(Carregando Índice)... (Carregando Índice)... |
Autor:
Daniel Soares Freire
Endocrinologista pelo Hospital das Clínicas da Faculdade de Medicina da USP;
Médico Colaborador do Serviço de Endocrinologia do Hospital das Clínicas da Faculdade de Medicina da USP
Última revisão: 29/09/2008
Comentários de assinantes: 0
Nódulos tireoidianos são extremamente comuns na população geral. A prevalência de nódulos palpáveis é de aproximadamente 4%-7%; contudo, se considerarmos os nódulos subclínicos, diagnosticados por ultra-sonografia cervical, essa prevalência ultrapassa 50% na população feminina com mais de 50 anos de idade.
Apenas 5%-10% dos nódulos representam risco de malignidade para o paciente. Assim, o principal desafio para o clínico é diferenciar um nódulo com potencial maligno de um nódulo tireoidiano inócuo. Para isto, utilizam-se dados de história e exame físico, características ultra-sonográficas e de Doppler, marcadores séricos e a citologia do nódulo, obtida por punção aspirativa com agulha fina (PAAF).
Com esses dados, é possível identificar com razoável sensibilidade e especificidade os pacientes portadores de carcinoma de tireóide, que podem ser carcinomas bem diferenciados da tireóide (papilífero ou folicular), carcinoma medular da tireóide (espontâneo ou familiar) e carcinoma anaplásico.
Histologicamente, os Nódulos tireoidianos são classificados como descrito na tabela 1.
Tabela 1: Classificação histológica dos tumores tireoidianos
|
Nódulos tireoidianos | |||
|
Tumores derivados de células foliculares |
Benignos |
Adenoma folicular | |
|
Malignos |
Diferenciados |
||
|
Pouco diferenciados |
Carcinoma insular | ||
|
Indiferenciados |
Carcinoma anaplásico | ||
|
Tumores derivados das células parafoliculares (C) |
carcinoma medular | ||
|
Outros tumores |
Linfomas Sarcomas | ||
Trata-se de um tumor benigno encapsulado formado por células foliculares. É o tumor de tireóide mais comum, encontrado em 4%-20% das glândulas em autópsias.
Uma variante é o adenoma de células de Hürthle, ou de células oxifílicas, composto por mais de 75% de células grandes com citoplasma granular eosinofílico.
Alguns adenomas foliculares adquirem autonomia após sofrerem mutações no receptor de TSH ou na subunidade alfa da proteína Gs, tornando-se hiperfuncionantes. O hipertireoidismo resultante pode ser subclínico, identificado apenas por supressão do TSH, ou determinar quadro de tireotoxicose franca, sem oftalmopatia e com presença de nódulo “quente” à cintilografia tireoidiana.
É a neoplasia maligna de tireóide mais comum, correspondendo a mais de 90% dos carcinomas bem diferenciados em áreas sem deficiência de iodo.
A Organização Mundial de Saúde (OMS) define como microcarcinoma papilífero os tumores com tamanho igual ou menor a 1 cm. Para essas lesões, a prevalência chega a ser cinco vezes maior do que a observada para o Carcinoma papilífero maior que 1 cm.
Caracteriza-se pelo arranjo das células foliculares tumorais em papilas e por características morfológicas típicas, como aumento do tamanho nuclear, presença de fissuras nucleares (aspecto em “grão de café”), aspecto em “vidro-fosco” dos núcleos (por presença de cromatina frouxa) e inclusões nucleares decorrentes de invaginação do citoplasma para o núcleo. Corpos psamomatosos são agregações concêntricas de material calcificado visíveis no interior das papilas e no estroma tumoral.
Como as alterações intra-celulares presentes no Carcinoma papilífero são muito típicas, sua identificação em material obtido a partir de PAAF permite o diagnóstico pré-operatório do Carcinoma papilífero.
Além da variante clássica do Carcinoma papilífero, há diversos subtipos definidos histologicamente: variante folicular, variante esclerosante difusa, variante de células altas e variante de células colunares. Evidências indicam que essas variantes (em especial as duas últimas) apresentam evolução clínica pior que a variante clássica.
O Carcinoma papilífero pode incidir em qualquer faixa etária, mas ocorre principalmente entre 30 e 50 anos de idade. As mulheres são mais acometidas do que os homens.
O quadro clínico é de nódulo cervical duro e indolor na topografia da tireóide. Rouquidão sugere acometimento do nervo laríngeo recorrente por invasão extratireoidiana. Muitos carcinomas papilíferos são diagnosticados por ultra-sonografia cervical solicitada para avaliação de doença extratireoidiana (“incidentalomas de tireóide”).
Os microcarcinomas papilíferos são assintomáticos na maioria absoluta dos casos, embora haja relatos de lesões menores que 1 cm associadas a doença metastática.
