(Carregando Índice)... (Carregando Índice)... |
Última revisão: 02/05/2016
Comentários de assinantes: 0
Jacob Levitt, MD, FAAD,
Vice Chairman, Residency Director, and Associate Professor, The Mount Sinai School of Medicine, New York, NY
Rachel Nazarian, MD*
Os autores e editores agradecem as contribuições dos autores da edição anterior, Elizabeth A. Abel, MD, e o finado Jean-Claude Bystryn, MD, pelo desenvolvimento e redação deste capítulo. Agradecemos a Dr. Robert Phelps por sua assistência editorial.
Artigo original: Levitt , Nazarian R. Vesiculobullous Diseases. ACP Medicine. 2011.
[The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2015 Decker Intellectual Properties Inc. All Rights Reserved.]
Tradução: Soraya Imon de Oliveira.
Revisão técnica: Dr. Lucas Santos Zambon.
Uma bolha se refere a uma fenda cheia de líquido na epiderme. Uma vesícula específica é uma lesão menor que 1 cm, enquanto uma bolha específica é uma lesão maior que 1 cm. As doenças vesicobolhosas podem ser mais facilmente caracterizadas pela localização anatômica da fenda da bolha, que pode resultar da disfunção de proteína de adesão autoimune-mediada, proteínas geneticamente defeituosas, necrose de queratinócitos induzida por fármaco e causas infecciosas ou traumáticas. Uma lista abrangente de doenças bolhosas é apresentada na Tabela 1, organizadas primariamente pela localização anatômica da bolha. Embora exista uma miríade de doenças bolhosas autoimunes e genéticas, este capítulo enfoca o diagnóstico e o tratamento das seguintes entidades mais frequentes—distúrbios de pênfigo, penfigoide bolhoso (PB), infecção disseminada pelo vírus varicela-zoster, herpes simples disseminado e erupções farmacológicas bolhosas—além de abordar brevemente outras entidades menos comuns—epidermólise bolhosa (EB), dermatite herpetiforme (DH), dermatose bolhosa de IgA linear (DBAL), porfiria cutânea tardia e edema bolhoso. Os distúrbios vesicobolhosos neonatais são abordados em outro capítulo.1
A maioria das doenças vesicobolhosas adquiridas comumente encontradas se manifestará com início agudo. A história é importante para excluir as hipóteses de (1) exacerbação pelo Sol (como na porfiria cutânea tardia), (2) imunossupressão concomitante (sugerindo varicela ou herpes disseminado) e (3) medicações (sugerindo eritema multiforme fármaco-induzido, necrólise epidérmica tóxica ou DBAL). O exame físico ajuda a distinguir (1) bolhas tensas versus bolhas flácidas (PB versus pênfigo vulgar [PV]); (2) distribuição das lesões (fotodistribuição para porfiria; envolvimento oral sugestivo de PV, pênfigo paraneoplásico (PPN) ou necrólise epidérmica tóxica mais que PB; envolvimento ocular sugestivo de penfigoide cicatricial mais que PV; e áreas gravidade-dependentes sugerindo edema bolhoso); e (3) predominância de lesões vesiculares (sugerindo, talvez, herpes intermitente ou varicela mais do que exacerbação de doença bolhosa autoimune primária em tratamento). As análises histológicas permitem diferenciar (1) causas infecciosas versus causas alérgicas versus causas imunológicas, e (2) alguns tipos de causas imunológicas entre si. Infelizmente, usando apenas a histologia, algumas entidades não podem ser prontamente distinguidas: (1) PV versus pênfigo foliáceo (PF) versus algumas formas de PPN, e (2) PB versus epidermólise bolhosa adquirida (EBA) versus lúpus bolhoso versus DBAL.
A biópsia deve incluir uma vesícula inteira e um pouco da pele circundante, onde for possível, contudo mais classicamente o tecido adjacente e, talvez, incluindo a própria bolha em si para possibilitar um ponto de aderência para o teto da lesão. No PV, a lesão frequentemente é uma erosão e não uma bolha, de modo que a probabilidade de a pele perilesional abrigar uma bolha subclínica é excelente. As condições bastante raras do penfigoide cicatricial e do PPN podem parecer clinicamente similares ao PV mais comum, e devem ser distinguidas histológica e imuno-histologicamente. O mesmo é válido para o penfigoide cicatricial e DBAL em relação ao PB.
A imuno-histoquímica é de dois tipos: imunofluorescência direta e imunofluorescência indireta. Na imunofluorescência direta, a pele do paciente é lavada com solução contendo anticorpos anti-IgA, anti-IgG, anti-C3, anti-IgM, e antifibrinogênio exógenos com marcação fluorescente, que localizam onde as imunoglobulinas patogênicas do paciente foram depositadas. Na imunofluorescência indireta, o soro do paciente é usado para lavar amostras de tecido ricas em um ou outro antígeno. Exemplificando, para excluir o PPN, o soro do paciente é aplicado ao epitélio da bexiga de rato, que contém a miríade de antígenos encontrados no PPN (membros da família de plaquinas e desmogleínas).1 O esôfago de macaco é rico em desmogleína 3, principal antígeno encontrado no PV. O esôfago de cobaia é rico em desmogleína 1, principal antígeno encontrado no PF. Anticorpos anti-anticorpos humanos com marcação fluorescente indicam, então, a presença ou ausência de anticorpos patogênicos. O título é determinado no ponto em que a reação desaparece com diluições seriadas. Recentemente, ensaios imunossorventes ligados a enzimas (ELISAs) usando antígenos purificados passaram a ser disponibilizados para a obtenção de títulos mais precisos.2 Para PB e EBA, a imunofluorescência direta resultará positiva ao longo da zona da membrana basal. Para distinguir estas duas entidades, realiza-se imunofluorescência indireta de pele humana normal. Essa pele é lavada em solução de NaCl a 1 M e isto leva à formação de uma bolha artificial ou divisão na zona da membrana basal, ao nível da lâmina lúcida. Os autoanticorpos IgG estão localizados no teto da bolha de pele partida por sais em casos de PB, e no assoalho da bolha em casos de EBA.
As imunoglobulinas perdem a antigenicidade quando em formalina, que é um fixador promotor de ligação cruzada. Sendo assim, ao submeter uma amostra de biópsia para imunofluorescência direta, é necessário transportá-la em solução conservante (p. ex., em solução de Michel, contendo sulfato de amônio).3 Alternativamente, a amostra pode ser colocada em gazes umedecidas com salina normal, mas deve ser mantida a 4°C para inativar as proteases e processada dentro de 24 horas, uma vez que as imunoglobulinas serão degradadas com o passar do tempo. Como a imunofluorescência direta requer meio de transporte diferente daquele usado na histologia de rotina, uma biópsia de 4 mm obtida por punção pode ser bisseccionada ou podem ser obtidas duas biópsias separadas por punção.
A meta final das análises histológicas e imuno-histoquímicas (e, às vezes, de microscopia eletrônica) é localizar o plano em que está havendo perda da adesão, com implicação de uma ou outra proteína de adesão cutânea (ou anticorpo dirigido contra essa proteína) como sendo o culpado fisiopatológico. Como sabemos que uma dada doença está associada fisiopatologicamente a uma ou outra proteína, estas análises nos permitem deduzir a doença em curso.
|
Tabela 1 Planos de bolha | ||
|
Nível |
Proteína |
Doença(s) |
|
Necrose dispersa ou em todo o queratinócito em todas as camadas da epiderme |
N/A |
Infecção pelo vírus varicela-zoster |
|
|
N/A |
Infecção pelo vírus do herpes simples |
|
Espongiose em todas as camadas da epiderme |
N/A |
Ponfolix |
|
|
N/A |
Dermatite de contato alérgica |
|
Necrose de queratinócito em todas as camadas da epiderme |
N/A |
Eritema multiforme |
|
|
N/A |
Síndrome de Stevens-Johnson |
|
|
N/A |
Necrólise epidérmica tóxica |
|
Subcorneal |
N/A |
Miliária cristalina |
|
Camada granular |
Desmogleína 1 |
Síndrome da pele escaldada estafilocócica |
|
|
Desmogleína 1 |
Pênfigo foliáceo |
|
|
Desmogleína 1 |
Pênfigo paraneoplásico |
|
Células espinhosas |
Queratina 1, 10 |
Hiperqueratose epidermolítica |
|
|
N/A |
Bolha por atrito |
|
|
|
Miliária rubra |
|
Camada suprabasal |
Desmogleína 3 |
Pênfigo vulgar |
|
|
Desmogleína 3, plectina, desmoplaquina I e II, envoplaquina, periplaquina, BPAg1 |
Pênfigo paraneoplásico |
|
|
Desmocolina 1 |
Pênfigo por IgA |
|
Células basais |
Queratina 5, 14; plectina; desmoplaquina; placofilina-1 |
Epidermólise bolhosa simples |
|
Subepidérmico |
N/A |
Bolha de edema |
|
|
N/A |
Porfiria cutânea tardia |
|
|
N/A |
Dermatite herpetiforme |
|
|
N/A |
Líquen plano bolhoso |
|
|
Fragmento proteolítico do antígeno BP180, uma proteína de 97 ou 120 kDa |
Dermatose bolhosa de IgA linear |
|
Hemidesmossomo |
Colágeno XVII |
Penfigoide bolhoso |
|
|
Colágeno XVII |
Epidermólise bolhosa juncional |
|
Lâmina lúcida |
Ligação mais fraca |
Pele partida com sal |
|
Lâmina densa |
Laminina 5 |
Penfigoide cicatricial |
|
|
Laminina 5 |
Epidermólise bolhosa juncional |
|
|
a6/b4 integrina |
Epidermólise bolhosa juncional |
|
Sublâmina densa |
Colágeno VII |
Epidermólise bolhosa adquirida |
|
|
Colágeno VII |
Epidermólise bolhosa distrófica |
NA = não aplicável.
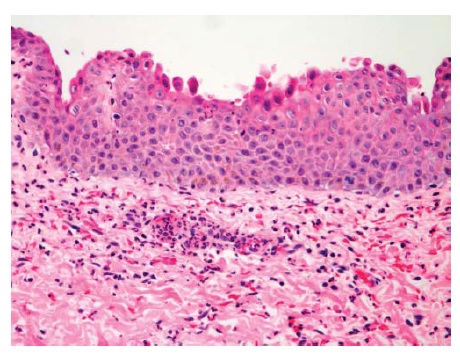
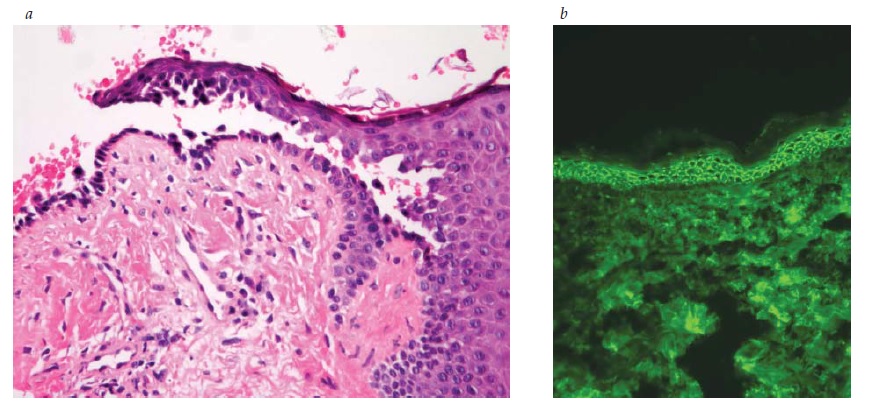
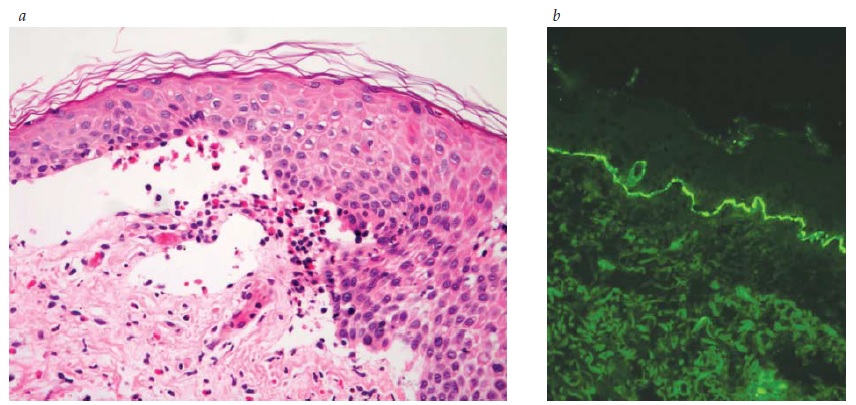
As Figuras 1, 2 e 3 ilustram os diferentes planos de clivagem vistos à coloração de hematoxilina-eosina para PF, PV e PB, respectivamente, e os anticorpos de fluorescência direta correspondentes para PV e PB. Em geral, o principal anticorpo observado por imunofluorescência direta nestas três condições é a IgG. na histopatologia do PV, a acantose intraepitelial é vista à medida que as células basais perdem o contato desmossômico com as células vizinhas, ainda que retenham a fixação hemidesmossômica à membrana basal. Como resultado, uma típica “fileira de lápides” é vista como fila única de queratinócitos ao longo da membrana basal [ver Figura 2]. Em contraste, no PB, toda a epiderme está solta em consequência da ruptura dos hemidesmossomos [ver Figura 3].
O pênfigo descreve um grupo de doenças bolhosas autoimunes crônicas caracterizadas por acantólise, ou seja, pela perda da coesão intercelular intraepidérmica. Os três subgrupos autoimune-mediados principais incluem PV, PF e PPN, dos quais PV é o mais comum. A doença de Hailey-Hailey, também denominada pênfigo familiar benigno, resulta de uma patologia de canais de cálcio cutânea hereditária e não da presença de autoanticorpos.

Figura 1 Pênfigo foliáceo com divisão na camada superior espinhosa/ranular (aqui falta o teto da bolha; coloração de hematoxilina-eosina; aumento de 200x do original).
