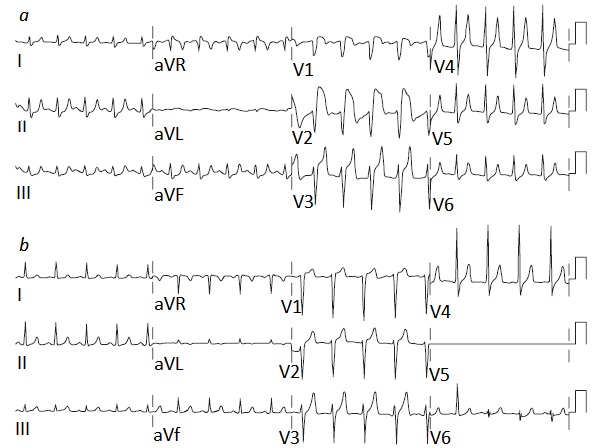
(Carregando Índice)... (Carregando Índice)... |
Última revisão: 25/10/2013
Comentários de assinantes: 0
Stuart L. Linas MD, FACP
Professor of Medicine, University of Colorado Denver, Denver Health Medical Center, Denver, CO
Artigo original: Linas SL. Disorders of acid-base and potassium balance. ACP Medicine. 2009;1-23.
[The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2011 Decker Intellectual Properties Inc. All Rights Reserved.]
Agradecimentos: O autor deseja agradecer a Robert M. Black, MD, por sua contribuições para a versão anterior deste capítulo, que nos serviu de base para esta atualização. Figura 1 – Janet Betries. Figuras 2 a 6 – Tom Moore. Figura 8 – Os eletrocardiogramas foram cordialmente fornecidos pelo dr. David Spodick, professor de medicina na University of Massachusetts Medical School, Worcester (Estados Unidos).
Tradução: Soraya Imon de Oliveira.
Revisão técnica: Dr. Rodrigo Antonio Brandão Neto.
Obs: O símbolo [] se refere a concentração.
O pH do sangue normalmente é mantido em 7,38 a 7,42. Qualquer desvio desta faixa indica uma alteração na concentração de íons hidrogênio ([H+]), pois o pH sanguíneo consiste no logaritmo negativo de [H+] que é expresso pela seguinte equação:
pH = -log10[H+]
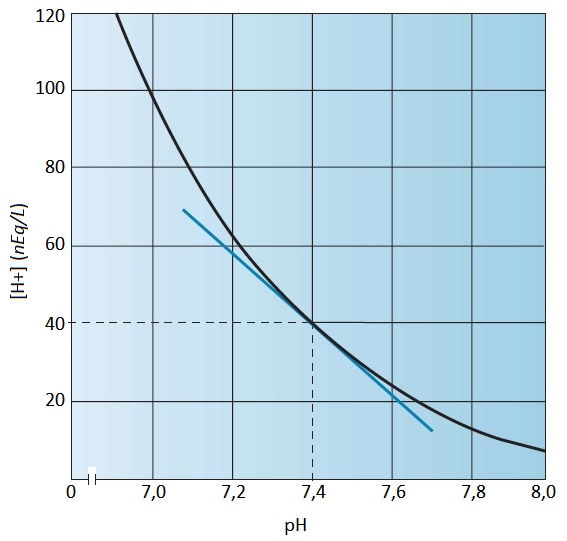
A [H+] no pH sanguíneo fisiológico de 7,4 é igual a 40 nEq/L [Figura 1]. Um aumento de [H+] (uma queda do pH sanguíneo) é denominado acidemia. Uma diminuição de [H+] (elevação do pH do sangue) é denominada alcalemia. Os distúrbios causadores destas alterações de pH sanguíneo são a acidose e a alcalose, respectivamente. Como as anormalidades do metabolismo ácido-base com frequência estão associadas ao desequilíbrio de potássio, as abordagens clínicas da hipo e da hipercalemia também serão discutidas neste capítulo.

Figura 1. Relação existente entre a concentração plasmática de íons hidrogênio ([H+]) e o pH sanguíneo (pH = –log10[H+]).
A dieta consumida por um adulto normal gera um excedente de 70 a 100 mEq de ácido que deve ser eliminado diariamente. A falha em eliminar este excedente resulta na queda persistente do pH sanguíneo, em decorrência da elevação da concentração plasmática de íons H+. O equilíbrio da homeostasia ácido-base é mantido, em parte, pela relação existente entre a tensão de dióxido de carbono arterial (PaCO2) e a concentração plasmática de bicarbonato ([HCO3–]), que pode ser demonstrada pela equação a seguir (uma expressão não logarítmica da equação de Henderson-Hasselbach):1
[H+] = 24 X PaCO2/[HCO3–]
Uma queda da [HCO3–] plasmática causada por perdas de bicarbonato gastrintestinais ou renais também aumenta a [H+] e diminui o pH sanguíneo.
A [HCO3–] plasmática é normalmente mantida em torno de 25 mEq/L por meio da reabsorção diária da carga de bicarbonato filtrado (cerca de 4.500 mEq) pelos rins. Se o bicarbonato filtrado não fosse reabsorvido, a [HCO3–] plasmática e o pH sanguíneo cairiam. Portanto, a manutenção de uma [HCO3–] plasmática normal requer a reabsorção de essencialmente todo o bicarbonato filtrado através dos capilares glomerulares a cada dia.
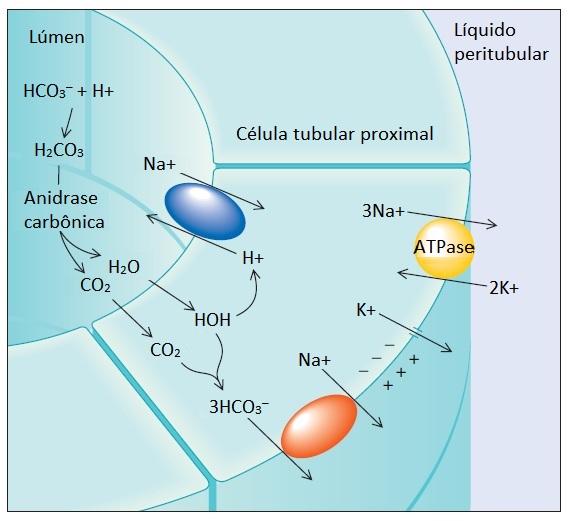
A maior parte da reabsorção do bicarbonato (quase 90%) ocorre no túbulo contornado proximal [Figura 2]. Em contraste, o néfron distal recupera muito pouco bicarbonato. A diferença resulta de um processo complexo, que é facilitado pela maior quantidade de anidrase carbônica existente no lúmen do túbulo proximal.
Figura 2. A reabsorção de bicarbonato ao nível tubular proximal é ativada pela ATPase de Na+/K+ existente na membrana celular peritubular. A troca de K+ peritubular por Na+ intracelular mantém a [Na+] intracelular baixa, permitindo que o Na+ se mova no sentido de seu gradiente de concentração menor e siga do lúmen tubular através do contratransportador de Na+/K+ para dentro da célula. O HCO3– combina-se ao H+ secretado para formar HCO3–. A rápida dissociação do H2CO3 em CO2 e H2O na presença de anidrase carbônica luminal permite a movimentação para dentro da célula, onde ocorre a dissociação. Por fim, o H+ reabsorvido é secretado em troca de Na+, enquanto o HCO3– segue o gradiente elétrico e se move do interior da célula para o espaço peritubular, onde é reabsorvido dentro da circulação sistêmica.
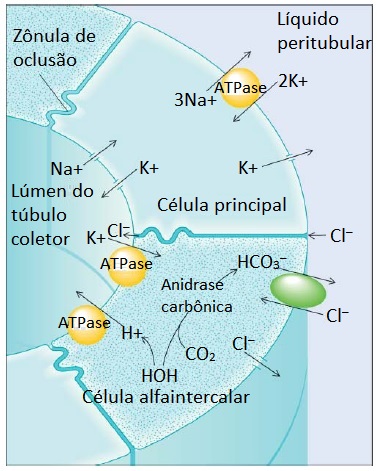
Além de reabsorverem essencialmente todo o bicarbonato filtrado, os rins excretam a carga diária de ácidos da dieta, derivada sobretudo dos aminoácidos que contêm enxofre. Os íons hidrogênio excretados na urina final são secretados principalmente ao nível dos túbulos coletores [Figura 3]. Este processo secretório é facilitado indiretamente pela aldosterona e, de forma direta, pelas células tubulares renais acidossensíveis.2
Figura 3. A secreção de H+ a partir do túbulo coletor cortical está indiretamente ligada à reabsorção de Na+. O potássio intracelular é trocado por sódio nas células principais, enquanto o H+ é ativamente transportado por uma ATPase a partir das células alfaintercalares. A aldosterona estimula a secreção de H+ ao entrar na célula principal, onde abre os canais de Na+ existentes na membrana luminal e aumenta a atividade da APTase de Na+/K+. Então, a movimentação de Na+ catiônico para dentro das células principais cria uma carga negativa junto ao lúmen tubular. O K+ move-se a partir das células principais, enquanto o H+ se desloca a partir das células alfa intercalares seguindo o gradiente eletroquímico e para dentro o lúmen. (Quando o K+ é depletado, a secreção de K+ pelas células principais é diminuída e a reabsorção do K+ através de uma ATPase existente nas células alfa intercalares é estimulada.) A aldosterona parece também estimular diretamente a ATPase de H+ nas células intercalares, intensificando a secreção de H+. O HCO3 é devolvido para o sangue através da membrana peritubular, em troca de Cl–, mantendo assim a eletroneutralidade.
A carga ácida diária é excretada dentro dos túbulos coletores pelas ATPases de H+, que estão localizadas na membrana luminal das células intercaladas. Este processo de secreção é inibido por uma quantidade trivial de íons hidrogênio livre que faz o pH da urina cair para baixo dos níveis críticos de 4,0 a 4,5. Esta limitação normalmente é superada pela presença de tampões urinários que se combinam aos íons hidrogênio livre e, assim, possibilitam a secreção contínua de ácido. Existem vários tampões urinários, dos quais o mais importante é a amônia, por ser o único tampão cuja concentração pode aumentar substancialmente na presença de uma carga ácida. As limitações da capacidade de gerar amônia urinária adequada, como se observa na insuficiência renal, geralmente levam ao desenvolvimento de acidose.
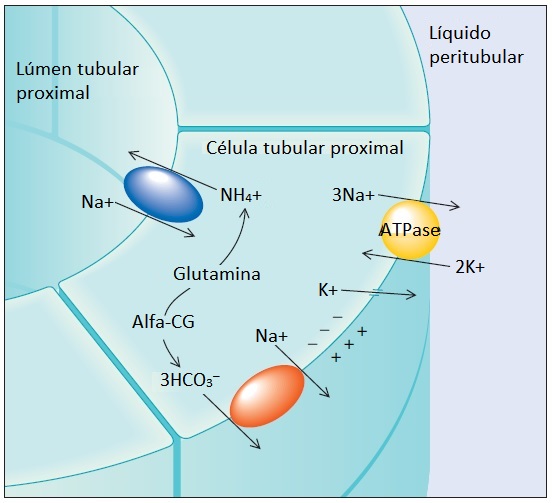
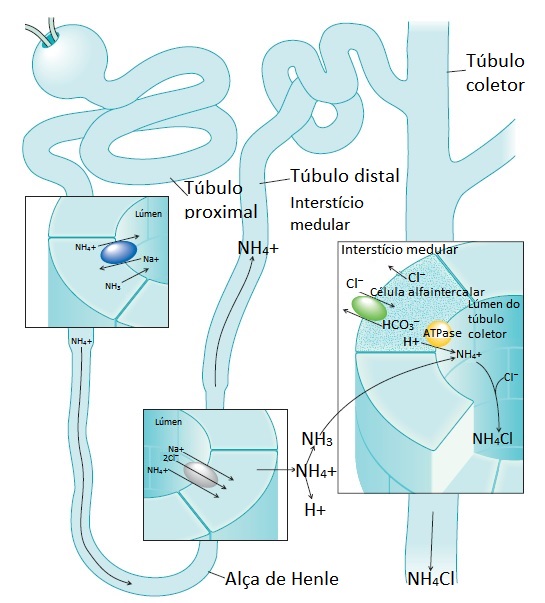
O principal sítio de produção de amônia no rim é o túbulo proximal [Figura 4]. A amônia move-se do túbulo proximal para o túbulo coletor, onde é eliminada [Figura 5]. A quantidade de amônia produzida é estimulada tanto pela acidemia como pela hipocalemia. Ao contrário, a alcalemia e a hipercalemia limitam a produção tubular renal de amônia e a excreção de ácido.
Figura 4. Toda a amônia usada no tamponamento do H+ urinário ao nível do túbulo coletor é sintetizada no túbulo contornado proximal. Acredita-se que a glutamina seja a principal fonte desta amônia. Conforme a glutamina é metabolizada, vai sendo formado alfacetoglutarato (alfa-CG) que, por sua vez, é quebrado em bicarbonato. O bicarbonato então é secretado no líquido peritubular por um contratransportador de Na+/HCO3–.
Figura 5. A amônia usada no tamponamento dos íons hidrogênio na urina é sintetizada no túbulo contornado proximal. Em seguida, sofre difusão para dentro do lúmen tubular proximal ou pode tornar-se acidificada junto à célula e formar amônio. O amônio pode entrar no lúmen tubular substituindo os íons hidrogênio no contratransportador de Na+/H+. O amônio flui pelo ramo ascendente espesso da alça de Henle, onde é transportado desde o túbulo até o interior do interstício medular, substituindo o potássio no transportador de Na+/K+/2Cl–. No interstício, o amônio dissocia-se em amônia, e esta sofre difusão seguindo seu gradiente de concentração para dentro do lúmen do túbulo coletor. Neste local, a amônia combina-se ao H+ secretado e forma amônio. O NH4+ então é excretado como NH4Cl para manter a eletroneutralidade. Uma molécula de bicarbonato é regenerada para cada H+ eliminado na urina.
A acidose metabólica ocorre sempre que a [HCO3–] plasmática diminui. Esta diminuição pode ser causada por diversos fatores: administração de ácido exógeno, produção endógena de ácido, comprometimento da secreção renal de hidrogênio e perdas de bicarbonato a partir dos rins ou secreções gastrintestinais. O cálculo do hiato aniônico plasmático é particularmente útil na identificação da causa específica da acidose metabólica e para estreitar o diagnóstico diferencial.
O hiato aniônico (medido em mEq/L) refere-se à diferença existente entre as concentrações plasmáticas medidas do principal cátion (sódio) e dos principais ânions (cloreto e bicarbonato), descrita pela seguinte equação:
Hiato aniônico = [Na+] – ([HCO3–] + [Cl–])
O hiato aniônico normal varia de 3 a 13 mEq/L, com uma média aproximada de 10 mEq/L. É constituído sobretudo por proteínas plasmáticas (principalmente albumina) que possuem uma carga negativa. No contexto de hipoalbuminemia severa, o hiato aniônico basal pode ser inferior a 3 mEq/L, pois sofre uma queda aproximada de 2,5 mEq/L para cada redução de 1 g/dL na concentração de albumina sérica.
O uso clínico mais importante do hiato aniônico consiste na identificação da etiologia da acidose metabólica.3 Os distúrbios causadores de acidose metabólica são classificados em 2 categorias: (1) distúrbios causadores de queda da [HCO3–] plasmática e aumento concomitante do hiato aniônico; e (2) distúrbios causadores de queda da [HCO3–] plasmática que não afetam o hiato aniônico. Neste último contexto, a [Cl–] plasmática aumenta e produz acidose metabólica hiperclorêmica.
Vários distúrbios, assim como a ingesta de toxinas podem causar acidose metabólica com hiato aniônico elevado [Tabela 1].
Tabela 1. Causas de acidose metabólica com hiato aniônico elevado
|
Insuficiência renal* |
|
Acidose láctica |
|
Cetoacidose* |
|
Rabdomiólise |
|
Toxinas e agentes ingeridos |
|
Salicilatos |
|
Etilenoglicol |
|
Metanol |
|
Tolueno* |
*Também pode estar associado à acidose com hiato aniônico normal.
Insuficiência renal. A insuficiência renal avançada é a causa mais comum de acidose metabólica com hiato aniônico, ou ânio-gap, aumentado no contexto ambulatorial. A retenção de íons hidrogênio acarreta a queda da [HCO3–] plasmática. Como o sulfato e o fosfato (que são os ânions acompanhantes) são excretados na urina, enquanto o cloreto é retido, o hiato aniônico permanece normal durante o curso inicial da acidose na insuficiência renal. Conforme a insuficiência renal progride (creatinina > 3 mg/dL), a excreção normal destes ânions ingeridos e produtos residuais metabólicos vai sendo comprometida. É neste momento que o hiato aniônico aumenta. Contudo, inexiste uma correlação linear entre o grau de acidemia (ou hipobicarbonatemia) e o nível de hiato aniônico. Na insuficiência renal sem complicação, a [HCO3–] plasmática raramente cai para menos de 12 mEq/L, ao passo que o hiato aniônico tipicamente permanece abaixo de 20 mEq/L.