Doença multifocal ou acometimento bilateral da tireóide são achados freqüentes. Invasão extratireoidiana está presente em aproximadamente 15% dos casos, e acometimento linfonodal em 30%. Contudo, em pacientes jovens (abaixo de 17 anos), a incidência de metástases ganglionares ao diagnóstico chega a 90%. Cerca de 1% dos pacientes apresentam metástases à distância ao diagnóstico.
Trata-se de uma doença rara em países onde não há deficiência iodo, correspondendo a cerca de 5% dos carcinomas bem diferenciados da tireóide. Contudo, a incidência aumenta em regiões com deficiência de iodo.
É um tumor bem diferenciado do epitélio folicular, que não apresenta as características típicas do Carcinoma papilífero. Nesse grupo também não estão incluídos a variante folicular do Carcinoma papilífero e os carcinomas pouco diferenciados (carcinoma insular) e indiferenciados (carcinoma anaplásico) da tireóide.
Para o diagnóstico histológico do Carcinoma folicular, o patologista deve demonstrar presença de invasão de cápsula, vaso ou extensão extratireoidiana da neoplasia; é por este motivo que a análise citológica não permite um diagnóstico pré-operatório do Carcinoma folicular como o faz para o Carcinoma papilífero. Assim, o reconhecimento de um “padrão folicular” na citologia aspirativa suscita três possibilidades diagnósticas: o Carcinoma folicular, o adenoma folicular e a hiperplasia folicular.
O carcinoma de células de Hürthle, ou de células oxifílicas, é reconhecido pela OMS como uma variante histológica do Carcinoma folicular.
A classificação histológica mais importante em termos de prognóstico divide o Carcinoma folicular em dois grupos:
Carcinoma folicular minimamente invasivo: observa-se invasão microscópica da cápsula ou de vasos em seu redor, mas não há invasão grosseira. Pode ser difícil diferenciar essa lesão de um adenoma folicular;
Carcinoma folicular extensamente invasivo: o tumor é apenas parcialmente encapsulado e a invasão é óbvia, muitas vezes visível macroscopicamente. Obrigatoriamente, deve-se observar um arranjo folicular para definir esse tipo de carcinoma, e quando este não está presente, o tumor pode ser classificado como um carcinoma pouco diferenciado (insular).
O Carcinoma folicular acomete indivíduos mais idosos que o papilífero, mas ainda com maior predileção pelo sexo feminino (aproximadamente 2:1).
Assim como no Carcinoma papilífero, a maioria dos pacientes se apresenta com nódulo cervical indolor. Raramente há envolvimento ganglionar cervical no Carcinoma folicular típico. A variante de células de Hürthle se apresenta com metástases ganglionares em 6% dos casos. Metástases à distância acometem principalmente os pulmões e os ossos, e podem ser observadas ao diagnóstico em 17% dos casos.
Trata-se de um tumor maligno derivado do epitélio folicular com características morfológicas e comportamento biológico intermediários entre o carcinoma bem diferenciado da tireóide o carcinoma indiferenciado.
Histologicamente, o carcinoma insular se caracteriza por arranjo das células em ninhos ou trabéculas. Os nódulos costumam ser grandes ao diagnóstico (> 5 cm) e a invasão local é nítida.
A idade média de acometimento é de 55 anos, e a incidência da doença atinge mais mulheres que homens (na proporção 2:1). O carcinoma pouco diferenciado é invasivo e muitas vezes letal. Metástases locais e à distância (pulmões, ossos, cérebro) são freqüentemente encontradas.
Constituem aproximadamente 1%-2% dos carcinomas de tireóide. Acredita-se que os carcinomas anaplásicos surjam a partir de carcinomas bem diferenciados, que sofrem mutações como a do gene p53.
Histologicamente caracterizam-se por células atípicas, que podem ser fusiformes, escamosas ou multinucleadas, com numerosas mitoses em diversos padrões de organização.
O pico de incidência situa-se entre 50-60 anos, e observa-se discreta predominância do sexo feminino (na proporção de 1,5:1). Cerca de um terço dos pacientes apresenta bócio de longa duração. O sintoma mais comum é de uma massa cervical com crescimento rápido, determinando sintomas compressivos (disfagia, dispnéia) e rouquidão por comprometimento laríngeo. Um terço dos pacientes apresenta dor cervical. A maior parte dos pacientes apresenta ao diagnóstico acometimento ganglionar cervical e invasão de estruturas adjacentes. Até 50% pode ter metástase à distância ao diagnóstico (pulmões, ossos, cérebro e fígado).
O carcinoma medular da tireóide (CMT), originário das células parafoliculares (ou células C), representa menos que 10% dos tumores malignos da tireóide. O CMT é esporádico em 75% dos casos, mas pode fazer parte de síndromes genéticas, como a síndromes das neoplasias endócrinas múltiplas dos tipos 2A e 2B, e a síndrome de carcinoma medular de tireóide familiar isolado. A mutação presente nos casos de CMT familiar ocorre no proto-oncogene RET, localizado no cromossomo 10q.
