(Carregando Índice)... (Carregando Índice)... |
Autores:
Danieli Castro Oliveira de Andrade
Doutora, Médica reumatologista do Centro Paulista de Investigação Clínica e do Instituto Fleury, São Paulo. (Ex-Residente da FMUSP - Reumatologia)
Eduardo Ferreira Borba
Professor Livre Docente da Disciplina de Reumatologia da Faculdade de Medicina da Universidade de São Paulo – FMUSP.
Sandra Regina Miyoshi Lopes
Professora Assistente da Disciplina de Reumatologia da Faculdade de Medicina de Catanduva. Especialista em Reumatologia pelo Serviço de Reumatologia do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo – FMUSP.
Samuel Katsuyuki Shinjo
Médico Assistente do Serviço de Reumatologia do Hospital das Clínicas da Faculdade de Medicina da USP
Clínico geral e Reumatologista pelo Hospital das Clínicas da Faculdade de Medicina da USP
Mestre e Doutor em Ciências pela Universidade Federal de São Paulo
André Regis
Especialista em Reumatologia pelo Serviço de Reumatologia do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo – FMUSP.
Última revisão: 28/02/2010
Comentários de assinantes: 0
Danieli Castro Oliveira de Andrade
Doutora em Reumatologia pela Disciplina de Reumatologia da Faculdade de Medicina da Universidade de São Paulo – FMUSP.
Eduardo Ferreira Borba
Professor Livre Docente da Disciplina de Reumatologia da Faculdade de Medicina da Universidade de São Paulo – FMUSP.
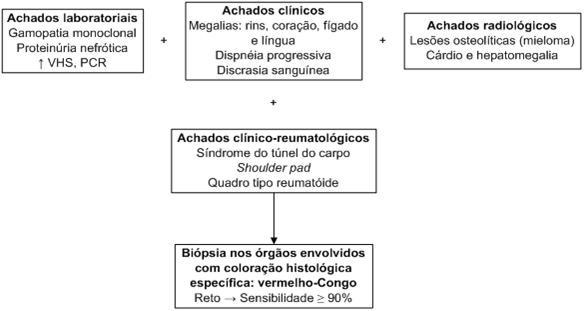
A amiloidose compreende um grupo heterogêneo de doenças que possuem em comum o depósito extracelular de uma substância proteica dita amiloide. É interessante que todas as formas de amiloide apresentam a birrefringência verde peculiar à microscopia após a coloração pelo vermelho Congo, e essa observação no material de biópsia continua sendo o padrão ouro para estabelecer o diagnóstico de amiloidose.
As doenças por amiloide são classificadas de acordo com a composição bioquímica das proteínas precursoras séricas que formam as fibrilas de amiloide e depósitos. Apesar destes avanços, sua história natural é ainda pouco conhecida e o seu diagnóstico, na maioria das vezes, realizado tardiamente, quando a doença já se encontra num estágio mais avançado.
As doenças amiloides são classificadas de acordo com o padrão e a extensão do depósito. Podem ser divididas em adquiridas ou hereditárias, ou ainda sistêmicas ou localizadas de acordo com a extensão do depósito (Tabela 1).
As formas mais frequentes de amiloidose são as adquiridas, onde encontramos as formas AL (imunoglobulinas de cadeia leve ou primária), as AA (reativa ou secundária) e as Ab2M (b2-microglobulina ou associada à diálise). A amiloidose do tipo AL é rara, com incidência estimada de 4,5/100000 indivíduos. Por sua vez, amiloidose secundária ou reativa (tipo AA) está principalmente associada a infecções e inflamações crônicas, e corresponde a
Por outro lado, as formas hereditárias são mais raras e variam de acordo com a região geográfica, acometendo 1/100000 indivíduos. Geralmente, o modo de transmissão é autossômico dominante, com a exceção da febre familiar do Mediterrâneo, que apresenta o padrão autossômico recessivo.
As apresentações clínicas variam de acordo com os tipos de fibrilas amiloides (Tabela 1).
A amiloidose primária sistêmica (tipo AL) é uma discrasia de célula plasmática com envolvimento multissistêmico, progressão rápida e sobrevida curta. As fibrilas de amiloide são compostas de cadeia leve monoclonal. A mais encontrada é a l, numa proporção de 2:1 em relação à k. Usualmente afeta indivíduos com mais de 40 anos de idade, é mais frequente em homens (65%) e os sintomas e sinais são frequentemente inespecíficos, tais como fraqueza, fadiga e perda ponderal.
As principais repercussões clínicas, que alertam para o diagnóstico, comprometem rins, pulmões e coração e são reconhecidas tardiamente, por isso apresentam pior evolução. A gravidade está relacionada ao aumento dos órgãos secundários ao depósito de amiloide. Nos rins, a proteinúria, por vezes nefrótica, é encontrada em 1/3 dos pacientes e frequentemente leva à insuficiência renal. Em aproximadamente 1/4 dos casos identifica-se insuficiência cardíaca congestiva, miocardiopatia restritiva, baixa voltagem ao eletrocardiograma e hipersensibilidade digitálica. O depósito pulmonar ocorre em aproximadamente 90% dos casos e determina quadro de dispneia progressiva. O comprometimento hepático normalmente não leva à perda de função, apesar de levar à hepatomegalia. Alterações gastrintestinais como má absorção, diarreia e obstrução também podem ser encontradas.
Um sinal fácil de observação e que apresenta um importante valor preditivo na doença é a presença de macroglossia, observada em até 20% dos casos, e também frente a sangramentos devido à deficiência de fator X. Além disso, o acometimento cutâneo caracterizado por púrpura, pápulas ou tumores pode ser encontrado em mais de 40% dos casos. Aproximadamente
Os achados reumatológicos mais significantes são o aparecimento de síndrome do túnel do carpo, observado em mais de 20% dos casos, e o sinal de shoulder pad, que corresponde ao depósito de amiloide nos ombros. A artropatia não é frequente, ocorre em menos de 5% dos casos e predomina nas grandes articulações.