Acidose láctica. A acidose láctica é a causa mais comum de acidose metabólica com hiato aniônico elevado em pacientes internados. É comum a produção de ácido láctico aumentar como resultado de hipotensão ou sepse, que são ambas causadoras de isquemia tecidual relativa ou verdadeira. As vias oxidativas do metabolismo do piruvato são acentuadamente comprometidas nas condições em que há disfunção mitocondrial, como aquelas induzidas por hipoxemia tecidual. Este contexto intensifica a conversão do piruvato em lactato. O fígado e (em menor grau) os rins são os principais órgãos removedores de lactato da circulação. Nestes órgãos, o lactato é convertido de volta em piruvato e, em seguida, em dióxido de carbono e água através do ciclo do ácido tricarboxílico. Quando a perfusão tecidual normal é restaurada, o metabolismo do lactato (CH3CHOHCOO-) nestes órgãos rapidamente regenera o bicarbonato usado a princípio no tamponamento da carga ácida – um processo bastante dependente da excreção ácida renal e que pode ser resumido pela seguinte equação:
CH3CHOHCOO–+3O2 ? 2CO2+2H2O+HCO3–
A velocidade normal de produção de lactato pode chegar a 320 mEq/h (p. ex., durante a prática de exercício). Esta velocidade geralmente é maior do que a velocidade de produção do lactato na acidose láctica. Este achado indica que, para haver acúmulo de ácido láctico, o metabolismo do lactato também precisa estar comprometido. No choque, por exemplo, a acentuada redução da perfusão hepática retarda a depuração do lactato.
Certos medicamentos também podem causar acidose láctica. São exemplos a terapia com metformina de pacientes diabéticos com insuficiência renal e a terapia antirretroviral de pacientes com síndrome da imunodeficiência adquirida (Aids).4
Na acidose láctica, o hiato aniônico quase sempre está acima dos níveis basais. Como o limiar de excreção renal para o lactato é 6 a 8 mEq/L e os níveis normais de lactato são menores que 1 mEq/L, o excesso de lactato acumula-se no sangue, em vez de ser eliminado na urina, contribuindo assim para um hiato aniônico aumentado.
A acidose D-láctica é uma forma incomum de acidose láctica observada com maior frequência em pacientes submetidos ao desvio ileal ou ressecção de intestino delgado.5 Em cada um destes casos, o paciente pode desenvolver a síndrome do intestino curto, com consequente aumento do metabolismo bacteriano de carboidratos em ácido D-láctico causado pela proliferação local excessiva. Na acidose D-láctica, o hiato aniônico inicialmente aumenta, mas pode cair ao longo do tempo, porque a reabsorção tubular renal de D-lactato é insuficiente quando comparada à reabsorção do L-lactato.
Cetoacidose. A cetoacidose causada pela produção excessiva de cetoácidos (ácido acetoacético e ácido beta-hidroxibutírico) ocorre quando a deficiência de insulina, jejum ou resistência à insulina comprometem o uso de glicose. Nestes contextos, os corpos cetônicos são produzidos em excesso (uma condição denominada cetose) e servem de fonte alternativa de energia para muitas células. A cetona inicialmente formada é o ácido acetoacético que, então, pode ser reduzido a ácido beta-hidroxibutírico ou sofrer descarboxilação não enzimática e se transformar em acetona. Embora a acetona seja quimicamente neutra, as outras cetonas são ácidos orgânicos e seu acúmulo acarreta acidose metabólica. A equação a seguir resume as reações:
Ácido acetoacético ? ácido beta-hidroxibutírico ? cetona
Na cetoacidose, o hiato aniônico caracteristicamente aumenta. O acetoacetato e o beta-hidroxibutirato são os principais ânions não medidos que se acumulam, embora uma acidose láctica concomitante possa ser observada em alguns pacientes. A [HCO3–] plasmática pode estar acentuadamente diminuída na cetoacidose diabética. Em contraste, a acidemia costuma ser leve e a concentração plasmática de bicarbonato raramente é inferior a 18 mEq/L na cetose por inanição ou jejum.
Em pacientes com cetoacidose, a reposição de líquidos isotônicos acarreta cetonúria. Quando a reposição de líquidos é iniciada, o hiato aniônico está alto. Entretanto, conforme a taxa de excreção urinária de acetoacetato e beta-hidroxibutirato excede a taxa de produção, o hiato aniônico passa a diminuir. Por fim, o hiato aniônico é normalizado no momento em que a [HCO3–] plasmática ainda está baixa.
Rabdomiólise. A quebra muscular maciça é uma causa importante de acidose metabólica.6 A retenção de ácidos metabólicos e de ânions inorgânicos como o fosfato parece contribuir para o aumento do hiato aniônico. O desenvolvimento de acidose metabólica é mais provável quando há insuficiência renal concomitante [ver 10:VI Insuficiência renal aguda].
Toxinas e agentes ingeridos. Os salicilatos e os álcoois etilenoglicol (componente de anticongelantes e solventes) e metanol, ou álcool da madeira (componente da goma-laca, verniz, soluções descongelantes e outras preparações comerciais, são as causas mais frequentes de acidose metabólica entre as toxinas e agentes ingeridos. A ingesta de álcool etílico não está associada a uma acidose com hiato aniônico elevado, exceto quando acompanhada de acidose láctica ou cetoacidose.
A causa mais comum de anormalidade ácido-base em indivíduos adultos intoxicados com salicilato é a alcalose respiratória causada pela estimulação direta do centro respiratório medular [ver Acidose e alcalose respiratória, adiante]. Em adultos, a presença de alcalose respiratória parece ser um pré-requisito para o desenvolvimento de acidose metabólica, uma vez que a acidose metabólica isolada é rara. A intoxicação por salicilatos de grau moderado a severo, por sua vez, promove alcalose respiratória mista e acidose metabólica com hiato aniônico elevado. O metanol e o etilenoglicol podem produzir intoxicação fatal e ambos podem causar um hiato osmolal plasmático, que se refere à diferença existente entre a osmolalidade plasmática (Posm) medida em laboratório e aquela calculada pela seguinte fórmula:
Posm (calculada) = (2 X [Na+] plasmática) + ([glicose]/18) + (ureia/2,8)
Um hiato osmolal plasmático alto (com níveis medidos sendo no mínimo 10 mOsm/kg maiores do que o valor calculado) pode ser detectado somente quando o Posm é medido por depressão do ponto de congelamento. Em contraste, a contribuição osmótica dos álcoois voláteis não é incluída quando se utiliza um osmômetro de vapor-pressão, pois este dispositivo considera que somente a água está em fase de vapor.7 Além disso, a existência de um hiato osmolal é inespecífica para estas ingestas, e a confirmação do diagnóstico requer ensaios com plasma para cada fármaco separadamente [ver Diagnóstico, adiante].
Um hiato aniônico normal no contexto da acidose metabólica pode ter diversas causas, como a administração de ácidos inorgânicos (p. ex., ácido hidroclorídrico), perda de bicarbonato a partir do trato gastrintestinal (p. ex., na diarreia) ou pelos rins (p. ex., acidose tubular renal [ATR] de tipo 2) e comprometimento da excreção renal de íons hidrogênio (p. ex., insuficiência renal e ATR de tipo 1) [Tabela 2]. Nas condições resultantes, a eletroneutralidade é mantida pela diminuição da concentração plasmática de bicarbonato e substituição por cloreto. Em consequência, estes distúrbios por vezes são conjuntamente denominados acidoses hiperclorêmicas.
Tabela 2. Causas de acidose metabólica com hiato aniônico normal
|
Administração de ácido e cloreto |
|
Hiperalimentação* |
|
NH4Cl, HCl para tratamento de alcalose metabólica severa* |
|
Perda de bicarbonato (ou de outros álcalis) |
|
Perda gastrintestinal de álcali† |
|
Diarreia, fístulas pancreáticas-intestinais-biliares, transplante de pâncreas com drenagem para dentro da bexiga urinária |
|
Desvios ureterais† |
|
Ureterossigmoidoscopia, bexiga ileal (quando obstruída) |
|
ATR de tipo 2† |
|
Recuperação da cetoacidose† |
|
Pós-hipocapnia† |
|
Diminuição da excreção renal e de hidrogênio |
|
Insuficiência renal* |
|
ATR de tipo 1 [Tabela 3] |
|
Formas hipocalêmicas* |
|
Formas hipercalêmicas† |
|
ATR de tipo 4* [Tabela 4] |
*Concentração plasmática de K+ normal ou aumentada.
†Concentração plasmática de K+ normal ou diminuída.
ATR = acidose tubular renal.
Administração de ácido e cloreto. A infusão de soluções de aminoácidos durante a nutrição parenteral fornece ácido hidroclorídrico em abundância. Em determinados contextos, a administração de uma solução de cloreto de sódio também pode diminuir a [HCO3–] plasmática por diluição (uma condição denominada acidose por expansão), com consequente diminuição do pH sanguíneo.
Perdas de bicarbonato e outros álcalis. O bicarbonato pode ser perdido pelo corpo através do trato gastrintestinal ou dos rins. Em comparação com o sangue, o conteúdo intestinal é alcalino e isto ocorre porque as secreções pancreáticas e biliares adicionam bicarbonato. Este bicarbonato, por sua vez, é trocado posteriormente por cloreto no íleo e no cólon, para manutenção de um estado acidobásico normal. As perdas gastrintestinais de bicarbonato (ou de precursores de bicarbonato, como lactato e acetato) são mais comumente observadas em pacientes com diarreia severa. O diagnóstico de diarreia como causa de acidose com hiato aniônico normal costuma ser evidenciado pela história do paciente [ver Diagnóstico, adiante]. A hipocalemia resultante de perdas de potássio através das fezes também sustenta o diagnóstico.
De forma menos comum, a acidose metabólica resultante de perdas gastrintestinais de álcali é causada por fístulas pancreáticas, drenagem biliar ou desvios urinários para o cólon ou intestino delgado.8 Esta condição também ocorre após o transplante pancreático em pacientes que perdem bicarbonato via anastomose pâncreas-bexiga.
As perdas renais de bicarbonato causam a acidemia observada na ATR de tipo 2 (ou proximal) e em pacientes pós-hipocapneicos. Na ATR de tipo 2, o limiar normal da reabsorção de bicarbonato está diminuído. Desta forma, o bicarbonato não pode ser reabsorvido a uma taxa adequada para a manutenção de níveis plasmáticos normais aproximados de 25 mEq/L. Durante esta fase, ocorre uma bicarbonatúria que resulta em um pH urinário acima de 5,3. Entretanto, a perda de bicarbonato cessa assim que a diminuição da [HCO3–] plasmática é estabilizada. Quando isto ocorre, o pH da urina pode ser inferior a 5.
A ATR proximal pode ser observada em várias circunstâncias, tais como na administração de inibidores de anidrase carbônica e em alguns pacientes com mieloma múltiplo. No mieloma múltiplo, o dano tóxico às células tubulares proximais causado pelas cadeias leves de mieloma filtradas frequentemente resulta na diminuição generalizada da reabsorção proximal (inclusive de glicose e fosfato), além do bicarbonato. Este grupo de enfermidades, que são decorrentes de uma disfunção tubular proximal, é chamado síndrome de Fanconi.9,10
A hipocapnia promove uma queda da reabsorção de bicarbonato ao nível tubular proximal. Decorridos 1 a 3 dias, os níveis plasmáticos de bicarbonato diminuem. Como este processo adaptativo renal demora o mesmo período para ser concluído, um aumento súbito da PaCO2 não promove uma alteração imediata após a reabsorção do bicarbonato. Esta acidose metabólica pós-hipocapneica é resolvida espontaneamente, dentro de 24 a 72 horas.
Excreção renal diminuída de íons hidrogênio. Uma excreção renal ácida diminuída é observada em 3 condições: insuficiência renal, ATR de tipo 1 (distal) e ATR de tipo 4 (hipoaldosteronismo) [Tabelas 3 e 4]. A acidose da insuficiência renal é causada principalmente pela diminuição do número de néfrons. Em contraste, a ATR de tipo 1 é caracterizada por uma redução da excreção renal ácida realizada por cada néfron. Como a quantidade total de amônia que pode ser sintetizada também é menor na insuficiência renal, o pH urinário está abaixo de 5,3 na maioria dos pacientes.
Tabela 3. Causas de ATR de tipo 1
|
Formas hipocalêmicas |
|
Primária |
|
Idiopática |
|
Genética |
|
Familiar |
|
Síndrome de Marfan |
|
Síndrome de Ehlers-Danlos |
|
Nefrocalcinose |
|
Hipercalcemia crônica |
|
Rim esponjoso medular |
|
Condições hipergamaglobulinêmicas |
|
Amiloidose* |
|
Crioglobulinemia |
|
Cirrose |
|
Fármacos e toxinas |
|
Anfotericina B |
|
Carbonato de lítio |
|
Tolueno† |
|
Doenças autoimunes |
|
Síndrome de Sjögren* |
|
Tireoidite |
|
Hepatite ativa crônica |
|
Cirrose biliar primária |
|
Formas hipercalêmicas |
|
Obstrução do trato urinário |
|
Anemia falciforme |
|
Lúpus eritematoso sistêmico |
|
Rejeição de transplante renal |
*Também pode causar ATR de tipo 2 [Tabela 5].
†Também pode causar acidose metabólica com hiato aniônico elevado [Tabela 1].
ATR = acidose tubular renal.
Tabela 4. Causas de ATR de tipo 4 e resistência à aldosterona
|
Distúrbio |
Causa |
|
ATR de tipo 4 |
Atividade reduzida do sistema renina-angiotensina ATR de tipo 4 hiporreninêmica (mais comum no diabetes) Fármacos AINH (com possível exceção do sulindaco) IECA Bloqueadores do receptor da angiotensina II Ciclosporina Aids* Diminuição da síntese de aldosterona Níveis de cortisol baixos Insuficiência adrenal primária Deficiências enzimáticas primárias (principalmente, hiperplasia suprarrenal) Níveis de cortisol normais Heparina Imediatamente após a remoção de um adenoma suprarrenal no aldosteronismo primário Deficiências enzimáticas |
|
Resistência à aldosterona (níveis de aldosterona normais ou aumentados) |
Diuréticos poupadores de potássio, trimetoprima Pseudo-hipoaldosteronismo (hereditário ou adquirido) ATR de tipo 1 hipercalêmica† |
*A adrenalite causadora de ATR de tipo 4 também pode ocorrer em indivíduos soropositivos para HIV.
†Neste contexto, a urina é alcalina (pH > 5,3).
AINH = anti-inflamatórios não hormonais; ATR = acidose tubular renal; IECA = inibidores da enzima conversora de angiotensina.
A ATR de tipo 1, que pode ser adquirida em associação a alguns distúrbios (p. ex., síndrome de Sjögren), é mais frequente quando os íons hidrogênio não podem ser bombeados para fora das células alfa intercaladas e dentro do lúmen do túbulo coletor ou quando vazam de volta para dentro da célula a partir do lúmen tubular [Figura 3]. As mutações subjacentes à ATR de tipo 1 congênita podem ser causadas por mecanismos similares àqueles atuantes na forma adquirida da doença ou via translocação do transportador Cl–/HCO3–, a partir da membrana basolateral seguindo para a membrana apical (luminal) das células alfaintercaladas.11 Como resultado, a urina não pode ser maximamente acidificada e o pH urinário permanece sempre acima de 5,3. Além disso, a hipocalemia está caracteristicamente presente, em parte devido à troca intensificada de Na+/K+ junto ao néfron distal, que é um processo necessário à manutenção do equilíbrio de sódio porque os íons hidrogênio não podem ser secretados em resposta à reabsorção de sódio.
Uma forma hipercalêmica de ATR distal também foi descrita. Esta condição é mais frequente em pacientes com obstrução do trato urinário. Caracteriza-se por uma extensa lesão tubular que envolve a secreção de hidrogênio aldosterona-sensível e aldosterona-independente. O pH da urina de pacientes com este distúrbio é maior que 5,3.
A complicação clínica mais importante da ATR de tipo 1 hipocalêmica é a formação e deposição de sais de fosfato de cálcio, que pode causar a formação de cálculos em todo o rim (nefrocalcinose). Um dos principais fatores contribuidores para a formação de cristais é a hipocitratúria decorrente do aumento da reabsorção de citrato ao nível do túbulo proximal. Como o citrato de cálcio é significativamente mais solúvel do que o fosfato de cálcio, uma diminuição da concentração urinária de citrato facilita a precipitação dos cristais de fosfato de cálcio no lúmen do túbulo coletor.
A ATR de tipo 4 pode ser causada por diversas medicações, incluindo os fármacos anti-inflamatórios não hormonais (AINH), inibidores da enzima conversora de angiotensina (IECA), bloqueadores de angiotensina II, ciclosporina e heparina. Entretanto, esta condição é observada com maior frequência em pacientes com diabetes melito [ver Distúrbios de potássio plasmático, e Hipercalemia, adiante]. A perturbação eletrolítica mais comum na ATR de tipo 4 é a hipercalemia ([K+] plasmática > 5 mEq/L), cuja causa é o comprometimento da reabsorção luminal dos íons sódio que resulta na diminuição da secreção de íons potássio pelas células principais do túbulo coletor [Figura 3]. Este defeito é causado pela diminuição da produção ou da ação da aldosterona. Em contrate com a hipocalemia, os níveis plasmáticos de potássio elevados comprometem a produção renal de amônia. O tamponamento urinário insuficiente realizado sob a forma de amônia limita a secreção de íons hidrogênio junto ao túbulo coletor. Apesar desta reduzida capacidade excretora de íons hidrogênio, a urina geralmente está acidificada (pH < 5,3). Este achado aparentemente paradoxal ocorre porque o limitado tamponamento urinário permite que a [H+] urinária livre exceda a capacidade secretora das células alfa intercaladas e, desta forma, limita o transporte adicional de íons hidrogênio a partir das células para dentro do lúmen do túbulo coletor. Em contraste com os pacientes com ATR de tipo 1, os indivíduos com ATR de tipo 4 conseguem excretar um pouco de ácido, ainda que em concentrações inadequadas. Em consequência, o pH da urina é tipicamente inferior a 5,3 nos pacientes que sofrem deste distúrbio.