No PV, os autoanticorpos IgG são dirigidos contra proteínas desmossômicas, desmogleína 3 e, às vezes, também contra a desmogleína 1, levando à perda da adesão entre as células na parte mais profunda da epiderme, acima da camada de células basais. Uma forma rara de pênfigo por IgA resulta de autoanticorpos dirigidos contra a desmocolina 1.1 A causa é mais frequentemente idiopática, todavia a hipótese de PV fármaco-induzido (por não tiois, como dipirona e enalapril) deve ser considerada.4
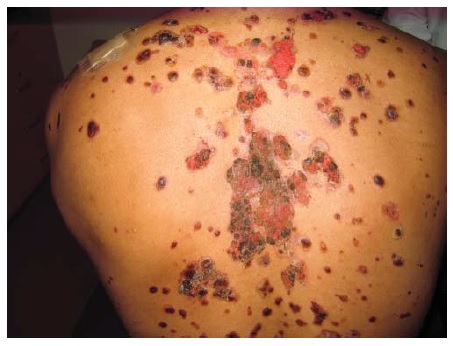
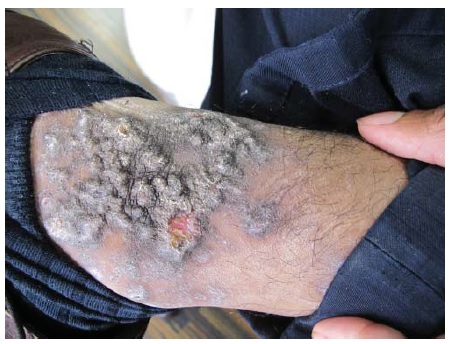
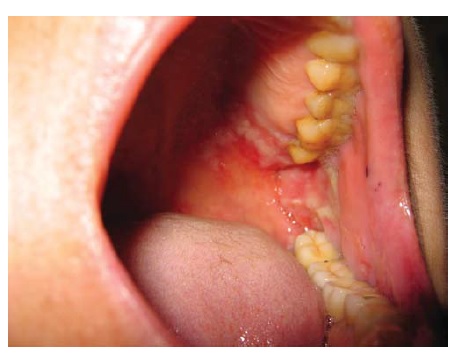
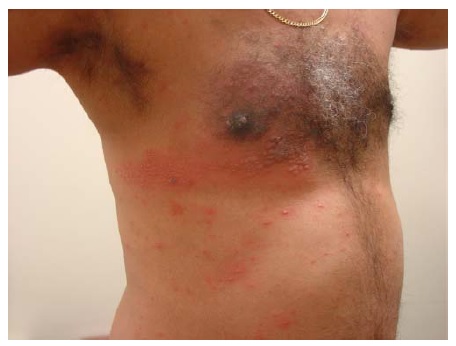
O PV se manifesta mais comumente com erosões, em vez de bolhas, porque estas são tipicamente frágeis e, assim, se tornam descobertas [ver Figura 4]. Ocasionalmente, é possível encontrar bolhas intactas [ver Figura 5]. Em alguns casos, há formação de uma crosta verrucosa, friável e delgada [ver Figura 6] como teto da bolha. Isto gera confusão clínica, porque a lesão não se parece com uma bolha clássica nem com uma erosão clássica. Quando esta crosta verrucosa se torna hipertrófica e úmida (muitas vezes, na virilha ou axilas), é descrita como pênfigo vegetante (mas não difere do PV quanto à imunopatogenética) [ver Figura 7]. O pênfigo vegetante pode se tornar superinfeccionado. As áreas comumente envolvidas no PV são a mucosa oral, especificamente os lábios, as gengivas no ponto de encontro com os dentes, a mucosa bucal e o palato [ver Figura 8]. O envolvimento das conjuntivas é infrequente.5 O envolvimento da laringe pode resultar em rouquidão, enquanto o envolvimento da cavidade nasal pode resultar em epistaxe. A parte superior do dorso é outro sítio comum. O couro cabeludo pode ser particularmente refratário ao tratamento. Lesões com envolvimento sacral e glúteo, assim como as lesões no dorso, frequentemente são as últimas a cicatrizar, devido à compressão e ao atrito produzidos pela posição de decúbito dorsal. O envolvimento da unidade ungueal resulta em distrofia da unha. A aplicação de compressão lateral ao teto da bolha (ativa ou subclínica) usando os dedos da mão protegidos com luva causa seu deslocamento lateral, deixando erosão em seu sulco (sinal de Nikolsky) [ver Vídeo 1 (disponível apenas online ou em CD)]. Raramente, o PV está associado com timoma e/ou miastenia grave.6,7 As exacerbações graves de pênfigo costumam ser fatais quando não são tratadas. Nestes casos, a taxa de desnudamento da pele excede a taxa de reparo cutâneo. A dor bucal impede uma ingesta nutricional adequada. A dor na pele limita o movimento expondo o paciente ao risco de pneumonia e trombose de veia profunda, enquanto extensivas áreas de erosão cutânea predispõem à superinfecção, perda de proteína e, menos comumente, a perturbações de equilíbrio hídrico.
As exacerbações do pênfigo podem ser (1) inicialmente leves e, com o passar dos dias, evoluir para exacerbações extensivas; (2) leves e permanecerem leves; ou (3) intensas desde o início. O curso é imprevisível e isto tem implicações no grau de agressividade da abordagem terapêutica. Mesmo com terapia, o curso da doença aumenta e diminui de intensidade. A remissão completa permanente é difícil de alcançar.
Figura 2 (a) Pênfigo vulgar, com divisão suprabasal, fazendo a camada basal parecer uma “fileira de lápides” (coloração de hematoxilina-eosina; aumento de 200x do original). (b) Pênfigo vulgar, imunofluorescência direta, com fluorescência máxima na camada espinhosa (imunofluorescência direta e aumento de 200x do original).
Figura 3 (a) Penfigoide bolhoso com divisão subepidérmica. Note a presença de eosinófilos (coloração de hematoxilina-eosina; aumento de 200x do original). (b) Penfigoide bolhoso, imunofluorescência direta, com revestimento fluorescente da junção dermoepidérmica (imunofluorescência direta e aumento de 200x do original). Figura 3b, cortesia de Beutner Laboratories.
Figura 4 Pênfigo vulgar, erosão.
Figura 5 Pênfigo vulgar, bolhas intactas.
Figura 6 Pênfigo vulgar, crosta friável.
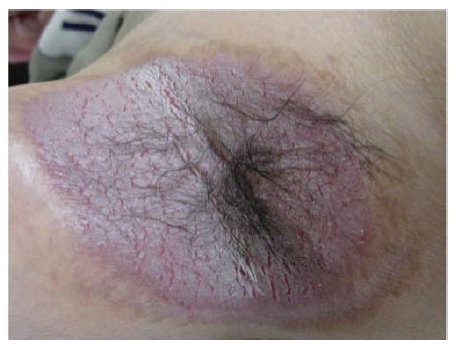
Figura 7 Pênfigo vegetante, visto com frequência na virilha e na axila. Cortesia de Daniel Carrasco, MD.
Figura 8 Pênfigo vulgar, erosão oral no palato.
Antes do advento dos esteroides, cerca de 75% dos pacientes com pênfigo morriam dentro de 1 ano.8 Entretanto, as complicações do tratamento continuam sendo responsáveis por uma taxa de mortalidade aproximada de 5-15%.9,10 A maioria dos pacientes com pênfigo entra em remissão parcial dentro de 2-5 anos. Em um estudo longitudinal dos resultados alcançados por 40 pacientes com PV, 45% entraram em remissão completa e prolongada após 5 anos, e 71% após 10 anos.11
A suspeita de pênfigo deve ser considerada em pacientes com erosões orais refratárias ou crônicas, ou em pacientes com erosões, crostas e bolhas no corpo. O diagnóstico costuma ser evidente no momento da apresentação. Para fins de confirmação, recomenda-se obter biópsia para coloração com hematoxilina-eosina e realização de imunofluorescência direta. Os títulos basais de PV devem ser determinados e enviados a um laboratório especializado (p. ex., nos EUA, Beutner Laboratories [http://www.beutnerlabs.com]), se possível, capacitado a distinguir simultaneamente o PV de PF, PPN e PB. Outros exames basais são determinados pelas medicações previstas para uso. Como os títulos de PV determinados por imunofluorescência indireta estão correlacionados com a atividade da doença, são úteis quando as erosões estão resolvidas e ainda há alto nível de imunossupressão.12 Títulos altos sugerem um afunilamento suave, enquanto títulos baixos podem permitir um afunilamento mais rápido.
A terapia será determinada pela gravidade inicial da condição no momento da apresentação, e pelas comorbidades do paciente. A proporção risco x benefício das diversas opções de tratamento também deve ser ponderada. Antigamente, um curso terapêutico típico poderia consistir na administração de uma dose alta de prednisona durante 1 mês (com os efeitos colaterais associados) aliada ou seguida de um agente poupador de esteroide (p. ex., micofenolato de mofetil).13 Quando o agente poupador de esteroide inicial falhava, tentava-se a administração contínua de prednisona com outro agente subsequente, como a azatioprina. Apesar destes regimes, um amplo subgrupo de pacientes continuava com doença inadequadamente controlada nos dois primeiros meses subsequentes à apresentação.
Nos últimos anos, duas classes de agentes muito caros e notavelmente efetivos têm se mostrado eficientes contra o pênfigo: imunoglobulina intravenosa (IVIg)14 e anticorpos anti-CD20 (p. ex., rituximabe).15,16 A IVIg induz diminuição rápida e seletiva dos títulos de autoanticorpos do pênfigo.17 Os possíveis mecanismos incluem o catabolismo aumentado de anticorpos patogênicos,18,19 interferência anti-idiótipo 20 e prevenção de acantólise por meio da interferência nas elevações autoanticorpo-induzidas de caspases pró-apoptóticas.21 O rituximabe é um anticorpo anti-CD20 citolítico que atua diminuindo a população hiperrepresentada de células B produtoras de autoanticorpo.
Os regimes de dosagem, monitoramento especial e principais riscos associados a cada fármaco são listados na Tabela 2. Não há consenso quanto ao(s) tratamento(s) a ser instituído primeiro e com quais combinações. Para os casos graves com necessidade de terapia urgente, uma estratégia defende a inicialização de um curso de prednisona em dose alta por via oral ou intravenosa, na dosagem de 1 mg/kg/dia, iniciando a IVIg a 2 g/kg divididas ao longo de 3 dias e uma única dose de 1 g de rituximabe por via intravenosa. Os ciclos de IVIg podem ser mantidos mensalmente, até um total de 2-3 ciclos. O rituximabe deve ser repetido em duas semanas, passando, então, a ser repetido a cada seis meses, conforme a necessidade (aquém do esquema de dosagem rotulado para artrite reumatoide). Decorridas duas semanas da primeira dose de rituximabe, os níveis de células B quantificados por CD19 devem ser quase nulos, mesmo assim uma segunda dose é administrada de forma rotineira na segunda semana. A prednisona pode ser continuada em dose alta, quando tolerada, até as lesões começarem a se reepitelizar e não haver formação de novas lesões (com frequência em 1-4 semanas), seguida de afunilamento lento com, digamos, 10 mg a cada 1-2 semanas. O avanço das bolhas com o afunilamento da prednisona necessitaria de um agente poupador de esteroide adicional, como micofenolato de mofetil ou azatioprina. A prednisona, então, seria aumentada em 5-10 mg acima da dose em que houve avanço das bolhas e mantida por 2-4 semanas, até que o novo agente poupador de esteroide pareça estar funcionando (evidenciado pela ausência de formação de novas bolhas e pela reepitelização das bolhas existentes). Neste momento, o afunilamento seria concluído. Medidas extremadas além das mencionadas anteriormente incluiriam plasmaférese ou ciclofosfamida.22 No evento da impossibilidade de descontinuar a prednisona, em geral é possível realizar a manutenção a longo prazo com uma dose baixa de 7,5 mg ou menos preferida, devido à falta de supressão do eixo hipotálamo-hipófise-suprarrenal com esta dose ou com doses menores.23,24
Dada a impossibilidade de prever se um caso brando evoluirá para um caso grave e devido à existência de equilíbrio com relação às toxicidades relativas de doses altas de prednisona versus micofenolato de mofetil versus rituximabe versus IVIg, alguém poderia argumentar facilmente em favor do uso de rituximabe e/ou IVIg como terapia de primeira linha (à parte das considerações referentes ao custo). As considerações referentes ao custo poderiam determinar a realização de triagem com prednisona e/ou micofenolato de mofetil como terapias iniciais, com um baixo limiar para mudar para IVIg e/ou rituximabe ao sinal de uma exacerbação grave.13 O momento de instituir o tratamento agressivo e definitivo com rituximabe não é após o afloramento de uma exacerbação grave e sim, imediatamente depois do início da progressão de doença branda para doença grave.
