(Carregando Índice)... (Carregando Índice)... |
Autor:
Jordana de Fraga Guimarães
Médica internista.
Última revisão: 16/12/2013
Comentários de assinantes: 0
Uma paciente do sexo feminino, 30 anos, negra, procura auxílio médico no serviço de emergência devido a fadiga e dispneia progressiva na realização de esforços, com início há dois meses. A paciente afirma não apresentar dor de peito, ortopneia, dispneia paroxística noturna e/ou edema de membros inferiores, tosse produtiva e/ou qualquer história de asma e doença pulmonar obstrutiva crônica. Durante a revisão de sistemas, ela relata menorragia por aproximadamente três meses. A partir de investigação preliminar, verificam-se eritrócitos de 2 M/ L, hemoglobina de 5 g/dL, volume corpuscular médio (VCM) de 50 fL, 204 plaquetas, leucócitos de 6 M/ L, ferritina de 7 ng/mL, ferro sérico de 22 g/dL e capacidade ferropéxica de 680 g/dL.
O termo anemia refere-se à redução dos níveis de hemoglobina no sangue. A Organização Mundial de Saúde (OMS) define os seguintes valores para caracterizar anemia: hemoglobina de menos de 12 g/dL em mulheres e menos de 13 g/dL em homens. O mecanismo fisiopatológico de desenvolvimento das anemias é a produção deficiente de hemácias ou o aumento da destruição dessas células (Quadro 50.1).
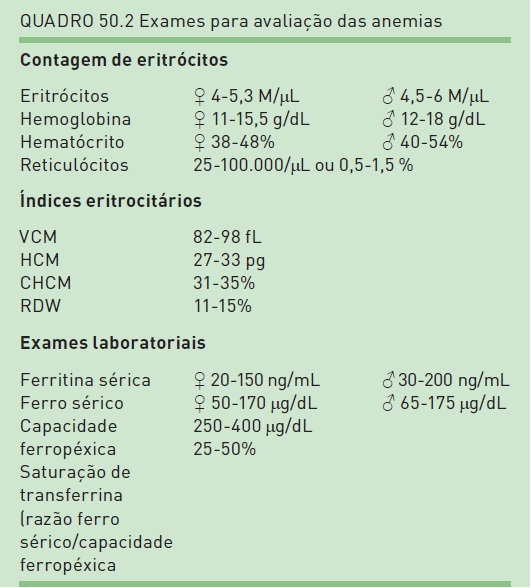
Antes da abordagem isolada de cada uma das anemias, é apresentada uma breve revisão dos principais parâmetros de hemograma e exames laboratoriais utilizados para avaliação. Os valores de normalidade para eles encontram-se no Quadro 50.1.
•Hemoglobina: indica a gravidade da anemia. Atualmente, com os equipamentos automatizados, proporciona mais fidedignidade do que o hematócrito para tal fim.
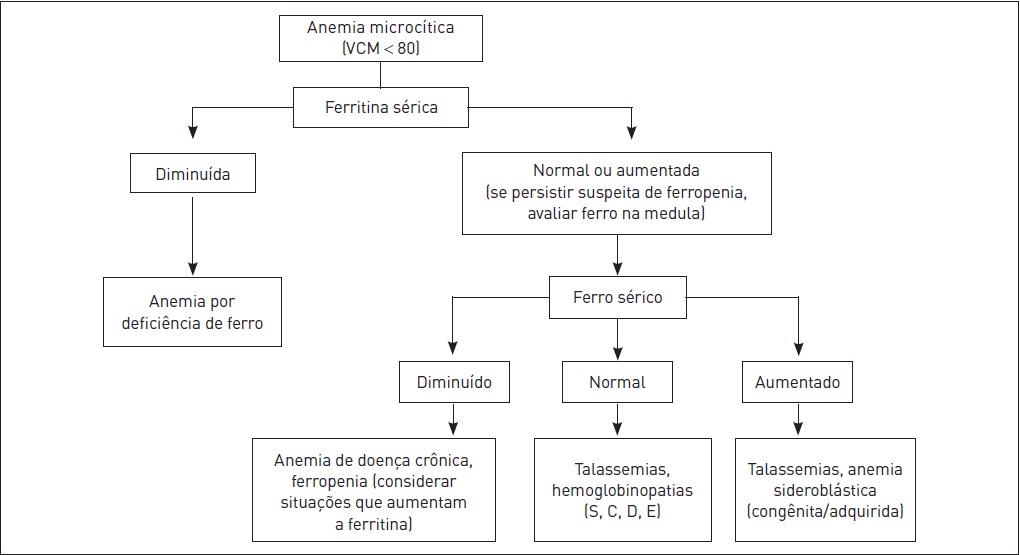
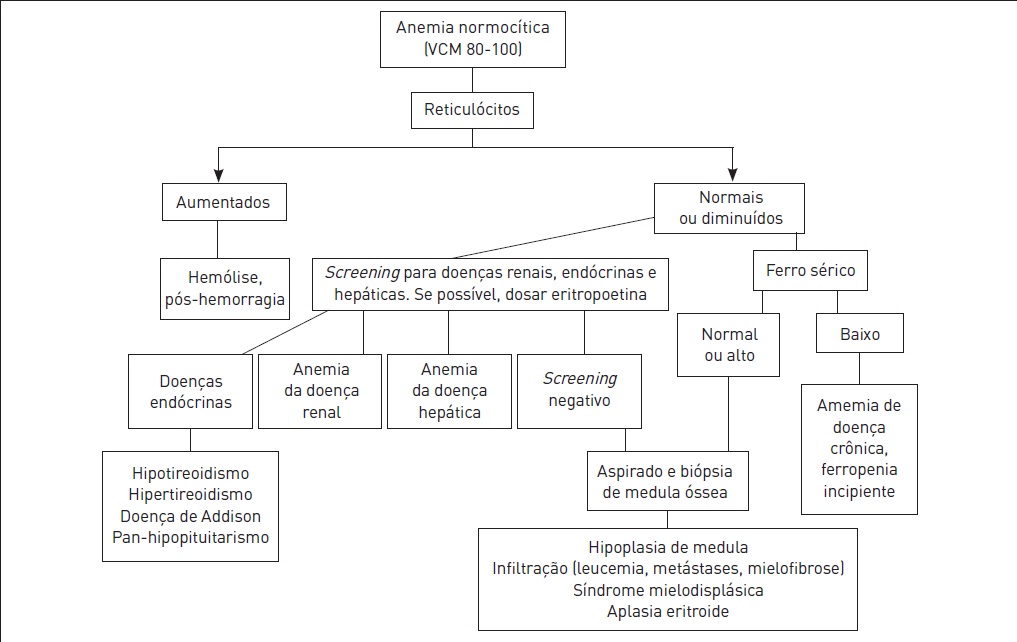
•VCM (volume corpuscular médio): é utilizado para classificar as anemias. Para uma melhor abordagem diagnóstica, as anemias podem ser classificadas de acordo com o resultado do VCM em microcíticas (VCM < 80 fL), normocíticas (VCM entre 80 e 100 fL) e macrocíticas (VCM > 100 fL) (ver Figs. 50.1, 50.3 e 50.4).