A hipercalemia pode ser observada em pacientes com acidose metabólica com hiato aniônico normal. Na acidose respiratória, há um menor deslocamento de potássio a partir das células para o plasma. Uma queda na [HCO3–] plasmática leva à troca de íons hidrogênio extracelulares por íons potássio intracelulares. Este mecanismo de defesa permite o tamponamento intracelular dos íons hidrogênio. A troca H+/K+ mantém a eletroneutralidade. Sendo assim, o paciente com diarreia que tem acidose metabólica com hiato aniônico normal e baixa [K+] plasmática sofre uma depleção de potássio significativamente maior do que o paciente que apresenta a mesma [K+] plasmática e pH sanguíneo normal.
Em contraste com a situação observada em pacientes com acidose metabólica com hiato aniônico normal, os deslocamentos de potássio causados pela acidemia são menos pronunciados em pacientes com acidoses orgânicas endógenas (p. ex., acidose láctica e cetoacidose).12 Neste contexto, quando há desenvolvimento de hipercalemia, esta é tipicamente causada por insuficiência renal, catabolismo celular que permite o vazamento de potássio a partir das células (acidose láctica) ou deficiência de insulina e hiperglicemia, que são ambas promotoras da saída do potássio das células.
Manifestações clínicas. A respirações de Kussmaul sugerem a ocorrência de acidose metabólica. O aumento do volume corrente, em vez da frequência respiratória, que caracteriza estas alterações ventilatórias resulta da estimulação do sistema respiratório junto ao tronco cerebral pelo baixo pH.
A hipotensão secundária também pode ser observada em indivíduos gravemente acidêmicos. Neste contexto, a diminuída pressão arterial resulta de uma contratilidade miocárdica deprimida e de vasodilatação arterial, que são induzidas pelo reduzido pH sanguíneo. Inicialmente, os nível altos de catecolaminas circulantes se opõem aos efeitos cardiovasculares da acidemia. Entretanto, os efeitos da acidemia podem predominar a um pH sanguíneo abaixo de 7,15 a 7,20.13 Podem ocorrer arritmias reentrantes e uma diminuição do limiar para fibrilação ventricular, enquanto o limiar de desfibrilação permanece inalterado.
Os sintomas de acidose metabólica produzidos pela insuficiência renal dependem da causa, da velocidade do desenvolvimento de insuficiência renal e da possível existência de outras condições concomitantes (p. ex., insuficiência cardíaca congestiva). Os sintomas clínicos podem estar ausentes, mesmo que o paciente apresente azotemia e acidemia.
A cetoacidose frequentemente está associada ao aumento da sede e poliúria. Costuma ser precipitada por uma agressão não relacionada, como uma infecção, que pode dominar o quadro clínico.
As características distintivas da rabdomiólise são a mialgia e a mioglobinúria. Esta última acarreta a produção de uma urina avermelhada na ausência de uma concentração significativa de hemácias. Em muitos pacientes, contudo, nenhum destes achados é encontrado. Apesar da falta de achados, os níveis séricos de creatinina quinase estão uniformemente elevados.
Os sinais e sintomas de acidose láctica são caracteristicamente aqueles produzidos pela perturbação subjacente causadora do distúrbio – a saber, hipotensão ou sepse. Mais comumente, os pacientes exibem evidências de hipoperfusão, como pressão arterial baixa e membros frios ou com livedo. Com menos frequência, uma medicação (p. ex., metformina) pode ser a causa. Os pacientes com acidose D-láctica podem apresentar encefalopatia. O ensaio de L-lactato desidrogenase é considerado padrão para o diagnóstico de acidose láctica. Como este teste não mede a concentração de ácido D-láctico, é necessário realizar um ensaio enzimático específico para D-lactato (isto é, um ensaio que use desidrogenase D-láctica) para confirmar o diagnóstico de acidose D-láctica.
Além das alterações ácido-base, os achados clínicos que podem acompanhar a intoxicação severa por salicilatos incluem zumbido, hiperpirexia, vasodilatação que leva ao choque, e edema periférico ou pulmonar. Os sintomas de acidose causados pela ingesta de metanol ou etilenoglicol [ver Toxinas e agentes ingeridos, anteriormente] podem se desenvolver em 12 a 36 horas após a ingesta. Além das alterações ácido-base, os sintomas iniciais que surgem após a ingesta de metanol incluem enfraquecimento, náusea, cefaleia e diminuição da visão, que podem progredir para cegueira, coma e morte. O exame fundoscópico pode revelar um brilho retinal produzido por edema retinal. Após a ingesta de etileno glicol, os primeiros achados são as anormalidades neurológicas que variam da embriaguez ao coma. Se o paciente não for tratado, estas alterações podem ser seguidas principalmente por sintomas cardiopulmonares (taquipneia e edema pulmonar) e, então, por uma dor no flanco e insuficiência renal decorrente da deposição de cristais de oxalato de cálcio, que podem ser encontrados no sedimento urinário [ver Figura 2 em 10: XII Nefrolitíase].
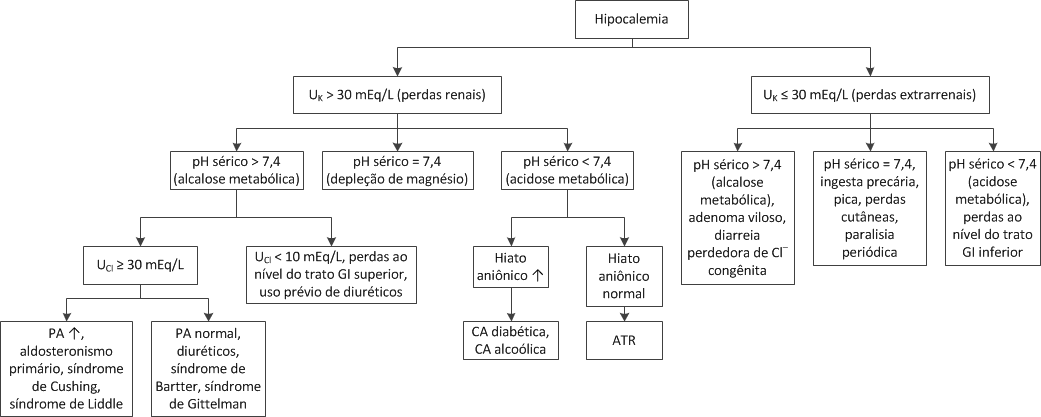
Exames laboratoriais. O diagnóstico de acidose metabólica é estabelecido com relativa facilidade na presença de um baixo pH sanguíneo e uma [HCO3–] plasmática diminuída. O hiato aniônico, assim, pode ser usado para identificar uma causa específica. O achado de hipo ou hipercalemia concomitante também pode ser útil.
Uma vez determinada a presença/ausência de um hiato aniônico sérico elevado, a defesa respiratória contra a acidemia pode ser avaliada. A resposta respiratória começa imediatamente, embora possa não ser máxima em 12 a 24 horas. A compensação respiratória apropriada pode ser calculada empregando-se a fórmula de Winter:14
PCO2 esperada = 1,5 [HCO3–] + 8 ± 2
A compensação respiratória é inadequada quando a PaCO2 é maior do que o valor esperado, e pode ser excessiva quando a PaCO2 é inferior ao valor esperado. Quando estas respostas inadequadas ou excessivas ocorrem, significa que uma acidose respiratória ou alcalose respiratória sobreposta, respectivamente, está presente [ver Acidose e alcalose respiratória, adiante]. Exemplificando, se a [HCO3–] sérica fosse igual a 16 mEq/L em um paciente com pH sanguíneo baixo (acidose metabólica), a PCO2 esperada seria de aproximadamente 32 [(1,5 X 1,6) + 8] mmHg. Uma PCO2 inferior a este valor indica a ocorrência de alcalose respiratória sobreposta.
Outro cálculo laboratorial, o hiato aniônico urinário, pode ser útil para definir a causa da acidose metabólica quando o hiato aniônico sérico está normal. Exemplificando, embora o diagnóstico de diarreia geralmente seja evidenciado pela história do paciente e pela presença de hipocalemia, um perfil de eletrólitos plasmáticos similar àquele apresentado por indivíduos com diarreia pode ser encontrado em pacientes com ATR de tipo 1. Estes distúrbios geralmente podem ser distinguidos pelo pH da urina. A urina tende a ser acidificada (pH < 5,3) em pacientes com diarreia, e tende a ser alcalinizada (pH > 5,3) em pacientes com ATR de tipo 1. Entretanto, em alguns pacientes com diarreia a urina pode ser alcalina, provavelmente porque a produção de amônia (induzida pela hipocalemia) aumenta de tal modo que o tamponamento urinário é produzido com excesso de secreção de íons hidrogênio.
O cálculo do hiato aniônico urinário, como mostra a equação a seguir, pode ser bastante útil para fornecer uma estimativa da secreção urinária de amônia:
Hiato aniônico urinário = ([Na+] urinária + [K+] urinária) – [Cl–] urinária
Sempre que os íons hidrogênio secretados são excretados sob a forma de cloreto de amônio (NH4+Cl–), a excreção urinária de cloreto aumenta. Este aumento diminuiu o hiato aniônico urinário, produzindo um valor negativo na maioria dos pacientes com diarreia. Em comparação, na ATR de tipo 1, o hiato aniônico é positivo. Citemos como exemplo um paciente com acidose metabólica com hiato aniônico normal (p. ex., [HCO3–] = 10 mEq/L), baixos níveis de potássio (hipocalemia) e pH urinário alcalino (6,0), que poderia ter diarreia ou ATR de tipo 1. Se a [Na+] urinária fosse igual a 50 mEq/L, a [K+] urinária fosse de 28 mEq/L e a [Cl–] urinária fosse igual a 55 mEq/L, o hiato aniônico urinário seria de +23, sustentando um diagnóstico de ATR.
Apesar do potencial valor do hiato aniônico urinário na estimativa da excreção urinária de NH4+Cl–, a presença de certos ânions (p. ex., beta-hidroxibutirato e ácido acetoacético) pode contribuir para um hiato urinário positivo. Isto pode sugerir erroneamente que a acidose metabólica é causada pela ATR, e não pela diarreia. O hiato osmolal urinário, que representa em grande parte os sais de amônio, pode ser usado para confirmar que o hiato aniônico urinário positivo resulta da ATR de tipo 1. O hiato osmolal urinário consiste na diferença entre a osmolalidade urinária (Uosm) medida pelo laboratório e aquela calculada empregando-se a seguinte fórmula:
Uosm calculada = 2 ([Na+] + [K+]) + (ureia/2,8)
O fator de multiplicação 2 representa os ânions que acompanham o sódio e o potássio, enquanto o fator de divisão 2,8 reflete o ajuste necessário à conversão dos achados de ureia expressos rotineiramente em mg/dL para mmol/L ou mOsm/kg. O hiato osmolal urinário normal vale 80 a 100 mmol/L, refletindo uma taxa de excreção de NH4+ aproximadamente igual à metade deste valor – a saber, 40 a 50 mEq/L – como resultado do ânion cloreto acompanhante. Diante de uma acidose metabólica, o valor deve aumentar de maneira significativa. Um valor normal ou mais baixo em um indivíduo com acidemia metabólica sustenta fortemente o comprometimento da excreção de NH4+Cl– (e, enfim, a ATR de tipo 1) como causa subjacente do hiato aniônico positivo.
O tratamento da acidose metabólica é voltado para a correção da acidemia e do distúrbio subjacente. A probabilidade de ser necessário administrar álcalis e de que estes venham a ser efetivos depende do pH sanguíneo, dos mecanismos compensatórios e da condição subjacente.
Até que o pH do sangue arterial caia abaixo de 7,15 a 7,20, os efeitos colaterais da acidemia geralmente são compensados pelos altos níveis plasmáticos de catecolaminas [ver Diagnóstico, anteriormente]. Para manter reservas de tamponamento adequadas, a terapia com álcalis deve ser considerada para manutenção da [HCO3–] acima de 10 a 12 mEq/L. A administração de álcalis geralmente é desnecessária, todavia, caso a acidose tenda a se resolver de maneira espontânea (como na acidose láctica subsequente a uma convulsão de grande mal). A terapia deve considerar a causa subjacente de acidose metabólica (ver adiante).
Insuficiência renal crônica. É interessante o achado de que a acidemia tende a ser mais severa em pacientes não diabéticos com insuficiência renal crônica do que em pacientes diabéticos com graus similares de insuficiência renal.15 Esta diferença pode resultar da geração mais eficiente de bicarbonato extrarrenal observada nos pacientes diabéticos.
Enquanto a acidose metabólica permanece leve, muitos adultos com insuficiência renal não são tratados à base de reposição com álcali, em parte devido à preocupação com a possibilidade de o bicarbonato de sódio exacerbar a expansão de volume e a hipertensão comumente presentes.
Contudo, alguns estudos sugerem que vários motivos justificam o uso da terapia de reposição com álcali em casos de pacientes com insuficiência renal.16-18 Entre estes motivos, estão a probabilidade de a acidemia intensificar a quebra de músculo esquelético, reduzir a síntese de albumina e (por meio da ativação do sistema complemento) contribuir para a lesão tubulointersticial, além da possibilidade de o tamponamento ósseo dos íons hidrogênio acarretar reabsorção óssea.19 Estes achados levaram alguns médicos a defenderem o uso antecipado da terapia à base de álcali para manutenção da [HCO3–] plasmática acima de 22 mEq/L. Estudos definitivos ainda se fazem necessários, todavia, para estabelecer os benefícios proporcionados por este tratamento.
Nota do editor: Um estudo recente demonstrou que na insuficiência renal crônica, a reposição de bicarbonato com alvo de manter PH sérico normal, alem de preservar o osso e manter o estado nutriciona adequado, conseguiu adiar a necessidade de terapia dialítica.
Os médicos devem ter em mente que os exames de eletrólitos de pacientes sob hemodiálise que usam unidades de diálise não hospitalares podem ser realizados em laboratórios centrais localizados a vários quilômetros de distância. Em comparação com os resultados dos exames que usam amostras obtidas localmente, as amostras testadas nos laboratórios centrais parecem mostrar diminuições consistentes e clinicamente significativas da [HCO3–] sérica.20 Esta intensificação in vitro da acidose metabólica precisa ser confirmada antes que o tratamento seja iniciado.
Acidose láctica. A correção do distúrbio subjacente constitui a terapia primária da acidose láctica. A reversão da falha circulatória, hipoxemia ou sepse diminui a taxa de produção de lactato e intensifica sua remoção.
Assim como na insuficiência renal crônica, o uso da terapia com álcali na acidose láctica é controverso.21,22 A principal lógica da administração de bicarbonato reside na potencial manutenção da hemostasia cardiovascular normal. Esta possível vantagem deve ser ponderada contra os efeitos colaterais deletérios, como a sobrecarga de volume, hipernatremia e alcalose (com a administração excessiva de bicarbonato).
Estudos clínicos também sugeriram que a terapia com bicarbonato de sódio pode não melhorar o pH sanguíneo nem a sobrevida dos pacientes com acidose láctica. Foi observado que a administração de bicarbonato de sódio diminui o desempenho cardíaco em pacientes com cardiopatia, insuficiência cardíaca congestiva ou infarto agudo do miocárdio.23 Esta ineficácia possivelmente resulta de um aumento associado da produção de ácido láctico, embora a hiperosmolalidade da solução de álcali administrada também possa ser importante. O achado de que o dicloroacetato (que promove a conversão do piruvato no ciclo de Krebs, em vez de permitir sua conversão em lactato) pode diminuir os níveis de lactato e aumentar o pH sanguíneo em pacientes com acidose láctica sem aparentemente melhorar a sobrevida confere sustentação adicional ao conceito de que o tratamento da causa subjacente de acidose láctica é mais importante do que tratar a acidemia. Em vista destas observações, uma abordagem razoável pode ser a administração de bicarbonato para manter o pH do sangue arterial acima de 7,15 e os níveis de bicarbonato plasmáticos acima de 10 mEq/L. Caso o paciente desenvolva uma das complicações associadas à terapia com bicarbonato, os benefícios proporcionados pela continuação da terapia com álcali precisam ser reavaliados.
A terapia para acidose D-láctica deve tratar ambos os fatores subjacentes que contribuem para a produção excessiva de ácido D-láctico: a presença de bactérias intestinais produtoras de ácido D-láctico e a distribuição aumentada de carboidratos para o cólon, secundária à síndrome do intestino curto. O tratamento geralmente consiste na instituição de uma terapia antibiótica para diminuir o número de organismos produtores de D-lactato. A terapia com álcali, ainda que seja usada, não foi avaliada por estudos controlados.
