(Carregando Índice)... (Carregando Índice)... |
Autores:
Letícia Schwerz Weinert
Médica internista e endocrinologista. Professora da Faculdade de Medicina da UCPel. Doutoranda do Programa de Pós-graduação
em Ciências Médicas: Endocrinologia da UFRGS.
Leonardo Rauber Schmitt
Acadêmico da Faculdade de Medicina da UFRGS. Bolsista PIBIC/CNPq/UFRGS do Serviço de Endocrinologia do HCPA.
Bárbara Simionato
Médica.
Paula Kalinka Menegatti
Médica residente do Serviço de Endocrinologia do HCPA.
Sandra Pinho Silveiro
Médica endocrinologista. Professora associada do Departamento de Medicina Interna da UFRGS. Preceptora do Programa de Residência
Médica em Endocrinologia do HCPA. Professora
do Programa de Pós-graduação em Ciências Médicas: Endocrinologia da UFRGS. Doutora em Medicina: Ciências Médicas pela UFRGS.
Última revisão: 09/07/2014
Comentários de assinantes: 0
Um paciente do sexo masculino, 46 anos, procura serviço de emergência devido a cefaleia, palpitação, dispneia, dor torácica e sudorese de início súbito há 20 minutos. Episódios semelhantes vêm ocorrendo há cerca de um ano, com aumento de intensidade dos sintomas no último mês. O paciente relata ser previamente hígido e não utilizar medicações cronicamente. Ao realizar exame físico, ele apresenta-se pálido, com tremores de extremidades, frequência cardíaca de 104 bmp, pressão arterial de 182/110 mmHg sentado e de 148/90 mmHg em ortostatismo, frequência respiratória de 20 rpm, afebril, sem outras particularidades no que diz respeito ao exame físico cardiovascular, respiratório ou abdominal. A avaliação inicial com eletrocardiograma evidencia taquicardia sinusal e troponinas dentro da normalidade; raio X de tórax com discreto aumento da área cardíaca; tireotrofina, glicemia, hemograma, função renal e hepática normais. O paciente é hospitalizado para investigação do quadro.
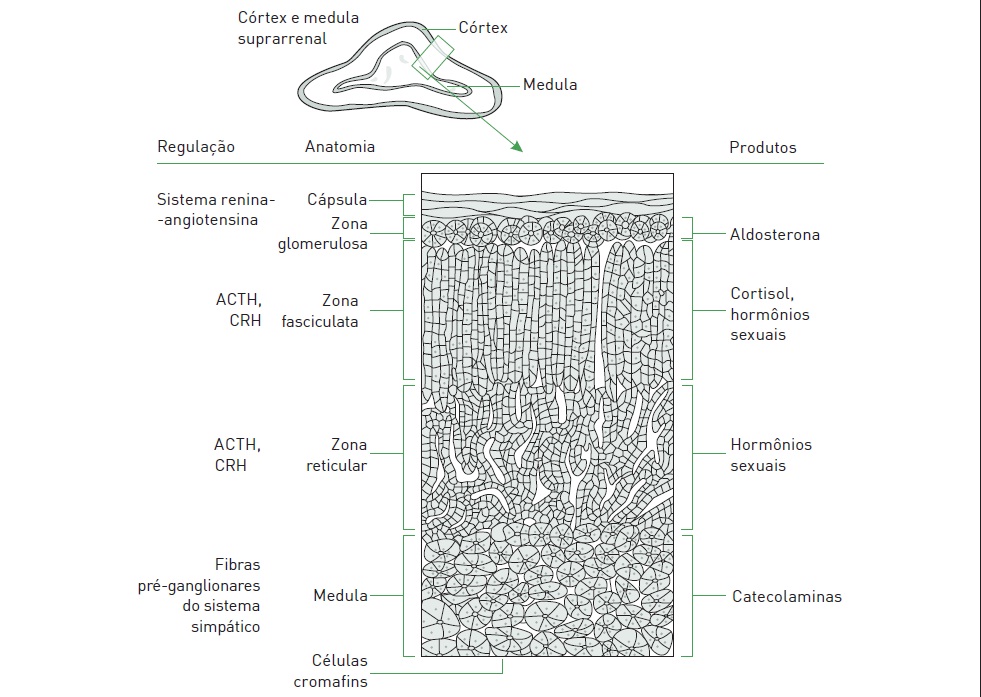
Os feocromocitomas são tumores secretores de catecolaminas formados pelas células cromafins da medula suprarrenal. A apresentação clínica desses tumores é semelhante à dos paragangliomas secretores de catecolaminas, e, por isso, alguns autores consideram estes como feocromocitomas extra-suprarrenais, apesar de serem provenientes dos gânglios simpáticos.