A presença de um dos sintomas descritos associado à proteína monoclonal sérica ou urinária constitui forte suspeita de amiloidose; a presença de proteína monoclonal no soro ou urina é observada em 90% dos casos.
A amiloidose associada ao mieloma múltiplo também apresenta fibrilas compostas de cadeia leve monoclonal, assim como a amiloidose primária. Porém, nesta forma, existe um predomínio da cadeia leve k em relação à cadeia l, numa relação de 2:1. As lesões osteolíticas encontradas no mieloma são de grande auxílio no seu diferencial, visto que as manifestações clínicas são semelhantes àquelas encontradas na forma primária.
A amiloidose secundária ou reativa ou adquirida (tipo AA) é uma complicação pouco comum de estados inflamatórios crônicos, infecções e, de forma mais rara, a determinados tipos de neoplasias. A forma reativa a processos inflamatórios crônicos tem sido relacionada principalmente às doenças reumatológicas, particularmente a AR, espondilite anquilosante, artrite reativa e artrite psoriásica. Interessante notar que, apesar disso, a amiloidose é rara nas doenças do tecido conectivo, principalmente no LES. Na doença de Crohn, a amiloidose ocorre em até 8% dos casos, em contraste com a retocolite ulcerativa, onde é extremamente rara.
A suspeita de amiloidose nestas doenças fundamenta-se basicamente na presença de proteinúria, má absorção intestinal ou envolvimento gastrintestinal como náusea, diarreia e hepato ou esplenomegalia. O depósito amiloide é o da proteína AA que é distinto das cadeias leves de imunoglobulinas, e os pacientes raramente apresenta acometimento cardíaco e de nervo periférico. A macroglossia não é uma característica da amiloidose AA. O tratamento desta forma secundária de amiloidose consiste no controle do processo inflamatório da doença de base, levando a estabilização ou resolução parcial do depósito amiloide.
A amiloidose heredofamiliar é identificada pelas várias regiões onde cada família e tipos de depósito amiloide foram descritos e estão associados a quadros clínicos distintos. O modo de transmissão habitualmente é autossômico dominante, com a exceção da febre familiar do Mediterrâneo, que é autossômica recessiva. Esta caracteriza-se por surtos recorrentes de febre, serosites (principalmente peritonite e pleurite) e artrite, que se inicia na infância/adolescência, principalmente nos de origem mediterrânea. O depósito amiloide é do tipo AA, semelhante ao das formas reativas, e envolve fígado, baço, supra-renal e principalmente rins, onde a proteinúria é inicialmente observada e, após
A amiloidose local pode estar presente em qualquer órgão, por vezes assemelhando-se a tumores, principalmente em pulmão, pele, laringe, olhos e vesícula.
A amiloidose associada à hemodiálise crônica é um grande desafio no tratamento da insuficiência renal crônica, pois, nesta forma, existe o depósito de fibrilas constituídas por b2 microglobulina intacta que normalmente não é removida durante a diálise. A sua incidência é superior a 50% após 5 anos do início desse procedimento. Caracteriza-se por quadro de poliartrite tipo reumatoide com formação de pannus, tenossinovite, síndrome do túnel do carpo e aparecimento de grandes cistos e erosões ósseas, eventualmente levando as fraturas patológicas. Até o momento, o transplante renal é o único tratamento efetivo desta condição, pois leva à rápida normalização dos níveis séricos de b2 microglobulina e resolução dos sintomas.
A amiloidose cerebral está estreitamente relacionada a doença de Alzheimer, síndrome de Down e amiloidose hereditária com hemorragia cerebral (holandesa), que compartilham o depósito da proteína amiloide b (Ab).
A amiloidose endócrina ocorre em glândulas secretoras de hormônios polipeptídicos, bem como em tumores secretores hormonais. No diabetes mellitus não insulino dependente, o depósito de proteína amiloide do tipo AIAPP (polipeptídeo amiloide das ilhotas de Langerhans) pode ser encontrado em até 95% dos pacientes com esta doença.
Tabela 1: Formas clínicas de amiloidose
|
Amiloidose |
Proteína amiloide |
Condições associadas |
|
Primária |
AL |
Sem doença prévia ou concomitante |
|
Do mieloma múltiplo |
AL |
Mieloma múltiplo |
|
Secundária ou reativa |
AA |
Infecções crônicas: Hanseníase, tuberculose, osteomielite |
|
AA |
Inflamações crônicas: AR, AR juvenil, espondilite anquilosante | |
|
Heredofamiliar |
AA |
Febre familiar do Mediterrâneo |
|
AA |
Síndrome de Muckle-Well | |
|
ATTR |
Neuropática | |
|
ATTR |
Cardíaca | |
|
AGel |
Distrofia corneana familiar amiloide | |
|
Localizada |
AL |
Sem comprometimento sistêmico |
|
Da hemodiálise |
Ab2M |
Hemodiálise crônica |
|
Cerebral |
Ab |
Doença de Alzheimer |
|
Ab |
Síndrome de Down | |
|
ACys |
Hereditária com hemorragia cerebral | |
|
AScr |
Doença de Creutzfeldt-Jakob | |
|
Endócrina |
ACal |
Carcinoma medular de tireoide |
|
AIAPP |
Diabetes mellitus |
A substância amiloide é depositada na articulação, na membrana e no líquido sinovial. A atropatia amiloide é encontrada principalmente na amiloidose secundária ao mieloma múltiplo, mas também na forma primária, na febre familiar do Mediterrâneo e na forma associada à diálise.
A artrite amiloide geralmente mimetiza aquela encontrada na AR, com quadro simétrico, de pequenas articulações, associado a rigidez matinal e fadiga. As articulações preferencialmente comprometidas são ombros, punhos, joelhos e dedos; elas se apresentam edemaciadas e firmes, mas sem rubor e dor importante. Neste sentido, um importante espessamento sinovial deve ser considerado como indicativo de amiloidose.
