(Carregando Índice)... (Carregando Índice)... |
Última revisão: 21/05/2014
Comentários de assinantes: 0
Um paciente do sexo masculino, 42 anos, branco, com sobrepeso, procura o serviço de emergência por ter acordado pela manhã com intensa dor no joelho direito, a ponto de não conseguir flexioná-lo ou apoiar o membro inferior no chão. Ao ser questionado sobre comorbidades, relata ser hipertenso em terapia com hidroclorotiazida e dislipidêmico sem tratamento. Afirma não ter apresentado episódios prévios semelhantes. Ao realizar exame, verificam-se tempermatura axilar de 37,5°C, frequência cardíaca de 101 bpm e pressão arterial de 160/100 mmHg. Não há particularidades no aparelho cardiovascular, no respiratório e no exame físico do abdome. A partir do exame osteoarticular, constata-se hipersensibilidade ao toque e à mobilização do joelho direito, associada a edema, hiperemia e elevação da temperatura local. Para investigação complementar, realiza-se raio X do joelho direito, o qual evidencia edema de partes moles, sem alterações na articulação. No hemograma, verificam-se 13.000 leucócitos (sem desvio), creatinina de 1,3 mg/dL e ácido úrico de 7,9 mg/dL. Efetua-se punção articular do joelho direito e o material, turvo, não viscoso e não purulento, é enviado para análise laboratorial.
Gota é o termo utilizado para a doença inflamatória, metabólica e de natureza heterogênea causada pelo depósito tissular de cristais de monourato de sódio (MUS) em indivíduos com hiperuricemia. Pode manifestar-se por ataques agudos e recorrentes de artrite, depósitos de MUS em tecidos periarticulares e doença renal. É importante ressaltar que o termo “gota” só pode ser utilizado quando há processo inflamatório tissular, não devendo ser usado em casos isolados de hiperuricemia ou nefrolitíase por cálculo de urato.
A epidemiologia da gota varia de acordo com fatores como idade e sexo, sendo mais frequente em homens (> 90% dos casos) e tendo pico de incidência em torno dos 40 anos. Em mulheres, apresenta-se geralmente após a menopausa. Na população geral, a ocorrência de casos é de 0,2 a 0,35/1.000 habitantes, com distribuição universal, e a incidência é diretamente proporcional aos níveis séricos de MUS, tendo uma incidência anual de 5% em indivíduos com mais de 9 mg/dL. A prevalência de hiperuricemia varia de 2 a 40%, dependendo da população estudada.
A partir do desenvolvimento natural da doença, pode-se dividi-la em quatro situações: hiperuricemia assintomática, artrite gotosa aguda, período intercrise e gota tofácea crônica.
Hiperuricemia assintomática. É o estágio no qual há níveis elevados de ácido úrico (> 7 mg/dL) sem a ocorrência de artrite, tofos ou nefrolitíase. A maioria dos pacientes hiperuricêmicos permanece assintomática por toda a vida (apenas 20% apresenta episódios da doença), e a primeira crise de artrite ou cólica renal ocorre, em média, após 20 anos de hiperuricemia assintomática (Quadro 123.1).
Artrite gotosa aguda. O quadro típico de uma crise de gota é uma monoartrite extremamente dolorosa, de apresentação inicial aguda em articulação de membro inferior, geralmente durante a madrugada ou pela manhã.
Em 50% dos casos, o primeiro ataque ocorre na primeira articulação metatarsofalângica (podagra), porém outras articulações também são afetadas com frequência: tornozelos, joelhos, punhos e cotovelos.
Ao longo da evolução, em pacientes não medicados adequadamente, pode haver comprometimento de qualquer articulação, e as crises podem ser oligoarticulares (até quatro articulações).
Concomitantemente ao quadro, pode ocorrer febre, e a articulação acometida apresenta evidentes sinais de flogose, podendo ocorrer descamação local da pele nos dias seguintes. Ressalta-se que, em um terço dos casos, o ácido úrico apresenta nível normal durante a crise.
Pode haver depósito de cristais de ácido úrico em tecidos periarticulares, não se configurando um quadro típico de artrite. Esse evento ocorre de forma isolada ou concomitantemente à artrite aguda.
Período intercrise. É o período entre uma crise e a seguinte. O intervalo é variável, tendendo a ser cada vez menor com o desenvolvimento da doença. Alguns indivíduos nunca apresentam uma segunda crise.