Esse tumor apresenta uma evolução mais agressiva que a maioria dos carcinomas bem diferenciados da tireóide. Os pacientes costumam se apresentar com nódulos palpáveis, muitas vezes com invasão de estruturas adjacentes (determinando rouquidão, disfagia e dispnéia) e de linfonodos cervicais. Metástases à distância costumam acometer pulmões, fígado e ossos.
Os níveis elevados de calcitonina são o marcador específico dessa doença. A calcitonina, quando muito alta, pode causar sintomas de diarréia e flushing. Raramente os carcinomas medulares de tireóide secretam ACTH, causando síndrome de Cushing ACTH-dependente. O antígeno cárcino-embriônico (CEA) é um marcador de extensão da doença, e, embora seja menos sensível que a calcitonina, pode funcionar como um marcador da massa tumoral.
O prognóstico do CMT é ruim quando o paciente apresenta metástases à distância ou quando a cirurgia radical não é curativa. A sobrevida em 10 anos é de 65%.
O linfoma primário de tireóide é um tipo de linfoma não-Hodgkin que se origina do tecido linfático da tireóide. Apesar de ser uma doença rara em comparação com as outras etiologias dos Nódulos tireoidianos, o risco de linfoma de tireóide é cerca de 70 vezes maior em portadores de tireoidite de Hashimoto em comparação com a população geral. O mecanismo proposto para explicar essa associação é a estimulação antigênica crônica dos linfócitos pela doença auto-imune; em um determinado momento, o tecido linfóide ativado sofre uma mutação e dá origem ao linfoma.
O linfoma primário de tireóide corresponde a 3% de todos os linfomas não-Hodgkin e a 2%-5% dos nódulos de tireóide. Seu reconhecimento é importante, pois o tratamento não envolve tireoidectomia como nos casos dos outros tumores malignos da tireóide.
O quadro clínico é de um tumor cervical de crescimento rápido, geralmente associado a linfadenopatia cervical, em paciente com mais de 60 anos de idade. Ocasionalmente, o tumor pode ter um crescimento mais indolente. O diagnóstico é feito com PAAF, mas uma biópsia incisional pode ser necessária para determinar o tipo histológico.
Os sarcomas primários da tireóide são tumores raros, derivados do estroma ou do tecido vascular da glândula. Sarcomas verdadeiros devem ser diferenciados dos carcinomas anaplásicos, que podem ter aspecto sarcomatoso na histologia.
O tratamento é cirúrgico associado ou não à radioterapia. A quimioterapia não costuma ter boa resposta.
Tabela 2: Características mais importantes dos tumores de tireóide
|
Adenoma folicular |
Benigno tumor de tireóide mais comum Quadro clínico: de hipertiroidismo subclínico a tireotoxicose |
|
Carcinoma papilífero |
Neoplasia maligna de tireóide mais comum (90% dos carcinomas bem diferenciados) Nódulo cervical duro e indolor que pode causar rouquidão Principalmente em mulheres entre 30 e 50 anos 15% dos casos com invasão local, 30% com acometimento de linfonodos, 1% com metástases à distância |
|
Carcinoma folicular |
5% dos carcinomas bem diferenciados da tireóide A maioria dos pacientes se apresenta com nódulo cervical indolor Raramente há envolvimento ganglionar cervical 17% com metástases ao diagnóstico (pulmões e ossos) |
|
Carcinoma insular |
Tumor maligno que pode ser letal Nódulos > 5 cm com invasão local Acomete mais mulheres com idade média de 55 anos Invasivo: metástases para pulmões, ossos, cérebro |
|
Carcinoma anaplásico |
1% a 2% dos carcinomas de tireóide Mais em indivíduos de 50 a 60 anos Massa cervical de rápido crescimento Gera dispnéia, disfagia e rouquidão Um terço dos pacientes apresenta bócio 50% com metástases no diagnóstico (pulmões, ossos, cérebro e fígado) |
|
carcinoma medular |
Menos que 10% dos tumores malignos da tireóide Associado com síndromes das neoplasias endócrinas múltiplas dos tipos 2A e 2B, e com a síndrome de carcinoma medular de tireóide familiar isolado Nódulos palpáveis com invasão de estruturas adjacentes e de linfonodos cervicais Causa rouquidão, disfagia e dispnéia Metástases à distância: pulmões, fígado e ossos Os níveis elevados de calcitonina são o marcador específico |
|
Linfoma |
linfoma não-Hodgkin O risco de linfoma de tireóide é cerca de 70 vezes maior em portadores de tireoidite de Hashimoto Tumor cervical de crescimento rápido, associado à linfadenopatia cervical Pacientes com mais de 60 anos de idade |
|
Sarcoma |
Sarcomas primários da tireóide são tumores raros Devem ser diferenciados dos carcinomas anaplásicos |
Dados semiológicos podem auxiliar consideravelmente na avaliação de risco de um nódulo tireoidiano (tabela 3). Contudo, esses achados de alto valor diagnóstico têm baixa sensibilidade, uma vez que a maior parte dos carcinomas de tireóide são nódulos assintomáticos.