A importância clínica do diagnóstico e do tratamento desses tumores é a possibilidade de haver hipertensão arterial potencialmente curável, paroxismos associados a risco de morte, malignidade e familiares afetados.
Os feocromocitomas são tumores considerados raros, sendo a incidência anual de casos de aproximadamente 0,80/100.000 pessoas a cada ano, embora muitos casos sejam diagnosticados apenas na autópsia. Entre os pacientes com hipertensão arterial sistêmica, a prevalência estimada de portadores de tumores produtores de catecolaminas encontra-se entre 0,1 e 0,6%.
As manifestações clínicas dos tumores produtores de catecolaminas geralmente são causadas devido ao excesso de catecolaminas circulantes e constam no Quadro 32.1. Hipertensão, cefaleia, sudorese e palpitação são os sinais/sintomas mais frequentemente relatados. Alguns sintomas podem estar presentes de forma contínua, embora outros sejam evidentes apenas durante os episódios de paroxismos. Os paroxismos podem ocorrer de forma espontânea ou após fator desencadeante, como ansiedade, exercício, medicações (p. ex., anestésicos) e aumento da pressão intra-abdominal. Eles são autolimitados e recorrentes. A frequência e a duração dos paroxismos são variáveis entre os pacientes, podendo ocorrer várias vezes ao dia ou apenas uma vez ao mês e durar de poucos minutos até várias horas, com duração média de 15 a 20 minutos.

A hipertensão arterial está presente de forma persistente em aproximadamente metade dos pacientes, de forma paroxística em um terço, e cerca de 20% dos pacientes podem apresentar-se normotensos. A hipotensão ortostática é um sinal característico no exame físico. Normo ou hipotensão são de frequente incidência em pacientes com tumor produtor de dopamina.
Tanto homens quanto mulheres podem ser afetados geralmente entre 40 a 60 anos. Cerca de 10% dos casos ocorrem em crianças.
A maioria dos tumores manifesta-se de forma isolada, embora possa haver associação a síndromes genéticas em 10 a 24% dos casos. Nesses casos, as manifestações clínicas típicas dessas síndromes podem predominar. As doenças hereditárias mais comumente associadas aos feocromocitomas são neoplasia endócrina múltipla tipo 2(NEM-2 A: associação com carcinoma medular de tireoide, hiperparatireoidismo, ou NEM-2 B: associação com ganglioneuromas de mucosas), doença de vonHippel-Lindau (VHL), neurofibromatose tipo 1 (NF-1) e paragangliomas familiares.