A artropatia amiloide pode manifestar-se como síndrome do túnel do carpo devido à infiltração de punhos, principalmente em indivíduos com mais de 50 anos de idade. Na amiloidose primária, a síndrome do túnel do carpo pode ser encontrada em até 20% dos casos e pode ser decisiva para o seu diagnóstico. Porém, esta é mais frequente na amiloidose secundária à hemodiálise crônica, onde a compressão do nervo mediano ocorre em mais de 70% dos pacientes. Outra articulação que merece destaque é o ombro. O shoulder pad ocorre principalmente na amiloidose primária em decorrência da intensa infiltração amiloide
Na maioria das vezes, a suspeita clínica é secundária ao depósito amiloide nos órgãos afetados. Portanto, a investigação deve ser dirigida mediante o sítio comprometido. As alterações nos exames de sangue e urina são, por vezes, inespecíficos, como um aumento de VHS e da PCR. A presença de proteinúria e hipergamaglobulinemia pode levar a suspeita de amiloidose AL, principalmente quando associadas a neuropatia periférica ou insuficiência cardíaca congestiva. Nas formas de amiloidose primária sistêmica e naquela associada ao mieloma múltiplo, a identificação de uma cadeia leve monoclonal pode auxiliar na suspeita diagnóstica, embora possa ainda ser negativa em
O diagnóstico de amiloidose baseia-se na demonstração histológica do depósito tecidual de amiloide por meio de coloração específica. A confirmação histológica do amiloide é classicamente demonstrada pela coloração com vermelho Congo, que, sob microscopia polarizada, apresenta uma birrefringência verde. Nem sempre as fibrilas amiloides são identificadas à microscopia eletrônica e, na verdade, seu achado isolado também não permite a confirmação diagnóstica.
A biópsia deve ser preferencialmente realizada no órgão suspeito de infiltração amiloide, considerando-se que existe o risco de complicações relacionadas a sangramento. A biópsia de reto ou de gordura subcutânea são as menos invasivas, com positividade de mais de 90% tanto nas formas primárias quanto secundárias. Outros sítios passíveis de biópsias são a gengiva e a medula óssea, que possuem sensibilidades de 80% e 50%, respectivamente. O uso de anticorpos dirigidos contra as proteínas fibrilares amiloides, por imunofluorescência e por imunoperoxidase, pode facilitar a sua identificação. Porém, a positividade à imuno-histoquímica do amiloide não constitui método isolado de diagnóstico, havendo também a necessidade da confirmação pela coloração com vermelho Congo.
Radiologicamente, a osteopenia generalizada é observada em até 80% dos casos, mesmo na ausência de lesões líticas. O fator reumatoide habitualmente é negativo, sendo de auxílio na sua diferenciação. A análise do líquido sinovial revela um líquido não-inflamatório com predomínio de mononucleares e presença de depósitos amiloides no vilo sinovial à coloração do vermelho Congo.
Os princípios da terapêutica da amiloidose baseiam-se na redução da formação do amiloide e, ao mesmo tempo, dissolução dos depósitos existentes.
A redução do precursor amiloide consiste na diminuição da síntese hepática das proteínas de fase aguda. De fato, tem sido demonstrado que a utilização de drogas citotóxicas na AR juvenil e do adulto reduziu a incidência de amiloidose e melhorou o prognóstico dessas doenças.
O clássico uso da colchicina no tratamento da febre familiar do Mediterrâneo, além de prevenir ataques febris, também se mostrou comprovadamente efetivo na redução do depósito amiloide, preservando função renal e aumentando a sobrevida ao longo do tempo. A colchicina também parece exercer efeitos adicionais na amiloidose AL, principalmente quando associada a melfalam e prednisolona. A realização de plasmaférese e o transplante hepático também parecem ser efetivos como redutores da amiloidose familiar por TTR, assim como o transplante renal para a amiloidose causada pela b2 microglobulina.
Medidas de suporte são particularmente importantes no seguimento destes pacientes, por retardar a piora da insuficiência de um determinado órgão, por exemplo, o controle rigoroso dos níveis pressóricos na amiloidose renal. O transplante hepático, renal e cardíaco pode ser necessário em casos extremos, sobretudo na amiloidose do tipo AA. A utilização de determinadas drogas, como digital e bloqueadores de cálcio, deve ser cuidadosa nos casos com envolvimento cardíaco, pois podem agravar a função miocárdica.
Até o momento, a história natural da amiloidose sistêmica apresenta prognóstico e desfecho ruins, principalmente devido ao início tardio do tratamento. O reconhecimento precoce dessa doença, prevenindo o depósito de amiloide nos órgãos, é um dos fatores mais importantes para sua boa evolução.
Algoritmo 1: Fluxograma para amiloidose.

VHS: velocidade de hemossedimentação; PCR: proteína C reativa.
1. Husby G. Amyloidosis. Semin Arthritis Rheum 1992;22:67-82.
2. Husby G. Nomenclature and classification of amyloid and amyloidoses. J Int Med 1992;232:511-512.
3. Skinner M. Protein AA/SAA. J Int Med 1992;232:513-514.
4. Kisilevsky R. Proteoglycans, glycosaminoglycans, amyloid-enhancing factor, and amyloid deposition. J Int Med 1992;232:515-516.
5. Gertz MA. Secondary amyloidosis (AA). J Int Med 1992;232:517-518.
6. Pepys MB. Amyloid P component and the diagnosis of amyloidosis. J Int Med 1992;232:519-521.
7. Kyle RA. Primary systemic amyloidosis. J Int Med 1992;232:523-524.
8. Benson MD. Familial amyloidosis. J Intl Med 1992;232:525-527.
9. Livneh A et al. Colchicine in the treatment of AA and AL amyloidosis. Semin Arthritis Rheum 1993;23:206-214.
10. Tan SY, Pepys MB. Amyloidosis. Histopathology 1994;25:403-414.
11. Gertz MA, Kyle RA. Amyloidosis: prognosis and treatment. Semin Arthritis Rheum 1994;24:124-138.
12. Cohen AS. Amyloidosis. In: McCarty DJ, Koopman WJ. Arthritis and allied conditions. 12 ed. Philadelphia: Lea & Febiger, 1993. p.1427-1447.
