50 A maior parte dos pacientes com EMPS permanece no tratamento que haviam iniciado para esclerose múltipla remitente-recorrente (EMRR), tendo em vista que é impossível prever quando deixarão de ter recidivas. Usualmente, pacientes com EMPP não estão fazendo qualquer tipo de terapia modificadora da doença.
Tratamento sintomático
Muitos dos sintomas comuns de esclerose múltipla respondem ao tratamento farmacológico [ver a Tabela 6].
Depressão e fadiga. A depressão ocorre em torno de 20% dos pacientes com esclerose múltipla, provavelmente com maior frequência do que a previsão de incidência de outras enfermidades crônicas. O tratamento de depressão em pacientes com esclerose múltipla não é diferente do tratamento de EM em outros tipos de pacientes. A fadiga ocorre em pelo menos 50% de todos os pacientes com esclerose múltipla e, às vezes, é o sintoma mais incapacitante. Geralmente ocorre nos casos de recidivas, porém também está presente entre ataques. A fadiga da esclerose múltipla é diferente da fadiga normal, seja pelo nível de gravidade ou pelo fato de que, com frequência, não está relacionada a qualquer tipo de atividade. O tratamento com amantadina produz pelo menos alívio parcial em aproximadamente 50% de pacientes com esclerose múltipla. Nos casos em que a amantadina não conseguir aliviar a fadiga, outros agentes que poderão ter alguma utilidade são modafinil, armodafinil, metilfenidato e inibidores seletivos de reabsorção da serotonina.
Espasticidade. Espasticidade é outro sintoma comum passível de tratamento. O aumento no tônus do extensor nas extremidades inferiores poderá compensar, até certo ponto, a fraqueza associada, embora a espasticidade excessiva iniba uma marcha fluida, resultando em espasmos dolorosos ou em contratura articular. De maneira geral, os agonistas do ácido y-aminobutírico (GABA, do inglês gamma-aminobutyric acid) com ação central, como o baclofeno, diminuem a espasticidade, porém preservam a marcha funcional ou a força dos membros inferiores, que é imprescindível para a transferência de peso. Usualmente, as doses efetivas de baclofeno variam de 30 a 120 mg por dia em doses divididas. Deve-se iniciar o tratamento com doses baixas que deverão ser tituladas para cima até um nível que maximize os benefícios. Outros medicamentos úteis incluem diazepam, clonazepam e tizanidina, um agonista alfa-adrenérgico. Pacientes selecionados com espasticidade grave, que não respondam ao tratamento por via oral, possivelmente se beneficiem com o tratamento à base de baclofeno intratecal ou de injeções de toxina botulínica seletiva.
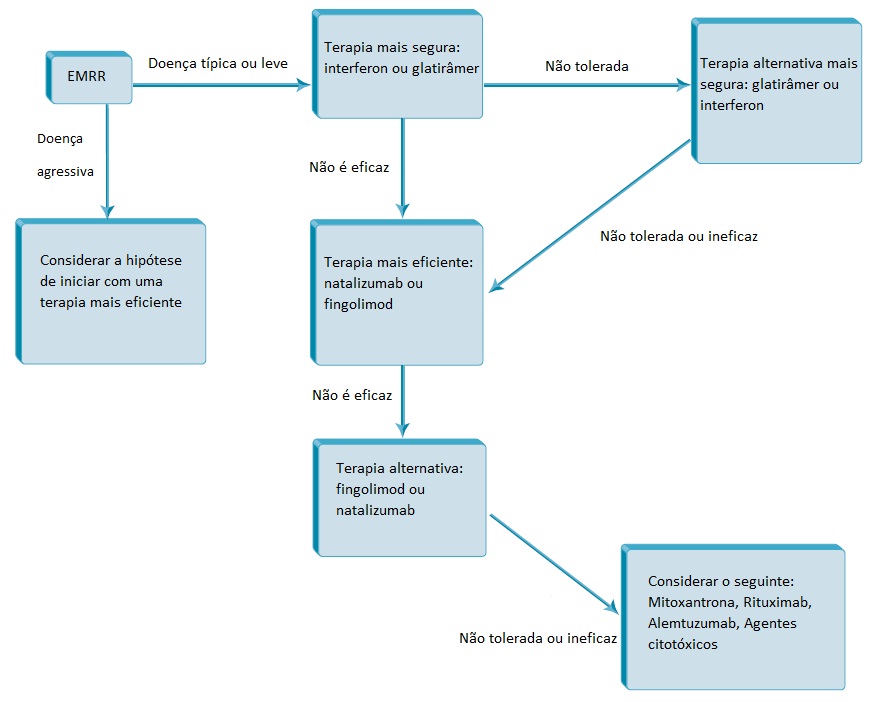
Figura 3 - Abordagem ao tratamento de esclerose múltipla remitente-recorrente (EMRR)
Disfunção da bexiga. A disfunção da bexiga é comum nos casos de esclerose múltipla. De maneira geral, os pacientes se apresentam com urgência e frequência, que poderão ser aliviadas com relaxantes específicos para músculos lisos e agentes anticolinérgicos. A determinação periódica de volumes urinários após a eliminação residual facilita a identificação de pacientes que poderão se beneficiar com os programas de autocateterização. Em alguns pacientes a patofisiologia pode ser mais complexa; os sintomas de dissinergia detrusor-esfíncter, a função hiperativa do detrusor e bexiga flácida podem ocorrer individualmente ou em diversas combinações e, em geral, oscilam ao longo do tempo. O encaminhamento para um urologista para a realização de estudos urodinâmicos possivelmente ajude a delinear esses vários padrões e sugira a adoção de abordagens racionais ao tratamento.