Outras terapias auxiliares [ver Tabela 2] que têm sido usadas com graus variáveis de sucesso são a dapsona,25 etanercept,26 ouro,27 metotrexato,25 ciclosporina28 e plasmaférese.29
|
Tabela 2 Fármacos sistêmicos usados no tratamento do pênfigo* | |||
|
Fármaco |
Dosagem |
Principais efeitos colaterais |
Monitoramento sugerido |
|
Rituximabe115–117 |
1 g, IV, semanas 0 e 2, repetir em 6 meses; ou 375 mg/m2 nas semanas 0-4 e, subsequentemente todo mês, × 4 meses |
Leucoencefalopatia multifocal progressiva, infecção, anafilaxia com a primeira dose, reativação de hepatite B, arritmias cardíacas, neutropenia tardia |
HC, PMC antes de cada, CD19 basal e AgHbs de hepatite; seguidos de PMC e HC em um mês após a segunda dose
|
|
IVIg115,118,119 |
2 g/kg divididas ao longo de 3 dias a intervalos mensais × 1-3 meses; ou 2 g/kg divididas ao longo de 3 dias por mês, até a cicatrização de todas as lesões, seguida de aumento do intervalo em 2 semanas até completar 16 semanas, para, então, descontinuar |
Trombose venosa profunda, acidente vascular encefálico, insuficiência renal, anemia hemolítica, meningite asséptica, edema pulmonar não cardiogênico, TRALI (lesão pulmonar aguda associada à transfusão), anafilaxia, pseudo-hiponatremia |
Níveis séricos basais de IgA, PMC, HC |
|
Prednisona (como exemplo para corticosteroide)1,120,121 |
1–2 mg/kg/dia, VO ou IV, até as lesões cicatrizarem ou mais nenhuma lesão nova aparecer; em seguida afunilar lentamente durante 3 meses; para controle agudo, administrar a megadose de 1 g/d × 5 dias (ao longo de 2-3 horas e com monitoramento cardíaco) |
Diabetes, osteoporose, hipertensão, supressão do eixo HHS, infecção, necrose avascular da cabeça femoral, catarata, glaucoma, síndrome de Cushing, dano fetal no 1º trimestre, ruptura de parede livre ventricular esquerda (quando administrada após um IM recente), psicose, sem vacinas vivas |
Glicose basal, pressão arterial, HC, potássio sérico, densitometria óssea, 25- Hidroxivitamina D; em seguida, pressão arterial mensal, glicose e potássio; pressão intraocular (se > 6 semanas) |
|
Micofenolato de mofetil122 |
1,0-1,5 g VO 2x/dia; começar com 500 mg VO, 2x/dia, então aumentar para a dose-alvo após 1 semana |
Linfoma e malignidade cutânea, leucoencefalopatia multifocal progressiva, neutropenia, aplasia pura de hemácias, hepatotoxicidade, infecção (varicela especialmente disseminada),arritmias, alterações GI incluindo enterocolite erosiva, defeitos inatos |
Níveis basais de HC, PMC, seguido de HC e PMC semanais após cada aumento de dose (e/ou durante o primeiro mês de terapia); subsequentemente, 2x/mês durante os próximos dois meses, e depois 1x/mês durante o primeiro ano |
|
Azatioprina123,124 |
50 mg, VO, 1x/dia na semana 1; em seguida, aumentar para a dose-alvo de 3 mg/kg dividida em 2 doses diárias |
Neoplasia (linfoma, pele, retículo), mielotoxicidade, infecção, perturbação gástrica, hepatotoxicidade, defeitos inatos |
Níveis basais de tiopurina metiltransferase, HC, PMC; em seguida HC e PMC semanalmente após cada aumento de dose (e/ou no 1º mês de terapia); subsequentemente, 2x/mês durante os próximos dois meses, e depois 1x/mês |
|
Metotrexato125 |
Até 25 mg/semana tomadas em um único dia (considerar a dose de teste de 7,5–15 mg na primeira semana, aumentando em seguida para a dose-alvo semanalmente, subsequentemente) |
Hepatotoxicidade e cirrose, fibrose pulmonar, infecção, mielotoxicidade, estomatite ulcerativa, câncer/linfoma, defeitos inatos |
Níveis basais de HC, PMC; então, uma semana após o aumento da dose e, depois, mensalmente |
|
Ciclosporina126 |
5 mg/kg, VO, dividida em duas doses diárias |
Hipertensão, nefrotoxicidade, infecção, hipertricose, linfoma, hipercalemia, hipomagnesemia, hiperuricemia, hipertrigliceridemia, hiperplasia gengival |
Basal: pressão arterial (em duas ocasiões distintas), creatinina sérica (considerar a média de duas medidas), ureia, HC, PMC, ácido úrico, triglicerídeos e magnésio; então, a cada duas semanas por 3 meses e, subsequentemente, a cada mês |
|
Dapsona127,128 |
Até 300 mg VO, 1x/dia (mais frequentemente 100 mg, VO, 1x/dia) |
Enzimas hepáticas elevadas, anemia hemolítica, metemoglobinemia, hipersensibilidade, agranulocitose, discrasias sanguíneas, hepatotoxicidade, neuropatia periférica (= 200 mg/dia) |
Níveis basais de G6PD, HC, PMC; então, HC, PMC semanalmente no 1º mês; em seguida, mensalmente por seis meses e, depois, semianualmente |
|
Aurotiomalato de sódio (como exemplo para ouro)129 |
Semana 1: dose de teste de 10 mg, IM; Semana 2: 25 mg, IM; então, 50 mg, IM semanalmente; quando não houver mais formação de novas bolhas, aumentar o intervalo entre as doses em 1 semana até chegar ao intervalo de 4 semanas, para então descontinuar; a dose total máxima é 0,8-1,0 g |
Reação nitroide (reação vasomotora com náusea, rubor, desmaio), nefropatia com proteinúria, trombocitopenia, leucopenia, anemia, dermatite/prurido, estomatite, depósitos corneais de ouro, crisíase, gosto metálico |
Basal: HC, PMC, urinálise; seguido de HC, PMC e urinálise antes de cada injeção durante o primeiro mês e, então, mensalmente |
|
Tabela 2 (Continuação) | |||
|
Fármaco |
Dosagem |
Principais efeitos colaterais |
Monitoramento sugerido |
|
Ciclofosfamida28,130,131 |
1,1-1,5 mg/kg/dia VO |
Malignidade (em especial, bexiga urinária, mieloproliferativa, linfoproliferativa), defeitos inatos, esterilidade, cistite hemorrágica, fibrose da bexiga urinária, infecção, cardiotoxicidade, anafilaxia, leucopenia, trombocitopenia, alopécia |
Níveis basais de HC, PMC, urinálise e RxT; em seguida, semanalmente por 2 meses e, depois, a cada mês; RxT a cada 6 meses |
|
Plasmaférese com imunossupressão (metilprednisolona e azatioprina ou ciclofosfamida isolada)132,133 |
4-6 trocas de plasma (50 mL/kg) ao longo de 5-15 dias com solução de albumina humana a 5% |
Síntese de autoanticorpo de rebote 7-14 dias após a plasmaférese, requerendo imunossupressão; sepse; desequilíbrios de volume e eletrólitos |
PMC, magnésio, e fósforo após o tratamento |
|
Etanercept134 |
50 mg, SC, 2x/semana |
Reativação de tuberculose, infecção (primariamente do trato respiratório), reativação de hepatite B, exacerbação de insuficiência cardíaca congestiva, esclerose múltipla, síndrome lúpus-símile, malignidade (linfoma, leucemia e câncer de pele não melanoma), pancitopenia |
PPD basal e, depois, anualmente; níveis basais de HC, PMC, então a cada 3-6 meses |
|
Tetraciclina e niacinamida135 |
500 mg, VO, 4x/dia (tetraciclina) e 750 mg, VO, 2x/dia (niacinamida) |
Perturbação GI, fotossensibilidade, pressão intracraniana aumentada, esofagite |
Nenhum |
HC = hemograma completo, com plaquetas e diferencial; PMC = painel metabólico completo, incluindo creatinina, ureia, aspartato aminotransferase, alanina aminotransferase, bilirrubina total, albumina, potássio, sódio, glicose e cálcio; RxT = raio X de tórax; GI = gastrintestinal; G6PD = glicose-6-fosfato desidrogenase; HHS = hipotálamo-hipófise-suprarrenal; IVIg = imunoglobulina intravenosa; IM = infarto do miocárdio; PPD = derivado proteico purificado.
*Somente riscos selecionados foram listados. O monitoramento é amplamente baseado na prática do autor com orientação a partir de toda informação de prescrição de fármacos. Os leitores devem rever a bula destas medicações antes da prescrição.
Como o uso prolongado de prednisona ainda é empregado com muita frequência no tratamento do pênfigo, os seguintes tratamentos e monitoramentos devem ser considerados: (1) densitometria óssea basal, repetida anualmente30; (2) níveis basais de vitamina D, supridos conforme a necessidade; (3) carbonato de cálcio (500 mg VO 2x/dia); (4) bisfosfonato para profilaxia contra osteoporose; (5) teriparatida, no evento em que a osteoporose induzida por prednisona já ocorreu; (6) triagem da pressão intraocular quando o curso terapêutico se estende por mais de 6 semanas; (7) profilaxia contra úlcera péptica com H2-antagonista ou inibidor de bomba de próton; e (8) profilaxia contra Pneumocistis jiroveci com trimetoprima-sulfametoxazol de potência única.31
Uma complicação séria que pode ocorrer com a terapia imunossupressora para PV é a infecção disseminada pelo vírus do herpes simples (e, às vezes, infecção disseminada pelo vírus varicela-zoster). Ocasionalmente, um paciente bem controlado, ainda que imunocomprometido, muitas vezes sob tratamento com micofenolato de mofetil, apresentará bolhas novas e não, necessariamente, com distribuição evidente. Neste momento, é preciso decidir se esta é uma exacerbação da doença que requer imunossupressão adicional ou se é varicela com necessidade de medicação antiviral e suspensão temporária da imunossupressão. O teste de imunofluorescência direta feito com uma vesícula ou um esfregaço de Tzank devem fornecer resultados rapidamente. A terapia empírica com 1 g de valaciclovir por via oral, 3 vezes ao dia durante 7 dias, raramente será prejudicial se houver qualquer dúvida quanto à possibilidade de varicela.32
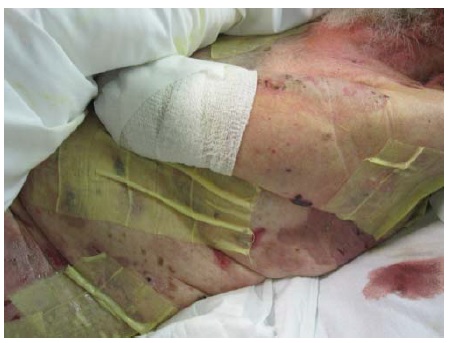
As medidas de suporte adotadas em casos de exacerbação grave do pênfigo incluem o controle adequado da dor durante as trocas de curativo e/ou mudanças de posição significativas; trocas diárias de curativos que consistem em gazes impregnadas com tribromofenato de bismuto a 3%/petrolato mantidas em posição com gazes enroladas (i.e., sem grudar fita adesiva na pele) [ver Figura 9]; e roupas de cama não aderentes (p. ex., lençóis Exu-Dry).
Figura 9 Gazes xeroformes sobre erosões de pênfigo, mantidas no lugar com gazes enroladas.
No PF, os pacientes abrigam autoanticorpos contra desmogleína 1, localizada na camada granular. O plano de clivagem, portanto, é ainda mais superficial do que no PV. Uma forma endêmica de PF encontrada no Brasil é chamada “fogo selvagem” e parece ser deflagrada por um antígeno ambiental ainda não identificado. A síndrome de Senear-Usher, também conhecida como pênfigo eritematoso, localiza as lesões do PF nas bochechas e compartilha achados imunológicos associados ao lúpus. É interessante notar que uma toxina esfoliativa produzida por Stafylococcus aureus cliva a desmogleína 1 e causa a síndrome da pele escaldada estafilocócica ou impetigo bolhoso, cujas bolhas estão no mesmo plano que no PF. (Em contraste, o vesicante cantaridina causa acantólise na camada basal via clivagem enzimática de desmossomos, comportando-se histologicamente de forma mais parecida com o PV.33) Os tiois, como captopril ou penicilamina, e os tiois mascarados, como as penicilinas e cefalosporinas, podem causar PF fármaco-induzido.4
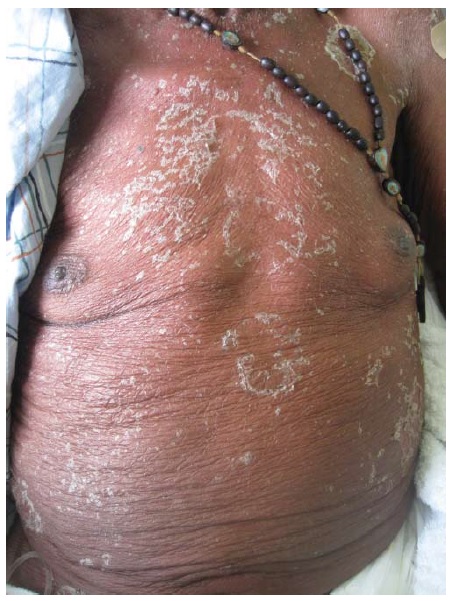
As lesões no PF raramente se manifestam como bolhas [ver Figura 10] e sim como placas ou pápulas achatadas aparentemente hiperqueratóticas que podem evoluir para erosões [ver Figura 11]. As lesões podem se generalizar em uma forte exacerbação e formar eritroderma esfoliativo envolvendo todo o integumento. Não há envolvimento da mucosa oral.
O diagnóstico é estabelecido por biópsia e imunofluorescência indireta de pele.
As estratégias terapêuticas para o PF são, na maior parte, as mesmas adotadas para o PV. Entretanto, há quem diga que o PF responde mais prontamente à terapia. Nos casos menos graves, tem sido demonstrado que abordagens mais brandas, como a aplicação tópica de corticosteroides superpotentes,34 dapsona35 e antimaláricos,36 são efetivas.
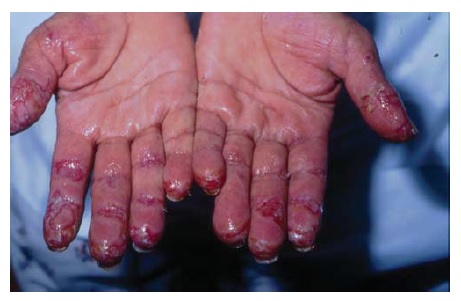
No PPN, os pacientes têm uma neoplasia subjacente, mais comumente um linfoma não Hodgkin ou leucemia linfocítica crônica (representando 70% dos casos).1 Em média, a idade do paciente no momento do aparecimento da condição é 59 anos.37,38 Diferente do observado no PV, a formação de crostas hemorrágicas nos lábios acompanhada de estomatite refratária é característica, sendo que o envolvimento conjuntival, genital e palmoplantar e frequente. O tratamento tipicamente é inefetivo, mas o rituximabe tem se mostrado promissor, a não ser pelo alerta de possibilidade de morte por sepse, de modo que sua coadministração com IVIg deve ser considerada.39 A condição está associada a uma taxa de mortalidade de 90% em um mês a dois anos após o estabelecimento do diagnóstico.40 Outras morbidades incluem a conjuntivite pseudomembranosa com formação de cicatriz e a bronquiolite obliterante.1
Figura 10 Pênfigo foliáceo; uma bolha intacta rara.
Figura 11 Pênfigo foliáceo; crostas aderentes constituem o teto da bolha.
Também conhecida como pênfigo familiar benigno, a doença de Hailey-Hailey é um distúrbio cutâneo autossômico dominante, em que surgem placas fissuradas e maceradas em áreas intertriginosas [ver Figura 13]. O defeito genético envolve uma adenosina trifosfatase dependente de cálcio (ATPase)—a ATP2C1.41 A doença costuma ser confundida com intertrigo de cândida ou bacteriano. A terapia inclui esteroides tópicos e tratamento de infecção concomitante, quando presente. Como a condição pode ser bastante refratária ao tratamento, outras modalidades que alcançam graus variados de sucesso são o tacrolimo tópico ou a ciclosporina tópica, injeção de toxina botulínica e ablação a laser de dióxido de carbono.42
Figura 12 Pênfigo paraneoplásico; erosões das palmas das mãos. Cortesia de Amit Pandiaa, MD.
Figura 13 Doença de Hailey-Hailey. Envolvimento característico de áreas intertriginosas.