Tabela 3: Achados clínicos sugestivos de carcinoma de tireóide em paciente eutireóideo com nódulo solitário (de acordo com o grau de suspeita)
|
Alto grau de suspeita |
História familiar de neoplasia endócrina múltipla História familiar de carcinoma medular familiar Crescimento rápido do tumor Nódulo endurecido e aderido aos tecidos adjacentes Paralisia de cordas vocais Adenomegalia cervical |
|
Moderado grau de suspeita |
Idade < 20 anos ou > 70 anos Sexo masculino História prévia de irradiação cervical Nódulo > 4 cm ou com liquefação central |
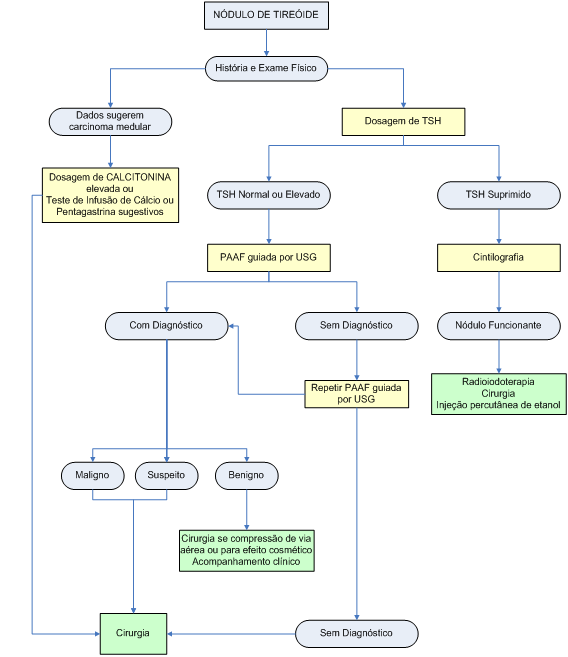
A dosagem de TSH está indicada em todo paciente portador de nódulo tireoidiano. O achado de TSH suprimido deve ser complementado com dosagem de T3 e T4 livres, para confirmar a tireotoxicose. Aproximadamente 10% dos nódulos palpáveis apresentam autonomia suficiente para reduzir os níveis de TSH, o que é um achado fortemente sugestivo de benignidade. Assim, para nódulos autônomos demonstrados no mapeamento tireoidiano (nódulos “quentes”), não há necessidade de PAAF.
Se as concentrações de TSH forem elevadas, os títulos de anticorpo antitireoperoxidase (anti-TPO) devem ser aferidos, para confirmar o diagnóstico de tireoidite de Hashimoto. O risco de linfoma de tireóide é 67 vezes maior em pacientes com tireoidite de Hashimoto, mas ainda assim, o linfoma é um diagnóstico incomum.
A dosagem de calcitonina em todos os Nódulos tireoidianos é um assunto controverso. De fato, o diagnóstico precoce e o tratamento apropriado (tireoidectomia total com esvaziamento cervical) reduzem substancialmente o risco de recidiva e disseminação, e a dosagem sérica de calcitonina tem sensibilidade superior ao exame citológico convencional em identificar o carcinoma medular de tireóide. Contudo, a raridade dessa etiologia (1 em cada 250 nódulos), a baixa sensibilidade da maioria dos ensaios de calcitonina disponíveis comercialmente e o alto custo da investigação tornam o rastreamento populacional impraticável.
Para pacientes de risco (parentes de pacientes com neoplasia endócrina múltipla do tipo 2 ou carcinoma medular familiar) justificam-se dosagem de calcitonina basal e teste de infusão de cálcio ou pentagastrina para confirmar o diagnóstico.
O teste de infusão de cálcio se baseia no fato de que as células C liberam calcitonina diante de uma elevação da calcemia. Nesse teste, a calcitonina é dosada antes e depois (2, 5 e 10 minutos) da injeção de 2 mg de íon cálcio por kg de peso do paciente. Elevação da calcitonina para valores acima de 100 pg/ml sugerem carcinoma medular de tireóide.
O teste da pentagastrina é feito com a injeção IV de 0,5 µg/kg de peso e coleta de calcitonina nos tempos 0, 2, 5 e 10 minutos. Elevação da calcitonina acima de 100 pg/ml sugere CMT.
A ultra-sonografia tireoidiana de última geração, com transdutores de alta freqüência (maior que 10 MHz), permite estudo anatômico detalhado dos nódulos e identificação de características que parecem estar associadas com maior risco de malignidade. Presença de microcalcificações, bordas irregulares e hipoecogenicidade são achados ultra-sonográficos sugestivos de neoplasia maligna. Porém, a especificidade dessas características é relativamente baixa (66%).
A adição do Doppler aumentou a acurácia diagnóstica: ausência de vascularização ou vascularização exclusivamente periférica são dados sugestivos de benignidade, ao passo que vascularização predominante ou exclusivamente central sugere doença maligna.