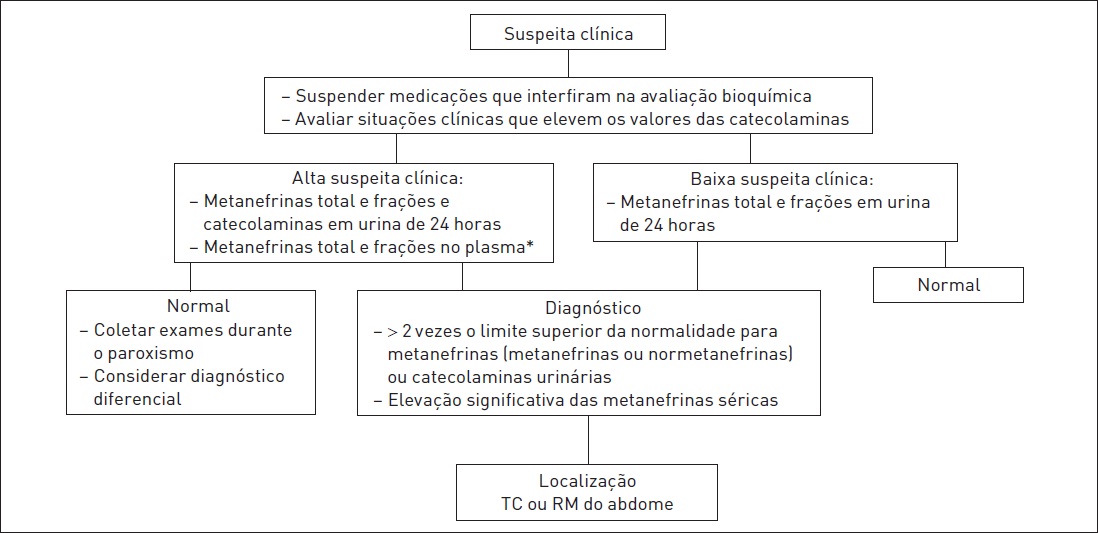
Deve-se suspeitar de diagnóstico de tumor produtor de catecolamina quando os pacientes apresentarem uma ou mais das características citadas no Quadro 32.2. A abordagem diagnóstica é sugerida na Figura 32.1.
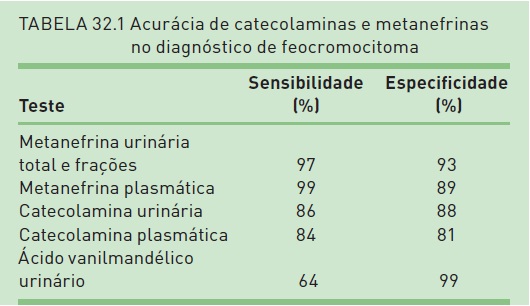
A suspeita clínica de feocromocitoma deve ser avaliada por meio de dosagem das catecolaminas (dopamina, norepinefrina e epinefrina) e seus metabólitos (metanefrinas e normetanefrinas) na urina ou no plasma. Embora ainda em debate, hoje em dia a melhor estratégia para investigação de pacientes considerados de baixa probabilidade para feocromocitoma (pacientes em investigação por hipertensão ou incidentaloma suprarrenal) éadosagemdemetanefrinas (metanefrina e normetanefrina) e catecolaminas em urina de 24 horas, devido à elevada sensibilidade e especificidade (98% quando ambos os exames são realizados). A dosagem de metanefrinas plasmáticas é considerada, por alguns autores, como o melhor exame diagnóstico, e sua dosagem está indicada em situações de alta suspeição clínica (p. ex., síndrome genética associada a feocromocitoma, feocromocitoma prévio, lesão suprarrenal característica), devido à alta sensibilidade, porém moderada especificidade. No entanto, a dosagem de metanefrinas plasmáticas está disponível apenas em alguns centros, limitando sua utilização. A dosagem de dopamina urinária pode ser realizada quando houver suspeita de tumores produtores de dopamina, os quais apresentam dosagem normal de metanefrinas. A sensibilidade e a especificidade dos diferentes exames são descritas na Tabela 32.1.
A condição clínica do momento da avaliação laboratorial deve ser considerada, uma vez que diversos fármacos e algumas situações clínicas acarretam resultados falso-positivos para feocromocitoma, conforme descrito no Quadro 32.3. Fármacos como os antidepressivos tricíclicos devem ser suspensos por duas semanas antes da avaliação hormonal se possível. Outros cuidados para a coleta de exames constam no Quadro 32.4.