Cetoacidose. A tendência à acidose metabólica com hiato aniônico normal durante a recuperação da cetoacidose exerce efeito importante sobre a velocidade em que a acidemia pode ser corrigida. Uma velocidade máxima de 500 mL de líquido/hora parece ser efetiva como terapia de reposição para pacientes com cetoacidose. Velocidades significativamente maiores de administração de líquido podem resultar em um hiato aniônico diminuído sem aumento da [HCO3–] plasmática. Quando isto ocorre, a correção da acidose exige regeneração do bicarbonato pelos rins, em um processo que pode demorar 2 a 3 dias.24 Como consequência, uma administração de líquidos mais vigorosa (acima de 500 mL/hora) pode retardar a recuperação a partir da acidose. A velocidade da administração de líquido deve ser diminuída após a correção do comprometimento do volume intravascular – que se manifesta como pressão arterial reduzida ou aumento das concentrações plasmáticas de creatinina e ureia.
Apesar das exceções, a administração de bicarbonato de sódio geralmente é desnecessária nos casos de cetoacidose, pois aparentemente não há diferença em termos de mortalidade entre pacientes tratados com bicarbonato de sódio e indivíduos-controle. Neste contexto, a terapia com bicarbonato de sódio pode atuar como fator de risco de desenvolvimento subsequente de edema cerebral.25
Nota do editor: Na primeira hora de tratamento a reposição de liquido pode ser maior, sendo recomendado 1 litro ou mais neste momento. Quanto a reposição de bicarbonato um estudo demonstrou ausência de benefício com reposição de bicarbonato em pacientes com PH entre 6,9 e 7,1, a literatura ainda tem Duvida se o ponto de corte para reposição de bicarbonato na cetoacidose é o PH de 6,9 ou 7,0.
Intoxicação por salicilato, etilenoglicol e metanol. A remoção do salicilato é intensificada pela alcalinização da urina (para manter o pH sanguíneo entre 7,45 e 7,50), que aumenta a excreção urinária até mesmo no contexto da diálise.26 O tratamento inicial da intoxicação por etilenoglicol ou metanol consiste em administrar um agente que diminui a conversão do álcool não tóxico em seus produtos metabólicos tóxicos, bem como em utilizar a diálise em presença de dano tecidual ou acidemia. Embora o etilenoglicol seja historicamente administrado, o fomepizol (4-metilpirazol) é o único inibidor potente da álcool desidrogenase que foi estudado de maneira prospectiva e teve o uso aprovado pelo Food and Drug Administration (FDA) para tratamento desta condição.27
Acidoses hiperclorêmicas. A abordagem terapêutica do paciente com diarreia e acidose metabólica depende da severidade de ambos os distúrbios. A administração de álcalis é desnecessária nos casos de diminuição leve a moderada da [HCO3–], se a diarreia estiver controlada (minimizando, assim, as perdas adicionais de bicarbonato) e a função renal estiver normal (permitindo, então, que a excreção de ácido aumente). Entretanto, alguns pacientes necessitarão de tratamento com bicarbonato.
A acidemia na ATR de tipo 1 pode ser corrigida com bicarbonato ou algum precursor de bicarbonato, como o citrato. Os requerimentos usuais são de 1 a 3 mEq/kg/dia. A correção da acidemia diminui a reabsorção tubular do citrato, que, por sua vez, resulta no aumento da excreção urinária e diminuição da tendência à nefrolitíase e nefrocalcinose. Um sal de potássio, como o citrato de potássio, geralmente é administrado porque também corrige o déficit de potássio.
Em adultos, o tratamento da ATR de tipo 2 é voltado para a causa subjacente (p. ex., mieloma múltiplo). Como a acidemia é tipicamente leve, a terapia com álcali pode ser desnecessária. Em crianças, a correção da acidemia é apropriada, pois nesta faixa etária a acidemia tende mais a comprometer o crescimento e contribuir para o desenvolvimento de doença óssea metabólica.
A ATR de tipo 4, quando leve, pode dispensar tratamento. A reposição da aldosterona com fludrocortisona (0,1 a 1 mg/dia) pode aumentar a secreção ácida ao diminuir a [K+] sérica. Entretanto, muitos pacientes são intolerantes aos efeitos colaterais (p. ex., edema e hipertensão) associados a esta terapia. Nestes indivíduos, a correção da [K+] sérica pode ser efetivada por meio da administração de um diurético de alça, restrição do potássio da dieta ou eliminação de fármacos promotores de hipercalemia. Quando a terapia com fludrocortisona se faz necessária, a administração concomitante de um diurético de alça pode limitar o desenvolvimento de pressão arterial alta e retenção de líquido.
A alcalose metabólica primária é caracterizada por uma [HCO3–] plasmática elevada e um pH arterial acima de 7,42. Contudo, havendo acidose metabólica concomitante, o pH sanguíneo pode estar aumentado, diminuído ou normal. Além disso, a hiperbicarbonatemia isolada não é diagnóstica de alcalose metabólica primária, pois também pode representar a resposta fisiológica adequada à acidose respiratória crônica. Estas condições em geral podem ser facilmente distinguidas pela medida do pH do sangue arterial, que está diminuído na acidose respiratória.
A alcalose metabólica é um problema clínico relativamente comum, que é induzido com mais frequência pela terapia diurética ou pela perda de secreções gástricas, como resultado de vômito ou sucção nasogástrica.28 É necessário que 2 condições estejam presentes para que um indivíduo desenvolva e mantenha a alcalose metabólica. Primeiro, deve haver um aumento inicial da [HCO3–] plasmática decorrente da perda de hidrogênio nas secreções gastrintestinais ou na urina; movimentação do hidrogênio para dentro das células; administração de álcali; ou contração do volume em torno de uma concentração extracelular de bicarbonato relativamente constante (denominada alcalose por contração) [Tabela 5]. Em segundo lugar, é preciso que 1 dentre 3 fatores (na ausência de insuficiência renal avançada) esteja presente para manter a alta [HCO3–] plasmática após o término do evento iniciador: depleção efetiva do volume circulante, depleção de cloreto e hipocloremia ou hipocalemia [ver Fatores adicionais que contribuem para a alcalose metabólica, adiante].
Tabela 5. Causas de alcalose metabólica
|
Distúrbio |
Causa |
|
Perda de hidrogênio |
Perdas gastrintestinais Remoção de secreções gástricas (vômitos ou sucção nasogástrica)* Condições com diarreia perdedora de cloreto Perdas renais Diuréticos de alça ou tiazidicos* Excesso de mineralocorticoides Após a correção rápida da hipercapnia crônica Hipercalcemia (incluindo a síndrome do leite álcali) Doses altas IV de derivados da penicilina Síndrome de Bartter |
|
Movimentação de hidrogênio para dentro das células |
Hipocalemia |
|
Retenção de bicarbonato |
Administração de álcali (como bicarbonato ou sob a forma de precursor de bicarbonato com transfusões sanguíneas em massa ou antiácidos absorvíveis)† |
|
Alcalose por contração |
Diuréticos* Perda de secreções gastrintestinais ricas em [Cl–] e pobres em [HCO3–], em comparação ao plasma (habitualmente por vômito)* |
*Causas mais comuns.
†Para que a alcalose seja mantida, a excreção renal de bicarbonato também deve estar comprometida, seja por diminuição da filtração ou aumento da reabsorção junto ao túbulo proximal.
IV = via intravenosa.
Perda gastrintestinal de hidrogênio. O suco gástrico contém uma alta concentração de ácido clorídrico e uma concentração menor de cloreto de potássio. Cada 1 mEq de hidrogênio perdido gera 1 mEq de bicarbonato. Em indivíduos normais, a secreção gástrica de hidrogênio não promove alcalose metabólica, pois é equiparada pela secreção de bicarbonato estimulada pela entrada de ácido no duodeno. Entretanto, a secreção de bicarbonato não é estimulada quando o vômito ou uma drenagem via tubo nasogástrico impedem que os íons hidrogênio atinjam o duodeno. O vômito pode ser sub-reptício em alguns casos, como em pacientes com distúrbios alimentares. Há casos em que o uso abusivo de laxantes também pode acarretar alcalose metabólica, mais provavelmente resultante de um aumento hipocalemia-induzido da produção de amônia urinária, que, por sua vez, promove aumento da excreção de cloreto de amônio na urina.
Perda renal de hidrogênio. Uma perda renal ácida indevida pode ocorrer diante da secreção aumentada de íons hidrogênio pelos néfrons distais. Nesta situação, os mineralocorticoides (inclusive a aldosterona) atuam promovendo a estimulação direta da ATPase de H+ secretora e tornando o lúmen tubular mais eletronegativo via estimulação da reabsorção do sódio [Figura 6]. A secreção de potássio distal também é intensificada neste contexto e resulta em hipocalemia concomitante.
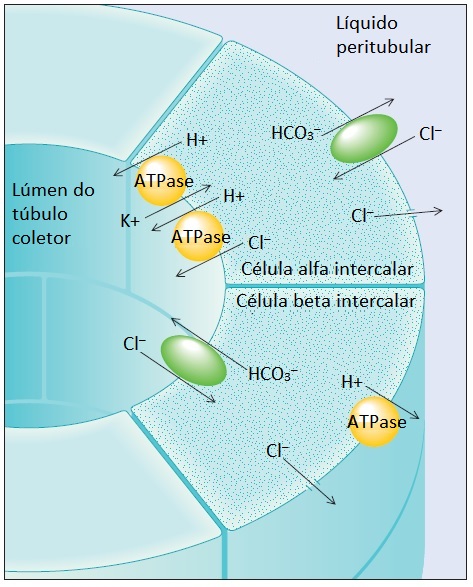
Figura 6. Na alcalose metabólica, a alta proporção de células alfa intercalares para células beta intercalares diminui. Diferente das alfa intercalares, que regeneram bicarbonato e o adicionam ao sangue venoso, as células beta intercalares promovem excreção urinária de HCO3– trocando-o por Cl– no filtrado glomerular. Como resultado, a administração de cloreto tem função importante no tratamento da maioria dos indivíduos com alcalose metabólica.
A alcalose metabólica associada à perda renal de hidrogênio pode ser causada pelo excesso de mineralocorticoides, administração de diuréticos de alça ou tiazídicos, alcalose pós-hipercapneica ou hipercalcemia (p. ex., síndrome do leite-álcali) [Tabela 5]. A alcalose metabólica também ocorre nas síndromes de Bartter e Gitelman. Estes 2 distúrbios produzem anormalidades eletrolíticas similares àquelas causadas pela terapia diurética, pois estão associados aos defeitos de transportadores ao nível da alça de Henle e do túbulo distal, respectivamente, que inibem os mecanismos também inibidos pelos diuréticos de alça e tiazidas [ver Síndrome de Bartter e Síndrome de Gitelman, adiante]. Qualquer causa de excesso mineralocorticoide, como o aldosteronismo primário, pode resultar em alcalose metabólica. O aldosteronismo primário em geral é acompanhado de hipertensão e hipocalemia. Em contraste, os pacientes com aldosteronismo secundário não tratados, cuja condição é decorrente de insuficiência cardíaca congestiva ou cirrose, geralmente não apresentam alcalose metabólica nem hipocalemia. Nestes casos, o efeito da aldosterona é contraposto pela reduzida distribuição distal de sódio (exceto com a administração de diuréticos) e pelo volume de urina diminuído. Estes fatores limitam a quantidade de ácido e de potássio secretada e também excretada na urina final.
Quando os pacientes são tratados à base de diuréticos tiazídicos ou diuréticos de alça, observa-se uma distribuição distal adequada de cloreto de sódio e o aumento da secreção de aldosterona. O aumento da secreção distal de íons hidrogênio e a contração do volume, diante de uma diurese significativa, contribuem para o desenvolvimento de alcalose metabólica.
A acidose respiratória crônica promove o aumento adequado da secreção de hidrogênio, à medida que a elevação da [HCO3–] plasmática aumenta o pH até normalizá-lo. A rápida diminuição da PaCO2, em geral por ventilação mecânica, promove alcalose metabólica quando a [HCO3–] plasmática do paciente permanece elevada. Esta anormalidade é denominada alcalose metabólica pós-hipercapneica.
Hipercalcemia. A hipercalcemia aumenta a reabsorção tubular renal de bicarbonato. Contudo, uma alcalose metabólica significativa em pacientes hipercalcêmicos é mais comumente observada em indivíduos com síndrome do leite-álcali. Nesta síndrome, uma carga alcalina aumentada (produzida pela ingesta de carbonato de cálcio) e a insuficiência renal hipercalcemia-induzida aumentam a produção e diminuem a excreção de bicarbonato. A alcalose metabólica pode ser observada em qualquer condição hipercalcêmica que seja causada pela contração de volume e pelo comprometimento da absorção de cloreto de sódio e cloreto de potássio resultante de lesão tubular.
Deslocamento intracelular de hidrogênio. Além de ser causada pela perda de hidrogênio, a alcalose metabólica pode ser produzida pelo deslocamento do hidrogênio para dentro das células. Tanto o vômito como a terapia diurética induzem diretamente as perdas de potássio e hidrogênio. A hipocalemia produz um deslocamento transcelular, em que o potássio sai das células para aumentar as reservas extracelulares. Para manter a eletroneutralidade, o hidrogênio entra nas células. Este deslocamento não só aumenta o pH extracelular como também diminui o pH intracelular. A diminuição do pH intracelular promove reabsorção tubular proximal do bicarbonato e secreção distal de íons hidrogênio.
Administração de álcali. A administração de bicarbonato de sódio a uma dosagem de até 1.000 mEq/dia normalmente não induz alcalose metabólica em indivíduos normais, pois o excesso de bicarbonato é rapidamente excretado na urina. Se a capacidade de excretar bicarbonato estiver comprometida, porém, o indivíduo pode desenvolver alcalose metabólica com a administração de uma quantidade muito grande de bicarbonato ou de precursor de bicarbonato (p. ex., lactato, citrato ou acetato). Isto ocorre com o citrato nas transfusões de grandes volumes de sangue.
Alcalose por contração. A alcalose por contração desenvolve-se com a perda de volumes relativamente amplos de líquido isento de bicarbonato. Neste contexto, a [HCO3–] plasmática sobe, pois o volume extracelular se contrai em torno de uma quantidade relativamente constante de bicarbonato extracelular.
A causa mais comum de alcalose por contração é a administração de um diurético de alça para induzir uma rápida remoção de líquido em pacientes com edema acentuado. De modo semelhante, a alcalose por contração ocorre diante de outras condições que envolvem perda de líquido contendo alta concentração de cloreto e perda de uma pequena [HCO3–]. Entre estas causas, estão a prescrição de diuréticos tiazídicos, perda de secreções gástricas (até mesmo em pacientes com acloridria), perdas por sudorese em pacientes com fibrose cística, e diarreia em alguns pacientes com adenomas vilosos ou cloridorreia congênita.
Fatores adicionais que contribuem para a alcalose metabólica. Na ausência de insuficiência renal avançada, 1 dentre 3 fatores deve estar presente para manter uma alta [HCO3–] plasmática: depleção efetiva do volume circulante, depleção de cloreto ou hipocalemia.
Tanto a queda da taxa de filtração glomerular (TFG) como a associada avidez do sódio observadas com a hipovolemia limitam a excreção do bicarbonato de sódio. A maior parte do bicarbonato de sódio é reabsorvida no túbulo proximal [Figura 2]. Um estímulo importante para a intensificação da reabsorção neste segmento do néfron é a atividade aumentada do contratransportador de Na+/H+ junto às membranas das células tubulares. A contração do volume promove troca de Na+/H+ neste segmento do néfron, em parte via liberação de angiotensina II. Os íons hidrogênio secretados dentro do lúmen combinam-se ao bicarbonato filtrado e, por fim, aumentam a taxa de transporte de volta para dentro das células tubulares. Mais bicarbonato é devolvido para o sangue venoso ao nível dos túbulos coletores, em parte sob a influência do aldosteronismo secundário. A depleção de volume não causa alcalose metabólica sem perdas de seletivas de cloreto. Exemplificando, a perda de sangue não aumenta a [HCO3–] porque as perdas de cloreto são idênticas às perdas em um volume de plasma igual.
A depleção do cloreto pode promover regeneração de bicarbonato e diminuição da secreção distal de bicarbonato. A regeneração do bicarbonato nas células alfa intercalares do túbulo coletor é mediada pela secreção de íons hidrogênio através das ATPases de H+ presentes na membrana luminal [Figura 6]. A cossecreção passiva do cloreto é requerida para a manutenção da eletroneutralidade. O bicarbonato intracelular é devolvido para a circulação sistêmica em troca de cloreto.
As células beta intercalares situadas no túbulo coletor cortical (que se tornam mais numerosas com o desenvolvimento de alcalose metabólica) conseguem secretar bicarbonato diretamente, ao reverterem a localização dos transportadores [Figura 6]. Desta forma, os trocadores de Cl–/HCO3– estão localizados na membrana luminal e promovem a secreção de bicarbonato dentro do lúmen tubular. As ATPases de H+ estão localizadas na membrana basolateral. Embora a atividade destas células seja devidamente intensificada pela alcalemia, em uma tentativa de excretar o excesso de bicarbonato, a queda associada da [Cl–] no líquido tubular diminui o gradiente de entrada favorável ao cloreto e, desta forma, diminui a secreção de bicarbonato. No entanto, a depleção de cloreto em seres humanos quase sempre está associada a uma depleção de volume efetiva.