Outro emprego da ultra-sonografia é a orientação para a PAAF, facilitando a coleta de material e reduzindo a necessidade de novas punções por coleta inadequada de material.
Atualmente, considera-se a PAAF o exame mais confiável e acurado no diagnóstico de doença nodular tireoidiana. Trata-se de um exame seguro, pouco invasivo, de baixo custo e fácil realização. A utilização desse método em ampla escala reduziu o número de tireoidectomias em 50%, ao mesmo tempo que dobrou a prevalência de câncer nos casos operados. Não há contra-indicações formais à PAAF (inclusive uso de antiagregantes plaquetários e anticoagulantes), e não há risco de disseminação neoplásica pelo trajeto da punção. O principal efeito colateral é desconforto local, que dura menos que 24 horas e pode ser aliviado com compressas frias ou analgésicos simples.
A PAAF deve ser realizada em todo nódulo com mais de 1 cm de diâmetro ou com características ultra-sonográficas sugestivas de malignidade ou história clínica de risco, exceto quando há suspeita de nódulo funcionante pelo mapeamento de tireóide.
Usualmente, os patologistas classificam os resultados em uma de quatro possibilidades: benigno, maligno, suspeito ou inconclusivo. O diagnóstico diferencial entre as neoplasias foliculares (adenoma e carcinoma) só é possível com demonstração de invasão de cápsula ou invasão vascular, ou seja, pelo exame histológico. Assim, quando se fala de uma citologia com “padrão folicular”, o clínico deve ter em mente que tal diagnóstico não é conclusivo.
Imunocitoquímica pode aumentar a especificidade da citologia. A calcitonina é específica do carcinoma medular de tireóide, derivado das células parafoliculares C. Outros marcadores como a galectina-3 e a ciclooxigenase-2 estão mais expressos em neoplasias tireoidianas malignas que benignas, mas a sensibilidade dessas técnicas não é suficientemente grande para justificar seu emprego de rotina.
Apesar do mapeamento de tireóide com isótopos tradicionais como 131I, 123I e 99mTc ser útil para avaliação funcional de bócios tóxicos à procura de áreas hipercaptantes, o achado de uma área hipocaptante (“fria”) tem baixa especificidade para neoplasia maligna da tireóide, uma vez que 90% dos nódulos de tireóide apresentam esse padrão de captação, e apenas 5% representam carcinomas.
Estudos recentes com outros radioisótopos (99mTc-metoxi-iso-butil-lisonitrila, 201Tl e 18F-desoxi-glicose) vêm mostrando resultados promissores no diagnóstico diferencial dos Nódulos tireoidianos.
Tabela 4: Avaliação laboratorial dos nódulos de tireóide
|
TSH |
Realizar em todo paciente portador de nódulo Tireotoxicose: TSH suprimido e T3 /T4 livre aumentados TSH suprimido sugere benignidade |
|
Calcitonina |
Alta sensibilidade para carcinoma medular |
|
USG |
Achados de neoplasia maligna (especificidade de 66%): o presença de microcalcificações; o bordas irregulares; o hipoecogenicidade. Achados de malignidade ao Doppler: o Vascularização predominante ou exclusivamente central |
|
PAAF |
Indicação: todo nódulo com mais de 1 cm de diâmetro ou com características ultra-sonográficas sugestivas de malignidade |
|
Medicina nuclear |
Útil para avaliação funcional de bócios tóxicos O achado de uma área hipocaptante (“fria”) tem baixa especificidade para neoplasia maligna |
Os nódulos com citologia benigna à punção devem ser acompanhados com ultra-sonografia 12-18 meses após a punção. Em caso de aumento do tamanho do nódulo acima de 20% do inicial, recomenda-se repetir a PAAF.
A maioria dos nódulos benignos é assintomática e não requer tratamento. Contudo, caso haja compressão de estruturas adjacentes à tireóide ou desconforto estético, pode-se indicar tratamento desses nódulos.
Para nódulos funcionantes, o tratamento pode ser medicamentoso, com drogas antitireoidianas (metimazol ou propiltiouracil), cirúrgico com o radioiodoterapia. Esse assunto será abordado com mais detalhes no capítulo HIPERTIREOIDISMO.
Lobectomia pode ser realizada para tratar lesões benignas da tireóide. Para os bócios multinodulares, principalmente os de grande volume, pode-se indicar tireoidectomia total.
A radioiodoterapia com 131I pode ser indicada como tratamento exclusivo ou complementar em Nódulos tireoidianos. Como tratamento exclusivo, o 131I está indicado nos nódulos funcionantes (com ou sem hipertireoidismo), em glândulas uninodulares ou multinodulares.
Após uma dose de 131I, obtém-se normalização da captação de iodo e dos níveis de TSH em 70% dos casos, e redução de 40% do volume tireoidiano inicial. O principal efeito colateral é o hipotireoidismo, que acomete 10% dos pacientes tratados em até cinco anos.