A avaliação por imagem deve ser realizada somente quando os testes bioquímicos confirmam o diagnóstico de tumor secretor de catecolaminas. Estes se localizam nas suprarrenais em 85% e no abdome em 95% dos casos (os paragangliomas localizam-se mais frequentemente na região abdominal para-aórtica superior e inferior). Os paragangliomas extra-abdominais podem estar localizados no tórax (10%), na ca beça e no pescoço (3%) ou na pelve (2%). Aproximadamente 10% dos tumores são múltiplos ou bilaterais.
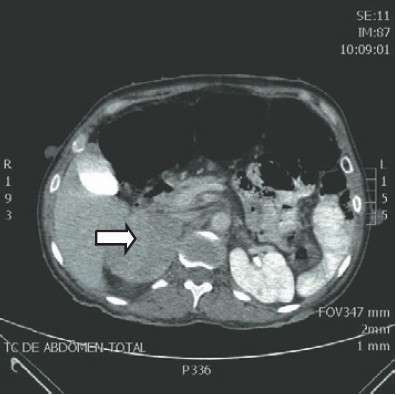
Ressonância nuclear magnética (RM) ou tomografia computadorizada (TC) de abdome podem ser utilizadas para a localização do feocromocitoma, com sensibilidade de 95%. A utilização de contraste com baixa osmolaridade na realização de TC é considerada segura, mesmo em pacientes que não estão alfa ou betabloqueados. As características radiológicas do feocromocitoma são descritas na Tabela 32.2 e demonstradas na Figura 32.2.
A cintilografia com I-metaiodobenzilguanidina(123I-MIBG) atua por meio do acúmulo do radiofármaco nos tumores produtores de catecolamina e está indicada em casos com diagnóstico bioquímico, porém exame de imagem abdominal negativo. Outras indicações para a realização são tumor maior do que 10 cm de diâmetro e presença de paraganglioma, já que esses fatores estão associados a risco elevado de malignidade e paragan gliomas adicionais. A sensibilidade da cintilografia é de aproximadamente 80%, inferior à da TC e à da RM, e não apresenta vantagens nos casos de feocromocitoma localizado por esses exames.
A cintilografia com 111In-DTPA-pentetreotide (octreoscan®) e a tomografia com emissão de pósitrons (PET) com 18F-fluorodeoxiglicose são exames menos utilizados, reservados geralmente para casos de doença metastática com imagem negativa. A biópsia dos tumores não está indicada devido à elevada taxa de complicações.
Abordagem diagnóstica dos tumores produtores de catecolaminas.
* Dosagem plasmática não é amplamente disponível.
Aproximadamente 40% dos casos de feocromocitomas e paragangliomas (isolados, familiares e associados a síndromes genéticas) apresentam mutação genética identificada. Os genes que conferem suscetibilidade a esses tumores são os associados às doenças genéticas, como os genes VHL, RET (associado à NEM-2) e NF1, mas também outros genes, como os SDHB eSDHD, da succinato desidrogenase e associados a tumores familiares. A testagem genética está indicada nos casos citados no Quadro 32.5.
O tratamento-padrão dos tumores secretores de catecolaminas é a cirurgia, que é potencialmente curativa.
O preparo pré-operatório deve ser realizado em todos os pacientes a fim de evitar complicações perioperatórias como hiper e hipotensão, isquemia cardíaca e arritmias. Os alfa- bloqueadores, preferencialmente a fenoxibenzamina, são utilizados para normalização da pressão arterial e expansão do volume vascular, devendo ser iniciados de 7 a 10 dias antes da cirurgia. A efetividade de seu uso, entretanto, é controversa. Como esse fármaco não é prontamente disponibilizado, outro alfabloqueador, como o prazosin, pode ser administrado. O uso de betabloqueadores é recomendado para o controle da taquicardia, porém deve-se iniciá-lo apenas após o alfabloqueio adequado, já que o uso isolado de betabloqueadores pode causar hipertensão grave. Em pacientes com alfa e betabloqueio ineficaz ou intolerável ou nos pacientes com previsão de grande manipulação ou destruição tumoral (ablação por radiofrequência ou presença de metástases), pode-se utilizar a metirosina, um inibidor da síntese de catecolaminas. A mortalidade cirúrgica é baixa, ocorrendo em apenas 2% dos casos, enquanto a morbidade pode alcançar 23% dos casos. Os níveis de metanefrinas e catecolaminas, a pressão arterial pré-operatória e a necessidade de reintervenção cirúrgica estão relacionados a taxas mais altas de complicações. Em casos de crise hipertensiva, antes ou após a cirurgia, deve-se escolher o tratamento intravenoso com nitroprussiato de sódio ou nicardipina. Se houver arritmias, administra-se esmolol ou lidocaína. Hipotensão pode ocorrer durante ou após a retirada tumoral e deve ser manejada com reposição vigorosa de fluidos, iniciando no pré-operatório.