Gota tofácea crônica. Essa condição pode ocorrer quando crises recorrentes de gota e hiperuricemia não são tratadas, havendo deposição de cristais de urato em cartilagens, membranas sinoviais, tendões e tecidos moles, formando nodulações indolores, denominadas tofos. Os locais de ocorrência mais típicos são sobre face extensora de cotovelos, mãos, tendões de Aquiles e pavilhão auricular. A rigor, tofos podem ser localizados em qualquer órgão ou tecido (laringe, miocárdio, valva mitral) e podem regredir se a hiperuricemia for tratada.
As complicações renais que podem surgir em decorrência da hiperuricemia crônica são a nefrolitíase e a nefropatia por urato.
A urolitíase afeta 10 a 25% dos indivíduos gotosos e apresenta relação direta com grau de acidez da urina, diminuição do volume urinário e concentração sérica de MUS. Os cálculos de ácido úrico são geralmente radiolucentes.
Em casos de nefropatia por urato, o comprometimento parenquimatoso ocorre devido à deposição de MUS no interstício medular, causando consequentemente proteinúria leve e perda de função renal; porém, na maioria dos casos, o principal dano renal é resultado de doenças associadas, como hipertensão, aterosclerose e diabetes.

Fisiopatologicamente, como se desencadeia uma crise de gota?
A crise aguda de gota ocorre quando existe uma quantidade suficiente de cristais de urato no líquido sinovial que estimule a fagocitose destes pelos polimorfonucleares. Estes, quando ingeridos, por meio de sua superfície pontiaguda, causam a ruptura da membrana do fagolisossomo, liberando enzimas líticas para o líquido sinovial e desencadeando, assim, o processo inflamatório articular.
A alteração bioquímica da gota é a hiperuricemia, e esta ocorre devido a duas condições básicas: excesso de produção (10 a 15%) e/ou diminuição na excreção de ácido úrico (85 a 90%), podendo a gota ser classificada em primária e secundária.
Na hiperuricemia primária, cerca de 90% dos casos são decorrentes de um defeito na eliminação renal do MUS e, em 10%, devido à hiperprodução endógena de ácido úrico.
Na hiperuricemia secundária, fatores externos estão associados, sendo os principais ingestão de bebidas alcoólicas, dieta rica em purina, obesidade e uso de medicamentos, como diuréticos e ácido acetilsalicílico em dose baixa.
Um quadro típico de artrite gotosa aguda associada à hiperuricemia pode sugerir diagnóstico presuntivo com alto índice de acerto; porém, como relatado previamente, há indivíduos normouricêmicos durante a crise, assim como hiperuricêmicos sem gota. Portanto, nas crises iniciais em pacientes sem tofos ou alterações radiológicas típicas, o diagnóstico de certeza é estabelecido apenas mediante constatação de cristais de MUS no líquido sinovial.
O líquido sinovial apresenta cristais de MUS (em formato de agulha), evidenciando birrefringência negativa à microscopia com luz polarizada. Nesse líquido, há entre 2.000 e 75.000 células/mm³, sendo os neutrófilos as predominantes.
A radiologia convencional auxilia pouco o diagnóstico nos casos iniciais de crise aguda, podendo evidenciar, em casos crônicos, as típicas lesões em “saca-bocado”que representam erosão óssea.
Os principais objetivos do tratamento de pacientes com gota são os seguintes:
•Eliminar a crise aguda;
•Prevenir a recorrência das crises;
•Prevenir e reverter as complicações do depósito de cristais nas articulações, nos rins e em outros órgãos;
•Combater fatores associados, como obesidade, diabetes, dislipidemia e hipertensão.
Deve-se tratar episódios de crise com colchicina ou anti-inflamatórios não esteroides (AINEs).
Pode-se utilizar a colchicina com a administração de 0,5 mg, a cada 2 horas, até que o paciente melhore ou desenvolva sintomas gastrintestinais (diarreia ou vômito). Não se deve ultrapassar a dose de 6 mg/dia.
Em geral, o uso de AINEs também é efetivo no controle da crise, e eles devem ser utilizados durante o período em que perduram os sintomas. Não há evidências da superioridade de um AINE sobre outro, e, no geral, são mais tolerados do que a colchicina.
Corticosteroides sistêmicos (prednisona ou dexametasona) ou intra-articulares podem ser administrados em casos selecionados, para os quais há contraindicação ao uso da colchicina ou de AINEs.
Os principais esquemas utilizados para prevenir novas crises são os seguintes:
•Uso profilático diário de colchicina, na dose de 0,5 a 1 mg/dia, até que a uricemia atinja valores menores do que 6 mg/dL;
•Redução de peso no paciente obeso;
•Evitar consumo de álcool e alimentos ricos em purinas;
•Uso de medicação hipouricemiante.
Tem-se como objetivo manter os níveis de urato sérico em valores menores do que 6 mg/dL, o que se pode conseguir por meio de dois mecanismos principais:
•Redução da síntese (alopurinol);
•Aumento da excreção renal (uricosúricos).