No PB, os autoanticorpos têm como alvo os antígenos hemidesmossômicos, especificamente o antígeno PB 1 (também conhecido como BP230) e o antígeno PB 2 (também conhecido como BP180 ou colágeno XVII). Raramente, o PB pode ser induzido por fármacos e ter como causadores a espironolactona,43 furosemida,44 captopril,45 penicilamina,46 iodeto de potássio,47 cipro?oxacina,48 amoxicilina e ibuprofeno.49 A descontinuação do fármaco agressor resulta em melhora.1
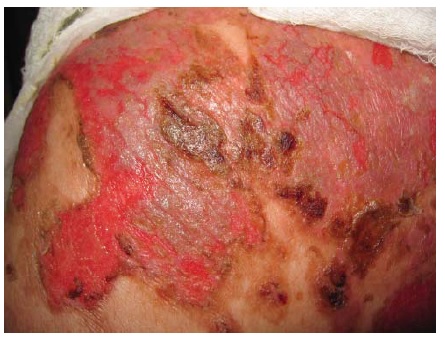
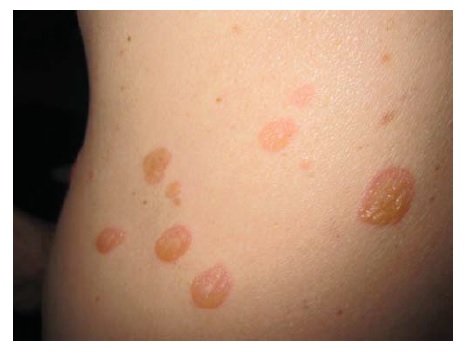
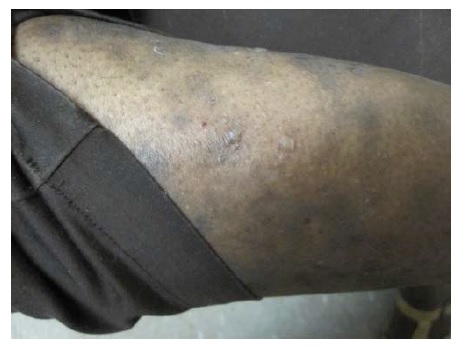
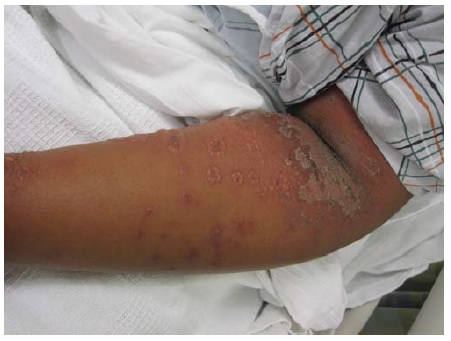
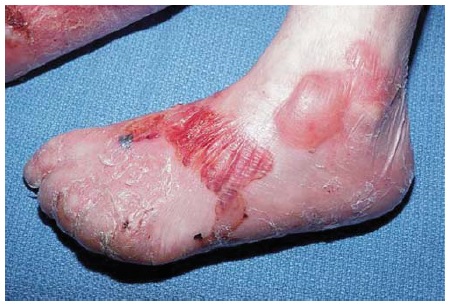
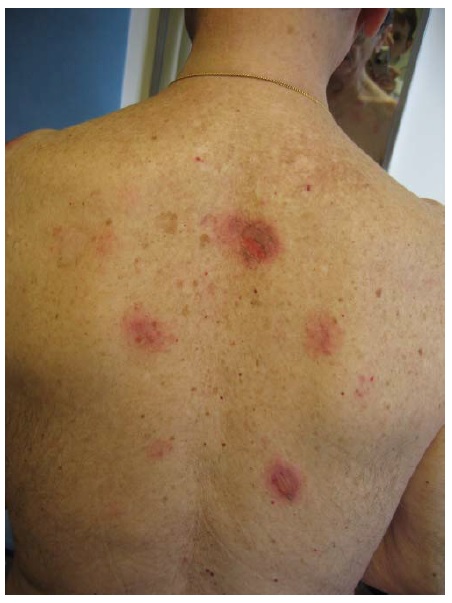
O PB ocorre, principalmente, em indivíduos com mais de 60 anos.1 Entretanto, crianças pequenas podem ser afetadas, ainda que raramente. As bolhas são tensas [ver Figura 14] devido à origem na junção dermoepidérmica, de modo que o sinal de Nikolsky será negativo. Estas bolhas não deixam cicatrizes, mas podem causar hiperpigmentação pós-inflamatória. As bolhas podem ocorrer perifericamente e serem pouco numerosas ou generalizadas e resultarem em erosões amplamente disseminadas ao se romperem [ver Figura 15]. De modo não incomum, as bolhas são hemorrágicas. Em alguns casos, a manifestação original e/ou isolada do PB é o prurido com erupção urticariforme na ausência de bolhas francas [ver Figura 16]. Em outros casos, vesículas generalizadas podem mimetizar o herpes disseminado. O envolvimento mucoso e, mais ainda, o envolvimento ocular são raros. histologicamente, eosinófilos são vistos na derme superficial associados com bolhas subepidérmicas [ver Figura 3a]. Ocorrem exacerbações e remissões espontâneas. Na gravidez, o PB é chamado penfigoide gestacional ou herpes gestacional e pode ocorrer a partir do 2º trimestre até o pós-parto. Os autoanticorpos transplacentários podem produzir bolhas no recém-nascido. Esses anticorpos recorrem nas gestações subsequentes.50
Figura 14 Penfigoide bolhoso, bolha tensa.
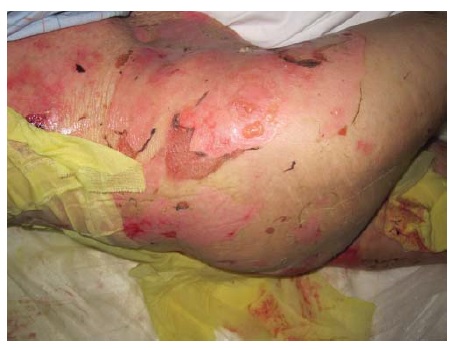
Figura 15 Penfigoide bolhoso grave com erosões amplamente disseminadas.
O diagnóstico é estabelecido por biópsia usando imunofluorescência direta. A imunofluorescência indireta também pode ser usada para fins diagnósticos, porém os títulos não se correlacionam com a atividade da doença, diferentemente do que ocorre no PV.51 O diagnóstico diferencial inclui erva venenosa, ataque de artrópode, eritema multiforme, varicela zoster disseminada e outros distúrbios bolhosos autoimunes (p. ex., EBA, DBAL, penfigoide cicatricial e PV).
A extensão da doença no momento da apresentação e a velocidade da progressão determinarão o qual agressiva a terapia deverá ser. Alguns pacientes nunca apresentam mais do que algumas bolhas, as quais podem ser tratadas com clobetasol tópico de acordo com a necessidade. Se a manifestação é amplamente disseminada ou progressiva, tipicamente é possível começar com prednisona a 1 mg/kg durante alguns dias seguida de afunilamento de uma a duas semanas.52 Os autores frequentemente usam 60 mg/dia por três dias, que então são desmamados em 10 mg a cada 2 dias. Havendo recidiva das bolhas com o afunilamento, torna-se necessário adotar uma opção poupadora de esteroide, além de voltar a acrescentar prednisona em uma dose pouco acima daquela em que houve exacerbação. As opções que poupam esteroides sistêmicos variam de acordo com a gravidade do PB. Tetraciclina (500 mg, VO, 4x/dia) e niacinamida (750 mg, VO, 2x/dia) são um regime que pode ser tentado até mesmo em paralelo ao afunilamento da prednisona inicial. Entretanto, apesar de seguro, esse regime não é consistentemente efetivo.53 Uma segunda opção é o micofenolato de mofetil, na dose de até 1 g por via oral, duas vezes ao dia. Quando estes regimes falham, pode ser necessário usar IVIg e rituximabe nas doses usadas no PV.54–57 Outros são a entender que a terapia intensiva administrada duas vezes ao dia usando apenas clobetasol tópico tem eficácia comparável e menos morbidade do que os esteroides sistêmicos no PB.58 Doses baixas de metotrexato e dapsona também são outras opções adicionais.59 Para os casos refratários, qualquer terapia listada na Tabela 2 para PV pode ser tentada para o PB.
Figura 16 Penfigoide bolhoso manifestado como lesões urticariformes.
O penfigoide cicatricial, também conhecido como penfigoide de membrana mucosa, é um raro distúrbio bolhoso autoimune crônico caracterizado pelo envolvimento e subsequente formação de cicatrizes nas superfícies mucosas, além de formação de bolhas na cabeça e na parte superior do tronco.1 Vários epítopos, como a laminina-5 ou o colágeno XVII, presentes na membrana basal são alvejados. A formação de cicatrizes nas mucosas pode levar à cegueira, à obstrução de vias respiratórias e à disfonia. Na variante de Brunsting-Perry, há envolvimento da cabeça e do pescoço com alopécia cicatricial, porém o envolvimento de mucosas é mínimo.1 O diagnóstico de pênfigo cicatricial requer a combinação de imunofluorescência direta positiva e envolvimento de mucosa com formação de cicatriz. A imunofluorescência indireta ou o ELISA específico para antígenos do pênfigo cicatricial não são disponibilizados na clínica. Em casos brandos, o tratamento pode ser feito com até 150 mg de dapsona/dia.60 Considerando a extrema morbidade associada aos casos graves, a IVIg e/ou o rituximabe são razoáveis como alternativas de primeira linha.61 Outras opções incluem ciclofosfamida, azatioprina, micofenolato de mofetil, metotrexato e plasmaférese.62–64 Os esteroides orais isolados tipicamente requerem períodos estendidos com doses altas e são usados como parte da terapia combinada e como terapia de pulso. O tratamento da ferida, evitação de traumatismo e tratamento multidisciplinar com oftalmologia, otolaringologia e odontologia são úteis.1 As sequelas a longo prazo da doença ocular incluem entrópio, triquíase, formação de catarata e glaucoma, que podem ser tratadas cirurgicamente. A cirurgia deve se seguir ao tratamento sistêmico para controlar a inflamação, uma vez que pode aumentar o risco de reicidência.65 Complicações sistêmicas prejudiciais à vida adicionais, como estreitamento de laringe ou envolvimento traqueal, podem requerer dilatação e traqueostomia para prevenir disfagia ou asfixia, respectivamente.1
EBA é uma doença bolhosa autoimune adquirida caracterizada por autoanticorpos IgG circulantes e ligados a tecidos, dirigidos contra o domínio não colagenoso (NC1) do colágeno de tipo VII, um dos principais componentes das fibrilas de ancoragem que conectam a membrana basal a estruturas dérmicas. A EBA afeta primariamente adultos. As bolhas subepidérmicas cicatrizam com formando escaras atróficas ou milhetes, frequentemente sobre os membros em sítios de traumatismo mecânico (i.e., parte dorsal das mãos, cotovelos e joelhos).66 Como resultado, pode haver distrofia da unha e pseudossindactilia. O envolvimento do couro cabeludo, ocular, laríngeo e esofagiano pode resultado em alopécia cicatricial, cegueira, estenose de laringe e disfagia. A enteropatia inflamatória é encontrada em cerca de 30% dos casos.67 A EBA deve ser distinguida do PB, penfigoide cicatricial, lúpus eritomatoso bolhoso, porfiria cutânea tardia e epidermólise bolhosa distrófica dominante (particularmente em crianças). O diagnóstico é estabelecido por biópsia e imunofluorescência indireta de pele separada com sal. Autoanticorpos IgG são vistos na lâmina densa e sublâmina densa na EBA (versus hemidesmossomo e lâmina lúcida superior no PB). Ocasionalmente, a imunofluorescência indireta fracassa em identificar a EBA devido à ausência de anticorpos circulantes, tornando a microscopia imunoeletrônica o exame diagnóstico ideal. Considerando a natureza recalcitrante do distúrbio, é razoável adotar uma abordagem terapêutica similar a usada para PV, com uma triagem inicial usando prednisona e dapsona, e um limiar baixo para mudar para IVIg68,69 e/ou rituximabe antecipadamente, em casos moderados a graves.70 Outras terapias que têm sido tentadas incluem dapsona, infliximabe, ciclosporina e colchicina. O lúpus bolhoso responde drasticamente à dapsona, na qual se pode apoiar do ponto de vista diagnóstico.1,71
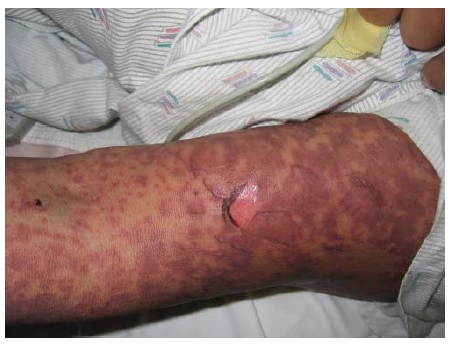
A DH é uma doença bolhosa crônica rara, atribuível à sensibilidade ao glúten que frequentemente está associada à enteropatia glúten-sensível (doença celíaca). Tipicamente, a condição afeta os descentes de norte europeus, principalmente os homens, e se manifesta entre 20 e 50 anos de idade. A gliadina é a fração solúvel em álcool do glúten, sendo que a condição é atribuída aos autoanticorpos IgA dirigidos contra a gliadina em ligação cruzada com a transglutaminase endomisial.1,72 Os relatos de caso de DH fármaco-induzida implicam o acetato de leuprolida, inibidores de fator de necrose tumoral-a, medicações anti-influenza e anticoncepcionais contendo progesterona.73,74
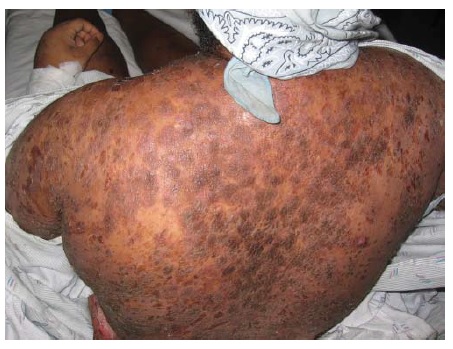
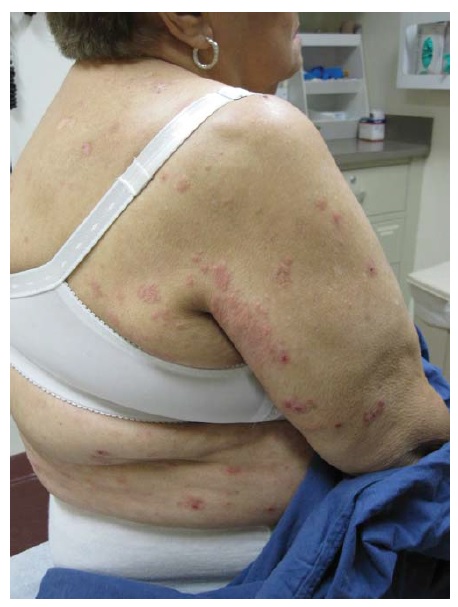
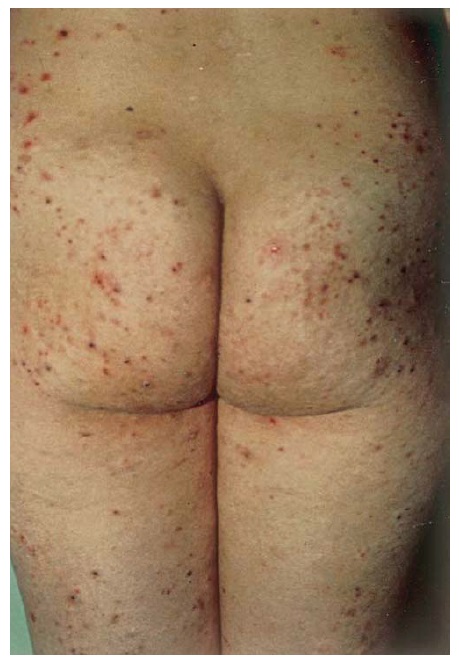
Os pacientes apresentam aglomerados pruriginosos de vesículas ou pápulas urticariformes, frequentemente escoriadas e simetricamente distribuídas sobre as superfícies extensoras dos membros, nádegas, couro cabeludo e dorso [ver Figura 17]. As membranas mucosas tipicamente são poupadas. A condição piora e melhora, muitas vezes conforme a ingesta dietética de glúten. A ingesta de iodo inorgânico causa exacerbações graves. As principais associações são a tireoidite de Hashimoto e o linfoma de células T associado a enteropatia.75 O risco deste linfoma está correlacionado com o grau de exposição ao glúten.