Sandra Regina Miyoshi Lopes
Professora Assistente da Disciplina de Reumatologia da Faculdade de Medicina de Catanduva. Especialista em Reumatologia pelo Serviço de Reumatologia do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo (HCFMUSP).
Samuel Katsuyuki Shinjo
Professor Colaborador da Disciplina de Reumatologia da Faculdade de Medicina da Universidade de São Paulo (FMUSP). Médico Assistente Doutor do Serviço de Reumatologia do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo (HCFMUSP).
A policondrite recidivante (PCR) é uma doença do tecido conectivo, autoimune, rara e de causa desconhecida. Caracteriza-se por inflamação recorrente de tecidos cartilaginosos.
Não tem predileção racial ou por sexo. A incidência é de 3,5/1 milhão de pessoas (EUA). Há um pico de maior incidência entre 40 e 50 anos de idade. Em aproximadamente 30% dos casos, encontra-se associada a outras doenças, como vasculites sistêmicas, cirrose biliar primária, doença inflamatória intestinal, artrite reumatoide (AR), outras colagenoses.
A etiologia ainda permanece desconhecida. Pode-se encontrar anticorpos anticolágeno tipo II no plasma destes pacientes. Supõe-se que uma agressão ao tecido cartilaginoso (por trauma ou infecção) poderia expor alguns autoantígenos da matriz, ativando a resposta autoimune.
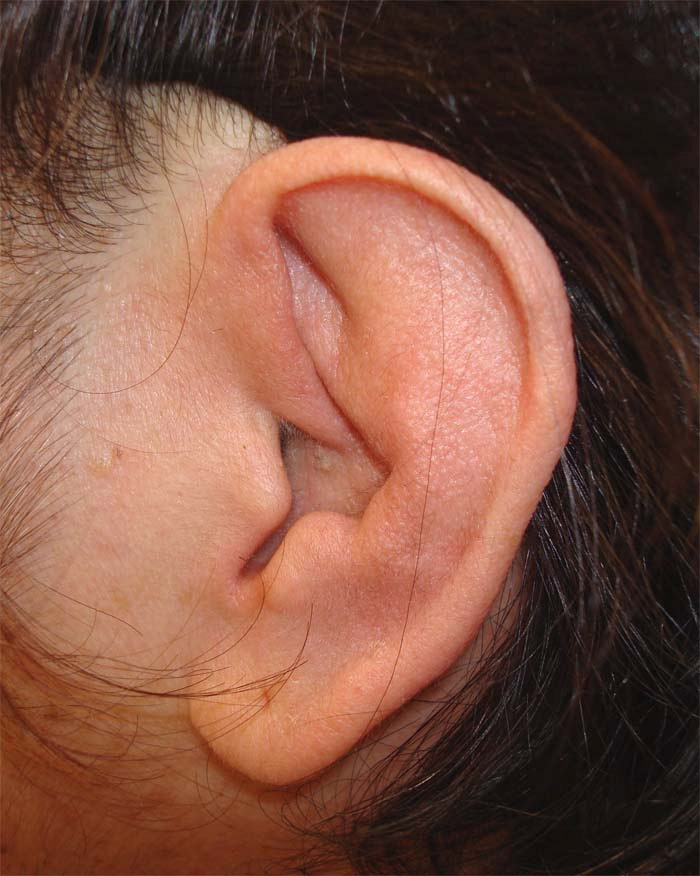
A manifestação clássica é a condrite auricular, caracterizada por acometimento uni ou bilateral, edema do pavilhão auricular (polpa ou lóbulo da orelha), vermelhidão e dor (intensidade variável) durando de dias a semanas. A condrite auricular pode ocorrer em 85% dos casos. O processo inflamatório pode levar a destruição da arquitetura auricular (Figura 1). O acometimento auditivo (hipoacusia) e vestibular (vertigem, tinido) pode ocorrer em 30% dos pacientes. A condrite nasal pode ocorrer em 55% dos casos, sendo que a cartilagem nasal fica edemaciada, vermelha e dolorosa. Também pode ocorrer obstrução nasal, rinorreia, epistaxe e nariz em sela (por degeneração cartilaginosa).
Figura 1: Policondrite recidivante com acometimento auricular. O lóbulo da orelha é poupado do processo inflamatório.
A artrite é autolimitada, assimétrica, não-erosiva e não deformante, podendo ocorrer em 50 a 70% dos casos. Pode ser oligo ou poliartrite, ocorrendo em grandes e pequenas articulações, geralmente tornozelos, punhos, pés, mãos, esternoclaviculares, costocondral e esternomanubrial.
A condrite laringotraqueobrônquica pode acometer quase metade dos pacientes. Tem importância devido ao potencial de gravidade. Podem ocorrer: rouquidão, tosse seca, odinofagia, estridor, dispneia, sibilância, disfonia, asfixia súbita (colapso de vias aéreas), atelectasias e pneumonias de repetição (estas últimas devido à obstrução brônquica).
Episclerite (40%), esclerite (15%), conjuntivite, proptose, vasculite retiniana, edema periorbital, ceratite, tarsite, uveíte anterior, catarata, coriorretinite, paralisia dos músculos extraoculares, úlceras de córnea. Dependendo do grau de acometimento, pode haver perda visual.
Podem haver vasculite de pequenos, médios e grandes vasos, aneurisma aorta (torácica e/ou abdominal), pericardite, arritmias, distúrbios de condução, valvulite (causando posterior insuficiência). Dentre as insuficiências, a mais encontrada é a insuficiência aórtica, sendo esta uma das mais graves complicações da PCR.