•HCM (hemoglobina corpuscular média) e CHCM (concentração hemoglobínica corpuscular média): são índices eritrocitários geralmente paralelos ao VCM.

•RDW (red distribution width): determina o coeficiente de variação do VCM, avaliação objetiva da heterogeneidade das hemácias em relação ao seu tamanho (anisocitose). Valores acima de 14,6% são considerados elevados.
•Contagem de reticulócitos: evidencia a capacidade regenerativa da medula. Valores inferiores a 2% ou contagem absoluta de menos de 50.000/mm³ indicam incapacidade da medula para responder ao estímulo anêmico. Os valores também proporcionam a verificação de adequada função medular.
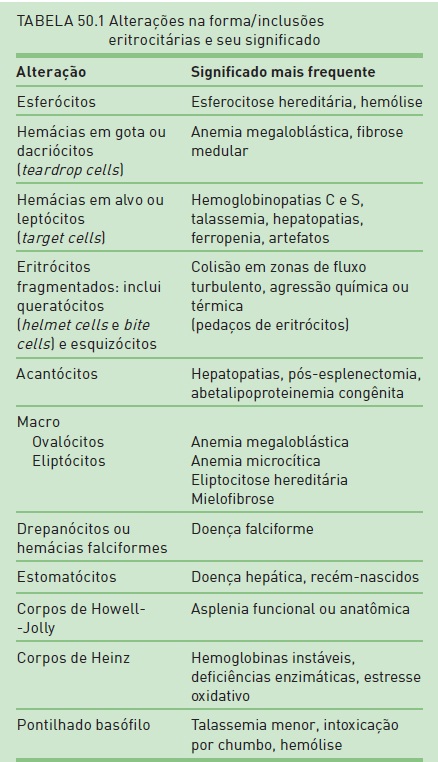
•Microscopia: no esfregaço do sangue periférico, é possível identificar diversas formas eritrocitárias e inclusões nessas células, as quais fornecem importantes indícios no diagnóstico da anemia (Tab. 50.1).
Metabolismo do ferro: a diferença entre transferrina, ferritina e hemossiderina
O ferro é um elemento essencial para a função de todas as células do organismo. A sua principal função é transportar o O2 (como porção heme da hemoglobina). A única fonte de ferro para um indivíduo saudável é a dieta alimentar. O ferro é absorvido no jejuno, um processo facilitado pela acidez estomacal. O transporte por meio da membrana é realizado pelo transportador 1 de metal divalente (DMT1). A partir da célula intestinal, o ferro liga-se à transferrina, a proteína responsável pelo seu transporte no plasma e que é produzida no fígado, pois, quando livre, é altamente tóxico. Em estados de deficiência de ferro, o fígado reage com um aumento da síntese dessa proteína. Geralmente, a transferrina encontra-se saturada, ou seja, ligada ao ferro, em 20 a 33%. Em casos de anemias com deficiência relativa ou absoluta de ferro, a saturação da transferrina apresenta-se diminuída porque há menos ferro para realizar a ligação com essa proteína.
Esse complexo transferrina-ferro circula até haver interação com receptores de transferrina, estando estes presentes em maior número nas células eritroides da medula óssea. Nessas células, o complexo é fagocitado e processado, e o ferro torna-se disponível para a síntese do heme, a transferrina retorna para a circulação, e os receptores novamente se acoplam à membrana plasmática. Dentro das células eritroides e de outras células de depósito, o excesso de ferro que não é utilizado imediatamente para a síntese de heme é armazenado na forma de ferritina. Outra proteína de depósito é a hemossiderina, que, ao contrário da ferritina, libera o ferro lentamente. Os órgãos de depósito de ferro são o fígado, o baço e a medula óssea. A produção eritrocitária é diretamente regulada pela eritropoietina. Esta é um hormônio produzido pelo rim em resposta à pressão parcial de O2 no sangue. As hemácias jovens lançadas na circulação contêm RNA e, por isso, são chamadas de reticulócitos. Após um dia, elas perdem o RNA e tornam-se hemácias.
As hemácias apresentam uma meia-vida média de 120 dias. Após esse período, elas são consideradas senescentes pelo sistema reticuloendotelial do baço e sofrem fagocitose. O ferro é, então, reciclado e volta a circular com a transferrina no plasma até ser utilizado novamente pelas células eritroides da medula óssea. Não existe via excretora de ferro. As únicas formas de perder ferro são por meio de sangramento ou da perda de células epiteliais da pele e do intestino.
•Deficiência de ferro
•Talassemias
•Anemia sideroblástica
•Anemia da doença crônica
•Hemoglobinopatias (S, C, D, E)
•Toxicidade pelo alumínio
A diminuição dos níveis de ferro no organismo é a deficiência nutricional mais comum em todo o mundo e a causa mais frequente de anemia. Entre as causas de anemia microcítica, a anemia ferropriva (ou ferropênica) é a de ocorrência mais prevalente. Esta somente se manifesta quando os estoques de ferro já foram esgotados, o que geralmente demora anos para ocorrer.
O teste de diminuição nos níveis séricos de ferritina é o mais sensível e específico para verificação de anemia ferropriva. Níveis menores do que 15 ng/mL são altamente específicos para diagnóstico de anemia ferropriva. Entretanto, níveis normais não descartam a possibilidade de ferritina, pois a ferritina também é uma proteína de fase aguda. Entre as condições que aumentam a ferritina estão hepatopatias agudas e crônicas, alcoolismo, neoplasias, infecções, doenças inflamatórias e hipertireoidismo. Uma regra simples para estimar as reservas de ferro em estados que interferem na contagem é dividir o seu número por três.
Outras alterações laboratoriais que podem ser apresentadas em casos de anemia ferropriva são as seguintes: concentração de ferro sérico diminuída (< 40 g/dL), saturação da transferrina diminuída, capacidade ferropéxica elevada.
O teste padrão-ouro para evidenciar deficiência de ferro é a coloração de Perls (azul de Prússia) em material aspirado de medula óssea, a qual possibilita verificar diminuição ou ausência nos estoques de ferro do organismo.