Corticoide deve ser utilizado se houver planejamento de suprarrenalectomia bilateral. Nesses casos, também é possível optar por cirurgia com preservação do córtex suprarrenal para prevenção da deficiência glicocorticoide (Fig. 32.3).
Após 1 a 2 semanas da retirada tumoral, os níveis de metanefrinas e catecolaminas urinárias devem ser reavaliados. Se houver normalização, a ressecção é considerada completa, porém deve haver reavaliação bioquímica anual para vigilância de recorrência tumoral, metástases ou aparecimento tardio de múltiplos tumores primários. Se os níveis laboratoriais mantiverem-se elevados após a cirurgia, há doença residual.
Os tumores malignos devem ser manejados, reduzindo os efeitos de massa e sintomas associados ao excesso de catecolaminas, com ressecção do tumor primário e metástases e tratamento medicamentoso semelhante ao do preparo pré-operatório. Radioterapia para metástases ósseas, tratamento trombótico e ablação com radiofrequência para metástases hepáticas, octreotida, irradiação com 131I-MIBG e quimioterapia são outras opções terapêuticas que podem ser utilizadas.
Tomografia computadorizada de abdome evidenciando feocromocitoma na glândula suprarrenal direita (seta).
Durante a internação hospitalar do paciente, foi realizada investigação para feocromocitoma com dosagem de metanefrina e normetanefrina em urina de 24 horas, com resultado de 1.300 e 455 g respectivamente (valores de referência: 200 e 428 g). Após o diagnóstico laboratorial, foi solicitada TC abdominal que evidenciou lesão nodular de 4,5 cm na suprarrenal direita, heterogênea, com áreas de hemorragia, 25 HU na fase pré-contraste, 50 HU na fase de impregnação pelo contraste e 30% de washout em 10 minutos. Iniciou-se, então, preparação cirúrgica com alfabloqueador, seguida de betabloqueador e hidratação intravenosa. A retirada tumoral foi realizada por meio de videolaparoscopia e houve episódio de hipotensão no período pós-operatório imediato, revertida com reposição volêmica vigorosa. O paciente recebeu alta hospitalar normotenso, com orientação de realizar dosagem de metanefrina urinária após 15 dias.
Suspeita clínica.
Fonte: Adaptada de Le e colaboradores.¹
1. Le T, Bhushan V, Tolles J. First Aid for the USMLE Step 1 2011. New York: McGraw-Hill; 2011.
Baid SK, Lai EW, Wesley RA, Ling A, Timmers HJ, Adams KT, et al. Brief communication: radiographic contrast infusion and catecholamine release in patients with pheochromocytoma. Ann Intern Med.2009;150(1):27-32.
Beard CM, Sheps SG, Kurland LT, Carney JA, Lie JT. Occurrence of pheochromocytoma in Rochester, Minnesota, 1950 through 1979. Mayo Clin Proc. 1983;58(12):802-4.
Edwin B, Kazaryan AM, Mala T, Pfeffer PF, Tonnessen TI, Fosse E. Laparoscopic and open surgery for pheochromocytoma. BMC Surg. 2001;1:2.