Cefaleias, neuropatias cranianas (II, VI, VII, VIII), encefalopatia, ataxia, hemiplegia, convulsões, meningoencefalite.
As lesões cutâneas podem ocorrer em 30% dos pacientes, sendo as de maior importância às lesões vasculíticas: púrpura, livedo reticular, vasculite urticariforme, tromboflebite superficial migratória, paniculite, eritema nodoso, eritema multiforme. Alteração no crescimento ungueal e alopecia também podem estar presentes.
Pode haver glomerulonefrite proliferativa focal e segmentar (cilindrúria, hematúria microscópica e proteinúria). Insuficiência renal é rara.
Quanto ao prognóstico, é relativamente bom. A sobrevida em 5 anos é de 75% e em 10 anos é de 55%. O prognóstico é pior quando há vasculite sistêmica associada, casos em que a sobrevida em 5 anos é de 45%.
Os critérios classificatórios da PCR são:
condrite auricular bilateral;
condrite nasal;
poliartrite não-erosiva;
inflamação ocular;
condrite laringotraqueobrônquica;
disfunção vestibulococlear;
biópsia da cartilagem compatível com condrite.
Presença de 3 ou mais destes critérios confirma PCR.
AR, lúpus eritematoso sistêmico (LES), espondiloartropatias, granulomatose de Wegener, sífilis, pericondrite infecciosa, condropatia degenerativa hereditária, micoses, tuberculose, hanseníase.
Não há exame laboratorial marcador específico para esta patologia. Os anticorpos anticolágeno tipo II não são positivos em todos os casos, assim como não são solicitados rotineiramente.
Pode haver alterações hematológicas inespecíficas que indiquem processo inflamatório (leucocitose, anemia de doença crônica, aumento do VHS, trombocitose, hipergamaglobulinemia).
Tomografia computadorizada é utilizada para avaliar acometimento de laringe e traqueia. As provas de função pulmonar podem estar alteradas.
A biópsia da cartilagem auricular pode confirmar o diagnóstico, sendo fundamental nos casos duvidosos.
Nos casos de condrite ou artrite leves, pode-se utilizar antiinflamatórios não hormonais ou corticosteroides em baixas doses.
Nos casos potencialmente graves, utiliza-se corticosteroides, p. ex., prednisona 1 mg/kg/dia e, às vezes, imunossupressores (azatioprina, ciclofosfamida, ciclosporina, clorambucil).
1. Imboden J, Hellmann D. Current Rheumatology. Lange, 2004.
2. Kelley WN. Textbook of rheumatology. Filadélfia, 1997.
3. Hochberg MC, Silman AJ. Rheumatology. 4.ed, Mosby, 2007.
4. Herman JH. Curr Opin Rheumatol 1991;3:28-31.
5. Issak BL. Ophtalmology 1986;93:681-689.
6. Cohen PR. Int J Dermatol 1986;25:280-285.
7. Sharma A et al. Scand J Rheumatol 2007;36:462-465.
8. Marchiori E et al. J Bras Pneumol 2008;34:47-54.
9. Kao KT et al. Clin Rheumatol 2007;26:1985-1988.
Sandra Regina Miyoshi Lopes
Professora Assistente da Disciplina de Reumatologia da Faculdade de Medicina de Catanduva. Especialista em Reumatologia pelo Serviço de Reumatologia do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo (HCFMUSP).
Samuel Katsuyuki Shinjo
Professor Colaborador da Disciplina de Reumatologia da Faculdade de Medicina da Universidade de São Paulo (FMUSP). Médico Assistente do Serviço de Reumatologia do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo (HCFMUSP).
A hemocromatose hereditária (HH) é autossômica recessiva, aonde ocorre distúrbio do metabolismo do ferro, caracterizado pelo aumento da absorção intestinal de ferro. Há depósitos progressivos nos órgãos e tecidos, resultando em lesões teciduais e comprometimento funcional. Os órgãos mais afetados são fígado, articulações, coração, pâncreas e hipófise.
Ocorre com maior frequência entre 40 - 60 anos de idade, acometendo 1 mulher : 4 homens. Tem incidência anual 2 a 4/100000 habitantes. É mais prevalente no norte da Europa (1 a 10/1000 habitantes).
A HH é a doença hereditária mais comum nos países caucasianos.
O gen responsável pela hemocromatose hereditária (gen HFE regula a absorção do ferro, suas mutações ® C282Y e H63D, levam à HH) está intimamente ligado ao locus HLA-A (cromossomo 6).
Na HH, ocorre um aumento na absorção de ferro da dieta e, como a excreção é mínima, há acúmulo de ferro nos tecidos. As principais células envolvidas são as células epiteliais intestinais (absorção do ferro) e os macrófagos (liberam o ferro no plasma).
Várias transfusões sanguíneas, pós anastomose portocava, anemias com sobrecarga de ferro, anemias hemolíticas crônicas, cirrose alcoólica e sobrecarga de ferro alimentar podem acarretar hemocromatose adquirida ou secundária.
As manifestações clínicas e a gravidade são variáveis. Pode haver:
diabetes;
hepatopatia (hepatomegalia e aumento das transaminases). Nessa fase, a maioria é assintomática, mas sintomas como desconforto e dores abdominais inespecíficas podem ocorrer. A hepatopatia geralmente progride para cirrose hepática nos casos não tratados. Cerca de 1/3 dos pacientes cirróticos podem evoluir para carcinoma hepatocelular);
artropatia (acomete até 80% dos pacientes). Há edema e dor, mas geralmente sem rubor e calor. As articulações mais afetadas são: metacarpofalângicas – 2ª e 3ª, punhos, ombros, joelhos, quadris, tornozelos, metatarsofalângicas. Episódios de artrite podem ocorrer quando há associação com condrocalcinose, o que ocorre em 30 a 60% dos casos;
astenia;
hiperpigmentação cutânea (marrom-acinzentada);
arritmia (cardiomiopatia pelo ferro);
insuficiência cardíaca congestiva (ocorre em 30% dos pacientes sendo a principal cauda de óbito nos não tratados);
hipogonadismo (por insuficiência gonadotrófica, causando impotência, diminuição da libido, amenorreia, rarefação de pêlos corpóreos, atrofia testicular).