As infecções na bexiga são comuns em mulheres com esclerose múltipla. Com a perda da sensibilidade perineal, as infecções agudas na bexiga provavelmente não produzam disúria, porém, em vez disso, podem se tornar evidentes como uma deterioração global da função neurológica, que, eventualmente, poderá ser confundida com uma recidiva aguda. A avaliação de pacientes em recidiva deve incluir urinálise e cultura. A bacteriúria assintomática poderá exigir o tratamento concomitante de recidivas agudas com uma combinação de metilprednisolona e um antibiótico apropriado. Os casos de retenção urinária significativa, com incontinência por transbordamento, podem ser acompanhados por exagero de reflexo de espasticidade nos membros inferiores. De maneira geral, a instituição de um programa de autocateterização para pacientes afetados é mais eficiente para diminuir a espasticidade do que a farmacoterapia.
Dor. A dor nos casos de esclerose múltipla tem muitas causas. Pode ser secundária a estresses mecânicos atípicos resultantes de fraqueza ou espasticidade assimétrica. Medidas como uso de órteses apropriadas, agentes antiespasmódicos e rotinas de prática de exercícios, suplementadas por analgésicos simples, costumam ser bastante úteis. As síndromes de dor paroxística, que se caracterizam pela presença de neurologia do trigêmeo, geralmente respondem a baixas doses de carbamazepínicos ou de outros medicamentos antiepilépticos, principalmente a gabapentina. As disestesias distais são comuns e possivelmente também reajam a esses medicamentos. Medicamentos como pregabalina, duloxetina ou antidepressivos tricíclicos também podem ser extremamente úteis.
Ataxia e tremor de intenção. Essas manifestações são particularmente difíceis de gerenciar.
|
Tabela 5: Terapias Sintomáticas Selecionadas para Esclerose Múltipla* | ||
|
Indicação |
Medicamento |
Dosagem |
|
Fadiga |
Amantadina |
100 mg duas vezes ao dia ou três vezes ao dia |
|
|
Modafinil |
100 a 20mg duas vezes ao dia |
|
|
Armodafinil |
150 a 250 mg diariamente |
|
|
Metilfenidato |
5 mg duas vezes ao dia
|
|
Bexiga |
|
|
|
Urgência |
Oxibutinina |
2 mg duas a quatro vezes ao dia |
|
|
Tolerodina |
2 mg duas vezes ao dia |
|
|
Solifenacina |
5 a 10 mg todos os dias |
|
|
Darifenacina |
7,5 a 15 mg todos os dias
|
|
Dissinergia |
Fenoxibenzamina |
10 mg duas vezes ao dia ou 20 mg três vezes ao dia |
|
|
Clonidina |
0,1 mg duas vezes ao dia ou 0,2 mg três vezes ao dia |
|
|
Terazosina |
1 a 5 mg todos os dias
|
|
Retenção |
Cateterização intermitente |
Quatro ou mais vezes por dia |
|
|
Betanecol
|
10 mg três vezes ao dia ou 50 mg quatro vezes ao dia |
|
Espasticidade |
|
|
|
|
Baclofeno |
5 mg três vezes ao dia a 20 mg quatro vezes ao dia |
|
|
Tizanidina |
4 mg todos os dias a 12 mg três vezes ao dia |
|
|
Diazepam |
2 mg três vezes ao dia a 10 mg quatro vezes ao dia |
|
|
Clonazepam |
0,5 mg três vezes ao dia a 5 mg quatro vezes ao dia |
|
|
Clonidina (adjuvante do baclofeno)
|
0,1 mg duas vezes ao dia a 0,2 mg três vezes ao dia |
|
Dor |
|
|
|
Disestética |
Gabapentina |
100 a 600 mg três vezes ao dia |
|
|
Pregabalina |
50 mg três vezes ao dia a 200 mg três vezes ao dia |
|
|
Duloxetina |
20 a 60 mg diariamente |
|
|
Amitriptilina
|
50 a 150 mg todas as noites ao deitar |
|
Paroxismática |
Carbamazepina |
100 a 300 mg três vezes ao dia |
|
|
Fenitoína |
300 a 400 mg diariamente |
|
|
Misoprostol (neuralgia do trigêmeo) |
100 a 200 µg quatro vezes ao dia
|
|
Caminhada |
Dalfampridina |
10 mg duas vezes ao dia
|
|
Envolvimento pseudobulbar |
Dextrometorfan-quinidina |
20 mg/10 mg duas vezes ao dia
|
*Doses adultas usuais para medicações geralmente utilizadas no tratamento de síndromes de esclerose múltipla. Veja a referência apropriada para informações completas para prescrição, incluindo contraindicações, alertas, efeitos colaterais e início e término do tratamento.
Em geral, as intervenções farmacológicas são decepcionantes, embora, às vezes, nas doses máximas toleradas, o clonazepam ou a gabapentina produza alívio sintomático em pacientes com ataxia incapacitante nas extremidades superiores. Embora os riscos sejam significativos, a estimulação cerebral profunda possivelmente tenha alguma utilidade nos casos mais graves.
Caminhada. A dalfampridina bloqueia os canais de potássio e, teoricamente, deveria melhorar a condução de sinais nervosos através das áreas com mielina danificada. A velocidade de caminhada foi usada como medição de resultados primários em estudos clínicos que testaram a eficácia,51 embora a dalfampridina devesse também melhorar outros tipos de sintomas. A resposta ao tratamento é idiossincrática, sendo que alguns pacientes observam que há benefícios significativos e outros observam que não há nenhuma alteração. O efeito colateral mais preocupante é a possibilidade de provocar convulsões.