A hipocalemia aumenta diretamente a reabsorção de bicarbonato, via pelo menos 2 mecanismos diferentes. Primeiro, a depleção de potássio ativa a ATPase de K/hidrogênio no néfron distal. Este transporte reabsorve o potássio e secreta hidrogênio. Em segundo lugar, a secreção distal de potássio e hidrogênio é mediada em troca de sódio luminal [Figura 6]. Nos estados de depleção de potássio, a taxa de secreção de hidrogênio em troca de sódio aumenta. Como resultado, a hipocalemia e o hiperaldosteronismo, que estimulam a secreção de íons hidrogênio, podem produzir um efeito potencializador sobre o desenvolvimento e a manutenção da alcalose metabólica.
Manifestações clínicas. Alguns indivíduos com alcalose metabólica apresentam câimbras severas, parestesias ou até mesmo tetania, enquanto outros com níveis similares de eletrólitos não desenvolvem estas manifestações. O motivo desta diferença em termos de manifestação é obscuro. Outros achados clínicos resultam da etiologia subjacente (p. ex., hipertensão com hiperaldosteronismo primário).
O diagnóstico de alcalose metabólica geralmente é evidenciado por história de vômitos ou prescrição de terapia diurética. Em alguns casos, porém, a alcalose metabólica não tem nenhuma causa evidente. Neste contexto, o diagnóstico mais provável é o vômito sub-reptício provocado por um distúrbio alimentar, uso de diuréticos ou uma das causas de excesso de mineralocorticoides (p. ex., aldosteronismo primário). Os primeiros 2 fatores induzem uma depleção de volume efetiva, enquanto o hiperaldosteronismo primário geralmente está associado a uma discreta expansão de volume como resultado do efeito estimulador da aldosterona sobre a reabsorção renal de sódio.
Vários achados fornecidos pelo exame físico podem sugerir o vômito sub-reptício como causa, incluindo a erosão dental associada à exposição repetida às secreções gástricas ácidas, e a presença de úlceras e calos no dorso da mão produzidos pela introdução de um dedo na região posterior da garganta para induzir o vômito.
Exames laboratoriais. A quantificação da [Na+] em uma amostra de urina obtida ao acaso é usada em muitas condições para distinguir entre depleção de volume ([Na+] urinária geralmente < 20 mEq/L) e euvolemia ([Na+] > 40 mEq/L). Entretanto, a alcalose metabólica é uma das condições em que a depleção de volume pode não acarretar uma [Na+] urinária baixa. Neste contexto, a capacidade de reter sódio pode ser antagonizada pela necessidade de excretar bicarbonato (como sal sódico) em uma tentativa de corrigir a alcalose. Nestes casos, uma determinação aleatória da [Cl–] urinária é mais útil.
A perda de sódio é mais provável durante os primeiros dias de vômito, quando a [HCO3–] plasmática e, portanto, a carga de bicarbonato filtrado estão aumentadas. No início do curso dos vômitos, a capacidade de intensificar a reabsorção de bicarbonato ainda é estimulada. Os efeitos líquidos são as altas [Na+] e [K+] na urina e o pH urinário superior a 7,0 produzido pela bicarbonatúria.
Como resultado, a [Na+] urinária não constitui necessariamente um reflexo acurado da condição do volume do paciente na alcalose metabólica. A presença de hipovolemia subjacente pode ser detectada de modo mais acurado pelo achado de uma [Cl–] urinária abaixo de 25 mEq/L. A conservação apropriada do cloreto é promovida pela depleção do volume e pela hipocloremia induzida pelas perdas de cloreto via secreções gástricas. No entanto, a concentração urinária de cloreto pode estar indevidamente alta, se a reabsorção de cloreto estiver defeituosa. Este tipo de defeito ocorre mais comumente em pacientes que recebem terapia diurética. Portanto, em pacientes com alcalose metabólica decorrente de vômito, a concentração urinária de cloreto é tipicamente baixa. Esta concentração é mais alta diante da administração de diuréticos. Em ambas as circunstâncias, a [Na+] urinária pode estar elevada.
Síndrome de Bartter e síndrome de Gitelman. As síndromes de Bartter e Gitelman são distúrbios da reabsorção de cloreto de sódio que ocorre ao nível da alça de Henle e do túbulo distal, respectivamente.29 A síndrome de Bartter é um distúrbio raro que produz alcalose metabólica hipocalêmica. Como as [Na+] e [Cl–] urinárias costumam ser maiores que 25 mEq/L, o uso sub-reptício de diuréticos constitui o principal distúrbio a ser considerado no diagnóstico diferencial. A síndrome de Bartter clássica em geral se manifesta no início da vida e pode estar associada ao retardo do crescimento e ao retardo mental. O espectro de achados, incluindo a hipercalciúria, é mais compatível com a existência de um defeito primário de reabsorção de cloreto de sódio no ramo ascendente espesso medular da alça de Henle.
A síndrome de Gitelman é uma condição mais benigna, que pode ser herdada como doença recessiva autossômica. Esta condição pode não ser detectada antes da fase tardia da infância ou até mesmo somente na fase adulta. Em contraste com os pacientes com síndrome de Bartter, que possuem um defeito na capacidade de concentração da urina (a geração de um gradiente osmótico intersticial alto requer o funcionamento normal da alça de Henle), os indivíduos com síndrome de Gitelman podem apresentar capacidade de concentração da urina normal e ter hipocalciúria. Este achado sugere que o defeito reside no túbulo distal, pois achados similares são encontrados em pacientes tratados com diuréticos tiazídicos. As síndromes de Bartter e Gitelman diagnosticadas somente após o uso do diurético – que constitui a outra causa, bem mais comum, destes achados – são excluídas.
Em pacientes com depleção de volume verdadeira causada por vômito, sucção nasogástrica, adenomas vilosos ou terapia diurética, a alcalose metabólica pode ser corrigida com a administração de cloreto de sódio.30 A administração de cloreto de potássio aos pacientes com hipocalemia concomitante também contribui para a correção da alcalemia. A determinação da [Cl–] urinária pode ser útil como indicação de quando a depleção de volume tiver sido corrigida. Uma vez restaurada a perfusão renal, este valor deve ser maior que 40 mEq/L.
Condições edematosas. Terapias distintas são usadas em casos de pacientes edematosos com alcalose metabólica geralmente causada por insuficiência cardíaca, cor pulmonale ou doença hepática em estágio avançado. Nestes distúrbios, o cloreto de sódio é contraindicado, pois aumenta o grau de edema. Entretanto, a administração de um inibidor de anidrase carbônica, a acetazolamida (dosagem inicial de 250 mg, via oral [VO], 1 a 2 vezes/dia; ou 125 mg, via intravenosa [IV], 1 a 2 vezes/dia), pode ser particularmente efetiva. Este fármaco preferencialmente inibe a reabsorção tubular proximal do bicarbonato de sódio e, desta forma, corrige tanto a alcalose como a sobrecarga de líquido.
Um potencial efeito colateral da terapia com inibidor de anidrase carbônica é o desenvolvimento ou piora da hipocalemia. Embora seja possível tratar a hipocalemia com suplementação de potássio, uma abordagem terapêutica alternativa consiste em administrar um diurético poupador de potássio (p. ex., espironolactona) no lugar do inibidor de anidrase carbônica. Os diuréticos poupadores de potássio comprometem a reabsorção de sódio junto aos túbulos coletores e, como resultado, limitam a secreção adicional de potássio e hidrogênio [Figura 6]. Para os pacientes com hepatopatia avançada, a espironolactona (um antagonista de aldosterona) ou a eplerenona podem ser os diuréticos mais efetivos.
Em ocasiões raríssimas, a alcalose metabólica pode tão severa que se torna necessário administrar ácido hidroclorídrico para corrigir o problema. A solução-padrão de ácido hidroclorídrico a 0,1 N (decinormal) contém 100 mEq de H+/L. Dada a natureza corrosiva da solução de ácido hidroclorídrico, a administração do ácido somente deve ser realizada quando outras ações de correção da alcalose metabólica tiverem falhado. A solução de ácido hidroclorídrico deve ser sempre administrada em uma veia central.
Síndrome de Bartter e síndrome de Gitelman. O defeito tubular observado nos pacientes com estas síndromes não pode ser corrigido. Em consequência, o tratamento é voltado para a minimização das anormalidades eletrolíticas e metabólicas. A combinação de um AINH (inclusive de inibidores de ciclo-oxigenase-2, porque os níveis de prostaglandina estão secundariamente aumentados) a um diurético poupador de potássio pode elevar a [K+] plasmática até os níveis normais e promover uma ampla reversão da alcalose metabólica. A maioria dos pacientes, contudo, necessita de suplementação oral e contínua de potássio e magnésio, porque a terapia farmacológica é totalmente efetiva somente em raros casos.
A ventilação alveolar fornece o oxigênio necessário ao metabolismo oxidativo e elimina o dióxido de carbono produzido por estes processos metabólicos. Portanto, convém que os principais estímulos fisiológicos da respiração sejam uma tensão de oxigênio arterial (PaO2) reduzida, denominada hipoxemia, e uma PaCO2 alta. O dióxido de carbono estimula a ventilação ao atuar sobre o pH. Em contraste, a intensificação hipoxêmica inicial da ventilação é mediada principalmente pelos quimiorreceptores existentes nos corpos carotídeos, que estão localizados perto da bifurcação das artérias carótidas. Alguns distúrbios podem ser responsáveis pela alcalose e acidose respiratória crônica [Tabelas 6 e 7].
Tabela 6. Causas de acidose respiratória
|
Distúrbio |
Causa |
|
Supressão do centro respiratório medular |
Medicações sedativas Administração de oxigênio na doença pulmonar crônica Apneia do sono (também causada pela obesidade extrema) Lesões no sistema nervoso central (incomum) Parada cardiopulmonar |
|
Diminuição da função da musculatura respiratória |
Aguda Enfraquecimento muscular ou paralisia (miastenia grave, paralisia periódica, aminoglicosídeos intraperitoneais, síndrome de Guillain-Barré, botulismo, hipocalemia severa, hipofosfatemia severa) Crônica Enfraquecimento muscular: poliomielite, esclerose lateral amiotrófica, mixedema Cifoescoliose |
|
Obstrução das vias aéreas superiores |
Aspiração de corpo estranho ou vômito Obstrução na apneia do sono Laringoespasmo |
|
Distúrbios que afetam as trocas gasosas pulmonares |
Agudos Síndrome do sofrimento respiratório agudo Edema pulmonar cardíaco agudo Asma ou pneumonia severa Pneumotórax ou hemotórax Crônicos Doenças pulmonares obstrutivas |
|
Ventilação mecânica inadequada |
– |
Tabela 7. Causas de alcalose respiratória
|
Distúrbio |
Causa |
|
Hipoxemia |
Doença pulmonar: pneumonia, êmbolos, edema, fibrose intersticial Insuficiência cardíaca congestiva Anemia severa Exposição a altitudes elevadas |
|
Estimulação direta do centro respiratório medular |
Síndrome da hiperventilação Encefalopatia hepática Sepse ou febre Intoxicação por salicilato* Subsequente à correção rápida da acidose metabólica Gestação (isto é, aumento dos níveis de progesterona) Distúrbios neurológicos (acidente vascular cerebral, tumores da ponte) |
|
Ventilação mecânica excessiva |
– |
*A alcalose respiratória é o distúrbio inicialmente observado, embora a acidose metabólica se desenvolva posteriormente, caso a intoxicação seja severa.
Manifestações clínicas. A acidose respiratória severa pode produzir uma variedade de anormalidades neurológicas. Entre os sintomas iniciais, estão a cefaleia, visão turva, agitação e ansiedade, que podem progredir para tremores, asterix, delírio e uma sonolência denominada narcose por dióxido de carbono. Alguns destes sinais, incluindo o papiledema, parecem ser causados por um aumento do fluxo sanguíneo cerebral induzido pela acidemia. De modo geral, estes sinais parecem resultar da queda do pH do líquido cerebrospinal, e não das alterações ocorridas no pH arterial ou na PaCO2.
Os sintomas produzidos pela alcalose respiratória estão relacionados à aumentada irritabilidade dos sistemas nervoso central e periférico. Tais sintomas incluem tontura (“cabeça leve”), alteração da consciência, parestesias de membros e área circum-oral, câimbras, espasmos carpopodais (indistinguíveis dos espasmos causados pela hipocalcemia) e síncope. Os pacientes gravemente enfermos também podem apresentar diversas arritmias. Estas anormalidades parecem estar relacionadas à capacidade da alcalose de diminuir o fluxo sanguíneo cerebral e aumentar a excitabilidade da membrana. Uma diminuição dos níveis de magnésio ou cálcio ionizado também contribui para a excitabilidade aumentada da membrana.
Exames laboratoriais. A alcalose respiratória primária pode ser diagnosticada quando o pH sanguíneo é maior que 7,42 na presença de uma PCO2 reduzida. A condição também pode ser identificada como um segundo distúrbio primário em pacientes com acidose metabólica, por meio da aplicação da fórmula de Winter [ver Acidose metabólica, anteriormente].
Em geral, a insuficiência respiratória causa acidose respiratória aguda. Como resultado, o tratamento costuma ser indicado e o paciente pode necessitar de ventilação mecânica [ver 14:XI Insuficiência respiratória]. Em contraste, o tratamento da alcalose respiratória geralmente é dispensável. A avaliação deve ser voltada para o diagnóstico e correção do distúrbio subjacente.
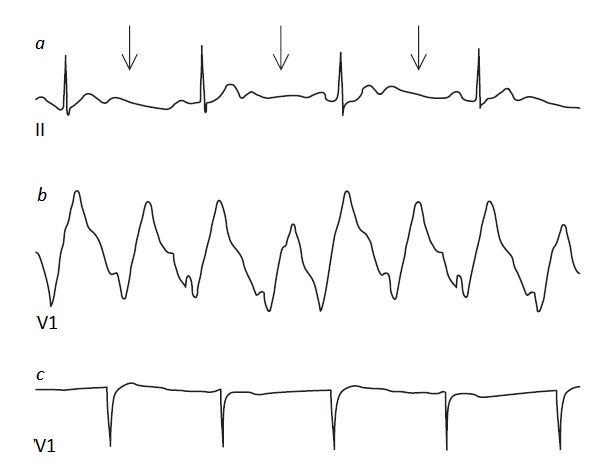
O potássio é o principal cátion intracelular, e sua concentração é significativamente maior dentro das células (123 a 140 mEq/L) do que no espaço extracelular (3,5 a 5 mEq/L). Esta diferença de concentração é preservada pela ATPase de Na+/K+, que transporta ativamente os íons sódio para fora e os íons potássio para dentro da maioria das células. A diferença de concentração de potássio dentro e fora da célula é o principal determinante da excitabilidade da membrana. Desta forma, apesar da quantidade comparativamente pequena de potássio extracelular, alterações sutis podem produzir efeitos drásticos sobre a contração muscular e a condução nervosa.
Nos Estados Unidos, a ingesta diária de potássio oriundo da dieta varia de 40 a 120 mEq (isto é, 1.560 a 4.680 mg). Sob condições normais, cerca de 90% do potássio da dieta é excretado na urina; a maior parte do restante é eliminada nas fezes. As perdas gastrintestinais de potássio aumentam nos pacientes com insuficiência renal, porém a importância desta adaptação é incerta.
Apenas cerca de 50% de uma carga VO ou IV de potássio aparece na urina durante as primeiras 4 horas subsequentes à administração. Uma hipercalemia acentuada e potencialmente prejudicial à vida pode ocorrer, se o potássio restante permanecer confinado no líquido extracelular. Este volume corresponde a apenas cerca de 14 L em um indivíduo de 70 kg do sexo masculino. O transporte da maior parte deste potássio para dentro das células antes da excreção na urina minimiza a elevação da [K+] plasmática.
Os fatores mais importantes associados ao transporte do potássio dietético para dentro das células são as influências exercidas pela insulina e pelos receptores beta-2-adrenérgicos. A insulina estimula a ATPase de Na+/K+ e acelera a entrada de potássio. Evidências recentes sugerem que a taxa de eliminação de uma carga de potássio IV é menor em norte-americanos afrodescendentes do que em norte-americanos brancos.31 A ativação dos receptores beta-2-adrenérgicos também promove movimentação de potássio para dentro das células. A aldosterona é o hormônio mais importante envolvido na secreção de potássio pelas superfícies epiteliais, incluindo as células epiteliais do túbulo renal. A aldosterona parece ser menos importante para o transporte de potássio para dentro de outras células.
O potássio é filtrado livremente no glomérulo. A concentração de íons potássio que entra pela porção inicial do túbulo proximal é de aproximadamente 4 mEq/L – igual à [K+] plasmática. Quando o filtrado glomerular chega ao túbulo distal, 90% do potássio filtrado já foi reabsorvido. Desta forma, a excreção renal de potássio ocorre quase exclusivamente por secreção ao nível do túbulo coletor.
A secreção de potássio dentro do túbulo coletor ocorre nas células principais [Figura 3]. A movimentação dos íons potássio partindo das células tubulares para dentro do lúmen é controlada pelas taxas de (1) ingesta de potássio oriundo da dieta; (2) reabsorção de potássio, que gera um gradiente elétrico negativo favorável à movimentação do potássio de dentro das células para o lúmen tubular; e (3) fluxo urinário distal, que mantém um elevado gradiente de concentração célula tubular:lúmen tubular por meio de eliminação por lavagem do potássio secretado.