Nos exames radiológicos, há alterações degenerativas semelhantes aos da osteoartrite, com margens escleróticas, lesões císticas, redução dos espaços articulares, osteófitos (forma de gancho característica). Osteopenia generalizada pode ocorrer (ferro sinovial aumentado inibe a formação óssea).
Osteoartrite, porfiria cutânea tardia, hepatopatia alcoólica.
Quando o diagnóstico é definido, todos os parentes de 1º grau devem ser investigados (saturação de transferrina, ferritina e genótipo) a cada 2 a 5 anos. Devido à forte associação genética, tipagem de HLA pode auxiliar na triagem.
Flebotomia 1 a 2 vezes/semana até valores de ferritina sérica menores do que 50mg/L e de saturação de transferrina menores do que 50%. Após, realizar flebotomias de acordo com a necessidade para mantê-las normais. Diabetes mellitus, hipogonadismo, artropatia e cirrose não melhoram com as flebotomias.
É essencial a abstinência de bebidas alcoólicas. Recomenda-se a interrupção do tabagismo.
É contraindicado o uso doses >500 mg/dia de vitamina C, não sendo necessário proibir o uso de frutas ou vegetais que possam conter vitamina C. Dieta pobre em ferro é desnecessária.
A terapia quelante (p. ex.: desferiprona, desferasirox) e antioxidante (p. ex.: alfatocoferol) pode trazer pouco benefício em pacientes nos quais as flebotomias são contraindicadas.
Os anti-inflamatórios não hormonais são úteis para os sintomas da artropatia.
O transplante hepático não é recomendado, devido a alta mortalidade e acúmulo progressivo de ferro no fígado transplantado.
1. Hochberg MC, Silman AJ. Rheumatology. 4. ed. Mosby,2007.
2. Imboden J, Hellmann D. Current Rheumatology. Lange,2004.
3. Kelley WN. Textbook of Rheumatology. Filadélfia,1997.
4. Franchini M, Veneri D. Ann Hematol 2005;84:347-352.
5. Franchini M, Veneri D. Hematology 2005;10:145-149.
6. Wheeler CJ, Kowdley KV. Compr Ther 2006;32:10-16.
7. Kowdley KV, Trainer TD.Gastroenterology 1997; 113-127.
8. Adams PC, Agnew S. Hepatology1006; 23:724-727.
9. Barton JC, McDonnell SM. Ann Intern Med 1998;129:932-939.
Sandra Regina Miyoshi Lopes
Professora Assistente da Disciplina de Reumatologia da Faculdade de Medicina de Catanduva. Especialista em Reumatologia pelo Serviço de Reumatologia do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo (HCFMUSP).
Samuel Katsuyuki Shinjo
Professor Colaborador da Disciplina de Reumatologia da Faculdade de Medicina da Universidade de São Paulo (FMUSP). Médico Assistente do Serviço de Reumatologia do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo (HCFMUSP).
A osteoartropatia hipertrófica (OH) é uma síndrome caracterizada pela proliferação anormal dos tecidos cutâneos, bem como ósseos, e, estes últimos, particularmente, das extremidades distais do corpo.
Pode ser primária ou secundária. A primária apresenta predisposição hereditária, com 1/3 dos casos com parentesco do 1º grau com mesmo quadro clínico. Ocorre predominantemente em indivíduos do sexo masculino, na razão de 9 homens:1 mulher. No caso de secundária, é classificada em:
1. Localizada: aneurismas, arterites infecciosas, ducto arterioso patente, hemiplegia.
2. Generalizada:
afecção pulmonar: fibrose cística, fibrose pulmonar, neoplasia, infecção crônica, mesotelioma, fístula arteriovenosa;
afecção cardíaca: doenças cianóticas congênitas, endocardite infecciosa;
afecção hepática: carcinoma, cirrose;
afecção intestinal: doença de Cröhn, colite ulcerativa, infecção crônica, polipose gastrintestinal, neoplasias;
afecção mediastinal: carcinoma esofágico, timoma, acalasia;
miscelânea: doença de Graves, talassemia, neoplasias, síndrome de POEMS, outros.
Quanto à fisiopatologia, ocorre edema dos dedos, acompanhado de hiperplasia endotelial e depósito excessivo de colágeno. Uma teoria para OH é que os megacariócitos provenientes da medula óssea são habitualmente fragmentados na microcirculação pulmonar. Em pacientes com OH secundária à afecção cardíaca cianótica, p. ex., os megacariócitos não participariam da circulação pulmonar. No entanto, participariam da circulação sistêmica, liberando fatores de crescimento nos territórios distais, ativando os endotélios e, portanto, induzindo o baqueteamento dos dedos. Em pacientes com neoplasia pulmonar, os fatores de crescimento produzidos pelos tecidos neoplásicos atingiriam a circulação sistêmica, induzindo o baqueteamento dos dedos.
O quadro clínico de OH é amplo. Pacientes podem ser totalmente assintomáticos, com mínimo de alteração dos seus dedos. Por outro lado, podem ter sensação de alodínea e deformidades significativas dos dedos, com incapacidade funcional.