Envolvimento pseudobulbar. Alguns pacientes com esclerose múltipla podem ter riso ou choro inapropriado, que poderá ser debilitante sob o ponto de vista social. A combinação de doses fixas de dextrometorfano e quinidina diminuem significativamente a frequência dos episódios de riso ou choro.52 Os efeitos colaterais mais comuns desse tipo de tratamento são diarreia, vômito, dor estomacal ou tontura. Há diversas contraindicações e interações medicamentosas.
Prognóstico
Embora o curso da esclerose múltipla (EM) varie de paciente para paciente, o efeito da doença foi determinado em grandes coortes de pacientes com EM. O tempo médio desde o início da doença até uma incapacitação suficientemente grave que implique na necessidade de ajuda para deambulação é de 23 anos.53 O efeito da esclerose múltipla é mínimo no tempo de vida.54 Talvez para 10 a 15% de pacientes a EM tenha um curso relativamente benigno, sendo que os indivíduos apresentam incapacidade mínima ou nenhuma incapacidade em um período de 20 anos após o início dos sintomas. Nos casos de pacientes com doença remitente-recorrente, a frequência média de recidiva é de aproximadamente uma a cada dois anos. Não há fatores conhecidos que sejam preditivos do curso clínico em pacientes individuais, embora sexo feminino, idade mais jovem de início da doença e neurite óptica ou sintomas sensoriais sejam os sintomas mais frequentes que se apresentam e tendem a estar associados a prognósticos mais favoráveis. Recidivas frequentes nos primeiros dois anos são preditoras de incapacitação precoce.55 Imagens por ressonância magnética normais do cérebro em pacientes que se apresentam com condições como neurite óptica, mielite transversa parcial e síndromes no tronco cefálico associadas a esclerose múltipla são preditoras de um curso mais brando para a primeira década ou mais da doença. Provavelmente, o prognóstico seja melhor para pacientes diagnosticados com base em critérios mais recentes e que forem tratados na fase inicial da doença, em comparação com coortes históricos diagnosticados com base em evidências clínicas.56
Neuromielite óptica
Neuromielite óptica (NMO), ou doença de Devic, é uma síndrome rara e geralmente grave. A NMO tem muitas semelhanças com esclerose múltipla, embora recentemente tenha sido reconhecida como uma doença separada com descobertas clínicas e laboratoriais distintas. A neuromielite óptica se caracteriza pelo envolvimento sequencial ou simultâneo dos nervos ópticos e da medula espinhal, com preservação relativa do cérebro, ausência de faixas oligoclonais e lesões longitudinais extensivas na medula espinhal nos estudos por IRM [ver a Figura 2, “k” e “l”].58 De maneira geral, o curso clínico é agressivo, com acúmulo precoce de incapacidade significativa.58 A IgG da neuromielite óptica, um anticorpo sérico que se liga à aquaporina-4, uma proteína abundante do canal de água associada aos processos do pé astrocítico, é um teste laboratorial muito útil com especificidade excelente e sensibilidade moderada.59 O tratamento de NMO se baseia em relatos clínicos de séries de casos sem controle. Doses elevadas de corticosteroides e, caso seja necessário, plasmaferese de salvamento, são terapias que poderão ser usadas nos casos de ataques agudos. Possivelmente a imunossupressão sistêmica com azatioprina e corticoseteroides orais, rituximabe, mitoxantrona ou micofenolato evitem a ocorrência de recidivas. Aparentemente, o interferon e o natalizumabe não são medicamentos eficazes.
Neurite óptica
Neurite óptica é uma neuropatia óptica inflamatória aguda. Os sintomas principais são perda unilateral da visão e dor retrobulbar com o movimento dos olhos. Recomenda-se a IRM das órbitas para confirmar a presença de inflamação no nervo óptico; os resultados da IRM do cérebro são úteis para alertar os pacientes sobre o risco do desenvolvimento de esclerose múltipla. Indica-se o encaminhamento dos pacientes para um oftalmologista para excluir a eventual presença de uveíte, glaucoma ou outras causas oftalmológicas de perda visual. O diagnóstico diferencial inclui neuropatia óptica isquêmica anterior, que usualmente é indolor e em geral ocorre em pacientes com idade acima de 50 anos; doenças hereditárias, como a neuropatia óptica hereditária de Leber; neuropatia óptica tóxica ou nutricional; esclerose múltipla; e neuromielite óptica. O tratamento com administração intravenosa de 1 g/dia de metilprednisona, durante três dias, acompanhado por prednisona oral por 11 dias, agiliza a recuperação da visão, embora tenha pouco benefício residual depois de um ano.60 Mesmo sem tratamento, quase todos os pacientes recuperam a visão dentro de quatro semanas.
A relação entre neurite óptica e esclerose múltipla é controversa. Alguns especialistas consideram a neurite óptica uma entidade distinta, embora outros a considerem como parte de uma transição clínica gradual de esclerose múltipla. Mais da metade de todos os pacientes com esclerose múltipla têm neurite óptica em algum momento durante o curso da doença. Entre os pacientes que se apresentam com neurite óptica e sem nenhuma outra deficiência neurológica, quase 40% têm uma ou mais lesões cerebrais ovoides ou periventriculares visíveis nas imagens por ressonância magnética; ao final, a esclerose múltipla definitiva sob o ponto de vista clínico desenvolve em 50% de casos depois de 15 anos, com o risco de esclerose múltipla fortemente relacionada com a presença de lesões na IRM inicial do cérebro.61 Os pacientes com resultados totalmente normais na IRM ou na avaliação do líquido cerebroespinhal raramente progridem para esclerose múltipla.