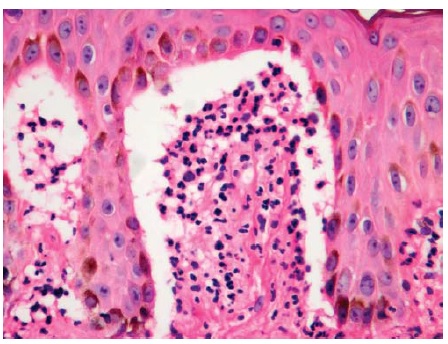
A histologia, a imunopatologia, a sorologia e a correlação clínica contribuem, todas, para o diagnóstico da DH. A coloração de hematoxilina-eosina revela a presença de microabscessos de neutrófilos nas pontas das papilas dérmicas e vesiculação ao nível da junção dermoepidérmica [ver Figura 18]. Este achado pode ser encontrado no lúpus bolhoso e na DBAL. A imunofluorescência direta de uma biópsia de pele obtida nas adjacências de uma vesícula deve mostrar a presença de depósitos granulares de IgA na papila dérmica. Também é possível observar deposição granular de IgA na zona da membrana basal. Quando encontrados isoladamente na membrana basal, os depósitos granulares de IgA são bastantes sensíveis e específicos para DH.76 Quando estes depósitos são encontrados IgM, IgG ou C3, torna-se necessário considerar as hipóteses de vasculite por imunocomplexo e lúpus. Sorologicamente, haverá positividade para anticorpos IgA antiendomísio ou anticorpos IgA anti-transglu- taminase tecidual. Os títulos estão correlacionados com o grau de restrição ao glúten na dieta.77-79 Em populações deficientes de IgA, os anticorpos IgG anti-transglutaminase tecidual devem ser quantificados.80 O diagnóstico diferencial da DH inclui escabiose, herpes, DBAL, PB e lúpus bolhoso.
Figura 17 A dermatite herpetiforme, uma erupção extremamente pruriginosa, comumente se manifesta como papulovesículas agrupadas e escoriadas, muitas vezes com distribuição simétrica (na foto, aparece nas nádegas, que são um sítio comum de envolvimento).
A DH deve levar ao encaminhamento ao gastrenterologista. A restrição ao glúten é a base da terapia. O glúten está presente no trigo, centeio e cevada, mas está ausente na aveia. O Gluten Intolerance Group (http://www.glúten.net) fornece uma lista de opções isentas de glúten. A dapsona, em doses de 50-200 mg/dia, trata a pele e promove resultados drásticos, mas não tem efeito sobre a enteropatia. Os pacientes pediátricos devem ser dosados em 0,5 mg/kg.1 A glicose-6-fosfato desidrogenase deve ser checada com antecedência, a fim de evitar hemólise grave e metemoglobinemia. A neuropatia periférica pode ser vista com doses maiores e com a terapia prolongada.81 A agranulocitose é um efeito colateral raro, mas perigoso. Quando a dapsona não é uma opção, a sulfassalazina (forma com revestimento entérico) é uma alternativa na dose de 2 g/dia.82 Ambas as medicações devem ser afuniladas até a dose mínima supressora de prurido e de formação de novas lesões.
A DBAL ocorre em crianças como doença bolhosa autoimune idiopática (conhecida como doença bolhosa crônica da infância ou DBCI) ou em adultos, nos quais é um fenômeno mais comumente induzido por fármaco. Classicamente, a vancomicina é o agente responsável, contudo as penicilinas, cefalosporinas e captopril, entre outros fármacos, têm induzido DBAL. 83–85
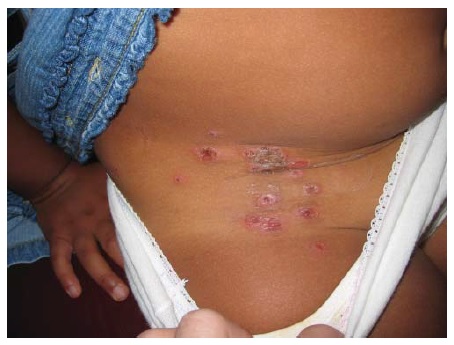
A IgA linear tipicamente é autolimitada em crianças, durando em média 2-4 anos,85 e tende mais a ser crônica em adultos. Clinicamente, aparecem bolhas que seguem uma distribuição anular ou surgem como lesões com característica de alvo, cuja borda externa é uma bolha anular, que são responsáveis pela descrição de “coroa de joias” que lhes é atribuída [ver Figura 19]. Raramente, as bolhas podem se tornar extensivas o bastante para mimetizarem a necrólise epidérmica tóxica. 86 Na DBCI, as lesões são encontradas na parte inferior do tronco, coxa e virilha. A DBAL pode se manifestar como variante do penfigoide cicatricial, com formação de cicatriz mucosa e ocular. Histologicamente, neutrófilos são encontrados nas papilas dérmicas e, a imunofluorescência direta, IgA é encontrada em forma de depósitos lineares ao longo da zona da membrana basal. O diagnóstico diferencial inclui DH, PB, penfigoide cicatricial, EBA, eritema multiforme bolhoso e, nos casos extensivos, necrólise epidérmica tóxica. A monoterapia com 100 mg de dapsona/dia ou com prednisona resolve a maioria dos casos.87,88
Figura 18 Dermatite herpetiforme (coloração de hematoxilina-eosina; aumento de 400x do original) com microabscessos neutrofílicos nas pontas das papilas dérmicas e vesiculação na junção dermoepidérmica. Cortesia de Steve Mercer, MD.
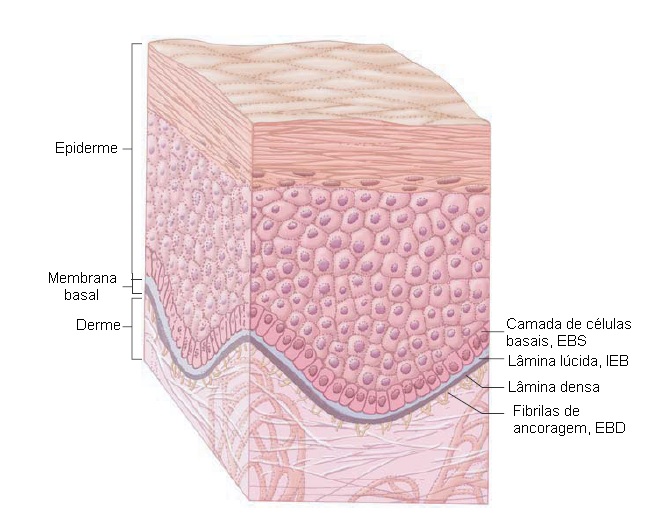
A epidermólise bolhosa abrange um grupo de doenças bolhosas hereditárias extremamente raras. As principais subdivisões são caracterizadas pela localização do plano das bolhas: epidermólise bolhosa simples (intraepidérmico), epidermólise bolhosa juncional (EBJ, lâmina lúcida) e epidermólise bolhosa distrófica (sublâmina densa) [ver Figura 20]. Junto a estas subdivisões principais, existem diversas variantes clínicas. Cada subdivisão está associada a um defeito genético envolvendo uma única proteína, que leva à formação de bolha ao nível da pele onde essa proteína está localizada. A Tabela 3 resume os aspectos pertinentes de cada variante de doença.
A maioria das formas de epidermólise bolhosa resulta em formação de bolha como consequência de forças de cisalhamento. O reconhecimento antecipado de uma doença permite a adoção de medidas preventivas adequadas para evitar a morbidade excessiva a partir da formação de bolhas. Além das erosões e bolhas amplamente disseminadas, as complicações adicionais incluem contraturas limitadoras da amplitude de movimento, com frequência na epidermólise bolhosa distrófica recessiva (EBDR). O carcinoma de células escamosas em feridas crônicas que não cicatrizam é uma das principais causas de morte na EBDR, mas raramente na EBJ.89 A ocorrência de melanoma é rara. Na EBDR e na EBJ, há estreitamento do esôfago, má absorção atribuível ao envolvimento do intestino delgado, constipação atribuível ao envolvimento do intestino grosso e estenoses anais. 1 Ocorre hipoplasia de esmalte na EBJ.90 A distrofia muscular ocorre na epidermólise bolhosa simples-distrofia muscular (EBS-DM), enquanto a atresia pilórica ocorre na EBJ-AP (EBJ com atresia pilórica). Insuficiência renal crônica e miocardiopatia podem ocorrer na EBDR. A sepse bacteriana é outra possibilidade. O estreitamento traqueolaringiano requer traqueostomia. As ulcerações e erosões corneais exigem tratamento rápido com pomada antibiótica. O enxerto de pele de espessura integral da pálpebra superior é indicado para os casos de ectrópio cicatricial. Por fim, a depressão não deve ser ignorada. Nas formas mais graves, a expectativa de vida pode ser drasticamente curta. 1
Figura 19 Dermatose bolhosa de IgA linear. Note a configuração anular em “coroa de joias” das bolhas.
A facilidade de indução de bolha favorece a obtenção da biópsia destas lesões na forma intacta e logo que são formadas. A amostra deve ser obtida da borda de uma bolha, de modo a incluir tanto a pele envolvida como a pele sem envolvimento. Embora a microscopia eletrônica seja extremamente útil no processo diagnóstico, a falta de ampla disponibilidade impede seu uso de rotina na clínica. Fine e colaboradores definem os critérios diagnósticos usando microscopia eletrônica. 91 O mapeamento com imunofluorescência usando anticorpos “relevantes para epidermólise bolhosa” pode ser realizado submetendo a biópsia em solução de Michel a um laboratório com conhecimentos sobre este protocolo (p. ex., http://www.beutnerlabs.com). É necessário estabelecer a correlação clínica. A análise de mutação de DNA e o teste pré-natal também podem ser realizados (ver http://www.GeneDx.com), mas são caros e trabalhos. Os resultados negativos não excluem a doença. Fine e colaboradores fornecem uma lista útil de laboratórios diagnósticos. 91 O diagnóstico diferencial inclui a síndrome de Kindler, que consiste em bolhas acrais, poiquiloderma progressivo, fotossensibilidade e carcinoma de células escamosas tardio. 91
Estes pacientes são mais bem tratados nos centros de terapia especializados em cuidar de pacientes com epidermólise bolhosa. Embora algumas dicas terapêuticas valiosas sejam oferecidas adiante, quatro fontes excelentes de informação são duas publicações de Fine e colaboradores 91,92 e os websites da Dystrophic Epidermolysis Bullosa Research Association of America e da Stanford University, Department of Dermatology (que traz informação excelente sobre cuidados de ferida) [ver barra lateral, Websites úteis]. Uma abordagem multidisciplinar deve incluir pediatria, dermatologia, oftalmologia, gastroenterologia, cirurgia plástica, fisioterapia, odontologia, nutrição, psicologia e genética.
Os cuidados com a pele giram em torno da prevenção da bolha e de cuidados meticulosos com a ferida. A cicatrização da ferida tipicamente é comprometida como resultado de infecção local, deficiências nutricionais e defeitos proteicos subjacentes. Os pacientes estão primariamente expostos ao risco de infecção por Stafylococcus aureus e Streptococcus pyogenes, e devem ser acompanhados de perto para monitoramento da sepse. 93 A pele desnudada deve ser tratada com antibióticos tópicos e, em seguida, protegida com curativo semioclusivo não aderente (p. ex., gazes impregnadas de geleia de petróleo cobertas com Aquafor), colocação de material curativo absorvente que também servirá de acolchoamento (em especial nas proeminências ósseas) e, por fim, compressão suave (p. ex., tela de estiramento). Não se deve nunca aplicar fita adesiva diretamente na pele. (Os tipos de curativos e locais onde são vendidos podem ser encontrados em http://www.debra.org.) A pseudossindactilia e as contraturas [ver Figura 21], vistas com frequência na EBDR, podem ser retardadas envolvendo cuidadosamente os dedos das mãos em extensão e evitando o atrito (i.e., não abrir tampas com rosca de garrafas) (ver o vídeo em http://dermatology.stanford.edu/gsdc/eb_clinic/eb-resources. html).94 Os casos graves podem requerer procedimentos de degloving. As bolhas intactas devem ser rompidas com agulha calibre 18, na base, após serem limpas com swab de álcool. Isto evita a disseminação lateral no plano da bolha. O superaquecimento é uma preocupação a ser considerada com a cobertura extensiva da ferida, requerendo hidratação adequada e monitoramento frequente da temperatura, em especial nos climas quentes.94
Figura 20 Planos de bolha cutâneos de epidermólise bolhosa (EB). Existem três formas principais de EB reconhecidas: epidermólise bolhosa simples (EBS), em que a separação ocorre junto à camada de células basais; epidermólise bolhosa juncional, em que a separação se junto à lâmina lúcida; e epidermólise bolhosa distrófica (EBD), em que a separação ocorre na sublâmina densa.
Algumas dicas valiosas para bebês incluem o uso de bico de Haberman para alimentação, que requer menos sucção para retirada de líquido, prevenindo erosões orais. O sulcralfato deve ser aplicado topicamente nas erosões dolorosas. Em casos graves, pode ser necessário usar tubo G. As fraudas devem ser revestidas com gazes impregnadas de petrolato. Após a evacuação, o ânus deve ser limpo com material umedecido com óleo mineral, em vez de toalhas de papel comuns ou outros tipos de lenços. Evitar medir a temperatura corporal por via retal, em vez disso, use um termômetro de orelha. Evitar usar derivações eletrocardiográficas adesivas normais. Caso essas derivações adesivas sejam necessárias, use as derivações Galicare obtidas em lojas de suprimentos médicos (são à base de silicina e não grudam forte demais). O prurido é comum à medida que as feridas cicatrizam e pode ser aliviado com hidratação. A anemia por perda de sangue crônica pode requerer suplementação de ferro ou transfusão. 94 A constipação pode requerer uso regular de amolecedores de fezes. A natação é uma atividade física ideal. A higiene oral cuidadosa com escova de espuma e o uso de enxaguatórios previnem a formação de cicatrizes intraorais (a língua pode se fundir com o assoalho da boca). Os assentos de carro e as roupas de cama e colchões são mais adequados quando confeccionados com seda ou cetim sobre couro de carneiro ou espuma de caixa de ovos. Por fim, evite levantar os bebês pelas axilas.