Aproximadamente 2% dos homens adultos e até 10% das mulheres apresentam anemia ferropriva. A deficiência de ferro, um estágio mais precoce, pode ocorrer ainda mais frequentemente. A probabilidade de que a deficiência de ferro seja a causa da anemia em uma mulher saudável é de 80 a 90%.
A deficiência de ferro desenvolve-se quando a exigência metabólica não é suprida pela absorção, devido a perda de ferro ou diminuição da absorção (Quadro 50.3). A principal causa dessa condição é a perda sanguínea. Em mulheres pré-menopausa, a causa mais comum é a perda por meio do sangramento menstrual; naquelas na pós-menopausa, o risco de doenças malignas do trato gastrintestinal como causa da anemia aumenta substancialmente.
Sintomas como pica (perversão do hábito alimentar, como geofagia, hábito de comer gelo) e sinais como queilite angular, esplenomegalia leve, atrofia de papilas linguais, glossite e alterações nas unhas, além dos próprios da anemia podem ocorrer.
Figura 50.1
Classificação das anemias conforme VCM (anemia microcítica).
Uma unidade de 300 mL de concentrado de hemácias promove o aumento de aproximadamente 1 g/dL na hemoglobina (Hb). Uma resposta não satisfatória, principalmente em pacientes críticos, requer investigação adicional para perda sanguínea (sangramento ou hemólise) em curso.
Em nível primário, justifica-se tratar crianças que apresentam história de alimentação inadequada e mulheres multíparas ou com hipermenorreia, com baixo risco para doenças graves, encaminhando para investigação os casos de falha terapêutica. Quando possível, a doença de base deve ser tratada. Para reposição das reservas de ferro, o sulfato ferroso (300 mg de sulfato ferroso contém 60 mg de ferro elementar), administrado oralmente, é a preparação menos dispendiosa e o tratamento de escolha para a deficiência de ferro. A dose que deve ser utilizada é de 150 a 200 mg de ferro elementar (2 a 4 cps/dia), preferencialmente em horário afastado das refeições. Com essa conduta, os níveis de hemoglobina começam a aumentar por volta do quinto dia. Deve haver um aumento de pelo menos 1 g/dL nas primeiras duas a três semanas de tratamento. Após correção da hemoglobina, deve-se manter o tratamento por quatro a seis meses para reposição das reservas de ferro. Ácidos elevam a absorção, mas produtos como chá, café, cereais, leite, ovo e medicamentos que diminuem a acidez estomacal (inibidores H2, inibidores da bomba de prótons e antiácidos) reduzem a sua absorção. Os principais efeitos adversos são desconforto abdominal, náusea/vômito, diarreia e constipação. Havendo esses efeitos, reduzir a dose e/ou administrar junto com as refeições. Em caso de constipação, a troca da preparação oral para líquida (5 mL contém 44 mEq de ferro elementar) pode aliviar esse sintoma. Caso não tenha sido resolvida a causa do sangramento, pode ser necessário tratamento de manutenção. O ferro parenteral (IV ou IM) é feito com ferro dextrano. Esse deve ser utilizado restritamente em casos de intolerância (após tentativa de redução de dose, administração com as refeições e formas alternativas, como ferro quelato ou sacarato) ou má absorção, devido ao risco de choque anafilático com probabilidade de ocorrência de 1%. A transfusão com concentrado de hemácias deve ser considerada para pacientes com hemoglobina entre 7 a 8 g/dL, de acordo com a perda sanguínea, doença cardiopulmonar subjacente ou estado clínico.
As anemias sideroblásticas compreendem um grupo heterogêneo de distúrbios em que a síntese da hemoglobina está reduzida devido à dificuldade de incorporar o heme à protoporfirina para formar hemoglobina.
Entre as possíveis causas para essa anemia, estão as formas congênitas e as adquiridas, sendo essas últimas as que ocorrem mais comumente devido à administração de certas drogas, como isoniazida, pirazinamida e cloranfenicol.
Não há sintomas específicos para anemia sideroblástica além dos da anemia.
Embora, na maioria das vezes, este tipo de anemia apresente normocitose ou até mesmo macrocitose, também pode haver hipocromia e microcitose grave, principalmente nas formas congênitas. O diagnóstico dessa anemia é confirmado pela existência de sideroblastos em anel no aspirado de medula óssea, observados com a coloração azul da Prússia. A medula encontra-se expandida com hiperplasia eritroide, mas esta é ineficaz, uma vez que não resulta em um aumento de reticulócitos no sangue periférico. Podem existir duas populações dimórficas de hemácias na periferia: uma normal e outra hipocrômica. Outras alterações laboratoriais apresentadas são níveis de ferro sérico e ferritina elevados, saturação de transferrina alta e capacidade ferropéxica (CFP) baixa. Há uma sobrecarga de ferro, principalmente nas mitocôndrias, pois a reabsorção desse elemento está inapropriadamente aumentada, podendo acarretar quadro de hemossiderose.
Não há um tratamento específico para essa anemia, e os pacientes não respondem à administração de eritropoietina exógena; por isso, em casos graves, deve ser considerada a transfusão com concentrado de hemácias. A reposição de ferro pode piorar o quadro ao aumentar os estoques de ferro.
As talassemias são distúrbios hereditários que se caracterizam pela redução da síntese de cadeias de globina (alfa ou beta), causando, concomitantemente, a redução da síntese de hemoglobina, sendo, por isso, considerada uma anemia hipoproliferativa.
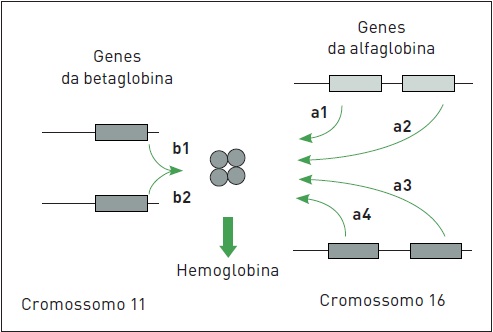
Durante as diversas etapas do desenvolvimento, são produzidas hemoglobinas diferentes. Cada uma delas consiste em um tetrâmero (2 dímeros) de cadeias de globina. Um adulto normal apresenta predominantemente hemoglobina do tipo A (98%), a qual é formada por duas cadeias alfa e duas cadeias beta (a2ß2). Cada gene da alfaglobina produz metade da quantidade de globina produzida por cada gene da betaglobina. Os dois genes da betaglobina produzem duas cadeias, cada uma com um heme próprio, para formar um dos dímeros. Já os quatro genes da alfaglobina produzem duas cadeias de globinas adicionais, cada uma com um heme próprio também, para formar o dímero restante. Os dois dímeros combinam-se,então, gerando a molécula da hemoglobina. Outras cadeias incluem os tipos delta e gama, as quais constituem as hemoglobinas A2 (a2d2), que representam 1 a 2% das hemoglobinas no adulto, e F (a2?2), que é a principal hemoglobina na vida intrauterina, representando menos de 1% das hemoglobinas no adulto (Fig. 50.2).