Encefalomielite aguda disseminada
Encefalomielite aguda disseminada (EMAD) é uma síndrome monofásica que, em geral, é precedida por exantema viral, infecção respiratória superior ou vacinação. Os vírus associados com maior frequência são sarampo, paramixovírus, varicela, rubéola e vírus de Epstein-Barr. Geralmente o início é rápido e se caracteriza pela presença de sinais meníngeos, cefaleia, convulsões e alteração no estado mental. As deficiências neurológicas associadas são variáveis e incluem hemiplegia, paraplegia, perda sensorial, perda da visão e mielite transversa. A encefalomielite aguda disseminada pode ser fatal, embora a maior parte dos pacientes comece a se recuperar dentro de 2 a 4 semanas. Tipicamente, a EMAD ocorre no grupo com idade pediátrica e pode ser mais grave em adultos.62 Encefalomielite hemorrágica aguda é uma variante fulminante de encefalomielite aguda disseminada
As caraterísticas patológicas principais de EMAD são múltiplas áreas de inflamação perivascular e de desmielinização, sem evidências de infecção viral ativa. A encefalomielite aguda disseminada possivelmente seja causada por alguma resposta autoimune contra os antígenos da mielina disseminados por proteínas virais com reação cruzada. Usualmente, observam-se múltiplas lesões na substância branca nas imagens por ressonância magnética, sendo que a maioria dessas lesões realça com contraste. Utiliza-se com frequência o tratamento à base de corticosteroides, embora a eficácia dessa abordagem ainda não tenha sido comprovada por testes clínicos. A plasmaferese provavelmente seja uma opção útil. O prognóstico varia de acordo com o vírus desencadeador, porém em uma série de casos 89% dos pacientes tiveram uma boa recuperação.63
Mielite transversa
Mielite transversa aguda é uma síndrome de disfunção da medula espinhal com início rápido. Assim como a encefalomielite aguda disseminada, a mielite transversa poderá ocorrer depois de alguma infecção ou vacina, ou eventualmente sem nenhum fator precipitante discernível. Possivelmente seja também a apresentação inicial de esclerose múltipla ou de neuromielite óptica. Os sintomas incluem paraparesias, que inicialmente são flácidas e depois espásticas; perda de sensibilidade com nível sensorial do tronco; e disfunção intestinal e da bexiga. A dor lombar pode preceder os sintomas neurológicos, sendo que os sintomas sensoriais podem iniciar no sentido distal e ascendente. A medula torácica é afetada com mais frequência. O diagnóstico diferencial inclui outras causas de mielopatia aguda, como compressão medular por alguma lesão estrutural extradural, neoplasmas na medula espinhal, isquemia e lúpus eritematoso sistêmico. A IRM é útil para excluir lesões estruturais e para confirmar a presença de alguma lesão intramedular no nível da medula espinhal correspondente aos sintomas. As lesões causadas por mielite transversa aguda tipicamente são hiperintensas nas imagens ponderadas em T2; elas envolvem a maior parte da área transversal da medula e podem ser realçadas com contraste. As lesões podem produzir edema na medula espinhal. Embora nenhum tipo de tratamento seja comprovadamente benéfico, geralmente se utilizam corticosteroides, sendo que a ciclofosfamida e a plasmaferese foram algumas das sugestões apresentadas.64 O prognóstico é variável: um terço de pacientes apresenta bons resultados, um terço resultados razoáveis e um terço não consegue se recuperar. Choque espinhal, dor lombar e início catastrófico estão associados a resultados insatisfatórios.
Doenças desmielinizantes hereditárias
Adrenoleucodistrofia
Adrenoleucodistrofia é um distúrbio hereditário associado à desmielinização progressiva e à disfunção do córtex adrenal. O padrão de herança pode ser autossômico recessivo ou recessivo ligado ao X. A forma ligada ao X é causada pela mutação de gene que codifica uma proteína integral de membrana encontrada nos peroxissomos. A deficiência nesse tipo de gene provoca o acúmulo de ácidos graxos de cadeias muito longas (VLCFAs, do inglês very long chain fatty acids). O fenótipo pode variar consideravelmente, mesmo dentro da mesma família. Na forma infantil, os pacientes se apresentam com deficiências cognitivas, seguidas de uma deterioração neurológica rápida, sendo que a morte poderá ocorrer dentro de um período de 2 a 5 anos. A forma adulta, que se denomina adrenomieloneuropatia, se apresenta em pacientes com idade média de 28 anos como uma disfunção espinhal progressiva com paraparesia espástica, perda sensorial e sintomas nos intestinos e na bexiga. O envolvimento cerebral possivelmente seja mínimo. Somente a metade dos pacientes, cuja doença tenha iniciado na vida adulta, tem anormalidades nas imagens por ressonância magnética (IRM); com frequência essas anormalidades se localizam nos tratos corticoespinhais. A maior parte dos pacientes se apresenta com atrofia difusa na medula espinhal. O diagnóstico se baseia em uma combinação de envolvimento neurológico e adrenal, no histórico familiar e em níveis séricos elevados de VLCFAs. O tratamento dietético à base de ácidos graxos insaturados baixa o nível de VLCFAs, porém não afeta significativamente a progressão dos sintomas. O transplante de medula óssea possivelmente seja eficaz, caso seja feito antes do desenvolvimento de sintomas mais graves e com um doador compatível. Os resultados iniciais da terapia genética que utiliza células autólogas transfectadas com o gene para os alelos selvagens da enzima afetada são muito promissores.65 O prognóstico não é bom para pacientes com a forma infantil da doença. Usualmente, os pacientes com a doença de início adulto necessitam de ajuda para deambulação dentro de um período de tempo que varia de 10 a 15 anos; em um grande percentual de pacientes, as lesões cerebrais com progressão rápida se desenvolvem dentro do período de tempo de 5 a 10 anos após o início dos sintomas na medula espinhal.