A aldosterona produzida pelas glândulas suprarrenais entra nas células principais a partir da superfície antiluminal ou capilar. No meio intracelular, a aldosterona liga-se aos receptores que aumentam o número de canais de sódio abertos junto à membrana celular luminal. O número e a atividade das ATPases de Na+/K+ presentes na membrana celular também aumentam. A resultante elevação do potássio celular leva à secreção do íon dentro do lúmen do túbulo coletor até que sejam alcançados uma concentração favorável e um gradiente eletroquímico.
Nos estados de depleção de potássio, a secreção de íons potássio pelas células principais é reduzida (contanto que os rins não estejam atuando como fonte de perda de potássio), e a reabsorção de potássio é estimulada. Este processo ocorre nas células intercalares adjacentes [Figura 3]. Recentemente, foi identificada uma nova classe de proteínas intracelulares reguladoras da secreção de potássio ao nível do néfron distal: as quinases WNK. Em condições basais, várias destas quinases inibem a reabsorção de sódio e a secreção de potássio. Sob estimulação da aldosterona, o efeito inibitório das quinases WNK é revertido, com consequente reabsorção de sódio e secreção de potássio.32
A hipocalemia, que pode ser definida por uma [K+] plasmática abaixo de 3,5 mEq/L, pode ser causada por uma baixa ingesta de potássio, um desvio do potássio para dentro das células ou por perdas corporais de potássio [Figura 7]. Na maioria das pessoas, as perdas de potássio ocorrem a partir do trato gastrintestinal, pele ou rins.
Figura 7. Uma abordagem para as causas de hipocalemia.
ATR = acidose tubular renal; CA = cetoacidose; GI = gastrintestinal; PA = pressão arterial; UCl = conteúdo de cloreto da urina; UK = excreção urinária de potássio.
Baixa ingesta de potássio. Uma ingesta de potássio inadequada é incomum entre indivíduos saudáveis que vivem em países industrializados, pois o potássio é abundante na maioria dos alimentos. Além disso, se a ingesta diminui, as perdas urinárias e intestinais podem ser reduzidas a menos de 15 mEq/dia.
Distribuição de potássio alterada. Mesmo quando as reservas corporais de potássio estão normais, existem várias condições que podem promover o transporte de potássio para dentro das células e, assim, diminuir a [K+] plasmática.
A alcalose metabólica e, em grau significativamente menor, a alcalose respiratória aguda podem estar associadas à hipocalemia. Neste contexto, o principal mecanismo é a transferência de íons hidrogênio para fora das células como parte da resposta de tamponamento que minimiza a elevação do pH extracelular. A eletroneutralidade é mantida em parte pela entrada de potássio dentro das células. A relação existente entre o grau de hipocalemia e o aumento do pH sanguíneo varia amplamente. A perda de potássio pelo corpo também exerce papel importante na hipocalemia observada com a alcalose metabólica produzida pelo vômito ou uso de diurético.
Uma oscilação de catecolamina, que ocorre durante o infarto agudo do miocárdio ou no delirium tremens, pode resultar no deslocamento intracelular agudo de potássio causado pela estimulação de receptores beta-2-adrenérgicos. Este fenômeno, em que a adrenalina pode converter uma hipocalemia leve em hipocalemia severa, pode contribuir para o aumento evidente da mortalidade coronariana observado nos estudos sobre pacientes com hipertrofia ventricular esquerda e hipertensão leve tratados com doses moderadas a altas de diuréticos tiazídicos. A [K+] plasmática tipicamente diminui durante a administração de insulina em pacientes com cetoacidose diabética, mesmo que esteja normal ou aumentada no momento da apresentação. Esta queda resulta em parte da transferência insulina-estimulada de potássio para dentro das células. Um problema similar pode ocorrer com a administração IV de dextrose em pacientes não diabéticos. Exemplificando, uma solução de dextrose a 5% em água pode acarretar a piora temporária da hipocalemia e possivelmente provocar arritmias cardíacas.
Entre as raras causas de hipocalemia induzida por redistribuição, estão a administração de ácido fólico ou vitamina B12 no tratamento das anemias megaloblásticas (em que a rápida produção de células novas resulta na captação de potássio a partir do líquido extracelular) e o envenenamento com sais de bário (que bloqueia os canais situados na membrana celular pelos quais o potássio normalmente sai das células). Os pacientes submetidos a procedimentos radiográficos gastrintestinais não são expostos ao risco de envenenamento por sais de bário, pois o sulfato de bário usado nos exames gastrintestinais não é absorvido na circulação sistêmica. As superdosagens de teofilina severas também podem causar hipocalemia resultante dos deslocamentos de potássio catecolamina-induzidos ou liberação de insulina glicose-induzida.
A paralisia periódica hipocalêmica é um distúrbio raro de causa incerta, que é caracterizado por episódios potencialmente fatais de enfraquecimento muscular ou paralisia resultantes da movimentação súbita do potássio para dentro das células. Os ataques agudos frequentemente são precipitados pelo repouso subsequente ao exercício, estresse ou ingesta de uma refeição contendo carboidratos – eventos que muitas vezes estão associados ao aumento da liberação de adrenalina ou insulina. Os ataques tendem a durar 6 a 24 horas, mas podem ser mais longos.
O distúrbio pode ser familiar, e o gene anormal, na maioria dos pacientes, parece codificar a parte dos canais de cálcio presentes no músculo esquelético que é bloqueada pelos bloqueadores de canal de cálcio diidropiridínicos (p. ex., nifedipina). Entretanto, alguns pacientes com hipertireoidismo, em particular os homens asiáticos, adquirem a doença.33 Nestes indivíduos, o excesso de hormônio da tireoide pode predispor a episódios de paralisia por aumentar a atividade da ATPase de Na+/K+ nas células [ver Tratamento, adiante].
Perdas corporais de potássio. O trato gastrintestinal pode ser um sítio importante de perda de potássio, particularmente em indivíduos que apresentam vômito ou diarreia. Entretanto, a [K+] nas secreções gástricas (5 a 10 mEq/L) é significativamente menor do que nas secreções intestinais, em que pode chegar a 75 mEq/L. Como resultado, seria necessário perder grandes volumes de secreção gástrica para que uma depleção substancial de potássio fosse produzida. A [K+] diminuída observada em casos de vômito resulta primariamente das perdas urinárias aumentadas, e não das perdas gástricas (ver adiante). Por comparação, o fosfato de sódio administrado para fins de preparação do cólon para colonoscopia pode acarretar hipocalemia em idosos.34
Existem 2 condições que podem causar perdas cutâneas de potássio com consequente hipocalemia: (1) a prática de exercício em ambiente quente e úmido; e (2) queimaduras extensas. Indivíduos que praticam exercícios vigorosos podem perder mais de 10 L de suor em 1 dia. Esta perda pode acarretar uma depleção de potássio significativa, mesmo que a [K+] no suor seja de apenas 5 mEq/L. As perdas de potássio na urina são mais comuns em pacientes que vomitam ou recebem terapia diurética. A depleção do volume extracelular promove liberação de aldosterona, com consequente intensificação das trocas de Na+/K+ ao nível do túbulo coletor [Figura 3]. No entanto, o aldosteronismo isolado é insuficiente para permitir o aumento da excreção de potássio, porque o fluxo urinário distal deve ser mantido para que o potássio secretado seja excretado. O fluxo urinário distal é mantido na presença de vômitos ou terapia diurética, pois o sódio e a água escapam da reabsorção. Os diuréticos inibem a reabsorção de água e sódio tanto ao nível da alça de Henle como ao nível do túbulo distal. A aumentada carga tubular de bicarbonato presente durante o vômito permite que o sódio e a água escapem da reabsorção junto ao túbulo proximal. A inibição direta da reabsorção de potássio também contribui para a hipocalemia observada com o uso de diuréticos de alça.
Até 40% dos indivíduos hipocalêmicos também apresentam hipomagnesemia. A causa da perda de potássio, como também o tratamento com diurético ou cisplatina, às vezes também são responsáveis pelo comprometimento da reabsorção de magnésio. Contudo, a depleção de potássio geralmente não pode ser corrigida se o equilíbrio do magnésio não for restaurado. Esta conexão é possível porque o magnésio pode atuar como bloqueador de canal de potássio. Na alça de Henle, por exemplo, o potássio pode vazar através da membrana celular para dentro do lúmen tubular e, desta forma, contribuir para as perdas urinárias em curso.
Existem, ainda, várias causas menos comuns de disfunção tubular que podem acarretar perdas urinárias de potássio. Entre estas causas, estão a ATR de tipo 1 clássica; fármacos (p. ex., cisplatina, anfotericina B e aminoglicosídeos); as leucemias aguda e, menos frequentemente, crônica; e a administração de levodopa. A reabsorção diminuída de sódio, com consequente depleção de volume, estimula a secreção de aldosterona e atua em muitos destes distúrbios. A hipótese de aldosteronismo primário, todavia, deve ser considerada sempre que uma perda de potássio não provocada for detectada em um indivíduo hipertenso.
A síndrome de Liddle é uma causa rara de hipertensão e alcalose metabólica, que resulta de um defeito genético ou adquirido envolvendo o canal de sódio epitelial. Esta anormalidade provoca aumento da reabsorção de sódio ao nível do túbulo coletor. Este distúrbio está associado a níveis de aldosterona normais ou suprimidos. Em consequência, é possível tratá-lo com diuréticos poupadores de potássio, como a amilorida, porém os antagonistas de aldosterona não promovem resposta.
Manifestações clínicas. Na maioria das circunstâncias, uma hipocalemia leve ([K+] plasmática = 3 a 3,5 mEq/L) não produz sintomas. A maioria das perturbações associadas a deficiências de potássio mais sérias resulta de alterações na função cardiovascular, neuromuscular e renal. A toxicidade cardíaca pode manifestar-se como arritmias graves, que ocorrem porque a hiperpolarização da membrana celular miocárdica produz um período refratário prolongado e uma suscetibilidade aumentada a arritmias reentrantes. Outras alterações eletrocardiográficas (ECG) associadas à hipocalemia incluem a depressão da onda T e ondas U proeminentes [Figura 8].
Figura 8. Tanto a hipo como a hipercalemia podem causar alterações no eletrocardiograma (ECG) do paciente. (a) O ECG de paciente com hipocalemia moderada mostra ondas U proeminentes (setas). (b) Uma hipercalemia acentuada resulta em ondas T com picos e complexos QRS ampliados no ECG. Decorridos 10 minutos da administração de cálcio por via intravenosa (IV), porém, todas as manifestações ECG de hipercalemia do paciente foram resolvidas (c).
A hiperpolarização também retarda a condução nervosa e a contração muscular, e isto pode contribuir para o enfraquecimento muscular, câimbras e parestesias, ainda que estes sintomas em geral somente sejam observados quando a [K+] plasmática cai a menos de 2,5 mEq/L. Uma hipocalemia severa pode comprometer a função da musculatura respiratória e causar hipoventilação. Como o potássio normalmente permite que ocorra vasodilatação em resposta à contração muscular, a hipocalemia severa também predispõe à rabdomiólise.
As manifestações renais primárias da hipocalemia são a poliúria resultante da sede estimulada e a resistência à ação do hormônio antidiurético. Esta última condição é denominada diabetes insípido nefrogênico. A intensificação da sede é uma resposta apropriada à poliúria, mas também é causada pela estimulação direta do centro hipotalâmico da sede pela hipocalemia. A resistência ao hormônio antidiurético é causada por uma queda hipocalemia-induzida do número de canais de água existentes no túbulo coletor.35 A hipocalemia crônica pode levar ao desenvolvimento de nefrite intersticial crônica e causar declínio da filtração glomerular.
Quando a hipocalemia é severa ([K+] plasmática < 2 mEq/L), a capacidade dos túbulos renais de reabsorver sódio e cloreto pode ser comprometida. Esta condição pode levar à depleção de volume e à perda urinária de sódio e cloreto, mesmo nas condições de perfusão renal diminuída. O mecanismo pode envolver o transportador de Na+/K+/2Cl– presente no ramo ascendente da alça de Henle. Este transportador desloca sódio, potássio e cloreto do lúmen para dentro da célula. Uma parte do potássio absorvido é reciclada para o lúmen, para fornecer o potássio necessário ao funcionamento do transportador. No contexto da depleção severa de potássio, existe uma quantidade de potássio inadequada junto à célula para ser reciclada para o lúmen com a finalidade de manter o funcionamento do transportador. Isto resulta em perdas de cloreto de sódio similares àquelas observadas com a prescrição de furosemida, um diurético que atua no mesmo sítio junto ao néfron.
A história e o exame físico geralmente sugerem o diagnóstico do paciente hipocalêmico. A Figura 7 revisa uma abordagem para hipocalemia. Esta condição pode ser causada por um deslocamento do potássio do espaço vascular para o espaço intracelular (insulina, catecolaminas) ou por reduções do conteúdo corporal total de potássio. A excreção urinária de potássio é usada para distinguir a perda extrarrenal (< 30 mEq/dia ou 30 mEq/L) da perda renal (> 30 mEq/dia ou 30 mEq/L). As perdas extrarrenais são distinguidas também com base no pH sérico. A existência de acidose metabólica sugere perdas gastrintestinais menores, enquanto a presença de alcalose metabólica sugere que o paciente tem um adenoma viloso ou diarreia congênita perdedora de cloreto. O pH normal aponta uma ingesta oral precária, pica, perdas cutâneas ou paralisia periódica. As perdas urinárias de potássio também são caracterizadas com base no pH sérico. A acidose metabólica com hiato aniônico aumentado sugere uma cetoacidose diabética ou cetoacidose alcoólica. Na ausência de hiato aniônico, a acidose metabólica sugere ATR, sobretudo do tipo 1. Um pH normal é sugestivo de depleção de magnésio. No paciente com alcalose metabólica, a hipocalemia é distinguida com base no cloreto urinário: uma baixa concentração de cloreto na urina (< 10 mEq/L) sugere a ocorrência de perdas ao nível do trato gastrintestinal superior ou uso prévio de diurético. Uma alta concentração de cloreto na urina (> 30 mEq/L) aliada à hipertensão indicam aldosteronismo primário, síndrome de Cushing ou síndrome de Liddle, enquanto as pressões arteriais normais sugerem o uso corrente de diurético, síndrome de Bartter ou síndrome de Gittelman.
A normalização da [K+] plasmática é indicada para a maioria dos casos, embora o significado de “normalização” e a quantidade e via de administração de potássio variem amplamente. Na ausência de fatores causadores de deslocamentos transcelulares de potássio, existe uma relação bastante previsível entre o grau de hipocalemia e a extensão da depleção de potássio corporal total. Para cada queda de 1 mEq/L na [K+] plasmática, as reservas de potássio diminuem em 200 a 400 mEq, até que a [K+] plasmática cai a menos de 2 mEq/L. Neste ponto, o déficit total pode exceder 1.000 mEq. Com a reposição oral e parenteral, o potássio entra no plasma antes de ser transferido para dentro das células. Em consequência, a suplementação de potássio, em particular quando fornecida via IV, está associada ao risco de hipercalemia se a dose administrada for alta demais ou quando sua administração é feita muito rapidamente. Por este motivo, sempre que possível, o potássio deve ser administrado por via oral.
A reposição de potássio com o uso de alimentos ricos em potássio pode ser efetiva para pacientes com acidose metabólica ou insuficiência renal. Entretanto, o potássio oriundo da dieta é inefetivo para a correção do déficit de potássio observado na alcalose metabólica. Na maioria dos alimentos, o potássio está ligado a ânions fracamente absorvíveis, como o fosfato. Por este motivo, os sais de cloreto (p. ex., cloreto de potássio, que é usado nos casos de alcalose metabólica produzida por diuréticos) em geral são necessários para repor as perdas de potássio. Uma exceção poderia ser o tratamento preventivo de cálculos renais com diuréticos tiazídicos, em que o citrato de potássio consegue manter níveis séricos de potássio normais.36
O potássio pode ser administrado por via IV, dentro de uma veia periférica, a concentrações de até 40 mEq/L. Concentrações mais altas podem causar flebite e por isso devem ser infundidas somente em veias de grande calibre. Exceto em contextos incomuns, a velocidade da administração não deve exceder 40 mEq/hora, embora dosagens de até 100 mEq/hora sejam infundidas em pacientes selecionados com paralisia ou arritmias prejudiciais à vida.37 As soluções contendo glicose devem ser evitadas, pois a estimulação da insulina pode deslocar o potássio para dentro das células e, desta forma, exacerbar a hipocalemia.
Para pacientes que tomam diuréticos para tratamento de pressão arterial elevada, existem várias alternativas disponíveis para corrigir a hipocalemia sem o uso de suplementos de cloreto de potássio. Como a complacência do paciente diminui à medida que o número de medicamentos aumenta (e isto inclui os suplementos de potássio), a diminuição da dose de diurético (p. ex., 12,5 mg de hidroclorotiazida) ou a substituição por um agente alternativo devem ser consideradas em 1º lugar.
Os indivíduos edematosos ou pacientes com aldosteronismo primário que também apresentam hipocalemia podem ser tratados com diuréticos poupadores de potássio (amilorida ou espironolactona) até que seja possível instituir uma terapia mais definitiva (p. ex., remoção cirúrgica de um adenoma suprarrenal no aldosteronismo primário).