A administração de TSH humano recombinante (Thyrogen®) antes da dose de 131I aumenta a captação do radioisótopo e a eficácia do tratamento.
A radioiodoterapia está contra-indicada em gestantes e lactantes. Gestação deve ser evitada por no mínimo seis meses após a dose de 131I.
A injeção percutânea de etanol guiada por ultra-sonografia representa uma alternativa terapêutica para nódulos sólidos ou císticos com citologia benigna, inclusive os funcionantes. Após três ou quatro aplicações, atinge-se normocaptação de iodo e desbloqueio do TSH em até 75% dos casos.
O principal efeito colateral é a dor local, que pode impossibilitar o tratamento em alguns pacientes. Há relatos de complicações graves devido a extravasamento de etanol, causando necrose do subcutâneo e pele adjacente.
Por muito tempo, administração de levotiroxina causando supressão do TSH a níveis inferiores de 0,3 mU/l foi considerada uma opção terapêutica para nódulos benignos. Uma meta-análise recente, contudo, não demonstrou diferença significativa do tamanho de nódulos após seis ou 12 meses de tratamento com levotiroxina.
Além disso, hoje são bem reconhecidas as complicações da tireotoxicose subclínica sobre o sistema cardiovascular (fibrilação atrial) e esquelético (osteoporose), de forma que essa opção terapêutica deve ser evitada na maioria dos casos.
O carcinoma bem diferenciado da tireóide compreende o maior número dos casos de neoplasias malignas da tireóide. Divide-se em Carcinoma papilífero e Carcinoma folicular.
Cirurgia é o tratamento de escolha para nódulos com citologia sugestiva de Carcinoma papilífero ou para aqueles com citologia indeterminada (“padrão folicular”) quando se suspeita de Carcinoma folicular. A tireoidectomia total com ressecção de linfonodos aumentados no compartimento central é recomendada para o tratamento adequado dos carcinomas bem diferenciados da tireóide. Não se recomenda lobectomia para os carcinomas bem diferenciados de tireóide em virtude da possibilidade de multicentricidade da lesão e da impossibilidade de tratamento adjuvante com 131I e de seguimento com tireoglobulina plasmática.
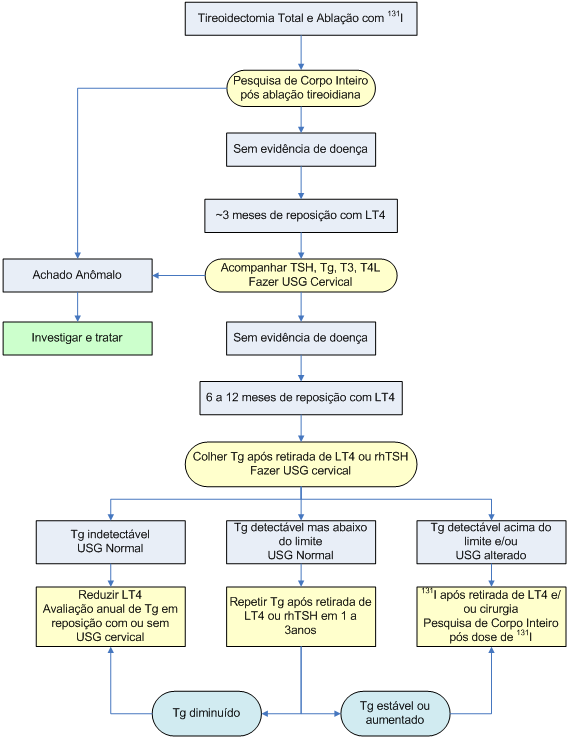
Após a tireoidectomia total, os pacientes com carcinomas bem diferenciados da tireóide (especialmente aqueles com tumores maiores que 2 cm e quando há invasão da cápsula tireoidiana ou metástases ganglionares) devem receber ablação dos remanescentes com 131I, a fim de reduzir o risco de recidivas e permitir o seguimento com dosagem de tireoglobulina sérica.
Antes da dose ablativa de 131I, recomenda-se um período de aproximadamente quatro semanas sem reposição com levotiroxina para permitir elevação do TSH (para níveis iguais ou maiores que 30 mU/l). Além disso, o paciente deve ser instruído a evitar consumo excessivo de iodo (sal iodado, frutos-do-mar, medicações com iodo e contrastes radiológicos) para não prejudicar a captação do 131I. As mulheres devem ser orientadas a procurar um método anticoncepcional, pois a gestação é contra-indicada por seis meses.
Após a dose, faz-se uma pesquisa de corpo inteiro (PCI) à procura de áreas de captação.
Após a radioiodoterapia, a reposição com levotiroxina deve ser iniciada, com dose em torno de 2 µg/kg/dia. O objetivo é manter o TSH ao redor de 0,1 mU/l, com T3 livre e T4 livre dentro da faixa da normalidade.