O alopurinol (assim como o febuxostat – este ainda não comercializado no Brasil) inibe a síntese de ácido úrico por meio de sua interferência no metabolismo das purinas e geralmente é utilizado nas doses de 100 a 300 mg/dia, podendo atingir 600 mg/dia.
Os agentes uricosúricos (benzbromarona, probenecida e sulfimpirazona) interferem na reabsorção tubular proximal do urato filtrado. São drogas de segunda linha no tratamento da hiperuricemia, e os principais efeitos adversos são erupção cutânea, precipitação de crise aguda de gota, intolerância gastrintestinal e formação de cálculos de urato.
Recomenda-se um período de espera de 15 a 20 dias entre a resolução da crise aguda e o início do tratamento hipouricemiante. Deve-se ressaltar que, se o paciente já utiliza essas drogas, não convém suspender o uso nas crises. A razão para tal é que mudanças bruscas nos níveis séricos de urato precedem a crise aguda de gota.
A dieta tradicionalmente recomendada para os pacientes com gota, com restrição estrita de proteínas e purinas, além de ser de difícil adesão, reduz apenas cerca de 1 mg/dL da uricemia. Esse objetivo pode ser atingido com o uso de agentes hipouricemiantes.
Atualmente, deve-se dirigir a orientação dietética à restrição calórica geral, e recomenda-se realizar ingestão proteica principalmente de derivados do leite com baixo teor de gordura e evitar grandes quantidades de carne e peixe. Quanto ao consumo de álcool, sabe-se que cerveja confere maior risco de crise de gota do que bebidas destiladas e que vinho tinto é fator protetor ao desenvolvimento de gota.
A artropatia por pirofosfato de cálcio é uma doença associada à precipitação de cristais de pirofosfato de cálcio (CPPC) no tecido conectivo. Pode ser assintomática ou associada a diversas síndromes clínicas. Esses distúrbios, incluindo artropatia inflamatória aguda e crônica e calcificação radiográfica, compõem o espectro da doença de deposição de cristais de pirofosfato de cálcio.
Terminologia. Os nomes tradicionalmente utilizados para doenças por depósito de CPPC são pseudogota, condrocalcinose e artropatia por pirofosfato. Um recente consenso elaborado pela Liga Europeia Contra o Reumatismo (EULAR, do inglês European League Against Rheumatism) sugeriu uma terminologia alternativa e revisou abordagens diagnósticas para essas condições. As síndromes clínicas, os achados e as limitações desses termos, junto com a nomenclatura proposta pelo EULAR, são:
Pseudogota: descreve com precisão os ataques agudos de sinovite induzida por CPPC, que clinicamente se assemelham às crises de gota por urato. O consenso da EULAR prefere o termo “artrite aguda por depósito de cristais de pirofosfato de cálcio” em vez de “pseudogota”.
Condrocalcinose: refere-se à calcificação radiográfica de fibrocartilagens. Ocorre comumente em pacientes com doença de deposição de CPPC, mas não é absolutamente específico por CPPC, nem universal entre os pacientes afetados. O grupo da EULAR designa esse achado como “calcificação da cartilagem” (CC) (Fig. 123.1).
Artropatia por pirofosfato: é o termo utilizado para a doença clínica articular ou para as anormalidades radiográficas que ocorrem concomitantemente à deposição de CPPC.
O depósito intra-articular de CPPC ocorre mais frequentemente em mulheres (3:1) e em pessoas com mais de 60 anos.
Esses cristais podem depositar-se em cartilagem articular, sinóvia, tendões e ligamentos periarticulares, provavelmente associados a alterações bioquímicas e estruturais relacionadas à idade. Algumas das condições associadas ao depósito de CPPC são hipotireoidismo, hiperparatireoidismo, hemocromatose, hipofosfatasia, hipomagnesemia e hipermobilidade articular.
Em relação à classificação clínica da doença por deposição de CPPC, deve-se atentar para a semelhança desse distúrbio com praticamente qualquer tipo de artrite. Quase todas as articulações podem ser acometidas, mas os joelhos, os punhos e as articulações metacarpofalângicas são as mais comumente afetadas. O espectro clínico da doença inclui:
•Doença assintomática
•Pseudogota
•Pseudoartrite reumatoide
•Pseudoartrose, com ou sem ataques agudos
•Doença articular pseudoneuropática
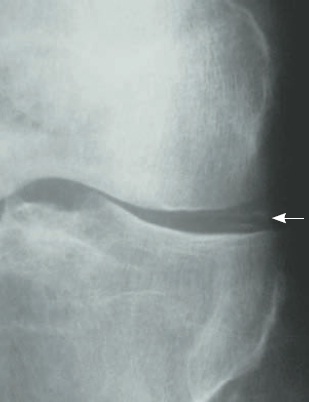
Figura 123.1
Raio X evidenciando condrocalcinose.