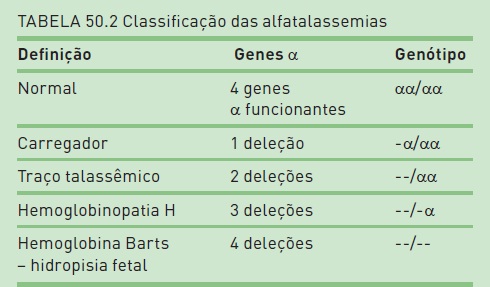
As alfatalassemias são caracterizadas pela deleção de um ou mais dos quatro genes existentes no cromossomo 16, responsáveis pela produção da cadeia alfa. Como há essa cadeia de globina em todos os tipos de hemoglobina, não ocorre alteração na proporção de distribuição entre os tipos de hemoglobina A, A2 e F. Indivíduos normais apresentam as quatro cópias dos genes para a cadeia alfa. Havendo três cópias, os indivíduos são denominados carreadores silenciosos, sendo uma condição assintomática e um pouco mais frequente em negros. Existindo duas cópias apenas, o paciente apresenta traço talassêmico (um tipo de talassemia menor). Esse indivíduo é saudável, e o único dado para que se deve atentar é uma anemia leve microcítica. Quando há apenas um gene, o paciente evidencia hemoglobinopatia H, um quadro com gravidade variável (talassemia menor/intermediária). Nesses pacientes, podem ocorrer hemólise, pois corpúsculos com excesso das cadeias beta são reconhecidos no baço, principalmente em situações de estresse, como nas infecções. Caso os quatro genes da cadeia alfa inexistam, desenvolve-se o quadro de hidropisia fetal com asfixia intrauterina e morte fetal (Tab. 50.2).
Figura 50.2
Representação da síntese de cadeias de hemoglobina.
361
362
As betatalassemias são causadas por diversos tipos de mutações nos genes responsáveis pela produção dessa cadeia, localizados no cromossomo 11. O resultado dessas mutações é a diminuição da síntese de cadeias beta. Defeitos que ocasionam ausência dessa cadeia são denominados ß0, enquanto os que reduzem a síntese, ß+. Ao contrário das alfatalassemias, há um aumento da proporção de hemoglobina A2 e F comparada à da hemoglobina A, já que as cadeias delta e gama substituem a cadeia beta ausente. Indivíduos homozigóticos (ß+, ß+ ou ß0, ß0) apresentam betatalassemia maior, um quadro que se caracteriza clinicamente por baixa estatura, deformidades ósseas, fraturas patológicas, cálculos biliares, hepatoesplenomegalia e icterícia. Fisiopatologicamente, a medula óssea encontra-se expandida com hiperplasia dos precursores eritroides; entretanto, essa eritropoiese é ineficaz. Há também hemólise no baço, que reconhece as hemácias com os corpúsculos de inclusão formados pelo precipitado de cadeias alfa em excesso. A realização de transfusões sanguíneas de repetição aumentam a sobrevida desses pacientes, mas a sobrecarga de ferro e a consequente hemocromatose causam insuficiência cardíaca e óbito aproximadamente entre 20 a 30 anos de idade. A desferoxamina é um agente quelante do ferro utilizado para o controle desses casos. Os pacientes com betatalassemia intermediária apresentam um fenótipo mais leve e podem não necessitar de transfusão para viver. Indivíduos heterozigóticos ( ß+, ß ou ß0, ß ) são considerados betatalassêmicos menor e não apresentam manifestações clínicas, exceto uma leve anemia microcítica.
•Anemia por doença crônica
•Anemia da doença renal crônica
•Hemólise
•Perda sanguínea aguda
•Anemia aplásica
•Aplasia eritroide pura
•Endocrinopatias
•Infiltração da medula óssea
Essa anemia é a segunda mais comum e manifesta-se um a dois meses após o início de doenças que ativam o sistema imunológico/inflamatório. É comum a associação da anemia de doença crônica à anemia ferropriva, e o diagnóstico diferencial entre elas pode ser problemático.
São etiologias dessa anemia infecções crônicas (p. ex., tuberculose, endocardite, osteomielite, Aids, etc.), neoplasias, doenças reumáticas inflamatórias (p. ex., artrites reumatoides, lúpus eritematoso sistêmico, sarcoidose, vasculite, doença inflamatória intestinal), doença hepática crônica, insuficiência cardíaca congestiva, trauma grave, diabetes melito.
Em caso de anemia de doença crônica, há uma diminuição da produção de hemácias pela medula óssea, ao mesmo tempo em que a meia-vida dessas células está reduzida. Os mecanismos que ocorrem são diversos: deficiência absoluta ou relativa de eritropoietina e bloqueio da absorção e utilização de ferro (acúmulo no sistema reticuloendotelial). As citocinas inflamatórias (IL-1, IL-6, TNF-alfa e IF-gama) produzidas pelos macrófagos e os linfócitos ativados são os responsáveis por essas alterações no metabolismo do ferro. Uma proteína de fase aguda recentemente identificada, a hepcidina, produzida no fígado, bloqueia a absorção de ferro no intestino e impede a liberação do ferro pelos macrófagos para o consumo na eritropoiese, induzindo um estado de hipoferremia.
Alterações no hemograma em caso de anemia de doença crônica
Essa anemia geralmente não é grave (Hb 10-11 g/ dL), com VCM normal (normocítica) ou baixo (mi crocítica). Ela é considerada hipoproliferativa, pois os reticulócitos estão diminuídos ou normais, e o RDW apresenta-se normal ou um pouco diminuído.
Alterações laboratoriais
Os níveis de ferro sérico estão baixos, e os da ferritina podem estar normais ou elevados. A saturação de transferrina, em geral, está normal ou diminuída, e a capacidade ferropéxica é baixa. A dosagem de outras proteínas de fase aguda, como velocidade de sedimentação globular, fibrinogênio plasmático e proteína C-reativa, encontra-se elevada. No exame da medula, os macrófagos apresentam-se com uma quantidade normal ou elevada de ferro.
Figura 50.3
Classificação das anemias conforme VCM (anemia normocítica).