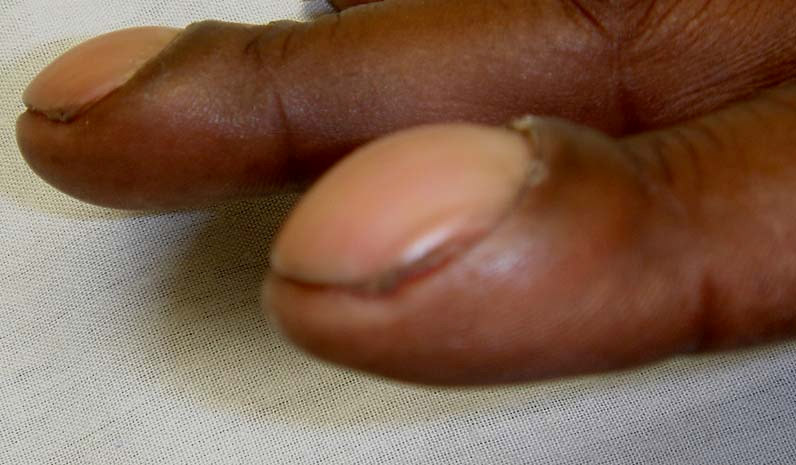
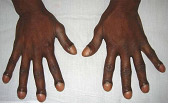
O diagnóstico é clínico e radiológico. Ao exame físico, podemos observar unhas usualmente convexas (unhas em “vidro de relógio”), como mostradas na Figura 2. Os dedos apresentam-se na forma de baqueteamento (Figura 3). Ao estudo de imagens, podemos visualizar periostite que, por sua vez, pode estar acompanhada de dor à palpação local. Ocorre remodelamento ósseo, cursando com processo de acro-osteólise e, mas raramente, neoformação óssea local. Em geral, tais alterações podem ser visualizadas inicialmente nos dedos dos pés e, depois, das mãos. No caso de periostose, apresentam uma distribuição simétrica, preservando espaços articulares sem causar erosões ósseas nem osteopenia periarticular. Estudo com cintilografia óssea é um método sensível para demonstração de envolvimento periosteal. Sinovite com edema articular geralmente pode ser vista em grandes articulações, como joelhos e punhos.
Ocorre também a presença de espessamento cutâneo, particularmente na forma primária de OH. Neste caso, recebe o termo de paquidermoperiostose. Outras alterações que podem cursar em OH são hiperidrose, seborreia e acnes, e combinações destas.
Para o diagnóstico de OH, é necessária a presença de baqueteamento dos dedos e evidência radiológica de periostose em ossos longos. A presença de sinovite não é essencial para o diagnóstico.
Figura 2: Unhas em “vidro de relógio”.
Figura 3: Baqueteamento dos dedos.
No caso de espessamento cutâneo, considerar a acromegalia. Atentar para os casos de OH secundária, cuja etiologia é ampla conforme visto anteriormente.
Baqueteamento dos dedos geralmente é assintomático e, portanto, não requer nenhuma intervenção terapêutica. No caso de pacientes com sintomas de dor, podemos considerar antiinflamatórios não-hormonais para a analgesia.
No caso de OH secundária a neoplasia ou afecção cardíaca (malformação cardíaca ou endocardite infecciosa), é reversível após a remoção dos mesmos.
1. Hochberg MC, Silman AJ. Rheumatology. 4 ed, Mosby, 2007.
2. Martínez-Lavín MD. J Rheumatol 1987;14:6-8.
3. Martínez-Lavín MD, Pineda C, Valdez T, et al. Semin Arthritis Rheum 1988;17:156-162.
4. Segal A, Mackenzie A. Semin Arthritis Rheum 1982;12:220-232.
5. Dickinson CJ, Martin JF. Lancet 1987; ii:1434-1435
André Regis
Especialista em Reumatologia pelo Serviço de Reumatologia do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo – FMUSP.
Sarcoidose é um distúrbio inflamatório sistêmico caracterizado patologicamente pela presença de granulomas não caseosos nos órgãos afetados.
De etiologia desconhecida, ocorre tipicamente em adultos jovens e, embora possa afetar qualquer órgão, é muito comum a presença de pelo menos 3 dos seguintes achados clínicos:
adenopatia hilar bilateral;
infiltrados pulmonares;
lesões de pele e oculares.
Doença musculoesquelética é o achado menos comum na sarcoidose (em torno de
O envolvimento muscular na sarcoidose costuma ser assintomático. Existem três apresentações clínicas bem conhecidas: nodular, miosítica aguda e miopática crônica. A apresentação nodular é a menos comum e envolve as junções musculotendíneas. A miosite aguda inflamatória é rara, sendo observada com mais frequência em mulheres. A forma mais comum é a miopatia crônica, manifestada por início insidioso de fraqueza muscular proximal simétrica. Tais formas de envolvimento muscular devem ser diferenciadas por enzimas musculares, eletroneuromiografia e biópsia muscular.
Artrites e periartrites ocorrem em
Geralmente está presente no curso inicial da sarcoidose, isoladamente ou como parte da síndrome de Lofgren. Esta síndrome caracteriza-se pela tríade adenopatia hilar, poliartrite aguda e eritema nodoso. As articulações mais envolvidas são os joelhos e os tornozelos, e podem, muitas vezes, ser confundidas com artrite reativa. Geralmente esta síndrome é autolimitada. O eritema nodoso desaparece em poucos meses e os sintomas articulares com 2 anos. Entretanto, 1/3 dos pacientes podem apresentar artrite persistente, e a recorrência é rara.
O envolvimento articular crônico começa cerca de 6 meses ou mais após o início dos sintomas da doença. Geralmente é menos severo e comum do que o envolvimento agudo. Joelhos, tornozelos e interfalângicas proximais das mãos são as articulações mais envolvidas, enquanto as sacroilíacas e temporomandibulares podem ser afetadas mais raramente.
Diferentes formas de artrite crônica podem ocorrer entre os pacientes com sarcoidose:
artrite não deformante com sinovite granulomatosa;
deformidade tipo Jaccoud (não erosiva);
edema articular adjacente a lesão óssea pela sarcoidose;
dactilite (edema de partes moles em um ou mais digitais).
A presença de artrite crônica é frequentemente associada com doença pulmonar parenquimatosa e elevação da concentração sérica da enzima de conversão da angiotensina (ECA). A correlação entre os níveis altos de ECA e manifestações extratorácicas da sarcoidose, incluindo artrite crônica, provavelmente refletem o alto peso corpóreo de granulomas.