A substituição de gene ex vivo tem levado à correção da doença,95 sendo que um estudo mostrou que fibroblastos cutâneos manipulados por engenharia molecular foram bem-sucedidos em fornecer colágeno VII ao serem injetados por via intravenosa.96,97 Mais recentemente, foi demonstrado que transplantes de medula óssea congênico selvagem e alogênico foram bem-sucedidos na promoção de melhora de sintomas e aumento de colágeno tipo VII em pacientes com EBDR e em modelos murinos.97,98 Entretanto, não houve normalização das fibrilas de ancoragem em nenhum dos pacientes.99
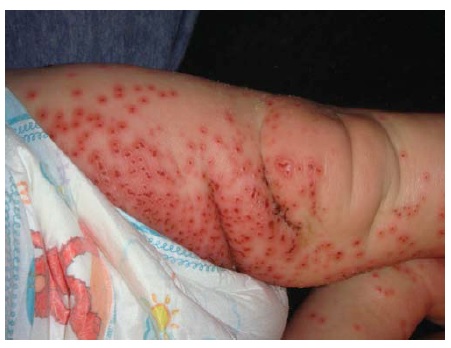
Os responsáveis mais comuns pelas doenças desta categoria são o impetigo bolhoso estafilocócico, a infecção pelo vírus do herpes simples e a infecção pelo vírus varicela-zoster. O impetigo bolhoso [ver Figura 22] é causado por uma toxina esfoliativa elaborada por Stafylococcus aureus, que clica a desmogleína 1 na camada granular. As formas localizadas podem ser tratadas topicamente. Lesões mais generalizadas requerem antibióticos orais. A síndrome da pele escaldada estafilocócica [ver Figura 23] resulta da disseminação hematogênica da toxina esfoliativa elaborada a partir de outra fonte localizada de infecção estafilocócica, como a faringite estafilocócica. O herpes simples disseminado é mais comum na pele eczematosa de pacientes com dermatite atópica, resultando em eczema herpético [ver Figura 24]. Pacientes com imunocomprometimento grave seja por infecção por HIV ou iatrogenicamente, também podem apresentar vesículas disseminadas. A infecção pelo vírus varicela-zoster disseminada terá um dermátomo identificável em adição às vesículas disseminadas [ver Figura 25], ocorrendo também em indivíduos imunocomprometidos. A terapia do herpes simples disseminado ou do zoster tipicamente requer administração intravenosa de aciclovir na concentração de 10 mg/kg a cada 8 horas.
|
Tabela 3 Epidermólise bolhosa*91 | ||||
|
Doença |
Subtipo da doença |
Defeito proteico |
Padrão de herança |
Achados clínicos |
|
EBS |
|
|
|
|
|
|
EBS, acantolítica letal |
Desmoplaquina |
AR |
Erosões com vazamento, sem bolhas; unhas distróficas; alopécia; Dentes neonatais; morte precoce. |
|
|
EBS, deficiência de placofilina-1 |
Placofilina-1 |
AR |
Erosão; unhas distróficas; hipotricose; ceratoderma focal; fissuras periorais; fissura da língua; ausência de cílios; retardo do crescimento. |
|
|
EBS superficial |
? |
AD |
Erosões, sem bolhas; unhas distróficas. |
|
|
EBS, localizada |
Queratina 5 ou 14 |
AD |
Bolhas nas palmas e solas; erosões orais; ceratoderma focal em alguns adultos. |
|
|
EBS, Dowling-Meara |
Queratina 5 ou 14 |
AD |
Bolhas; ceratoderma difuso; bolhas arciformes (“herpetiforme”); anemia; constipação. |
|
|
EBS, outras generalizadas |
Queratina 5 ou 14 |
AD |
Bolhas; ceratoderma focal; achados oculares raros |
|
|
EBS com pigmentação mosqueada |
Queratina 5 |
AD |
Bolhas generalizadas; pigmentação mosqueada ou marrom reticulada. |
|
|
EBS–distrofia muscular |
Plectina |
AR |
Bolhas ao nascimento; distrofia muscular na infância até a fase adulta; ptose; tecido de granulação e estenose do trato respiratório; morte precoce. |
|
|
EBS–atresia pilórica |
Plectina ou a6/b4-integrina |
? AR |
Início ao nascimento; ausência de pele congênita disseminada; anemia; retardo do crescimento; erosões orais; atresia pilórica; asas nasais e aurículas deformadas; contraturas articulares; criptorquidismo; morte precoce. |
|
|
EBS-AR |
Queratina 14 |
AR |
Bolhas generalizadas, inclusive anogenitais; placas ictióticas; erosões orais; constipação. |
|
|
EBS-Ogna |
Plectina |
AD |
Bolhas acrais; onicogrifose; tendência à contusão. |
|
|
EBS–migratória circinada |
Queratina 5 |
AD |
Bolhas generalizadas; eritema migratório circinado; hiperpigmentação pós-inflamatória marrom.
|
|
EBJ |
|
|
|
|
|
|
EBJ-Herlitz |
Qualquer uma das três subunidades de laminina-332 (também conhecida como laminina-5) |
AR |
Aparecimento ao nascimento; bolhas generalizadas; unhas distróficas; tecido de granulação; pseudossindactilia; anemia; retardo do crescimento; erosões orais; cáries dentais; envolvimento dos tratos gastrintestinal e geniturinário;carcinoma de células escamosas [raramente]; morte precoce. |
|
|
EBJ–não-Herlitz, generalizado |
Qualquer uma das três subunidades de laminina-332 ou colágeno tipo XVII (também conhecido como antígeno do penfigoide bolhoso-2) |
AR |
Aparecimento ao nascimento; bolhas generalizadas; unhas distróficas; anemia; retardo do crescimento; erosões orais; cáries dentais; envolvimento dos tratos gastrintestinal e geniturinário;carcinoma de células escamosas [raramente]; morte precoce. |
|
|
EBJ, não–Herlitz, localizada |
Colágeno tipo XVII |
AR |
Aparecimento ao nascimento; localizada; unhas distróficas; cáries dentais. |
|
|
EBJ–atresia pilórica |
Qualquer uma das duas subunidades de a6/b4-integrina |
AR |
Aparecimento ao nascimento; bolhas generalizadas; amplas áreas de aplasia cutânea; atresia pilórica; múltiplas deformações geniturinárias; orelhas rudimentares; morte precoce. |
|
|
EBJ, inversa |
Laminina-332 |
AR |
Bolhas intertriginosas; unhas distróficas; cáries dentais; envolvimento do trato gastrintestinal. |
|
|
EBJ, início tardio |
? |
AR |
Aparecimento na fase de adulto jovem; unhas distróficas; hiper-hidrose; ausência de dermatóglifos. |
|
|
Síndrome laringonicocutânea (também conhecida como Shabbir) |
Cadeia a3 da laminina-332 |
AR |
Aparecimento ao nascimento; erosões faciais e cervicais; unhas distróficas; tecido de granulação; envolvimento da laringe; cáries dentais; envolvimento da conjuntiva e do trato respiratório; incidência aumentada no Punjab; morte precoce. |
|
Tabela 3 (continuação) | ||||
|
Doença |
Subtipo da doença |
Defeito proteico |
Padrão de herança |
Achados clínicos |
|
EBDD |
|
|
|
|
|
|
EBDD, generalizada |
Colágeno tipo VII |
AD |
Aparecimento ao nascimento; bolhas generalizadas; unhas distróficas; “lesões albopapuloides”; erosões orais; Envolvimento do trato gastrintestinal; anemia. |
|
|
EBDD, acral |
Colágeno tipo VII |
AD |
Aparecimento na infância; bolhas nas mãos e pés; unhas distróficas. |
|
|
EBDD, pretibial |
Colágeno tipo VII |
AD |
Aparecimento ao nascimento ou na infância; bolhas nas bochechas, mãos, pés e unhas; unhas distróficas; lesões do tipo líquen plano; cáries dentais; constipação. |
|
|
EBDD, pruriginosa |
Colágeno tipo VII |
AD |
Aparecimento na infância; bolhas generalizadas ou localizadas; prurido; constipação. |
|
|
EBDD, apenas nas unhas |
Colágeno tipo VII |
AD |
Aparecimento ao nascimento ou infância; apenas unhas distróficas. |
|
|
EBDD, dermólise bolhosa do recém-nascido |
Colágeno tipo VII |
AD |
Aparecimento ao nascimento ou infância; bolhas generalizadas; unhas distróficas; cáries dentais. |
|
|
EBDR, generalizada grave |
Colágeno tipo VII |
AR |
Aparecimento ao nascimento; bolhas generalizadas; unhas distróficas; anemia; retardo do crescimento; erosões orais; cáries dentais; envolvimento gastrintestinal e ocular; pseudossindactilia; insuficiência renal crônica; miocardiopatia; osteoporose; carcinoma de células escamosas; melanoma; morte precoce. |
|
|
EBDR, outro generalizado |
Colágeno tipo VII |
AR |
Aparecimento ao nascimento; bolhas generalizadas; unhas distróficas; anemia; retardo do crescimento; erosões orais; envolvimento do trato gastrintestinal; pseudossindactilia; carcinoma de células escamosas; morte precoce. |
|
|
EBDR, inversa |
Colágeno tipo VII |
AR |
Aparecimento ao nascimento; bolhas intertriginosas, acrais, lombossacrais e axiais; unhas distróficas; anemia; retardo do crescimento; erosões orais; cáries dentais; envolvimento dos tratos gastrintestinal e genitourinário; pseudossindactilia; estenose do canal auditivo externo. |
|
|
EBDR, pretibial |
Colágeno tipo VII |
AR |
Aparecimento ao nascimento ou infância; bolhas nas bochechas, mãos, pés e unhas; unhas distróficas; lesões similares ao líquen plano; cáries dentais; constipação. |
|
|
EBDR, pruriginosa |
Colágeno tipo VII |
AR |
Aparecimento na infância; bolhas generalizadas ou localizadas; prurido; constipação. |
|
|
EBDR, centrípeta |
Colágeno tipo VII |
AR |
Aparecimento ao nascimento ou infância; bolhas nas bochechas e unhas; erosões orais. |
|
|
EBDR–dermólise bolhosa do recém-nascido |
Colágeno tipo VII |
AR |
Aparecimento ao nascimento ou infância; bolhas generalizadas; unhas distróficas; cáries dentais. |
AD = autossômica dominante; AR = autossômica recessiva; EBDD = epidermólise bolhosa distrófica dominante; EBS = epidermólise bolhosa simples; EBJ = epidermólise bolhosa juncional; EBDR = epidermólise bolhosa distrófica recessiva.
*Esta tabela pode não incluir todos os subtipos, cuja identificação e caracterização estão em contínua evolução.
Diferentemente das doenças bolhosas autoimunes, que tipicamente exibem um plano de clivagem particular na pele, as erupções do eritema multiforme, síndrome de Stevens-Johnson e necrólise epidérmica tóxica envolvem necrose dispersa ou completa dos queratinócitos em todas as camadas da epiderme. Estes padrões de ração constituem um espectro de atividade de doença contendo sobreposição histológica e clinicamente. A eliminação do fármaco causal é, talvez, a consideração terapêutica mais importante para estes distúrbios. As formas puras de cada distúrbio são caracterizadas conforme descrito a seguir.
O eritema multiforme pode ser causado por uma miríade de agressores, contudo os fármacos e as infecções crônicas pelo vírus do herpes simples (latente ou ativa) e por Mycoplasma pneumoniae ainda são os responsáveis mais frequentes.100,101 As lesões têm características de alvo e, por vezes, são centralmente bolhosas [ver Figura 26]. Pode haver envolvimento de mucosa causando erosões.102 O valaciclovir e os esteroides tópicos são as terapias padrão. Os casos graves ou recorrentes podem necessitar de corticosteroides orais ou talidomida.42
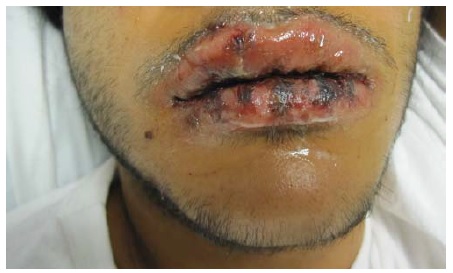
Por definição, esta síndrome envolve menos de 10% da área de superfície corporal e duas membranas mucosas (lábios/oral, conjuntiva, narinas/vias aéreas, mucosa vaginal, prepúcio e ânus). Clinicamente, as lesões podem ser morbiliformes ou do tipo alvo, com a clássica formação de crosta hemorrágica nos lábios [ver Figura 27]. Os responsáveis são os fármacos e a infecção. Dentre os fármacos, ibuprofeno, naproxeno, sulfonamidas, alopurinol, anticonvulsivos (os seguintes apresentam reação cruzada: carbamazepina, fenitoína, fenobarbital) e penicilinas são os mais comuns.103 A terapia é controversa, com prednisona oral considerada para casos em estágio inicial e os casos em estágio mais tardio sendo tratados do mesmo modo que a necrólise epidérmica tóxica.104,105 Recomenda-se consultar o oftalmologista para prevenção da formação de cicatriz conjuntival.
|
Websites úteis |
|
Beutner Laboratories http://www.beutnerlabs.com Excelente recurso para teste de imunofluorescência com amostras de sangue e pele, para diagnóstico e tratamento de doença bolhosas.
International Pemphigus and Pemphigoid Foundation http://www.pênfigo.org Recurso excelente para médicos e pacientes que lidam com pênfigo e penfigoide bolhoso.
Dystrophic Epidermolysis Bullosa Research Association of America http://www.debra.org Recurso excelente para médicos e pacientes que lidam com epidermólise bolhosa.
Department of Dermatology, Stanford University http://dermatologia.stanford.edu/gsdc/eb_clinic/eb-resources.html Recurso excelente para informação sobre tratamento de feridas de pênfigo e epidermólise bolhosa.
Gluten Intolerance Group http://www.gluten.net Recurso excelente para pacientes com dermatite herpetiforme, que fornece listas de alimentos isentos de glúten. |
Figura 21 Epidermólise bolhosa distrófica recessiva com bolhas, contratura e pseudossindactilia.
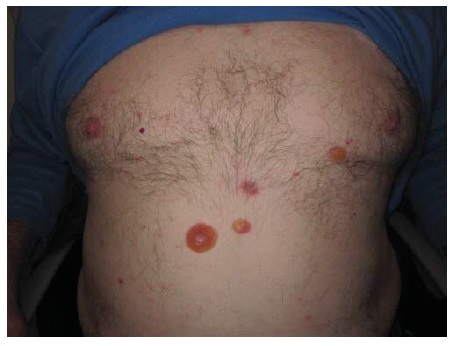
Figura 22 Impetigo bolhoso, com erosões atribuíveis a toxinas esfoliativas que clivam a camada granular.
Figura 23 Síndrome da pele escaldada estafilocócica em adulto com insuficiência renal. Cortesia de Lauren Geller, DM.
Figura 24 Eczema herpético, mostrando vesículas e erosões disseminadas da infecção pelo vírus do herpes simples no contexto de comprometimento da barreira cutânea, neste caso, por dermatite atópica. Cortesia de Caroline Halverstam, DM.
Figura 25 Infecção pelo vírus varicela-zoster disseminada em paciente infectado por HIV.