Leucodistrofia metacromática
Leucodistrofia metacromática é um distúrbio autossômico recessivo que provoca a desmielinização de axônios no sistema nervoso central e periférico. Esse tipo de distúrbio é causado por mutações no gene da arisulfatase A, provocando um acúmulo de sulfatídeos com coloração metacromática. De maneira geral, o início ocorre na infância, sendo raro o início na vida adulta. Os sintomas da doença com início na vida adulta são anormalidades comportamentais progressivas, demência, ataxia e neuropatia. As imagens por ressonância magnética e a tomografia computadorizada do cérebro demonstraram a presença de atrofia e de anormalidades difusas na substância branca, principalmente nos lobos temporais. Confirma-se o diagnóstico pela medição dos níveis de atividade da arisulfatase A nos leucócitos do sangue periférico ou nos fibroblastos da pele. É extremamente importante fazer a distinção entre deficiência real de arisulfatase e um estado comum de pseudodeficîencia causado por um alelo com baixa atividade enzimática. Os sintomas de leucodistrofia metacromática são implacavelmente progressivos, sendo que o início precoce está associado a uma progressão mais rápida. A sobrevida média de indivíduos portadores da doença com início na vida adulta é de aproximadamente 12 anos. Nos casos em que for viável, o transplante alogênico de medula óssea pode ser muito eficaz; encontram-se em fase de investigação várias outras abordagens ao tratamento, incluindo transplante de células-tronco e terapia genética.
Doenças metabólicas desmielinizantes
Mielinólise pontina central (MPC) é uma síndrome em que ocorrem deficiências neurológicas após uma correção rápida da hiponatremia. Usualmente, a MPC ocorre na vida adulta entre adultos jovens até adultos na meia idade e, na maioria dos casos, está associada ao abuso de bebidas alcoólicas, má nutrição e transplante de órgãos.66 Geralmente os sinais e sintomas começam a surgir após o início da terapia de reposição de sódio e coexistem com alterações no estado mental, disartria e outros sinais de disfunção corticobulbar, e quadriplegia espástica. Embora, de maneira geral, as melhoras se manifestem aproximadamente duas semanas após o início dos sintomas, o grau de recuperação é variável. A descoberta mais impressionante no exame patológico é a presença de lesões desmielinizadas simétricas na ponte central. As lesões desmielinizadas podem ocorrer também em padrão relativamente simétrico em locais como os gânglios basais, tálamo, cápsula interna, substância branca subcortical e endotélio. Geralmente a IRM ponderada em T2 mostra a presença de lesões hiperintensas. Normalmente essas lesões não realçam com contraste. A mielinólise pontina poderá ocorrer também depois de transplantes de fígado. Não há tratamento específico após o desenvolvimento dos sintomas. A correção de longa duração e rápida da hiponatremia aumenta o risco de incidência de MPC; a velocidade recomendada de correção da hiponatremia não poderá exceder 10 a 12 mEq por um período de 24 horas.
Deficiência de vitamina B12
A deficiência de vitamina B12 resulta na desmielinização dos axônios do sistema nervoso central e periférico. Os tratos dorsal e lateral da substância branca da medula espinhal são mais afetados – característica que deu origem ao termo “degeneração subaguda combinada da medula espinhal”. Os sintomas mais comuns que se apresentam são parestesias, perda sensorial que inicia nos pés e progride no sentido proximal, e ataxia sensorial. O estado de fraqueza sempre inicia logo após a perda sensorial. Fatores como dificuldades de memória, irritabilidade e confusão ocorrem em uma pequena minoria de pacientes. De maneira geral, por ocasião do exame, os pacientes apresentam uma redução na sensação de vibração e de posição, que é pior nos pés que nas mãos, e poderão ter também paraparesia espástica. O exame patológico revela a presença de perda simétrica de mielina nas colunas posterior e lateral da medula espinhal e, eventualmente, desmielinização irregular na substância branca do cérebro. Com frequência, a IRM da medula espinhal mostra a presença de lesões na substância branca, que acabam desaparecendo com o tratamento. O diagnóstico se baseia nas descobertas clínicas e num nível sérico baixo de cobalamina. Macrocitose ou anemia é uma presença comum na maior parte dos pacientes, embora não possa ser utilizada no lugar da medição do nível de cobalamina como uma medida diagnóstica. A demonstração de níveis séricos elevados de ácido metilmalônico e de homocisteína total em pacientes com sintomas e um nível normal baixo de cobalamina confirma a presença de deficiência funcionalmente significativa de cobalamina.
A etiologia subjacente deverá ser investigada na eventual presença de deficiência de cobalamina. Aproximadamente 80% de pacientes com deficiência de cobalamina têm anemia perniciosa. A administração de cobalamina impede a progressão dos sintomas e produz melhoras clínicas na maior parte dos pacientes. O óxido nitroso impede o metabolismo da cobalamina e pode produzir sintomas semelhantes depois de exposições prolongadas; após uma única exposição, o óxido nitroso poderá revelar a presença de uma deficiência subclínica de cobalamina.