No seguimento do carcinoma bem diferenciado de tireóide, o clínico utiliza dois importantes instrumentos diagnósticos: a ultra-sonografia de alta resolução e a dosagem sérica de tireoglobulina com TSH elevado (por retirada da reposição com levotiroxina ou por administração de TSH humano recombinante), que, juntos, atingem sensibilidade próxima a 100% na detecção de recidiva local. Quando um ou outro indicarem doença persistente, a localização topográfica deverá ser feita para o tratamento apropriado, que poderá ser cirúrgico ou com nova administração de 131I em altas doses. O algoritmo 2 ilustra o seguimento do carcinoma bem diferenciado da tireóide.
Quanto aos microcarcinomas unifocais (tumores menores que 1 cm), não há consenso em relação à realização de tratamento adjuvante com 131I ou necessidade de tireoidectomia total.
Tanto o carcinoma medular esporádico quanto o familiar são tratados cirurgicamente. Para esse tumor, preconiza-se a tireoidectomia total com dissecção do compartimento central (nível VI) profilática e das cadeias laterais na presença de sinais de comprometimento dos linfonodos. Se houver prejuízo à irrigação das paratireóides, estas devem ser retiradas e implantadas no antebraço.
Após a tireoidectomia, não há necessidade de radioiodoterapia ou terapia supressiva com levotiroxina, pois as células parafoliculares não são iodo-captantes e não respondem ao TSH.
O seguimento deve ser feito com dosagem basal e estimulada de calcitonina, dosagem de CEA e ultra-sonografia de tireóide. Havendo elevação da calcitonina ou do CEA, exames de imagem (tomografia computadorizada ou ressonância nuclear magnética) de pescoço, tórax, abdome e pelve são úteis para estadiar a doença.
O CMT é a principal causa de mortalidade nos portadores dessas síndromes genéticas, e a sua ocorrência na maior parte dos pacientes é durante as primeiras décadas de vida. Assim, a tireoidectomia profilática com dissecção do compartimento central vem sendo realizada em crianças com o diagnóstico molecular, antes do desenvolvimento dos tumores, principalmente se a mutação do RET se encontra nos códons 883, 922 e 611.
Radioterapia e quimioterapia podem ser empregadas, contudo as respostas são baixas. O esquema quimioterápico mais usado é a combinação de dacarbazina com vincristina e ciclofosfamida.
A progressão rápida da doença geralmente impede algum tratamento curativo. Assim, a maior parte dos procedimentos é feita para paliação dos sintomas. Radioterapia externa pode permitir algum controle local. A resposta à quimioterapia costuma ser ruim.
O nódulo tireoidiano é um dos principais motivos de procura ao consultório do endocrinologista. Dada sua prevalência cada vez mais alta, motivada em grande parte por solicitação inadvertida de ultra-sonografias cervicais, o conhecimento das formas de avaliação é fundamental para qualquer clínico.
Apenas 5% dos Nódulos tireoidianos são palpáveis. Entretanto, a prevalência pode chegar a 50% da população feminina após os 50 anos, se considerados os nódulos subclínicos.
A classificação dos Nódulos tireoidianos inclui os tumores derivados de células foliculares, os tumores derivados de células parafoliculares e outros tumores (linfomas e sarcomas).
O adenoma folicular é o tumor de tireóide mais comum, sendo benigno e derivado de células foliculares. Pode gerar hipertireoidismo subclínico ou tireotoxicose franca.
O Carcinoma papilífero é a neoplasia maligna de tireóide mais comum, correspondendo a mais de 90% dos carcinomas bem diferenciados em áreas sem deficiência de iodo. Pode incidir em qualquer faixa etária, mas ocorre principalmente entre 30 e 50 anos de idade. As mulheres são mais acometidas. Manifesta-se como nódulo cervical duro e indolor.
O Carcinoma folicular é uma doença rara em países onde não há deficiência iodo. Acomete indivíduos mais idosos que o papilífero, mas ainda com maior predileção pelo sexo feminino. Manifesta-se como nódulo cervical indolor.
O carcinoma insular se manifesta com grandes nódulos que acometem indivíduos mais idosos. É invasivo e muitas vezes letal.
O carcinoma anaplásico tem maior incidência entre os 50 e 60 anos. O sintoma mais comum é de uma massa cervical com crescimento rápido, determinando sintomas compressivos. Até 50% pode ter metástase à distância ao diagnóstico.
O carcinoma medular representa menos de 10% dos tumores malignos da tireóide. Pode fazer parte das síndromes das neoplasias endócrinas múltiplas dos tipos 2A e 2B, e da síndrome de carcinoma medular de tireóide familiar isolado. Apresenta uma evolução mais agressiva. Os níveis elevados de calcitonina são o marcador específico dessa doença.
O linfoma primário de tireóide é um tipo de linfoma não-Hodgkin. É um tumor cervical de crescimento rápido, geralmente associado a linfadenopatia cervical, em paciente com mais de 60 anos de idade. O tratamento não envolve tireoidectomia.
Os sarcomas primários da tireóide são tumores raros.
A maior parte dos carcinomas de tireóide são nódulos assintomáticos.