O tratamento dessa anemia é direcionado à causa básica. Se a anemia tiver uma influência negativa em diversos desfechos associados às doenças, é importante o seu adequado manejo. As indicações para a utilização de eritropoietina são anemia nas seguintes condições: doença renal em estágio final, malignidades não mieloides em quimioterapia e Aids. Alguns pacientes podem responder a altas doses de eritropoietina exógena, especialmente os que apresentam níveis séricos de eritropoietina menores do que 500 um/mL, e à reposição de ferro. A administração de ferro para pacientes com Anemia de doença crônica (ADC) é contestada, mas deve ser utilizada quando houver deficiência e uma resposta insatisfatória à eritropoietina exógena devido à deficiência funcional de ferro. A resposta é avaliada pelo aumento de, pelo menos, 1 g/dL nos níveis de hemoglobina nas primeiras duas a quatro semanas de tratamento. Para evitar os efeitos adversos da eritropoietina, como hipertensão arterial sistêmica, devem-se manter os níveis de hemoglobina menores do que 12 g/ dL. O objetivo é manter a saturação de transferrina acima de 20% e a ferritina acima de 100 ng/dL.
A anemia da doença renal crônica (DRC) pode ser observada quando os valores da filtração glomerular apresentam-se inferiores a 70 mL/min e 50 mL/min em homens e mulheres, respectivamente, e agrava-se paralelamente à redução desse marcador de função renal. Por isso, essa anemia ocorre mais frequentemente nos estágios avançados da DRC.
364
A anemia é um fator de risco modificável para doença cardiovascular. Além disso, alguns estudos indicam que a anemia parece ser um fator de risco independente para a progressão da DRC. Os possíveis mecanismos pelos quais a anemia pode piorar a função renal parecem estar relacionados à hipoxia e ao aumento do estresse oxidativo no tecido renal.
Alterações no hemograma em caso de anemia da doença renal crônica
A anemia geralmente é grave (Hb < 10 g/dL), com valores de VCM normais (normocítica). Ela é considerada hipoproliferativa, pois os reticulócitos estão diminuídos ou normais. O RDW é normal ou um pouco diminuído.
Alterações laboratoriais
A dosagem de ferro sérico é baixo, e a de ferritina pode estar normal ou elevada. A saturação de transferrina geralmente está diminuída, e a capacidade ferropéxica é baixa.
A principal causa de anemia em caso de DRC é a diminuição de produção de poietina pelos fibroblastos peritubulares do córtex renal. Outros fatores também podem contribuir para a ocorrência de anemia nessa população, tais como deficiência de ferro, perdas sanguíneas, deficiência de vitamina B12 e folato, hiperparatireoidismo e inflamação.
Se não tratada, a anemia da DRC está associada a diversas anormalidades fisiológicas, incluindo aumento no débito cardíaco, hipertrofia ventricular, angina, insuficiência cardíaca congestiva, diminuição da acuidade mental e cognitiva.
Estando a saturação de transferrina menor do que 20% e/ou os níveis de ferritina sérica menores do que 100 ng/mL, deve-se iniciar suplementação com ferro. Nas fases iniciais da DRC, há resposta satisfatória à reposição oral de ferro; entretanto, se ocorrer intolerância ou não houver resposta, este deve ser administrado por via intravenosa (sacarato de hidróxido de ferro III), sendo essa a via de administração para indivíduos em diálise. A normalização dos estoques de ferro com persistência da anemia acarreta uso de eritropoietina, o que em geral ocorre apenas a partir do estágio 4. Os níveis-alvo para os pacientes em tratamento conservador e em diálise são hematócrito de 33 a 35% e hemoglobina de 11 a 12 g/dL. A saturação da transferrina deve ser mantida superior a 20%, e os níveis de ferritina sérica iguais ou superiores a 100 ng/dL.
Alterações laboratoriais comuns na maioria dos casos de anemias hemolíticas
A gravidade da anemia é variável. Observa-se reticulocitose em todos os tipos. As anormalidades no sangue periférico que sugerem hemólise são esferócitos, células fragmentadas ou esquizócitos, acantócitos, dacriócitos, drepanócitos e corpo de Heinz.
Nos quadros hemolíticos, são lançadas no plasma hemoglobina (na hemólise intravascular), bilirrubina indireta (em geral níveis < 4 mg/dL) e desidrogenase lática (LDH). Haptoglobina é uma proteína que se liga à hemoglobina livre sérica com a função de depurá-la, embora seja também um marcador de fase aguda; essa proteína também evidencia anemia hemolítica quando os seus níveis estão reduzidos (< 25 mg/dL). A hemoglobina é filtrada nos glomérulos e reabsorvida nos túbulos renais, podendo ser encontrada no exame de urina quando a capacidade de reabsorção estiver saturada. Hemossiderina dentro das células tubulares que se desprendem na urina pode ser observada passada uma semana do episódio de hemólise.