Desmielinização induzida por vírus
Leucoencefalopatia multifocal progressiva
A leucoencefalopatia multifocal progressiva (LMP) é uma doença desmielinizante grave causada por uma infecção viral oportunista de oligodendrócitos em pacientes imunocomprometidos. O agente causador é o vírus JC, um papovavírus onipresente que infecta a grande maioria da população antes de atingir a vida adulta e estabelece uma infecção latente nos rins. Em hospedeiros imunocomprometidos, o vírus pode ser reativado e infectar produtivamente os oligodendrócitos. A LMP pode ser observada em pacientes com lúpus sistêmico, AIDS e imunossupressão iatrogênica, incluindo o tratamento com natalizumabe e outros anticorpos monoclonais. Usualmente os pacientes se apresentam com deficiências neurológicas focais implacavelmente progressivas, como hemiparesia ou deficiência no campo visual, ou com alterações no estado mental. A IRM do cérebro mostra a presença de uma ou mais lesões; essas lesões são hiperintensas nas imagens ponderadas em T2 e hipointensas nas imagens ponderadas em T1. Não há nenhum efeito de massa e o realce por contraste é variável. A confirmação do diagnóstico pode ser feita por meio de biópsia do cérebro, com demonstração do vírus por uma hibridização in situ, por imuno-histoquímica ou pela amplificação da reação da cadeia de polimerase de sequências do vírus JC do líquido cerebroespinhal. Não existe nenhuma terapia antiviral que seja eficaz, de modo que o foco terapêutico é corrigir a causa da imunossupressão.
Panencefalite esclerosante subaguda
A panencefalite esclerosante subaguda (PEES) é uma complicação tardia rara de infecções causadas pelo vírus do sarampo. A PEES ocorre com mais frequência em pacientes cuja infecção inicial pelo vírus do sarampo ocorrer antes da idade de dois anos. O intervalo médio entre a infecção inicial e a PEES é de sete anos. O uso da vacina contra sarampo diminuiu drasticamente a incidência desse tipo de complicação nos países desenvolvidos. Usualmente o sintoma mais precoce é a deterioração cognitiva progressiva, que é acompanhada por disfunção motora e mioclônus associado a anormalidades eletroencefalográficas distintivas. O exame patológico revela a presença de infecção viral ativa no cérebro, sendo que o RNA e a proteína do vírus do sarampo podem ser detectados nos oligodendrócitos e nos neurônios, além de uma resposta inflamatória vigorosa. O curso da doença é progressivo, com remissões temporárias ocasionais. Não há nenhum tratamento satisfatório.
|
J. William Lindsay, MD, recebeu honorários de palestrante ou honorários de consultoria das empresas Pfizer, EMD Serono, Biogen Teva Pharmaceuticals. |
Referências
1. Orton SM, Herrera BM, Yee IM, et al. Sex ratio of multiple sclerosis in Canada: a longitudinal study. Lancet Neurol 2006;5:932–6.
2. Wallin MT, Culpepper WJ, Coffman P, et al. The Gulf War era multiple sclerosis cohort: age and incidence rates by race, sex and service. Brain 2012;135:1778–85.
3. Langer-Gould A, Brara SM, Beaber BE, Zhang JL. Incidence of multiple sclerosis in multiple racial and ethnic groups. Neurology 2013;80:1734–9.
4. Koch-Henriksen N, Sorensen PS. The changing demographic pattern of multiple sclerosis epidemiology. Lancet Neurol 2010;9:520–32.
5. Poppe AY, Wolfson C, Zhu B. Prevalence of multiple sclerosis in Canada: a systematic review. Can J Neurol Sci 2008;35:593–601.
6. Ascherio A, Munger KL. Environmental risk factors for multiple sclerosis. Part I: The role of infection. Ann Neurol 2007;61:288–99.
7. Ascherio A, Munger KL. Environmental risk factors for multiple sclerosis. Part II: Noninfectious factors. Ann Neurol 2007;61:504–13.
8. Sadovnick AD, Baird PA, Ward RH. Multiple sclerosis: updated risks for relatives. Am J Med Genet 1988;29:533–41.
9. O’Gorman C, Freeman S, Taylor BV, et al. Familial recurrence risks for multiple sclerosis in Australia. J Neurol Neurosurg Psychiatry 2011;82:1351–4.
10. Robertson NP, Fraser M, Deans J, et al. Age-adjusted recurrence risks for relatives of patients with multiple sclerosis. Brain 1996;119( Pt 2):449–55.
11. Ebers GC, Sadovnick AD, Risch NJ, Canadian Collaborative Study Group. A genetic basis for familial aggregation in multiple sclerosis. Nature 1995;377:150–1.
12. Ebers GC, Sadovnick AD, Dyment DA, et al. Parent-of-origin effect in multiple sclerosis: observations in half-siblings. Lancet 2004;363:1773–4.
13. Lindsey JW. Familial recurrence rates and genetic models of multiple sclerosis. Am J Med Genet A 2005;135:53–8.
14. Hafler DA, Compston A, Sawcer S, et al. Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med 2007;357:851–62.
15. Caillier SJ, Briggs F, Cree BA, et al. Uncoupling the roles of HLA-DRB1 and HLA-DRB5 genes in multiple sclerosis. J Immunol 2008;181:5473–80.
16. Sawcer S, Hellenthal G, Pirinen M, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011;476:214–9.
17. von Budingen HC, Bar-Or A, Zamvil SS. B cells in multiple sclerosis: connecting the dots. Curr Opin Immunol 2011;23:713–20.
18. Trapp BD, Nave KA. Multiple sclerosis: an immune or neurodegenerative disorder? Annu Rev Neurosci 2008;31:247–69.
19. Leray E, Yaouanq J, Le Page E, et al. Evidence for a two-stage disability progression in multiple sclerosis. Brain 2010;133:1900–13.