Os principais fatores que aumentam a suspeita de malignidade diante de um nódulo de tireóide são: história familiar de neoplasia endócrina múltipla ou de carcinoma medular familiar, crescimento rápido do tumor, nódulo endurecido e aderido aos tecidos adjacentes, paralisia de cordas vocais e adenomegalia cervical.
A dosagem de TSH está indicada em todo paciente portador de nódulo tireoidiano. Aproximadamente 10% dos nódulos palpáveis reduzem os níveis de TSH, o que é um achado fortemente sugestivo de benignidade.
A dosagem sérica de calcitonina tem sensibilidade superior ao exame citológico para identificar o carcinoma medular de tireóide.
Presença de microcalcificações, bordas irregulares e hipoecogenicidade são achados ultra-sonográficos sugestivos de neoplasia maligna. Ao Doppler, vascularização predominante ou exclusivamente central sugere doença maligna.
A PAAF deve ser realizada em todo nódulo com mais de 1 cm de diâmetro ou com características ultra-sonográficas sugestivas de malignidade ou história clínica de risco, exceto quando há suspeita de nódulo funcionante pelo mapeamento de tireóide.
Em medicina nuclear, o achado de uma área hipocaptante (“fria”) tem baixa especificidade para neoplasia maligna da tireóide.
A maioria dos nódulos benignos é assintomática e não requer tratamento. Os funcionantes podem ser tratados com medicações, cirurgia ou radioiodoterapia. Recomenda-se cirurgia se houver compressão de estruturas ou desconforto estético.
Cirurgia é o tratamento de escolha para nódulos com citologia sugestiva de Carcinoma papilífero ou para aqueles com citologia indeterminada (“padrão folicular”) quando se suspeita de Carcinoma folicular. Após a tireoidectomia total, os pacientes com carcinomas bem diferenciados da tireóide (especialmente aqueles com tumores maiores que 2 cm e quando há invasão da cápsula tireoidiana ou metástases ganglionares) devem receber ablação dos remanescentes com 131I, a fim de reduzir o risco de recidivas e permitir o seguimento com dosagem de tireoglobulina sérica. Após a radioiodoterapia, a reposição com levotiroxina deve ser iniciada, com dose em torno de 2 µg/kg/dia. O objetivo é manter o TSH ao redor de 0,1 mU/l, com T3 livre e T4 livre dentro da faixa da normalidade.
Tanto o carcinoma medular esporádico quando o familiar são tratados cirurgicamente. Após a tireoidectomia, não há necessidade de radioiodoterapia ou terapia supressiva com levotiroxina. Recomenda-se tireoidectomia profilática em portadores de MEN2A e MEN2B
Doença metastática é tratada com quimioterapia e radioterapia, contudo as respostas são baixas.
No carcinoma anaplásico, a maior parte dos procedimentos são feitos para paliação dos sintomas.
Algoritmo 1: Diagnóstico e tratamento dos nódulos de tireóide

Algoritmo 2: Seguimento do carcinoma bem diferenciado de tireóide
BIBLIOGRAFIA
1. Elisei R, Bottici V, Luchetti F, Di Coscio G, Romei C, Grasso L et al. Impact of routine measurement of serum calcitonin on the diagnosis and outcome of medullary thyroid cancer: experience in 10,864 patients with nodular thyroid disorders. J Clin Endocrinol Metab 2004;89:163-8.
2. Hegedus L. The thyroid nodule. N Engl J Med 2004;351:1764-71.
3. Maia AL, Ward LS, Carvalho GA, Graf H, Maciel RM, Maciel LM et al. Thyroid nodules and differentiated thyroid cancer: Brazilian consensus. Arq Bras Endocrinol Metabol 2007;51:867-93.
4. Pacini F, De Groot L. Thyroid nodules. In: De Groot L, ed. Thyroid disease manager. Disponível em: <www.thyroidmanager.org>.
5. Pacini F, De Groot L. Thyroid cancer. In: De Groot L, ed. Thyroid disease manager. Disponível em: <www.thyroidmanager.org>.
6. Schlumberger M, Pacini F, Wiersinga WM, Toft A, Smit JW, Sanchez Franco F et al. Follow-up and management of differentiated thyroid carcinoma: an European perspective in clinical practice. Eur J Endocrinol 2004;151:539-48.
7. Schlumberger MJ. Anaplastic thyroid carcinoma. In: Filetti S, ed. Orphanet encyclopedia. Disponível em: <http://www.orpha.net>.
8. Scollo C, Baudin E, Travagli JP, Caillou B, Bellon N, Leboulleux S et al. Rationale for central and bilateral lymph node dissection in sporadic and hereditary medullary thyroid cancer. J Clin Endocrinol Metab 2003;88:2070-5.
9. Thieblemont C, Mayer A, Dumontet C, Barbier Y, Callet-Bauchu E, Felman P et al. Primary thyroid lymphoma is a heterogeneous disease. J Clin Endocrinol Metab 2002;87:105-11.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.