Goldstein RE, O’Neill JA Jr, Holcomb GW 3rd, Morgan WM 3rd, Ne- blett WW 3rd, Oates JA, et al. Clinical experience over 48 years with pheochromocytoma. Ann Surg. 1999;229(6):755-64.
Kramer CK, Leitão CB, Azevedo MJ, Canani LH, Maia AL, Czepielewski M, et al. Degree of catecholamine hypersecretion is the most important determinant of intra-operative hemodynamic outcomes in pheochromocytoma. J Endocrinol Invest. 2009;32(3):234-7.
Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet. 2005;366(9486):665-75.
Lenders JW, Pacak K, Walther MM, Linehan WM, Mannelli M, Friberg P, et al. Biochemical diagnosis of pheochromocytoma: which test is best? JAMA. 2002;287(11):1427-34.
Munakata M, Aihara A, Imai Y, Noshiro T, Ito S, Yoshinaga K. Altered sympathetic and vagal modulations of the cardiovascular system in patients with pheochromocytoma: their relations to orthostatic hypotension. Am J Hypertens. 1999;12(6):572-80.
Omura M, Saito J, Yamaguchi K, Kakuta Y, Nishikawa T. Prospective study on the prevalence of secondary hypertension among hypertensive patients visiting a general outpatient clinic in Japan. Hypertens Res.2004;27(3):193-202.
Opocher G, Schiavi F. Genetics of pheochromocytomas and paragangliomas. Best Pract Res Clin Endocrinol Metab. 2010;24(6):943-56.
Perry CG, Sawka AM, Singh R, Thabane L, Bajnarek J, Young WF Jr. The diagnostic efficacy of urinary fractionated metanephrines measured by tandem mass spectrometry in detection of pheochromocytoma. Clin Endocrinol (Oxf). 2007;66(5):703-8.
Plouin PF, Duclos JM, Soppelsa F, Boublil G, Chatellier G. Factors as- sociated with perioperative morbidity and mortality in patients with pheochromocytoma: analysis of 165 operations at a single center. J Clin Endocrinol Metab. 2001;86(4):1480-6.
Sawka AM, Jaeschke R, Singh RJ, Young WF Jr. A comparison of biochemical tests for pheochromocytoma: measurement of fractionated plasma metanephrines compared with the combination of 24-hour urinary metanephrines and catecholamines. J Clin Endocrinol Metab.2003;88(2):553-8.
Sawka AM, Prebtani AP, Thabane L, Gafni A, Levine M, Young WF Jr. A systematic review of the literature examining the diagnostic efficacy of measurement of fractionated plasma free metanephrines in the biochemical diagnosis of pheochromocytoma. BMC Endocr Disord. 2004;4(1):2.
Strong VE, Kennedy T, Al-Ahmadie H, Tang L, Coleman J, Fong Y, et al. Prognostic indicators of malignancy in adrenal pheochromocytomas: clinical, histopathologic, and cell cycle/apoptosis gene expression analysis. Surgery. 2008;143(6):759-68.
Taieb D, Sebag F, Hubbard JG, Mundler O, Henry JF, Conte-Devolx B. Does iodine-131 meta-iodobenzylguanidine (MIBG) scintigraphy have an impact on the management of sporadic and familial phaeochromo- cytoma? Clin Endocrinol (Oxf). 2004;61(1):102-8.
Young WF Jr, Kaplan NM. Clinical presentation and diagnosis of pheo- chromocytoma [Internet]. Waltham: UpToDate; 2011 [capturado em 5 set. 2012]. Disponível em: http://www.uptodate.com/contents/clinical-presentation-and-diagnosis-of-pheochromocytoma. Acesso restrito.
Young WF Jr, Maddox DE. Spells: in search of a cause. Mayo Clin Proc.1995;70(8):757-65.
Young WF Jr. Pheochromocytoma and paraganglioma. In: Kronenberg HM, Melmed S, Polonsky KS, Larsen PR. Williams textbook of endocrinology. 11th ed. Philadelphia: W.B. Saunders; 2008. p. 507-21.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.