20. Lucchinetti CF, Popescu BF, Bunyan RF, et al. Inflammatory cortical demyelination in early multiple sclerosis. N Engl J Med 2011;365:2188–97.
21. Trapp BD, Peterson J, Ransohoff RM, et al. Axonal transection in the lesions of multiple sclerosis. N Engl J Med 1998;338:278–85.
22. Lucchinetti C, Bruck W, Parisi J, et al. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol 2000;47:707–17.
23. Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol 2011;69:292–302.
24. Poser S, Wikstrom J, Bauer HJ. Clinical data and the identification of special forms of multiple sclerosis in 1271 cases studied with a standardized documentation system. J Neurol Sci 1979;40:159–68.
25. Shepherd DI. Clinical features of multiple sclerosis in north-east Scotland. Acta Neurol Scand 1979;60:218–30.
26. Awad A, Hemmer B, Hartung HP, et al. Analyses of cerebrospinal fluid in the diagnosis and monitoring of multiple sclerosis. J Neuroimmunol 2010;219:1–7.
27. Link H, Huang YM. Oligoclonal bands in multiple sclerosis cerebrospinal fluid: an update on methodology and clinical usefulness. J Neuroimmunol 2006;180:17–28.
28. Swanton JK, Rovira A, Tintore M, et al. MRI criteria for multiple sclerosis in patients presenting with clinically isolated syndromes: a multicentre retrospective study. Lancet Neurol 2007;6:677–86.
29. Montalban X, Tintore M, Swanton J, et al. MRI criteria for MS in patients with clinically isolated syndromes. Neurology 2010;74:427–34.
30. Miller DH, Weinshenker BG, Filippi M, et al. Differential diagnosis of suspected multiple sclerosis: a consensus approach. Mult Scler 2008;14:1157–74.
31. Kelly SB, Chaila E, Kinsella K, et al. Using atypical symptoms and red flags to identify non-demyelinating disease. J Neurol Neurosurg Psychiatry 2012;83:44–8.
32. Burton JM, O’Connor PW, Hohol M, Beyene J. Oral versus intravenous steroids for treatment of relapses in multiple sclerosis. Cochrane Database Syst Rev 2012;(12):CD006921.
33. Goodin DS, Frohman EM, Garmany GP Jr, et al. Disease modifying therapies in multiple sclerosis: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology and the MS Council for Clinical Practice Guidelines. Neurology 2002;58:169–78.
34. The IFNB Multiple Sclerosis Study Group, the University of British Columbia MS/MRI Analysis Group. Interferon beta-1b in the treatment of multiple sclerosis: final outcome of the randomized controlled trial. Neurology 1995;45:1277–85.
35. Jacobs LD, Cookfair DL, Rudick RA, et al. Intramuscular interferon beta-1a for disease progression in relapsing multiple sclerosis. Ann Neurol 1996;39:285–94.
36. PRISMS Study Group. Randomised double-blind placebo-controlled study of interferon beta-1a in relapsing/remitting multiple sclerosis. Lancet 1998;352:1498–504.
37. Johnson KP, Brooks BR, Cohen JA, et al. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind placebo-controlled trial. Neurology 1995;45:1268–76.
38. Comi G, Filippi M, Wolinsky JS, European/Canadian Glatiramer Acetate Study Group. European/Canadian multicenter, double-blind, randomized, placebo-controlled study of the effects of glatiramer acetate on magnetic resonance imaging—measured disease activity and burden in patients with relapsing multiple sclerosis. Ann Neurol 2001;49:290–7.
39. Polman CH, O’Connor PW, Havrdova E, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med 2006;354:899–910.
40. Bozic C, Richman S, Plavina T, et al. Anti-John Cunnigham virus antibody prevalence in multiple sclerosis patients: baseline results of STRATIFY-1. Ann Neurol 2011;70:742–50.
41. Gorelik L, Lerner M, Bixler S, et al. Anti-JC virus antibodies: implications for PML risk stratification. Ann Neurol 2010;68:295–303.
42. Kappos L, Radue EW, O’Connor P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med 2010;362:387–401.
43. Cohen JA, Barkhof F, Comi G, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med 2010;362:402–15.
44. O’Connor P, Wolinsky JS, Confavreux C, et al. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med 2011;365:1293–303.
45. Fox RJ, Miller DH, Phillips JT, et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med 2012;367:1087–97.
46. Gold R, Kappos L, Arnold DL, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med 2012;367:1098–107.
47. Ermis U, Weis J, Schulz JB. PML in a patient treated with fumaric acid. N Engl J Med 2013;368:1657–8.
48. van Oosten BW, Killestein J, Barkhof F, et al. PML in a patient treated with dimethyl fumarate from a compounding pharmacy. N Engl J Med 2013;368:1658–9.
49. Hartung HP, Gonsette R, Konig N, et al. Mitoxantrone in progressive multiple sclerosis: a placebo-controlled, double-blind, randomised, multicentre trial. Lancet 2002;360:2018–25.
50. Marriott JJ, Miyasaki JM, Gronseth G, O’Connor PW. Evidence report: The efficacy and safety of mitoxantrone (Novantrone) in the treatment of multiple sclerosis: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 2010;74:1463–70.
51. Cohen JA, Coles AJ, Arnold DL, et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet 2012;380:1819–28.
52. Coles AJ, Twyman CL, Arnold DL, et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial. Lancet 2012;380:1829–39.
53. Durelli L, Verdun E, Barbero P, et al. Every-other-day interferon beta-1b versus once-weekly interferon beta-1a for multiple sclerosis: results of a 2-year prospective randomised multicentre study (INCOMIN). Lancet 2002;359:1453–60.