Em pacientes com paralisia periódica hipocalêmica [ver Distribuição de potássio alterada, anteriormente], a administração de cloreto de potássio pode abortar os ataques agudos em questão de minutos. No entanto, a suplementação prolongada com cloreto de potássio geralmente não previne os ataques. O hipertireoidismo, quando presente, deve ser tratado, e a administração de um bloqueador beta-adrenérgico não seletivo (p. ex., propranolol) pode evitar os episódios em pacientes com a forma familiar da condição.
A hipercalemia é um distúrbio eletrolítico comum. Como diversas medicações utilizadas com frequência podem interferir na homeostasia normal do potássio, a hipercalemia muitas vezes é iatrogênica e, portanto, evitável. A hipercalemia severa está associada à alteração da função neuromuscular ou cardíaca.
A hipercalemia pode ser causada por uma ingesta excessiva de potássio, por aumento da liberação de potássio a partir das células ou por diminuição da excreção renal do potássio [Figura 9].
Figura 9. Abordagem para as causas de hipercalemia.
AINH = anti-inflamatório não hormonal; ATR = acidose tubular renal; BRA = bloqueador do receptor da angiotensina; DRC = doença renal crônica; IECA = inibidor da enzima conversora de angiotensina; IRD = inibidor de renina direto; TFG = taxa de filtração glomerular; TMP-Sulfa = trimetoprima-sulfametoxazol.
Ingesta aumentada de potássio. O desenvolvimento da [K+] plasmática aumentada subsequente a uma carga de potássio IV ou VO aguda depende de 4 fatores: (1) quantidade de potássio administrada; (2) capacidade de uma parte do excesso de potássio entrar nas células; (3) excreção urinária de potássio; e (4) nível anterior de ingesta de potássio – um processo denominado adaptação. A elevação da [K+] plasmática é minimizada quando a ingesta de potássio é aumentada lentamente. Durante um aumento lento, a excreção renal (e talvez a entrada celular) torna-se mais eficiente. Os níveis aumentados de aldosterona e o aumento da atividade da ATPase de Na+/K+ ao nível do túbulo coletor cortical exercem papel importante neste processo. Até mesmo na ausência de adaptação, o rim finalmente excreta o excesso de potássio, ainda que de forma mais lenta. Assim, a hipercalemia resultante de uma carga aguda é transiente, a não ser que a excreção renal do potássio sofra uma diminuição concomitante.
Liberação aumentada de potássio a partir das células. Tanto a deficiência de insulina como o bloqueio beta-2-adrenérgico podem aumentar a [K+] plasmática. Em pacientes com doença renal em estágio terminal sem hemólise, por exemplo, o propranolol ou labetalol (e não o bloqueador beta-1-adrenérgico seletivo atenolol) podem elevar os níveis da [K+] plasmática em cerca de 1 mEq/L. Contudo, doses maiores de atenolol podem acarretar a perda desta seletividade.
Uma elevação da osmolalidade plasmática resulta na movimentação osmótica da água das células para dentro do líquido extracelular. Este evento é acompanhado pela movimentação do potássio a partir das células através de 2 mecanismos. No primeiro mecanismo, a aumentada [K+] celular produzida pela perda de água cria um gradiente favorável à saída passiva do potássio através dos canais existentes na membrana celular. No segundo mecanismo, as forças de atrito atuantes entre solvente (água) e soluto transportam o potássio com a água através dos canais de água (aquaporinas) existentes na membrana celular.
A acidemia aguda também pode aumentar a [K+] plasmática ao deslocar o potássio das células para o plasma. Este deslocamento é provável durante a infusão de ácidos. Quando o hidrocloreto de arginina é infundido, por exemplo, o potássio é trocado pela arginina catiônica que entra nas células. Entretanto, este deslocamento parece ser importante somente nas acidoses inorgânicas. Em comparação, a hipercalemia observada na acidose láctica é causada primariamente pela quebra celular, enquanto a hipercalemia da cetoacidose resulta da insuficiência de insulina e da hiperglicemia.
A quebra celular em massa que ocorre durante a rabdomiólise, na formação de abscesso, com a necrose celular (p. ex., infarto intestinal) ou após a quimioterapia para tratamento da leucemia ou linfoma libera potássio para dentro do plasma. As causas menos comuns de [K+] plasmática aumentada incluem a intoxicação digitálica cardíaca, que resulta da inibição parcial de ATPase de Na+/K+; administração de succinilcolina, que despolariza a membrana celular e, assim, permite a saída do potássio das células; e o raro distúrbio de paralisia periódica hipercalêmica.
Excreção renal de potássio comprometida. Os rins, principal sítio de excreção do potássio, possuem a capacidade distintiva de aumentar a taxa excreção à medida que a [K+] plasmática aumenta. Desta forma, uma hipercalemia contínua quase sempre está associada a algum tipo de comprometimento da eliminação renal do potássio. A secreção de íons hidrogênio ao nível do néfron distal frequentemente também está comprometida, levando ao desenvolvimento de acidose metabólica com hiato aniônico normal, uma vez que o túbulo coletor é o local onde ocorre a secreção tanto de potássio como de íons hidrogênio. A excreção renal de potássio pode ser comprometida pela insuficiência renal oligúrica, diminuição do volume arterial efetivo, ATR de tipo 4 e ATR de tipo 1 hipercalêmica.
A insuficiência renal oligúrica compromete a excreção de potássio, mas não a faz cessar. Embora a insuficiência renal predisponha à retenção de potássio, o potássio dietético geralmente continua sendo excretado na doença renal crônica até que a TFG caia para menos de 5 a 10 mL/min. Embora seja menos eficiente na insuficiência renal oligúrica, a excreção renal ainda pode ocorrer, pois a maior parte do potássio urinário deriva da secreção tubular, e não da filtração glomerular.
Quando o volume arterial efetivo é diminuído, a distribuição de sódio e água ao túbulo coletor também diminui. A distribuição distal adequada de sódio e água é essencial para a excreção normal do potássio. A distribuição distal está caracteristicamente diminuída, por exemplo, em pacientes com insuficiência cardíaca congestiva em estágio avançado ou cirrose. Neste contexto, tanto a queda da TFG como o aumento da reabsorção proximal de sódio e água, mediadas em parte pela angiotensina II, atuam na diminuição da excreção de potássio. Embora a aldosterona aumente a reabsorção de sódio e a secreção de potássio nestas condições, este aumento é limitado em decorrência da menor disponibilidade de sódio para reabsorção. Além disso, o potássio secretado pode não ser eliminado totalmente diante de uma redução acentuada do fluxo de urina.
A deficiência ou resistência à aldosterona (isto é, ATR de tipo 4) é responsável pela hipercalemia em mais de 75 a 85% dos casos de pacientes com hipercalemia persistente na ausência de qualquer causa evidente, como insuficiência renal avançada, um fármaco agressor ou uma carga de potássio.
A ATR de tipo 4 também está associada à diminuição da secreção de íons hidrogênio, que é causada em parte pelo comprometimento da produção de amônia ao nível do túbulo proximal, possivelmente resultante de uma alcalose intracelular hipercalemia-induzida. O resultado é a diminuição do fornecimento de amônia junto aos segmentos mais distais do néfron. Em consequência, uma insuficiência de tamponamento na urina exerce papel importante na limitada secreção de íons hidrogênio e explica o motivo que faz com que o pH urinário seja caracteristicamente acidificado (< 5,3) neste distúrbio.
Várias condições causam ATR de tipo 4 [Tabela 4]. A ATR de tipo 4 hiporreninêmica é a forma mais comum do distúrbio e é mais frequente em pacientes com insuficiência renal leve. A nefropatia diabética ou a nefrite intersticial crônica frequentemente estão presentes. Medicações como os AINH também podem contribuir para este distúrbio.
A patofisiologia da ATR de tipo 4 em pacientes hiporreninêmicos ainda não está totalmente esclarecida. A produção reduzida de angiotensina II é nitidamente importante, pois o potássio e a angiotensina II constituem os principais estímulos fisiológicos para a liberação de aldosterona. Entretanto, ao menos alguns pacientes também devem ter um defeito adrenal concomitante, dadas as observações indicativas de que a glândula suprarrenal normal pode liberar angiotensina II. Também foi sugerido que, em alguns pacientes, a diminuição da renina pode ser secundária. Exemplificando, a expansão de volume, que aumenta os níveis de peptídeo natriurético atrial, inibe a liberação tanto de renina como de aldosterona.
Em outros casos de ATR de tipo 4, o defeito na síntese e liberação da aldosterona parece estar confinado às glândulas suprarrenais. A heparina (ou talvez seu conservante, o clorbutol) é um potente inibidor da produção de aldosterona, mesmo quando administrada em doses baixas por via subcutânea (SC). Todavia, o uso da heparina não está amplamente associado à hipercalemia, uma vez que a deficiência moderada de aldosterona não basta para causar uma retenção significativa de potássio na grande maioria dos pacientes cuja função renal continua normal. Pode haver hipercalemia, porém, caso haja doença renal ou diante da administração concomitante de agentes que comprometem o sistema renina-angiotensina-aldosterona (p. ex., AINH ou inibidores de ECA).
A possibilidade de insuficiência adrenal primária , (isto é cursando com, deficiência de aldosterona e cortisol, também conhecida como doença de Addison) deve ser avaliada antes de estabelecer o diagnóstico de deficiência seletiva de aldosterona. Quando o paciente tem insuficiência adrenal primária, o hipocortisolismo frequentemente compromete a excreção de água e leva ao desenvolvimento de hiponatremia em adição à hipercalemia resultante da deficiência de aldosterona.
Embora a ATR de tipo 1 clássica esteja associada à hipocalemia, alguns pacientes com comprometimento da secreção de íons hidrogênio ao nível do túbulo coletor também são incapazes de secretar potássio normalmente. Esta condição – ATR de tipo 1 hipercalêmica – é observada com maior frequência em casos de obstrução do trato urinário, mas também pode ser causada por danos tubulointersticiais decorrentes de distúrbios como nefropatia da célula falciforme, nefrite lúpica ou rejeição de transplante. A hipercalemia e a acidose metabólica são causadas pelo dano tanto às células principais (secretoras de potássio) como às células intercalares (secretoras de ácido). A ATR de tipo 1 hipercalêmica pode ser distinguida da ATR de tipo 4 pela detecção de uma urina persistentemente alcalina (pH > 5,3) e de níveis plasmáticos normais ou aumentados de aldosterona [Tabela 8].
Tabela 8. Avaliação do defeito renal na hipercalemia
|
Distúrbio |
pH da urina |
UNa (mEq/L) |
Níveis plasmáticos de aldosterona |
|
Insuficiência renal |
< 5,3 |
> 20 |
Normais ou aumentados |
|
Depleção de volume acentuada |
Variável |
< 20 |
Aumentados |
|
ATR de tipo 4 |
< 5,3 |
> 20 |
Baixos* |
|
ATR de tipo 1 hipercalêmica |
> 5,3 |
> 20 |
Normais ou aumentados |
*Níveis normais de aldosterona são inadequados na presença de hipercalemia.
ATR = acidose tubular renal; UNa = concentração urinária de sódio.
Enfim, é importante lembrar os efeitos das medicações que podem limitar diretamente a secreção de potássio.38 Os agentes evidentes são os diuréticos poupadores de potássio: amilorida, triamtereno e espironolactona. Outro agente menos comum é o trimetroprim-sulfametoxazol. O trimetoprim, seja nesta combinação ou isolado, pode comprometer a secreção de potássio ao bloquear o transporte de sódio do lúmen tubular para dentro da célula do túbulo coletor, em um mecanismo similar ao da amilorida.
Evidências sugerem que a aldosterona pode ser vasculotóxica, cardiotóxica e nefrotóxica.39 Uma das consequências disto é o aumento do uso combinado de inibidores de ECA ou bloqueadores do receptor da angiotensina (BRA) com a espironolactona. Os estudos iniciais sobre a terapia combinada à base de inibidores de ECA e BRA demonstraram uma toxicidade mínima por hipercalemia. Entretanto, foi comprovado que o risco de hipercalemia está associado à terapia combinada.40,41 Quando múltiplas medicações são administradas, e cada uma delas pode reduzir a secreção urinária de potássio, é preciso ter o cuidado de evitar a morbidade e a potencial mortalidade associadas à hipercalemia severa.42 Isto é particularmente importante no caso de pacientes com função renal comprometida.
Manifestações clínicas. Os sintomas de hipercalemia estão relacionados ao comprometimento da transmissão neuromuscular. Contudo, as manifestações neuromusculares são inespecíficas. Os primeiros achados são as parestesias e o enfraquecimento, que podem evoluir para paralisia e afetar os músculos respiratórios. Estes sintomas são similares àqueles observados com a hipocalemia. Contudo, a função do nervo craniano caracteristicamente permanece inalterada.
A facilidade de geração de um potencial de ação está relacionada à magnitude do potencial de membrana em repouso e ao estado de ativação dos canais de sódio existentes na membrana. A abertura destes canais de sódio, com consequente difusão passiva do sódio extracelular para dentro das células, constitui a etapa primária deste processo.
De acordo com a equação de Nernst, o potencial de membrana em repouso é regulado pela proporção [K+] intracelular/[K+] extracelular. Uma elevação da [K+] plasmática (extracelular) diminui esta proporção e, portanto, despolariza a membrana celular (ou seja, torna o potencial de membrana em repouso menos eletronegativo). Esta alteração a princípio aumenta a excitabilidade da membrana, pois um estímulo despolarizante menor passa a ser necessário à geração do potencial de ação. Entretanto, a despolarização persistente inativa os canais de sódio existentes na membrana celular e, desta forma, produz uma diminuição líquida da excitabilidade da membrana que pode se manifestar clinicamente como comprometimento da condução cardíaca, enfraquecimento muscular ou paralisia.
Em geral, os sintomas severos de hipercalemia não ocorrem enquanto a [K+] plasmática estiver abaixo de 7,5 mEq/L. Existe uma ampla variabilidade entre os paciente, contudo, porque fatores como hipocalcemia concomitante, acidose metabólica e velocidade do desenvolvimento da hipercalemia podem aumentar a toxicidade do excesso de potássio.43
Exames fisiológicos. O monitoramento cuidadoso do ECG e da força muscular se faz necessário para avaliar as consequências funcionais da hipercalemia. Diante de uma [K+] plasmática acima de 7,5 a 8 mEq/L, o enfraquecimento muscular severo ou as alterações marcantes de ECG são potencialmente prejudiciais à vida e requerem tratamento imediato com quase todas as modalidades terapêuticas disponíveis [ver Tratamento, adiante].
A anormalidade de ECG mais precoce é o pico simétrico das ondas T, que é seguido de diminuição da voltagem da onda P e ampliação dos complexos QRS [Figura 8]. Se a hipercalemia severa não for tratada, pode, enfim, produzir um padrão de ECG sinusoidal, em que uma oscilação representa um amplo complexo QRS e uma oscilação complementar representa uma onda T anormal. As alterações de ECG não costumam ocorrer antes de a [K+] plasmática ultrapassar 6,5 mEq/L e tendem mais a se desenvolver quando a elevação do potássio ocorre rapidamente. Entretanto, não existe nenhuma relação absolutamente preditiva entre a severidade do distúrbio eletrolítico e o padrão de ECG. Em casos raros, o padrão de ECG pode permanecer inalterado mesmo em pacientes com [K+] plasmática maior que 9 mEq/L. Em outros, um padrão de pseudoinfarto pode ser observado [Figura 10].44
Figura 10. Padrão de pseudoinfarto causado por hipercalemia. (a) Eletrocardiograma (ECG) de um paciente com níveis séricos de potássio iguais a 7,9 mEq/L. As derivações torácicas anteriores mostram elevação do segmento ST, além das ondas T com pico, mais notavelmente nas derivações V3 e V4. (b) Resolução das alterações ECG após a correção dos níveis séricos de potássio.
Quando a hipercalemia é sustentada, a causa geralmente é o clearence renal reduzido [Figura 9]. Quando uma ATR de tipo 4 é comprovada, a concentração plasmática de cortisol deve ser medida para determinar se o paciente tem insuficiência suprarrenal total.
A hipótese de pseudo-hipercalemia deve ser considerada diante de evidências de hemólise em uma amostra e quando a contagem de plaquetas ou de leucócitos sanguíneos (LEU) está acentuadamente aumentada. Em contraste com a hipercalemia verdadeira, que está associada a uma função neuromuscular ou cardíaca alterada, a pseudo-hipercalemia não impõe risco aos pacientes.