1. Laboratoriais: anemia típica de doença crônica e linfopenia são observadas na maioria dos pacientes; elevação de VHS, PCR e hipergamaglobulinemia são comuns durante a fase aguda.
2. Radiografia convencional de mãos e outras articulações: são normais, podendo-se observar edema de partes moles.
3. Imagem com radioisótopo Gálio67: pode revelar padrões característicos da evolução da sarcoidose, porém não é útil para correlacionar o envolvimento articular entre agudo e crônico.
4. Biópsia sinovial: é nescessária para confirmação diagnóstica de casos duvidosos, com poucos sintomas clínicos. Pode ser obtido por meio de artroscopia com histologia típica de granulomas não caseosos.
Algumas doenças podem apresentar clínica semelhante à sarcoidose e com presença de granulomas na biópsia sinovial, como infecção por micobactérias (p. ex.: Mycobacterium tuberculosis), infecção fúngica (p. ex.: Histoplasma capsulatum), vasculites sistêmicas (granulomatose de Wegener), doença de Cröhn, policondrite recidivante, tofos (cristais de ácido úrico ou pirofosfato de cálcio) e corpos estranhos.
Desde que a artrite aguda seja autolimitada, a maioria dos pacientes é inicialmente tratada com antiinflamatórios não hormonais (AINH) por pelo menos 2 semanas. Caso haja contraindicações ou ineficácia dos AINH, outros medicamentos podem ser efetivos:
colchicina (0,5 mg
sulfato de hidroxicloroquina (< 6,5 mg/kg/dia) ou difosfato de cloroquina (< 3~4 mg/kg/dia);
corticosteroides (p. ex., prednisona 10 a 15 mg/dia).
Em pacientes não-responsivos a tais terapias, metotrexato pode ser efetivo nas doses de
Sinovectomia pode ser indicada em casos mais resistentes.
1. Hochberg M, Silman A. Rheumatology 4 ed, Mosby, 2007.
2. Abril A, Cohen MD. Rheumatologic manifestations of sarcoidosis. Curr Opin Rheumatol 2004;16:51-55.
3. Gendel BR et al. Sarcoidosis: a review with twenty-four additional cases. Am J Med 1952;12:205-218.
4. Visser H et al. Sarcoid arthritis: clinical characteristics, diagnostic aspects, and risk factors. Ann Rheum Dis 2002;61:499-504.
5. Spilberg I et al. The arthritis of sarcoidosis. Arthritis Rheum 1969;12:126-137.
6. Siltzbach LE, Duberstein JL. Arthritis in sarcoidosis. Clin Orthop Relat Res 1968;57:31-50.
7. Glennas A et al. Acute sarcoid arthritis: occurrence, seasonal onset, clinical features and outcome. Br J Rheumatol 1995;34:45-50.
8. Isdale AH, Iveson JM. Synovial cysts and sarcoid synovitis. Br J Rheumatol 1992;31:497-499.
A amiloidose compreende um grupo heterogêneo de doenças que possuem em comum o depósito extracelular de uma substância proteica dita amiloide.
As doenças amiloides podem ser divididas em adquiridas (mais comuns) ou hereditárias, ou ainda sistêmicas ou localizadas de acordo com a extensão do depósito.
A amiloidose primária sistêmica é uma discrasia de célula plasmática com envolvimento multissistêmico, progressão rápida e sobrevida curta. Os principais órgãos acometidos são os rins, pulmões e coração. Outros achados são síndrome do túnel do carpo, macroglossia, hepatomegalia e má absorção. A presença de um dos sintomas descritos associado à proteína monoclonal sérica ou urinária constitui forte suspeita de amiloidose.
Outras formas de amiloidose são a amiloidose associada ao mieloma múltiplo, amiloidose secundária ou reativa ou adquirida (complicação pouco comum de estados inflamatórios crônicos, infecções e determinados tipos de neoplasias), amiloidose heredofamiliar, amiloidose local, amiloidose associada à hemodiálise crônica, amiloidose cerebral e amiloidose endócrina.
O diagnóstico de amiloidose baseia-se na demonstração histológica do depósito tecidual de amiloide, que classicamente é demonstrada pela coloração com vermelho Congo. A biópsia deve ser preferencialmente realizada no órgão suspeito de infiltração amiloide, considerando-se que existe o risco de complicações relacionadas a sangramento. A biópsia de reto ou de gordura subcutânea são as menos invasivas.
A policondrite recidivante (PCR) é uma doença do tecido conectivo, autoimune, rara e de etiologia desconhecida. Caracteriza-se por inflamação recorrente de tecidos cartilaginosos. A manifestação clássica é a condrite auricular, ocorrendo em 85% dos casos.
Outras manifestações são condrite nasal, poliartrite não erosiva, inflamação ocular, condrite laringotraqueobrônquica e disfunção vestibulococlear.
O tratamento de casos leves é com anti-inflamatórios ou corticoesteroides em baixas doses e os casos graves são tratados com doses maiores de corticosteroide e/ou imunossupressores.
As principais manifestações clínicas da hemocromatose são hepatopatia, artropatia, arritmias cardíacas, insuficiência cardíaca, e hipogonadismo.
É importante o reconhecimento precoce da doença, visto que o tratamento é muito eficaz (flebotomias), mas não reverte as manifestações já estabelecidas da doença.
A manifestação clássica da osteoartropatia hipertrófica é o baqueteamento dos dedos, e costuma ser acompanhado de periostose em ossos longos visualizada pela radiografia.
Aproximadamente 1/4 dos pacientes com sarcoidose tem artropatia associada.
Artrite aguda envolve, com frequência, as articulações das extremidades inferiores e pode estar acompanhada de adenopatia hilar e eritema nodoso (síndrome de Lofgren) que costuma se resolver espontaneamente, embora 1/3 destes pacientes possam desenvolver artrite persistente.
Artrite crônica é frequentemente associada com doença pulmonar parenquimatosa e outras manifestações extrapulmonares.
O envolvimento muscular em geral é assintomático.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.