54. Panitch H, Goodin DS, Francis G, et al. Randomized, comparative study of interferon beta-1a treatment regimens in MS: The EVIDENCE Trial. Neurology 2002;59:1496–506.
55. Mikol DD, Barkhof F, Chang P, et al. Comparison of subcutaneous interferon beta-1a with glatiramer acetate in patients with relapsing multiple sclerosis (the REbif vs Glatiramer Acetate in Relapsing MS Disease [REGARD] study): a multicentre, randomised, parallel, open-label trial. Lancet Neurol 2008;7:903–14.
56. O’Connor P, Filippi M, Arnason B, et al. 250 microg or 500 microg interferon beta-1b versus 20 mg glatiramer acetate in relapsing-remitting multiple sclerosis: a prospective, randomised, multicentre study. Lancet Neurol 2009;8:889–97.
57. Hawker K, O’Connor P, Freedman MS, et al. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Ann Neurol 2009;66:460–71.
58. Goodman AD, Brown TR, Krupp LB, et al. Sustained-release oral fampridine in multiple sclerosis: a randomised, double-blind, controlled trial. Lancet 2009;373:732–8.
59. Pioro EP, Brooks BR, Cummings J, et al. Dextromethorphan plus ultra low-dose quinidine reduces pseudobulbar affect. Ann Neurol 2010;68:693–702.
60. Confavreux C, Vukusic S, Moreau T, Adeleine P. Relapses and progression of disability in multiple sclerosis. N Engl J Med 2000;343:1430–8.
61. Kingwell E, van der Kop M, Zhao Y, et al. Relative mortality and survival in multiple sclerosis: findings from British Columbia, Canada. J Neurol Neurosurg Psychiatry 2012;83:61–6.
62. Scalfari A, Neuhaus A, Degenhardt A, et al. The natural history of multiple sclerosis, a geographically based study 10: relapses and long-term disability. Brain 2010;133:1914–29.
63. Fisniku LK, Brex PA, Altmann DR, et al. Disability and T2 MRI lesions: a 20-year follow-up of patients with relapse onset of multiple sclerosis. Brain 2008;131:808–17.
64. Lucchinetti CF, Gavrilova RH, Metz I, et al. Clinical and radiographic spectrum of pathologically confirmed tumefactive multiple sclerosis. Brain 2008;131:1759–75.
65. Elenein RG, Sharer LR, Cook SD, et al. A second case of Marburg’s variant of multiple sclerosis with vasculitis and extensive demyelination. Mult Scler 2011;17:1531–8.
66. Wingerchuk DM, Lennon VA, Pittock SJ, et al. Revised diagnostic criteria for neuromyelitis optica. Neurology 2006;66:1485–9.
67. Collongues N, Marignier R, Zephir H, et al. Neuromyelitis optica in France: a multicenter study of 125 patients. Neurology 2010;74:736–42.
68. Lennon VA, Wingerchuk DM, Kryzer TJ, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 2004;364:2106–12.
69. Jacob A, McKeon A, Nakashima I, et al. Current concept of neuromyelitis optica (NMO) and NMO spectrum disorders. J Neurol Neurosurg Psychiatry 2013;84:922–30.
70. Beck RW, Cleary PA, Anderson MM Jr, et al. A randomized, controlled trial of corticosteroids in the treatment of acute optic neuritis. The Optic Neuritis Study Group. N Engl J Med 1992;326:581–8.
71. Optic Neuritis Study Group. Multiple sclerosis risk after optic neuritis: final optic neuritis treatment trial follow-up. Arch Neurol 2008;65:727–32.
72. Ketelslegers I, Visser I, Neuteboom R, et al. Disease course and outcome of acute disseminated encephalomyelitis is more severe in adults than in children. Mult Scler 2011;17:441–8.
73. Tenembaum S, Chamoles N, Fejerman N. Acute disseminated encephalomyelitis: a long-term follow-up study of 84 pediatric patients. Neurology 2002;59:1224–31.
74. Greenberg BM, Thomas KP, Krishnan C, et al. Idiopathic transverse myelitis: corticosteroids, plasma exchange, or cyclophosphamide. Neurology 2007;68:1614–7.
75. Akman-Demir G, Serdaroglu P, Tasci B. Clinical patterns of neurological involvement in Behcet’s disease: evaluation of 200 patients. The Neuro-Behcet Study Group. Brain 1999;122(Pt 11):2171–82.
76. Al-Araji A, Kidd DP. Neuro-Behcet’s disease: epidemiology, clinical characteristics, and management. Lancet Neurol 2009;8:192–204.
77. Pawate S, Moses H, Sriram S. Presentations and outcomes of neurosarcoidosis: a study of 54 cases. QJM 2009;102:449–60.
78. Segal BM. Neurosarcoidosis: diagnostic approaches and therapeutic strategies. Curr Opin Neurol 2013;26:307–13.
79. Costello DJ, Eichler AF, Eichler FS. Leukodystrophies: classification, diagnosis, and treatment. Neurologist 2009;15:319–28.
80. Kohler W. Leukodystrophies with late disease onset: an update. Curr Opin Neurol 2010;23:234–41.
81. Gladstone JP, Dodick DW. Migraine and cerebral white matter lesions: when to suspect cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). Neurologist 2005;11:19–29.
82. Pantoni L, Pescini F, Nannucci S, et al. Comparison of clinical, familial, and MRI features of CADASIL and NOTCH3-negative patients. Neurology 2010;74:57–63.