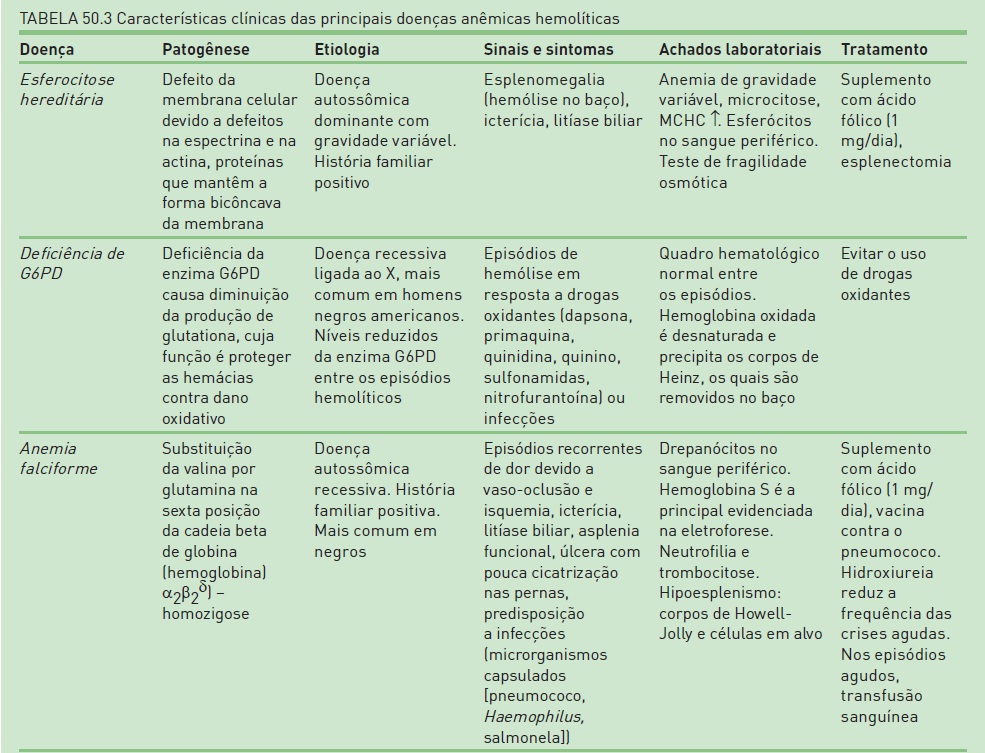
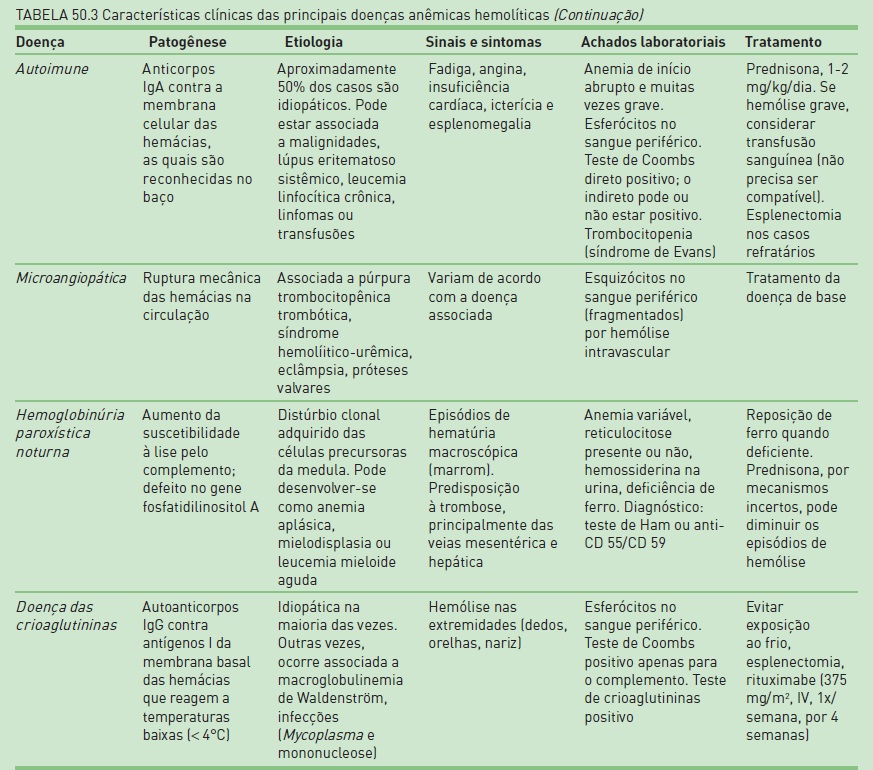
A anemia hemolítica compreende um grupo de distúrbios em que a meia-vida da hemácia, geralmente de 120 dias, está reduzida, esporádica ou continuamente. Genericamente, as anemias hemolíticas são classificadas em hereditárias (mais comumente devidas a defeitos intrínsecos às hemácias) e adquiridas (envolve fatores externos às hemácias) – ver Tabela 50.3 e Quadro 50.4. A hemólise intravascular em geral ocorre em situações de dano grave às hemácias; em casos mais leves, essas células são destruídas no tecido reticuloendotelial do baço, fígado e medula óssea.
A definição da epidemiologia da anemia hemolítica depende da doença de base.
A medula óssea responde à redução da meia-vida das hemácias aumentando a produção dessas células (hiperplasia eritroide) por meio do estímulo da eritropoietina.
Por isso, a reticulocitose (normal: 0,5-1,5%) é uma importante característica desse grupo de anemias, estando em geral acima de 4 a 5%.
Somente quando a destruição das hemácias é superior à produção eritroide é desenvolvida anemia ou, ainda, em situações em que a função medular está comprometida, como em caso de deficiência de ferro concomitante.
Em geral, a anemia hemolítica é normocítica, embora a reticulocitose possa causar um leve aumento do VCM, uma vez que o VCM dos reticulócitos é de cerca de 150 fL. Sintomas e sinais comuns à maioria dessas anemias são icterícia, esplenomegalia e litíase biliar. A ocorrência de linfadenopatia e esplenomegalia sugerem doença neoplásica.
O tratamento para a anemia hemolítica depende da causa subjacente da doença.
Teste de Coombs
Direto: esse teste é realizado misturando-se as células vermelhas do sangue com o reagente de Coombs que contém anticorpos IgM contra IgG ou complemento humano. Se houver aglutinação, isso indica a existência de anticorpos na superfície das hemácias. Um teste de Coombs direto positivo pode estar associado a várias condições; entre elas, estão reação hemolítica transfusional, doença autoimune e doença hemolítica do recém-nascido. Esse teste também pode ser secundário ao uso de determinados medicamentos, como metildopa, levodopa, ácido mefenâmico, penicilina, cefalosporinas, quinidina, digitálicos e insulina.
Indireto: nesse teste, o plasma do paciente é misturado a um painel de hemácias do tipo O. A aglutinação indica a existência de anticorpos livres no plasma. Ele pode ser utilizado para identificação de anticorpos antieritrocitários, acompanhamento de pacientes sensibilizados por antígenos de qualquer sistema imuno-hematológico, principalmente do sistema Rh, tipagem de antígenos eritrocitários e prova cruzada.
A perda de grandes volumes de sangue por meio de sangramento do trato gastrintestinal (varizes esofágicas, câncer de colo), ruptura de baço, fraturas, traumas, hemorragia em cavidades, entre outras condições causa perda de massa eritroide. Entretanto, um hemograma realizado instantes após essa perda estará normal, apresentando volemia. Realizada a reposição de volume, com a hemodiluição, os valores de hematócrito e concentração de hemoglobina são reduzidos. A medula óssea apresenta-se com hiperplasia dois a três dias após. Nesse momento, coexistem anemia normocítica e reticulócitos. Pode haver também leucocitose (desmarginação de granulócitos) e trombocitose (por um mecanismo ainda não compreendido). Não há tratamento específico para a anemia, exceto a transfusão de hemácias quando esta for grave.
A anemia aplásica é uma condição de falência medular que afeta as três séries celulares, ocasionando o quadro de pancitopenia.
Os picos de incidência dessa anemia ocorrem em indivíduos entre 20 e 30 anos de idade e em idosos.
Alterações no hemograma em caso de anemia aplásica
Inicialmente, apenas uma ou duas linhagens celulares podem estar reduzidas, sendo que o comprometimento das três séries pode ser evidenciado tardiamente. A anemia pode ser grave. Como a medula está hipoproliferativa, pode não haver reticulócitos ou estes apresentam-se em quantidade reduzida. O VCM pode estar normal ou discretamente elevado. Não há nenhuma forma morfológica dos eritrócitos no sangue periférico característica dessa condição.