Nesta condição, a [K+] plasmática medida pelo laboratório clínico está aumentada devido à liberação de potássio durante a coagulação que ocorre após a coleta da amostra de sangue. Sua causa mais frequente é a hemólise resultante de uma coleta de sangue traumática, mas também pode ser observada diante de uma trombocitose acentuada (contagem de plaquetas > 1 x 106/mcL) ou leucocitose (LEU > 1 x 105/mcL). A condição é encontrada, ainda, na pseudo-hipercalemia familiar, em que o potássio se desloca para fora das células à temperatura ambiente, após a coagulação, na ausência de hemólise evidente.45
O diagnóstico de pseudo-hipercalemia é estabelecido por meio da demonstração de uma [K+] normal em uma amostra de plasma não hemolisada. Esta amostra é obtida pela coleta de sangue em tubo heparinizado, que evita a coagulação. Nos casos em que a causa da hipercalemia não pode ser determinada (p. ex., função renal normal) e a [K+] sérica excede os níveis plasmáticos em mais de 1 mEq/L, a causa da hipercalemia é um fenômeno in vitro, caso o paciente apresente níveis séricos de potássio normais após a coleta de sangue sem auxílio de torniquete.46 O tratamento da pseudo-hipercalemia não é indicado, porque a [K+] in vivo (isto é, no plasma) está normal. A hipercalemia “verdadeira” pode ser causada pelo deslocamento de potássio para o espaço vascular, a partir das células normais (acidose) ou de células lesadas (rabdomiólise, isquemia); por incapacidade de transporte de potássio do espaço intravascular até as células (concentração baixa ou nula de insulina, terapia com betabloqueador não seletivo); ou pela diminuição na excreção renal. Sem dúvida, as causas renais da hipercalemia são as mais comuns. A Figura 9 destaca a abordagem para hipercalemia. Se a TFG for inferior a 30 mL/min, a hipercalemia é mais frequentemente causada por uma entrada de potássio excessiva (exógena, endógena) ou por fármacos. Quando a TFG for inferior a 10 a 15 mL/min, até mesmo uma ingesta de quantidades normais de potássio causa hipercalemia. Quando a TFG for superior a 30 mL/min, é útil medir os níveis séricos ou urinários de aldosterona. A deficiência de aldosterona pode estar associada a níveis de renina baixos (hipoaldosteronismo hiporreninêmico) ou altos. A quantificação do cortisol é útil em condições de níveis altos de renina e concentração baixa de aldosterona. Diminuições dos níveis de cortisol são indicativas de doença de Addison, enquanto uma concentração de cortisol elevada está associada ao uso de alguns fármacos (p. ex., heparina) ou à deficiência glomerulosa suprarrenal seletiva, como se observa na infecção por HIV/Aids. O achado de níveis altos de aldosterona no contexto de hipercalemia indica a ocorrência de pseudo-hipoaldosteronismo, conforme se observa no decorrer de algumas terapias farmacológicas ou na doença renal com envolvimento do interstício.
Na hipercalemia verdadeira, em particular nos casos graves ([K+] plasmática > 6 mEq/L), a descontinuação de todas as medicações que prejudicam o equilíbrio do potássio é obrigatória.47 Estas medicações incluem os betabloqueadores não seletivos, IECA, diuréticos poupadores de potássio, inibidores de renina diretos, AINH e trimetoprim. Os substitutos do sal, que contêm cloreto de potássio, também devem ser evitados. Indivíduos com hipercalemia leve ([K+] plasmática < 6 mEq/L) geralmente podem ser tratados de maneira conservadora, com redução da ingesta diária para menos de 2 g e, havendo indicação, adição de um diurético de alça.
O tratamento ativo para diminuir a [K+] plasmática ou antagonizar seus efeitos sobre a membrana celular deve ser iniciado diante da elevação da [K+] plasmática para 6 mEq/L ou mais, particularmente se houver manifestações ECG de hipercalemia. Existem várias opções terapêuticas disponíveis [Tabela 9].
Tabela 9. Tratamento da hipercalemia
|
Agente |
Resultado |
|
Cálcio |
Antagonismo dos efeitos de membrana hipercalêmicos |
|
Infusão de glicose (com insulina em diabéticos) Bicarbonato de sódio Agonistas beta-2-adrenérgicos |
Movimentação de potássio para dentro das células |
|
Diuréticos de alça ou tiazida Resinas trocadoras de cátions Hemodiálise ou diálise peritoneal |
Remoção de potássio corporal |
Uma infusão de cálcio normaliza rapidamente o ECG. Um aumento da [Ca+] eleva o limiar de potencial e, desta forma, normaliza a excitabilidade da membrana. O cálcio deve ser administrado, contudo, na presença somente das manifestações de ECG de hipercalemia que podem preceder uma fibrilação ventricular. Como o cálcio pode exacerbar ou precipitar arritmias cardíacas induzidas por digitálicos, deve ser usado apenas quando necessário e com bastante cautela no tratamento de pacientes que estejam recebendo digoxina ou outras preparações à base de digital.
O cálcio deve ser administrado via IV sob a forma de gliconato ou sal de cloreto (10 mL de uma solução de gliconato de cálcio a 10%, durante 2 a 3 minutos, sob monitoramento por ECG). É possível que a normalização do ECG persista por menos de 60 minutos. Além disso, este tratamento não altera a [K+] plasmática nem as reservas corporais de potássio totais. Em consequência, as intervenções que diminuem os níveis plasmáticos e removem o potássio do corpo devem ser iniciadas simultaneamente. O bicarbonato de sódio, a glicose (com ou sem insulina) e a estimulação do receptor beta-2-adrenérgico diminuem a [K+] plasmática ao promoverem a entrada do potássio do líquido extracelular dentro das células. A administração de bicarbonato de sódio diminui a concentração de íons hidrogênio no líquido extracelular. Teoricamente, se a bomba de troca de Na+/H+ estivesse no modo ativado, a administração de bicarbonato de sódio favoreceria a movimentação de H+ para fora das células, enquanto o Na+ entraria. A saída subsequente de Na+ por ação da ATPase de Na+/K+ deslocaria o potássio para dentro da célula. Entretanto, no estado estável, a bomba de troca de Na+/H+ parece estar inativa.48 Comparativamene, a bomba de troca de Na+/H+ é ativada pela acidose intracelular.
Vários estudos comprovaram que a terapia com bicarbonato de sódio é ineficaz para o tratamento da hipercalemia aguda, apesar da elevação da [HCO3–] sérica. Além disso, o bicarbonato aparentemente não potencializa a ação hipocalêmica dos agonistas beta-2-adrenérgicos ou da combinação de glicose com insulina, que são mais eficazez neste contexto.49 Por causa destes achados, o bicarbonato de sódio, quando usado no tratamento da hipercalemia, deve ser combinado a outros agentes.48
Os agonistas beta-2-adrenérgicos também podem promover uma diminuição transitória dos níveis séricos de potássio. O albuterol pode ser administrado a uma dosagem de 10 a 20 mg em 4 mL de salina por inalação nasal, no decorrer de 10 minutos, ou pela infusão IV de 0,5 mg. É comum os níveis sanguíneos de potássio sofrerem uma redução de 0,5 a 1,5 mEq/L em 30 minutos após a infusão IV. Contudo, é necessário esperar mais de 1 hora para obter o efeito de pico com a inalação de albuterol.50
O efeito hipocalêmico da insulina é observado dentro de 30 a 60 minutos e pode ser alcançado com a simples administração de 25 g de glicose IV, pois uma infusão de glicose estimula rapidamente a liberação de insulina. Como alternativa, em pacientes diabéticos, é possível administrar 10 unidades de insulina regular por via IV com esta carga de glicose, embora este regime possa causar hipoglicemia em indivíduos normais. Independentemente do método usado, é preciso ter cautela para evitar que a infusão de glicose provoque desenvolvimento de hiperglicemia severa. Uma alta concentração plasmática de glicose pode exacerbar a hipercalemia ao promover o deslocamento da água (que contém potássio) do compartimento intracelular para dentro do plasma.
Assim como na administração de cálcio, a administração de glicose e insulina, bicarbonato e agonistas beta-2-adrenérgicos é uma medida temporária, uma vez que as reservas corporais totais de potássio não diminuem. Por este motivo, é necessário instituir uma terapia adicional para garantir que a [K+] plasmática não volte aos níveis pré-tratamento. Além disso, em alguns pacientes, a diminuição da [K+] plasmática com o uso destes agentes pouco antes da diálise pode levar a uma menor remoção do potássio por diálise, possivelmente resultando em hipercalemia de rebote após a conclusão da terapia de diálise.50
Em alguns casos, aumentar o fluxo urinário com diuréticos pode ser útil, embora a insuficiência renal frequentemente limite a efetividade dos diuréticos. Contudo, uma resina trocadora de cátions Na+/K+ à base de sulfonato de poliestireno de sódio pode ser administrada em sorbitol (para provocar diarreia) por VO ou como enema de retenção. O uso de sulfonato de poliestireno de sódio permite trocar 4 mEq de Na+/g por 4 mEq de K+. Desta forma, teoricamente, 30 g de sulfonato de poliestireno de sódio poderiam remover 120 mEq de potássio. Entretanto, como a quantidade de potássio gastrintestinal disponível para troca pode ser de 5 mEq/dia, a efetividade desta resina foi questionada.48 Em alguns estudos, o sulfonato de poliestireno de sódio não se mostrou mais efetivo do que o laxante com o qual foi administrado. Alguns especialistas já não recomendam o uso da resina em sorbitol. Contudo, a diarreia induzida por este agente pode ser útil por si só para a diminuição da [K+] sérica. O uso de sulfonato de poliestireno de sódio é recomendado para pacientes com débito urinário baixo e diante da indisponibilidade imediata da diálise.
A diálise deve ser considerada diante de casos severos ou refratários de hipercalemia, em particular se houver insuficiência renal avançada. A hemodiálise, quando disponível, é preferível no contexto agudo, pois remove o potássio bem mais rapidamente do que a diálise peritoneal.
Se a deficiência de aldosterona tiver sido comprovada em casos de hipercalemia crônica inadequadamente controlada com dieta ou diuréticos, a reposição da aldosterona com acetato de fludrocortisona pode ser útil. A combinação de fludrocortisona e um diurético de alça pode limitar o desenvolvimento de hipertensão ou edema. No entanto, como o início de sua ação pode demorar vários dias ou mais, esta terapia não é suficientemente efetiva para ser usada no tratamento da hipercalemia aguda prejudicial à vida.
O autor não mantém relações comerciais com os fabricantes de produtos e prestadores de serviços mencionados neste capítulo.
1. Kraut JA, Madias NE. Approach to patients with acid-base disorders. Respir Care 2001;46:392.
2. Gluck SL. Acid sensing in renal epithelial cells. J Clin Invest 2004;114:1696.
3. Lolekha PH, Vanavanan S, Lolekha S. Update on value of the anion gap in clinical diagnosis and laboratory evaluation. Clin Chim Acta 2001;307:33.
4. Shaer AJ, Rastegar A. Lactic acidosis in the setting of antiretroviral therapy for the acquired immunodefi ciency syndrome: a case report and review of the literature. Am J Nephrol 2000;20:332.
5. Uribarri J, Oh MS, Carroll HJ. D-Lactic acidosis: a review of clinical presentation, biochemical features, and pathophysiologic mechanisms. Medicine (Baltimore) 1998;77:73.
6. Vanholder R, Sever MS, Erek E, et al. Rhabdomyolysis. J Am Soc Nephrol 2000;11:1553.
7. Sweeney T, Beuchat C. Limitation of methods of osmometry: measuring the osmolality of biological fluids [editorial]. Am J Physiol 1993;264:R469.
8. Cruz DN, Huot SJ. Metabolic complications of urinary diversions: an overview. Am J Med 1997;102:477.
9. Messiaen T, Deret S, Mougenot B, et al. Adult Fanconi syndrome secondary to light chain gammopathy: clinicopathologic heterogeneity and unusual features in 11 patients. Medicine (Baltimore) 2000;79:135.
10. Igarashi T, Sekine T, Inatomi J, et al. Unraveling the molecular pathogenesis of isolated proximal renal tubular acidosis. J Am Soc Nephrol 2002;13:2171.
11. Rungroj N, Devonald AW, Cuthbert F, et al. A novel missense mutation in AE1 causing autosomal dominant distal renal tubular acidosis retains normal transport functions, but is mistargeted in polarized epithelial cells. J Biol Chem 2004;279:13833.
12. Graber M. A model of hyperkalemia produced by metabolic acidosis. Am J Kidney Dis 1993;22:436.
13. Adrogue HJ, Madias N. Management of life-threatening acid-base disorders: fi rst of two parts. N Engl J Med 1998; 338:26.
14. Winter SD, Pearson R, Gabow PA, et al. The fall of the serum anion gap. Arch Intern Med 1990;150:311.
15. Caravaca F, Arrobas M, Pizarro JL, et al. Metabolic acidosis in advanced renal failure: differences between diabetic and nondiabetic patients. Am J Kidney Dis 1999;33:892.
16. Gadola L, Noboa O, Marquez MN, et al. Calcium citrate ameliorates the progression of chronic renal injury. Kidney Int 2004;65:1224.
17. Mehrotra R, Kopple J, Wolfson M. Metabolic acidosis in maintenance dialysis patients: clinical considerations. Kidney Int Suppl 2003;88:S13–25.
18. Eustace J, Astor B, Muntner P, et al. Prevalence of acidosis and infl ammation and their association with low serum albumin in chronic kidney disease. Kidney Int 2004;65: 1031.
19. Boirie Y, Broyer M, Gagnadoux MF, et al. Alterations of protein metabolism by metabolic acidosis in children with chronic renal failure. Kidney Int 2000;58:236.
20. Kirschbaum B. Spurious metabolic acidosis in hemodialysis patients. Am J Kidney Dis 2000;35:1068.
21. Forsythe SM, Schmidt GA. Sodium bicarbonate for the treatment of lactic acidosis. Chest 2000;117:260.
22. Luft FC. Lactosis acidosis update for critical care clinicians. J Am Soc Nephrol 2001;12 Suppl 17:S15.
23. Kraut J, Kurtz I. Use of base in the treatment of severe acidemic states. Am J Kidney Dis 2001;38:703.
24. Okuda Y, Adrogue HJ, Field JB, et al. Counterproductive effects of sodium bicarbonate in ketoacidosis. J Clin Endocrinol Metab 1996;81:314.
25. Glaser N, Barnett P, McCaslin I, et al. Risk factors for cerebral edema in children with diabetic acidosis. N Engl J Med 2001;344:264.
26. Higgins RM, Connolly JO, Hendry BM. Alkalinization and hemodialysis in severe salicylate poisoning: comparison of elimination techniques in the same patient. Clin Nephrol 1998;50:178.
27. Brent J, McMartin K, Phillips S, et al. Fomepizole for the treatment of methanol poisoning. N Engl J Med 2001;344: 424.
28. Galla JH. Metabolic alkalosis. J Am Soc Nephrol 2000;11: 369.
29. Kurtz I. Molecular pathogenesis of Bartter’s and Gitelman’s syndromes. Kidney Int 1998;54:1396.
30. Adrogue HJ, Madias NE. Management of life-threatening acid-base disorders: second of two parts. N Engl J Med 1998;338:107.
31. Suh A, DeJesus E, Rosner K, et al. Racial differences in potassium disposal. Kidney Int 2004;66:1523.
32. McCormick JA, Yang C-L, Ellison DH. WNK kinases and renal sodium transport in health and disease. Hypertension 2008;51:588–96.
33. Manoukian MA, Foote JA, Crapo LM. Clinical and metabolic features of thyrotoxic periodic paralysis in 24 episodes. Arch Intern Med 1999;159:601.
34. Beloosesky Y, Grinblat J, Weiss A, et al. Electrolyte disorders following oral sodium phosphate adminstration for bowel cleansing in elderly patients. Arch Intern Med 2003;163:803.
35. Marples D, Frokiaer J, Dorup J, et al. Hypokalemia-induced downregulation of aquaporin-2 water channel expression in rat kidney medulla and cortex. J Clin Invest 1996;97: 1960.
36. Odvina C, Preminger G, Lindberg J, et al. Long-term combined treatment with thiazide and potassium citrate in nephrolithiasis does not lead to hypokalemia or hypochloremic metabolic alkalosis. Kidney Int 2003;63:240.
37. Kruse J, Carlson R. Rapid correction of hypokalemia using concentrated intravenous potassium chloride infusions. Arch Intern Med 1990;150:613.
38. Perazella MA. Drug-induced hyperkalemia: old culprits and new offenders. Am J Med 2000;109:307.
39. Hollenberg N. Aldosterone in the development and progression of renal injury. Kidney Int 2004;66:1.
40. Juurlink D, Muhammad M, Lee D, et al. Rates of hyperkalemia after publication of the Randomized Aldactone Evaluation Study. N Engl J Med 2004;351:543.
41. McMurray J, O’Meara E. Treatment of heart failure with spironolactone: trial and tribulations. N Engl J Med 2004;351: 526.
42. Palmer B. Managing hyperkalemia caused by inhibitors of the renin-angiotensin-aldosterone system. N Engl J Med 2004;351:585.
43. Charytan D, Goldfarb DS. Indications for hospitalization of patients with hyperkalemia. Arch Intern Med 2000;160: 1605.
44. Wang K. Images in clinical medicine. Pseudoinfarction pattern due to hyperkalemia. N Engl J Med 2004;351:593.
45. Iolascon A, Stewart GW, Ajetunmobi JF, et al. Familial pseudohyperkalemia maps to the same locus as dehydrated hereditary stomatocytosis (hereditary xerocytosis). Blood 1999;93:3120.
46. Wiederkehr MR, Moe OW. Factitious hyperkalemia. Am J Kidney Dis 2000;36:1049.
47. Weiner ID, Wingo CS. Hyperkalemia: a potential silent killer. J Am Soc Nephrol 1998;9:1535.
48. Kamel K, Wei C. Controversial issues in the treatment of hyperkalaemia. Nephrol Dial Transplant 2003;18:2215.
49. Allon M, Shanklin N. Effect of bicarbonate administration on plasma potassium in dialysis patients: interactions with insulin and albuterol. Am J Kidney Dis 1996;28:508.
50. Allon M, Shanklin N. Effect of albuterol treatment on subsequent dialytic potassium removal. Am J Kidney Dis 1995; 26:607.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.