Figura 26 Eritema multiforme bolhoso, ainda retendo alguns resquícios de aparência alvo-símile.
Por definição, a histopatologia mostra necrose epidérmica de espessura integral e envolvimento de mais de 30% da área de superfície corporal [ver Figura 28]. O sinal de Nikolsky é positivo e a pele se torna bastante dolorida. Os fármacos amplamente responsáveis são similares àqueles que causam a síndrome de Stevens-Johnson. O tratamento envolve a adoção de medidas de suporte, vigilância contra infecção, monitoramento de síndrome de hipersensibilidade farmacológica e anulação do processo inflamatório. Para fins de suporte, deve ser feita a aplicação de gazes Xeroform na pele erodida, a perda inconsciente de líquidos deve ser reposta adequadamente e a alimentação por tubo nasogástrico deve ser instituída quando houver envolvimento oral grave. O envolvimento extensivo pode requerer tratamento na unidade de queimados. Devem ser realizadas culturas de amostras de pele, narinas, urina e sangue, além da obtenção de raiox torácico basal. A síndrome da hipersensibilidade farmacológica envolve hepatite, eosinofilia acentuada, leucocitose leucemoide e hipotireoidismo tardio. A terapia pode incluir IVIg, em doses de 2-3 g/kg igualmente divididas ao alongo de 3 dias, ou ciclosporina, na dosagem de 5 mg/kg por via oral em doses divididas (os autores preferem a administração de IVIg na dose de 2 g/kg).106,107
Figura 27 Síndrome de Stevens-Johnson, com a clássica formação de crostas hemorrágicas nos lábios.
O uso de prednisona é controverso, exceto em casos de síndrome de hipersensibilidade farmacológica, assim como a adição de antibióticos profiláticos. A morte por sepse pode chegar a 35%.103
O edema bolhoso ocorre em áreas de edema dependente, mais frequentemente braços e pernas, muitas vezes no contexto de anasarca ou estase venosa aguda grave, como se observa na insuficiência cardíaca congestiva, insuficiência renal ou cirrose hepática. 108 A localização em áreas de edema e um resultado negativo de imunofluorescência direta ajudam a distinguir esta entidade do PB, DBAL ou da doença bolhosa fármaco-induzida.
Figura 28 Necrólise epidérmica tóxica, com sinal de Nikolsky positivo.
A porfiria cutânea tardia é a mais comum das porfirias e representa um defeito de uroporfirinogênio descarboxilase (UROD).109 Pode ser deflagrada por hepatite C ou HIV por motivos desconhecidos, e está associada à hemocromatose, porque o ferro inibe a UROD.110,111 O acúmulo de uroporfirinas causa formação de bolhas na pele com a exposição à luz azul. Clinicamente, é possível ver bolhas cuja distribuição segue a exposição solar (face e mãos), milhetes na região dorsal das mãos, hipertricose da face e fotossensibilidade [ver Figura 29]. O diagnóstico é estabelecido pela elevação dos níveis de uroporfirina no soro e na urina de 24 horas. A urina exibirá fluorescência rosa ao ser iluminada com lâmpada Wood. O tratamento consiste na administração de 200 g de hidroxicloroquina, duas vezes por semana, ou na realização de flebotomia de 500 mL a cada 1-2 semanas, para níveis de hemoglobina da ordem de 10 mg/dL ou níveis de ferritina abaixo de 25 µg/L.112,113
Figura 29 Porfiria cutânea tardia, mostrando hipertricose e erosões das áreas expostas à luz solar.
Outras condições bolhosas diversas incluem:
•Dermatite de contato alérgica, em especial, à erva venenosa, pode ser amplamente disseminada e associada à formação de bolhas. A inflamação extrema resulta no rápido acúmulo de líquido intersticial intraepidérmico, responsável pelo aparecimento das bolhas. O mesmo mecanismo está por trás da formação de vesículas palmoplantares no eczema de mão e pé (também conhecido como ponfolix).
•Líquen plano, raramente se manifesta com bolhas atribuíveis à inflamação intensa na junção dermoepidérmica.
•Bolhas por atrito ocorrem tipicamente nos pés. O plano de formação das bolhas é a camada espinhosa. 114
•Miliária, resulta do entupimento dos canais sudoríparos. Dependendo do nível de obstrução do canal sudoríparo, a condição é denominada miliária profunda (na junção dermoepidérmica), miliária rubra (na camada espinhosa) e miliária cristalina (no estrato córneo). Esta última pode se manifestar com vesículas minúsculas nos sítios de obstrução (i.e., no dorso de um paciente aquecido e confinado ao leito).
Para obter mais informação sobre doenças vesicobolhosas, consulte os websites listados na barra lateral.
Os autores não mantêm relações comerciais com os fabricantes de produtos ou prestadores de serviços mencionados neste capítulo.
1.Bolognia JL, Jorizzo JL, Rapini RP. Dermatology. 2nd ed. Vol 1. St. Louis (MO): Elsevier; 2008.
2.Beutner Laboratories Web Site. Available at: http://www. beutnerlabs.com (accessed Sept 11, 2011).
3.Michel B, Milner Y, David K. Preservation of tissue-?xedimmunoglobulins in skin biopsies of patients with lupus erythematosus and bullous diseases — preliminary report. J Invest Dermatol 1972;59:449–52.
4.Brenner S, Bialy-Golan A, Anhalt GJ. Recognition of pemphigus antigens in drug-induced pemphigus vulgaris and pemphigus foliaceus. J Am Acad Dermatol 1997;36 (6 Pt 1):919–23.
5.Mehta M, Dacey M, Stephen Foster C. Recurrent conjunctivitis and scleritis secondary to coexistent conjunctival pemiphigus vulgaris and cryptic herpes simplex infection: a case report. Ocul Immunol In?amm2010;18:454–6. [Epub 2010 Sep 2]
6.Cruz PD Jr, Coldiron BM, Sontheimer RD. Concurrent fea- tures of cutaneous lupus erythematosus and pemphigus erythematosus following myasthenia gravis and thymoma. J Am Acad Dermatol 1987;16(2 Pt2):472–80.
7.Rakocevi!-Stojanovi! V, Rakocevi! I, Peri! S, Lavrni! D. Intravenous immunoglobulin therapy in two patients with myasthenia gravis and pemphigus vulgaris. Acta Myol2009;28:101–2.
8.Bystryn JC, Steinman NM. The adjuvant therapy of pemphigus: an update. Arch Dermatol 1996;132:203–12.
9.Ahmed AR, Moy R. Death in pemphigus. J Am Acad Dermatol 1982;7:221–8.
10.Truhan AP, Ahmed AR. Corticosteroids: a review with emphasis on complications of prolonged systemic therapy. Ann Allergy 1989;62:375–91.
11.Herbst A, Bystryn JC. Patterns of remission in pemphigus vulgaris. J Am Acad Dermatol 2000;42:422–7.
12.Mortazavi H, Kiavash K. Correlation of pemphigus vulgaris antibody titers by indirect immuno?uorescence with activity of disease based on pemphigus area and activity score. Acta Med Iran 2008;46:239–44.
13.Strowd LC, Taylor SL, Jorizzo JL, Namzi MR. Therapeutic ladder for pemphigus vulgaris: emphasis on achieving complete remission. J Am Acad Dermatol2011;64:490–4.
14.Amagai M, Ikeda S, Shimizu H, et al. Pemphigus Study Group. A randomized double-blind trial of intravenous immunoglobulin for pemphigus. J Am Acad Dermatol2009;60:595–603.
15.Salopek TG, Logsetty S, Tredget EE. Anti-CD20 chimeric monoclonal antibody (rituximab) for the treatment of recalcitrant, life-threatening pemphigus vulgaris with implications in the pathogenesis of the disorder. J Am Acad Dermatol 2002;47:785–8.
16.Ahmed AR, Spigelman Z, Cavacini LA, Posner MR. Treat- ment of pemphigus vulgaris with rituximab and intrave- nous immune globulin. N Engl J Med 2006;355:1772–9.
17.Bystryn JC, Rudloph JL. IVIg treatment of pemphigus: how it works & how to use it. J Invest Dermatol 2005;125:1093–8.
18.Li N, Zhao M, Hilario-Vargas J, et al. Complete FcRn dependence for intravenous Ig therapy in autoimmune skin blistering diseases. J Clin Invest 2005;115:3440–50.
19.Aoyama Y. What’s new in i.v. immunoglobulin therapy and pemphigus: high-dose i.v. immunoglobulin therapy and its mode of action for treatment of pemphigus. J Dermatol 2010;37:239–45.
20.Mimouni D, Blank M, Payne AS, et al. Ef?cacy of intrave- nous immunoglobulin (IVIG) af?nity-puri?ed antidesmoglein anti-idiotypic antibodies in the treatment of an experimental model of pemphigus vulgaris. Clin Exp Immunol 2010;162:543–9.
21.Arredondo J, Chernyavsky AI, Karaouni A, Grando SA. Novel mechanisms of target cell death and survival and of therapeutic action of IVIg in pemphigus. Am J Pathol2005;167:1531–44.
22.Lolis M, Toosi S, Czernik A, Bystryn JC. Effect of intravenous immunoglobulin with or without cytotoxic drugs on pemphigus intercellular antibodies. J Am Acad Dermatol2011;64:484–9. [Epub 2010 Aug 7]
23.Hartzband PI, Van Herle AJ, Sorger L, Cope D. Assessment of hypothalamic-pituitary-adrenal (HPA) axis dysfunction: comparison of ACTH stimulation, insulin- hypoglycemia and metyrapone. J Endocrinol Invest 1988;11:769–76.
24.Methlyprednisolone. Solu-Medrol [package insert]. New York: P?zer; October 2009.
25.Gürcan HM, Ahmed AR. Analysis of current data on the use of methotrexate in the treatment of pemphigus and pemphigoid. Br J Dermatol 2009;161:723–31. [Epub 2009 Apr 22]
26.Fiorentino DF, Garcia MS, Rehmus W, Kimball AB. A pilot study of etanercept treatment for pemphigus vulgaris. Arch Dermatol 2011;147:117–8.
27.Salomon D, Saurat JH. Oral gold therapy (Aurano?n) in pemphigus vulgaris. Dermatologica 1986;172:310–4.
28.Olszewska M, Kolacinska-Strasz Z, Sulej J, et al. Ef?cacy and safety of cyclophosphamide, azathioprine, and cyclosporine (ciclosporin) as adjuvant drugs in pemphigus vulgaris. Am J Clin Dermatol 2007;8:85–92.
29.Aoyama Y, Nagasawa C, Nagai M, Kitajima Y. Severe pemphigus vulgaris: successful combination therapy of plasmapheresis followed by intravenous high-dose immunoglobulin to prevent rebound increase in pathogenic IgG. Eur J Dermatol 2008;18:557–60. [Epub 2008 Aug 8]
30.Lim LS, Hoeksema LJ, Sherin K. Screening for osteoporo- sis in the adult U.S. population: ACPM position statement on preventive practice; ACPM Prevention Practice Committee. Am J Prev Med 2009;36:366–75.
31.Khellaf M, Godeau B. Pneumocystis pneumonia among patients with systemic diseases. Presse Med2009;38:251–9. [Epub 2008 Dec 4]
32.Stevenson M, Levitt J. Acute zoster in known pemphigus vulgaris and bullous pemphigoid: avoiding the “disease ?are” trap. J Am Acad Dermatol 2011;64:e125–6.
33.Yell JA, Burge SM. Cantharidin-induced acantholysis in Darier’s disease: does acantholysis initiate dyskeratosis? Br J Dermatol 1994;130:2, 148–57.
34.Dumas V, Roujeau JC, Wolkenstein P, et al. The treatment of mild pemphigus vulgaris and pemphigus foliaceus with a topical corticosteroid. Br J Dermatol1999;140:1127–9.
35.Basset N, Guillot B, Michel B, et al. Dapsone as initial treatment in super?cial pemphigus. Report of nine cases. Arch Dermatol 1987;123:783–5.
36.Hymes SR, Jordon RE. Pemphigus foliaceus. Use of antimalarial agents as adjuvant therapy. Arch Dermatol 1992; 128:1462–4.
37.Chung VQ, Moschella SL, Zembowicz A, Liu V. Clinical and pathologic ?ndings of paraneoplastic dermatoses. J Am Acad Dermatol 2006;54:745–62.
38.Robinson ND, Hashimoto T, Amagai M, Chan LS. The new pemphigus variants. J Am Acad Dermatol 1999;40 (5 Pt 1):649–71; quiz 672–3.
39.Peterson JD, Chan LS. Effectiveness and side effects ofanti-CD20 therapy for autoantibody-mediated blistering skin diseases: a comprehensive survey of 71 consecutive patients from the initial use to 2007. Ther Clin Risk Manag 2009;5 (Feb):1–7.
40.Allen KJ, Wolverton SE. The ef?cacy and safety of rituximab in refractory pemphigus: a review of case reports. J Drugs Dermatol 2007;6:883–9.
41.Hu Z, Bonifas JM, Beech J, et al. Mutations in ATP2C1, encoding a calcium pump, cause Hailey-Hailey disease. Nat Genet 2000;24:61–5.
42.Lebwohl M, Heymann WR, Berth-Jones J, Coulson I. Treatment of skin disease. 3rd ed. London: Mosby; 2010.
43.Bastuji-Garin S, Joly P, Picard-Dahan C, et al. Drugs associ- ated with bullous pemphigoid. A case-controlstudy. Arch Dermatol 1996;132:272–6.
44.Lee JJ, Downham TF 2nd. Furosemide-induced bullous pemphigoid: case report and review of literature. J Drugs Dermatol 2006;5:562–4.
45.Mallet L, Cooper JW, Thomas J. Bullous pemphigoid associated with captopril. DICP 1989;23:63.
46.Popadic S, Skiljevic D, Medenica L. Bullous pemphigoid induced by penicillamine in a patient with Wilson disease. Am J Clin Dermatol 2009;10:36–8.
47.Piletta P, Rieckhoff L, Saurat JH. Triggering of bullous pemphigoid by iodine. Br J Dermatol 1994;131:145–7.
48.Kimyai-Asadi A, Usman A, Nousari HC. Cipro?oxacin- induced bullous pemphigoid. J Am Acad Dermatol 2000; 42(5 Pt 1):847.
49.Fellner MJ. Drug-induced bullous pemphigoid. Clin Dermatol 1993;11:515–20.
50.Engineer L, Bhol K, Ahmed AR. Pemphigoid gestationis: a review. Am J Obstet Gynecol 2000;183:483–91.
51.Leuci S, Gürcan HM, Ahmed AR. Serological studies in bullous pemphigoid: a literature review of antibody titers at presentation and in clinical remission. Acta Derm Venereol2010;90:115–21.
52.Cooper SM, Wojnarowska F. Treatments of choice for bullous pemphigoid. Skin Ther Lett 2002;7:4–6.