As principais causas de anemia aplásica estão no Quadro 50.5. Deve-se considerar que outras condições, além da anemia aplásica, podem ocasionar pancitopenia, tais como mielodisplasia, leucemia aguda, mielofibrose, doenças infiltrativas, anemia megaloblástica, hiperesplenismo, infecções (tuberculose, síndrome da imunodeficiência humana adquirida). A maioria dos casos é idiopático e, para estes, acredita-se que o principal mecanismo seja a supressão autoimune da hematopoiese pelas células T.
Os sintomas inicialmente podem manifestar-se de forma abrupta ou insidiosa. Eles são os decorrentes da anemia (fadiga, dispneia), neutropenia (infecções) e trombocitopenia adjacente (sangramento, púrpura, petéquias e equimoses). É importante salientar que linfadenopatia e hepatoesplenomegalia geralmente não ocorrem em caso de anemia aplásica, e o diagnóstico deve ser questionado caso hajam esses sinais. Queixas sistêmicas e perda de peso são incomuns e, por isso, devem levar à busca de outros diagnósticos.
É possível estabelecer o diagnóstico de anemia aplásica quando a biópsia de medula óssea mostrar menos de 25% de células hematopoiéticas. Caracteristicamente, essas células são substituídas por gordura. Essa anemia está associada à hemoglobinúria paroxística noturna e à mielodisplasia.
O tratamento de escolha para os casos graves (neutropenia < 500 cél/mm³, plaquetas < 20.000/mm³, reticulócitos < 1%, celularidade medular < 20%) de indivíduos jovens é o transplante alogênico de medula óssea. Para indivíduos com mais de 50 anos ou sem doador adequado, a principal abordagem é realizada com fármacos imunossupressores (globulina antitimócito e ciclosporina). O prognóstico desses pacientes é alterado drasticamente. Sem tratamento, a sobrevida é de apenas 20% em um ano, com o transplante alogênico de medula óssea, mais de 80% desses indivíduos sobrevivem após cinco anos.
A aplasia eritroide pura, uma doença rara, ocorre provavelmente por um mecanismo autoimune em que linfócitos T produzem anticorpos IgG contra os precursores eritroides. Embora essa doença seja idiopática na maioria dos casos, algumas drogas têm sido utilizadas, tais como fenitoína, carbamazepina, cloranfenicol, azatioprina, isoniazida e procainamida. Infecções por parvovírus B19 (agente da quinta moléstia em crianças) também causam episódios transitórios de aplasia eitroide em casos de anemias hemolíticas. A associação com timoma é frequente. A anemia é em geral grave, não havendo reticulócitos e formas anormais no sangue periférico. A medula ósseamostra-se normocelular na biópsia, entretanto os precursores eritroides estão muito reduzidos. O tratamento envolve a suspensão de possíveis drogas implicadas, a ressecção do timoma, quando houver, e a administração de altas doses de imunoglobulina humana IV. A utilização de fármacos imunossupressores (globulina antitimócito e ciclosporina) é a abordagem preferida.
•Anemia megaloblástica: deficiência de B12 e folato
•Doença hepática
•Alcoolismo
•Síndromes mielodisplásicas
•Drogas (p. ex., zidovudina, carbamazepina, ácido valproico, fenitoína, ciclofosfamida, azatioprina)
A anemia megaloblástica envolve as condições em que a síntese de DNA das células da medula óssea está limitada pela interferência no metabolismo da vitamina B12 e do ácido fólico, que são importantes cofatores para a síntese deste.
A vitamina B12, obtida por meio de alimentos de origem animal, precisa estar ligada ao fator intrínseco, um peptídeo secretado pelas células gástricas parietais, para que seja absorvida no íleo terminal. Amplo estoque dessa vitamina está disponível no fígado, por isso é necessário mais de três anos de consumo sem reposição para que a deficiência se desenvolva. O ácido fólico advém principalmente das frutas cítricas e dos vegetais folhosos verdes. Os estoques dessa substância são suficientes para dois a três meses sem seu consumo. Tanto a vitamina B12 quanto o ácido fólico são substâncias que podem ser obtidas por meio da dieta, por isso as principais causas dessa anemia são as etiologias com deficiência nutricional/absortiva (Quadro 50.6).
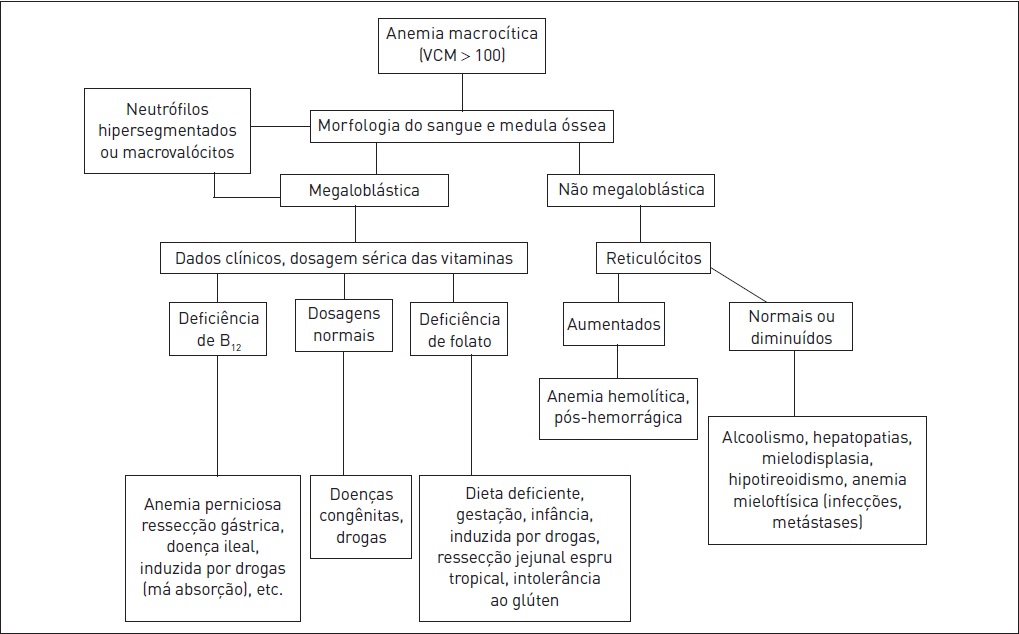
Figura 50.4
Classificação das anemias conforme VCM (anemia macrocítica).