53.Fivenson DP, Breneman DL, Rosen GB, et al. Nicotinamide and tetracycline therapy of bullous pemphigoid. Arch Dermatol 1994;130:753–8.
54.Schmidt E, Hunzelmann N, Zillikens D, et al. Rituximab in refractory autoimmune bullous diseases. Clin Exp Dermatol 2006;31:503–8.
55.Sami N, Bhol KC, Ahmed AR. Treatment of oral pemphigoid with intravenous immunoglobulin as monotherapy.Long-term follow-up: in?uence of treatment on antibody titres to human alpha6 integrin. Clin Exp Immunol 2002;129:533–40.
56.Segura S, Iranzo P, Martínez-de Pablo I, et al. High-doseintravenous immunoglobulins for the treatment of autoimmune mucocutaneous blistering diseases: evaluation of its use in 19 cases. J Am Acad Dermatol 2007;56:960–7.[Epub 2007 Mar 21]
57.Mazzi G, Raineri A, Zanolli FA, et al. Plasmapheresis therapy in pemphigus vulgaris and bullous pemphigoid. Transfus Apher Sci 2003;28:13–8.
58.Joly P, Roujeau JC, Benichou J, et al. A comparison of oral and topical corticosteroids in patients with bullous pemphigoid. Bullous Diseases French Study Group. N Engl J Med 2002;346:321–7.
59.Patton T, Korman N. Role of methotrexate in the treatment of bullous pemphigoid in the elderly. Drugs Aging 2008;25:623–9.
60.Knudson RM, Kalaaji AN, Bruce AJ. The management of mucous membrane pemphigoid and pemphigus. Dermatol Ther 2010;23:268–80.
61.Foster CS, Chang PY, Ahmed AR. Combination of ritux- imab and intravenous immunoglobulin for recalcitrant ocular cicatricial pemphigoid: a preliminary report. Ophthalmology 2010;117:861–9. [Epub 2010 Jan 4]
62.Sacher C, Hunzelmann N. Cicatricial pemphigoid (mucous membrane pemphigoid): current and emerging therapeutic approaches. Am J Clin Dermatol 2005;6:93–103.
63.Hashimoto Y, Suga Y, Yoshiike T, et al. A case of antiepiligrin cicatricial pemphigoid successfully treated with plasmapheresis. Dermatology 2000;201:58–60.
64.Chang JH, McCluskey PJ. Ocular cicatricial pemphigoid: manifestations and management. Cur Allergy Asthma Rep2005;5:333–8.
65.Meyer-ter-Vehn T, Schmidt E, Zillikens D, Geerlings G. Mucous membrane pemphigoid with ocular involvement. Part II: therapy. Ophthalmologe 2008;105:405–19.
66.Stein JA, Mikkilineni R. Epidermolysis bullosa acquisita. Dermatol Online J 2007;13:15.
67.Raab B, Fretzin DF, Bronson DM, et al. Epidermolysis bullosa acquisita and in?ammatory bowel disease. JAMA1983;250:1746–8.
68.Caldwell JB, Yancey KB, Engler RJ, et al. Epidermolysis bullosa acquisita: ef?cacy of high-dose intravenous immunoglobulins. J Am Acad Dermatol 1994;31:827–8.
69.Ko?er H, Wambacher-Gasser B, Topar G, et al. Intravenous immunoglobulin treatment in therapy-resistant epi- dermolysis bullosa acquisita. J Am Acad Dermatol 1997;36: 331–5.
70.Gürcan HM, Ahmed AR. Current concepts in the treatment of epidermolysis bullosa acquisita. Expert Opin Pharmacother 2011;12:1259–68. [Epub 2011 Jan 22]
71.Hughes AP, Callen JP. Epidermolysis bullosa acquisita responsive to dapsone therapy. J Cutan Med Surg 2001;5: 397–9.
72.Junkins-Hopkins JM. Dermatitis herpetiformis: pearls and pitfalls in diagnosis and management. J Am Acad Dermatol 2010;63:526–8.
73.Marakli SS, Uzun S, Ozbek S, Tuncer I. Dermatitis herpetiformis in a patient receiving in?iximab for ankylosing spondylitis. Eur J Dermatol 2008;18:88–9.
74.Hassan S, Dalle S, Descloux E, et al. Dermatitis herpetifor- mis associated with progesterone contraception. Ann Dermatol Venereol 2007;134(4 Pt1):385–6.
75.Freeman HJ. T cell lymphoma of the thyroid gland in celiac disease. Can J Gastroenterol 2000;14:635–6.
76.Preisz K, Sárdy M, Horváth A, Kárpáti S. Immunoglobu- lin, complement and epidermal transglutaminase deposition in the cutaneous vessels in dermatitis herpetiformis. J Eur Acad Dermatol Venereol 2005;19:74–9.
77.Kumar V, Beutner EH, Chorzelski TP. Antiendomysialantibody—useful serological indicator of dermatitis herpetiformis. Arch Dermatol Res 1987;279:454–8.
78.Accetta P, Kumar V, Beutner EH, et al. Anti-endomysialantibodies. A serologic marker of dermatitis herpetiformis. Arch Dermatol 1986;122:459–62.
79.Marietta EV, Camilleri MJ, Castro LA, et al. Transglutaminase autoantibodies in dermatitis herpetiformis and celiac sprue. J Invest Dermatol 2008;128:332–5.
80.Green PH, Cellier C. Celiac disease. N Engl J Med. 2007;357:1731–43.
81.Ahrens EM, Meckler RJ, Callen JP. Dapsone-inducedperipheral neuropathy. Int J Dermatol 1986;25:314–6.
82.Willsteed E, Lee M, Wong LC, Cooper A. Sulfasalazine and dermatitis herpetiformis. Australas J Dermatol 2005;24: 101–3.
83.Santos-Juanes J, Coto Hernández R, Trapiella L, et al. Amoxicillin-associated linear IgA bullous dermatosis. J Eur Acad Dermatol Venereol 2007;21:992–3.
84.Yawalkar N, Reimers A, Hari Y, et al. Drug-induced linear IgA bullous dermatosis associated with ceftriaxoneandmetronidazole-speci?c T cells. Dermatology 1999;199:25–30.
85.Friedman IS, Rudikoff D, Phelps RG, Sapadin AN.Captopril-triggered linear IgA bullous dermatosis. Int J Dermatol 1998;37:608–12.
86.Khan I, Hughes R, Curran S, Marren P. Drug-associatedlinear IgA disease mimicking toxic epidermal necrolysis. Clin Exp Dermatol 2009;34:715–7.
87.Khar? M, Khaled A, Karaa A, et al. Linear IgA bullous dermatosis: the more frequent bullous dermatosis of children. Dermatol Online J 2010;16:2.
88.Wolf R, Matz H, Orion E, et al. Dapsone. Dermatol Online J 2002;8(1):2.
89.Venugopal SS, Murrell DF. Treatment of skin cancers in epidermolysis bullosa. Dermtol Clin 2010;28:283–7.
90.Wright JT, Hall KI, Deaton TG, Fine JD. Structural and compositional alteration of tooth enamel in hereditary epidermolysis bullosa. Connect Tissue Res 1996;34:271–9.
91.Fine JD, Eady RA, Bauer EA, et al. The classi?cation of inherited epidermolysis bullosa (EB): report of the Third International Consensus Meeting on Diagnosis and Classi?cation of EB. J Am Acad Dermatol2008;58:931–50.
92.Fine J-D, Hintner H. Life with epidermolysis bullosa (EB): etiology, diagnosis, multidisciplinary care and therapy. New York: Springer-Verlag/Wien; 2008.
93.Mellerio JE. Infection and colonization in epidermolysis bullosa. Dermal Clin 2010;28:267–9.
94.Lapioli-Zafelt A, Morris EJ. Skin and wound care manage- ment for a child with epidermolysis bullosa. J Wound Ostomy Continence Nurs 1998;25:314–6.
95.Mavilio F, Pellegrini G, Ferrari S, et al. Correction of junctional epidermolysis bullosa by transplantation of genetically modi?ed epidermal stem cells. Nat Med 2006;12:1397–402. [Epub 2006 Nov 19]
96.Woodley DT, Remington J, Huang Y, et al. Intravenously injected human ?broblasts home to skin wounds, deliver type VII collagen, and promote wound healing. Mol Ther2007;15:628–35. [Epub 2007 Jan 23]
97.Remington J, Wang X, Hou Y. Injection of recombinant human type VII collagen corrects the disease phenotype in a murine model of dystrophic epidermolysis bullosa. Mol Ther 2009;17:26–33.
98.Tolar J, Ishida-Yamamoto A, Riddle M. Amelioration of epidermolysis bullosa by transfer of wild-type bone marrow cells. Blood 2009;29:1167–74.
99.Wagner JE, Ishida-Yamamoto A, McGrath JA, et al. Bone marrow transplantation for recessive dystrophic epidermolysis bullosa. N Engl J Med 2010;363:629–39.
100.Martire B, Foti C, Cassano N, et al. Persistent B-cell lymphopenia, multiorgan disease, and erythema multiforme caused by Mycoplasma pneumoniae infection. Pediatr Dermatol 2005;22:558–60.
101.Grosber M, Alexandre M, Poszepczynska-Guigne E, et al. Recurrent erythema multiforme in association with recurrent Mycoplasma pneumoniae infections. J Am Acad Dermatol 2007;56(5 Suppl):S118–9.
102.Sanchis JM, Began JV, Gavalda C, et al. Erythema multi- forme: diagnosis, clinical manifestations and treatment in a retrospective study of 22 patients. J Oral Pathol Med2010;39:747–52.
103.Harr T, French LE. Toxic epidermal necrolysis andStevens-Johnson syndrome. Orphanet J Rare Dis 2010;5: 39.
104.Halebian PH, Corder VJ, Madden MR, et al. Improved burn center survival of patients with toxic epidermal necrolysis managed without corticosteroids. Ann Surg1986;204:503–12.
105.Schneck J, Fagot JP, Sekula P, et al. Effects of treatments on the mortality of Stevens-Johnsonsyndrome and toxic epidermal necrolysis: a retrospective study on patients included in the prospective EuroSCAR Study. J Am Acad Dermatol 2008;58:33–40.
106.Molgó M, Carreño N, Hoyos-Bachiloglu R, et al. Use of intravenous immunoglobulin for the treatment of toxic epidermal necrolysis and Stevens-Johnson/toxic epidermal necrolysis overlap syndrome. Review of 15 cases. Rev Med Chil 2009;137:383–9.
107.Rajaratnam R, Mann C, Balasubramaniam P, et al. Toxic epidermal necrolysis: retrospective analysis of 21 consecu- tive cases managed at a tertiary centre. Clin Exp Dermatol 2010;35:853–62.
108.Rapini RP, Bolognia JL, Jorizzo JL. Dermatology. St. Louis: Mosby; 2007.
109.Kappas A, Sassa S, Galbraith RA, et al. The porphyrias. In: Scriver CR, Beaudet AL, Sly WS, et al, editors. The meta- bolic basis of inherited disease. New York:McGraw-Hill; 1995. p. 2103–59.
110.Lambrecht RW, Bonkovsky HL. Hemochromatosis and porphyria. Semin Gastrointest Dis 2002;13:109–19.
111.Bonkovsky HL, Poh-Fitzpatrick M, Pimstone N, et al. Porphyria cutantea tarda, hepatitis C and HFE gene mutations in North America. Hepatology 1998;97:972–81.
112.Malkinson FD, Levitt L. Hydroxychloroquine treatment of porphyria cutanea tarda. Arch Dermatol 1980;116:1147– 50.
113.Poh-Fitzpatrick MB, Honig PJ, Kim HC, Sassa S. Child-
hood-onset familial porphyria cutanea tarda: effects of therapeutic phlebotomy. J Am Acad Dermatol 1992;27 (5 Pt 2):896–900.
114.Elder DE, Elenitsas R, Bernett LJ. Lever’s histopathology of the skin. 10th ed. Philadelphia: Lippincott Williams & Wilkins; 2008.
115.Schiavo AL, Puca RV, Ruocco V, Ruocco E. Adjuvant drugs in autoimmune bullous diseases, ef?cacy versus safety: facts and controversies. Clin Dermatol 2010;28:337–43.
116.Rituxan [package insert]. Biogen Idec Inc. and Genentech USA, Inc; February 2010.
117.Kasperkiewicz M, Schmidt E. Current treatment of auto- immune blistering diseases. Curr Drug Discov Technol2009;6:270–80.
118.Gammagard [package insert]. Westlake Village (CA): Baxter Healthcare Corporation; December 2009.
119.Ahmed AR. Intravenous immunoglobulin therapy in the treatment of patients with pemphigus vulgaris unresponsive to conventional immunosuppressive treatment. J Am Acad Dermatol 2001;45:679–90.
120.Prednisone [package insert]. Corona (CA): Wastson Pharma, Inc.; April 2008.
121.Werth VP. Treatment of pemphigus vulgaris with brief,high-dose intravenous glucocorticoids. Arch Dermatol 1996;132:1435.
122.Cellcept [package insert]. San Francisco (CA): Genentech USA, Inc.; February 2010.
123.Azathioprine [package insert]. Morrisville (NC): Salix Pharmaceuticals, Inc.; October 2005.
124.Chams-Davatchi C, Esmaili N, Daneshpazhooh M, et al. Randomized controlled open-label trial of four treatment regimens for pemphigus vulgaris. J Am Acad Dermatol2007;57:622–8.
125.Methotrexate [package insert]. Lake Forest (IL): Hospira, Inc.; 2008.
126.Neoral [package insert]. East Hanover (NJ): Novartis Pharmaceuticals Corporation; October 2009.
127.McCarty M. How clinically relevant is dapsone-relatedperipheral neuropathy? An overview of available data with emphasis on clinical recognition. J Clin Aesthet Dermatol2010;3:19–21.
128.Dapsone [package insert]. Princeton (NJ): Jacobus Pharma- ceutical Company, Inc.; August 2009.
129.Myochrysine [package insert]. Lake Forest (IL): Akorn, Inc.; September 2008.
130.Wolverton SE. Comprehensive dermatologic therapy. New York: W.B. Saunders; 2001.
131.Cytoxan [package insert]. Princeton (NJ): Bristol-MyersSquibb Company; September 2005.
132.Turner MS, Sutton D, Sauder DN. The use of plasma- pheresis and immunosuppression in the treatment of pemphigus vulgaris. J Am Acad Dermatol 2000;43:1058– 64.
133.Harman KE, Albert S, Black MM. Guidelines for the management of pemphigus vulgaris. Br J Dermatol 2003;149:926–37.
134.Enbrel [package insert]. Thousand Oaks (CA): Immunex Corporation; September 2011.
135.Tetracycline [package insert]. Sellersville (PA): Teva Pharmaceuticals USA; March 2011.
Agradecimentos
Figuras 1, 2a, 3a, 4-6, 8-11, 13-17, 19-22 e 25-29 – direitos de copyright por Jacob Levitt, DM. Direitos de publicação e distribuição por Decker Intellectual Properties.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.