Alterações hematológicas na anemia megaloblástica
A presença de VCM > 130 fL é um forte indicativo da presença de anemia megaloblástica. A avaliação da medula óssea revela formas megaloblásticas (macrovalócitos) e diferenças no grau de maturação entre o núcleo e o citoplasma das células. O citoplasma cresce, mas o núcleo não se divide, dando os aspecto dos eritrócitos megaloblásticos. No sangue periférico, identificam-se neutrófilos hipersegmentados (com 5 ou 6 lóbulos).
Por meio da anamnese e do exame físico, notam-se coloração amarelo-esverdeada da pele, glossite (língua lisa, típica da anemia megaloblástica), alterações neurológicas (somente em caso de deficiência de vitamina B12) e queilite angular. A anemia megaloblástica pode estar associada a outras doenças autoimunes, como vitiligo, hipotireoidismo e síndrome endócrina poliglandular. Havendo suspeita de anemia perniciosa, a realização de endoscopia digestiva alta com biópsia é essencial para evidenciar atrofia gástrica.
As dosagens de ácido fólico e B12 são de grande utilidade. Elas podem estar normais mesmo existindo deficiência de tais vitaminas. Em algumas doenças, como na infecção pelo HIV, podem estar baixas, porém sem constituírem-se causa da anemia. Classicamente, havendo deficiência de vitamina B12, os níveis séricos dessa vitamina apresentam-se baixos (< 100 pg/mL), e os de folato sérico, elevados. A medida de folato eritrocitário é preferível (< 150 ng/mL), pois o folato sérico pode apresentar variações de acordo com as refeições. Outros exames que podem ser solicitados para a realização de um melhor diagnóstico diferencial são o ácido metilmalônico (AMM) e a homocisteína. Se os níveis de ambos estiverem acima dos limites normais, há uma grande sensibilidade no diagnóstico de deficiência de B12(AMM > 1.000 nmol/L), sendo que apenas os níveis de homocisteína elevados são compatíveis com deficiência de folato. Outros achados laboratoriais da anemia megaloblástica são bilirrubina indireta aumentada (hemólise intramedular), LDH elevado, evidenciando a hemólise intramedular e ferro sérico elevados.
Deve-se realizar reposição de vitamina B12 e ácido fólico quando houver deficiência. Nos casos de deficiência na absorção de vitamina B12, esta deve ser administrada via parenteral (100 g 1x/dia na primeira semana, 1x/semana no primeiro mês e, após, 1x/mês). Na administração VO, a dose é de 1.000 g/dia. A dose de ácido fólico é de 1 a 5 mg, uma vez ao dia. A quantidade de reticulócitos começa a aumentar em torno do quinto ao sétimo dia de reposição, e o quadro hematológico é normalizado em até dois meses. Havendo dúvida quanto ao diagnóstico carencial, deve-se repor vitamina B12, pois doses altas de ácido fólico podem até corrigir a anemia, mas os sintomas neurológicos não desaparecem e podem até piorar.
Nesse caso, há os componentes da anemia de doença crônica com ferropenia relativa e retenção dos depósitos de ferro nas células do sistema hematopoiético. Características peculiares, porém, manifestam-se nesse tipo de anemia. O VCH apresenta-se aumentado de forma precoce, mas dificilmente ultrapassa os 115 fL. Além disso, no sangue periférico, é possível observar inicialmente leptócitos e estomatócitos. Em fases mais tardias, com o hiperesplenismo, surgem caracteristicamente os acantócitos e as hemácias em alvo, podendo haver também anemia hemolítica. Essa anemia decorre também do hiperesplenismo, nessa fase, comprometimento das outras séries com o consequente desenvolvimento de pancitopenia.
A anemia do alcoolista ocorre basicamente devido à deficiência nutricional e à interferência direta do álcool na síntese das células hematopoiéticas. A deficiência de folato é de incidência comum no alcoolista, e o álcool pode interferir na atividade dessa substância. Com isso, a anemia do alcoolista apresenta características megaloblásticas, como macrocitose. Esse índice é um importante marcador do consumo crônico de álcool. Esse tipo de anemia não afeta os bebedores de cerveja, pois essa bebida é rica em folato.
369
A paciente do caso clínico apresenta um quadro clássico de anemia por deficiência de ferro, provavelmente devido à perda de sangue no período menstrual. Os testes preliminares confirmam a deficiência de ferro: níveis de ferritina, de ferro plasmático e saturação de transferrina baixos e capacidade ferropéxica elevada. Quanto à causa da anemia (como já mencionado) nas pacientes em pré-menopausa, a causa mais comum é a perda de sangue por meio de sangramento menstrual. Já nas pacientes em pós-menopausa, o risco de doenças malignas do trato ginecológico e gastrintestinal como causa da anemia aumenta substancialmente.
Alcindor T, Bridges KR. Sideroblastic anaemias. Br J Haematol.2002;116(4):733-43.
Capra M, Brun CP, Stefani SD. Hematologia. In: Stefani SD, Barros E, organizadores. Clínica médica: consulta rápida. 3. ed. Porto Alegre: Artmed; 2008. p. 255-82.
Costa C, Berdichevski R. Nefropatia isquêmica. In: Barros E, Gonçalves LF, organizadores. Nefrologia no consultório. Porto Alegre: Artmed; 2007. p. 307-26.
Dhaliwal G, Cornett PA, Tierney LM. Hemolytic anemia. Am Fam Physician. 2004;69(11):2599-606.
Failace R, organizador. Hemograma: manual de interpretação. 4. ed. Porto Alegre: Artmed; 2003.
Gertz MA. Management of cold haemolytic syndrome. Br J Haematol.2007;138(4):422-9.
Goddard AF, McIntyre AS, Scott BB. Guidelines for the management of iron deficiency anaemia. Gut. 2000;46 Suppl 3-4:IV1-IV5.
Kasper D, Fauci A, Longo DL, Braunwald E, Hauser SL, Jameson JL, editores. Harrison medicina interna. 16. ed. Rio de Janeiro: McGraw- Hill; 2006.
Killip S, Bennett JM, Chambers MD. Iron deficiency anemia. Am Fam Physician. 2007;75(5):671-8.
McPhee SJ, Papadakis MA, Tierney LM, editors. Current: medical diag- nosis and treatment. 47th ed. New York: McGraw-Hill; 2008.
Nurku S. Anemia in chronic kidney disease: causes, diagnosis, treat- ment. Cleve Clin J Med. 2006;73(3):289-97.
Stuart M, Nagel RL. Sickle-cell disease. Lancet. 2004;364(9442):1343- 60.
Weber CS, Bittar C. anemias. In: Xavier RM, Albuquerque GC, Barros E, organizadores. Laboratório na prática clínica: consulta rápida. Porto Alegre: Artmed; 2005. p. 335-46.
Weiss G, Goodnough LT. Anemia of chronic disease. N Engl J Med.2005;352(10):1011-23.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.