A maioria dos indivíduos com doença por depósito de CPPC perceptível nas radiografias é provavelmente assintomática com relação ao envolvimento articular.
A pseudogota ocorre em 25% dos pacientes e afeta principalmente homens. Os ataques podem ser tão graves quanto os da gota, permanecendo os pacientes assintomáticos entre as crises. Em geral, uma única articulação é afetada, sendo o joelho o local mais frequentemente acometido. Pode ocorrer febrícula e leucocitose.
Cerca de 5% dos pacientes apresentam esse quadro subagudo, caracterizado por um comprometimento poliarticular (incluindo mãos e punhos), mas em geral mais assimétrico do que o de casos de artrite reumatoide. Pode durar de semanas a meses e acarretar deformidades articulares por contraturas em flexão.
Aproximadamente 50% dos pacientes com doença por deposição de CPPC sintomática evidenciam degeneração articular progressiva, em geral envolvendo múltiplas articulações. A tendência é de que o envolvimento seja bilateral e simétrico, e o joelho é a articulação mais afetada. Em cerca de metade desses pacientes, episódios de artrite inflamatória aguda típica de pseudogota pontuam o curso.
A doença articular pseudoneuropática é caracterizada por degeneração articular grave, semelhante àquela observada em articulações neuropáticas, como a de Charcot; contudo não há anormalidade neurológica.
Pode-se estabelecer o diagnóstico por meio de análise do líquido sinovial ou de estudos radiológicos, associados ao quadro clínico.
O líquido sinovial apresenta cristais com birrefringência positiva, por meio de microscopia de luz polarizada, em articulações inflamadas durante um ataque de pseudogota, e a concentração total de leucócitos no líquido é tipicamente de 15.000 a 30.000 por mm³, 90% dos quais são neutrófilos.
Identifica-se a condrocalcinose como calcificações paralelas à superfície do osso subcondral, sendo observadas com mais frequência nos meniscos dos joelhos, no ligamento triangular dos punhos e na sínfise púbica.
A abordagem terapêutica da artropatia por deposição de CPPC varia conforme a apresentação clínica.
Em casos de pseudogota, uma abordagem ou, mais frequentemente, uma combinação delas são apropriadas para o tratamento de ataques agudos, as quais incluem punção aspirativa da articulação comprometida (no caso de crise monoarticular) com posterior infiltração local com glicocorticoide e a administração oral de colchicina ou AINEs nos mesmos parâmetros utilizados para a gota.
Para a pseudoartrite reumatoide, a EULAR formulou recomendações de tratamento em ordem de preferência, sendo suas sugestões o uso de AINEs e/ou colchicina como primeira escolha, seguido de baixas doses de metotrexato, glicocorticoides e hidroxicloroquina.
Em casos de pseudoartrose, pacientes nos quais a degeneração articular é a manifestação principal da doença por deposição de CPPC e sem pseudogota episódica, as abordagens atuais de tratamento são as mesmas utilizadas para a osteoartrose.
A análise do líquido sinovial evidencia celularidade de 20.000 leucócitos por mm³, com predomínio de neutrófilos e existência de cristais de monourato de sódio fagocitados. A análise de gram é negativa. Estabelece-se o diagnóstico de gota por meio da evidência de cristais de monourato de sódio.
A síndrome articular típica da gota é a monoartrite aguda muito dolorosa. Idade, sexo, ausência de traumatismo e pouca evidência para infecção sugerem o diagnóstico. Mesmo assim, principalmente nas primeiras crises, deve-se descartar infecção e outras doenças articulares com manifestações atípicas, como artrite reumatoide, artropatia psoriásica e artrite reativa.
A constatação de hiperuricemia favorece muito o diagnóstico, mas deve-se enfatizar que a maioria dos hiperuricêmicos não é portadora de gota. O diagnóstico definitivo, na primeira crise, é o achado dos cristais de ácido úrico no líquido sinovial. Também é importante ressaltar que, apesar de geralmente a artrite na pseudogota ser menos intensa, o quadro anteriormente descrito enquadra-se, da mesma forma, nessa doença.
O achado de cristais de pirofosfato de cálcio na análise do líquido sinovial é decisivo para o diagnóstico diferencial quando não há alterações radiológicas específicas.
Choi HK. Diet, alcohol, and gout: how do we advise patients given recent developments? Curr Rheumatol Rep. 2005;7(3):220-6.
Choi HK, Atkinson K, Karlson EW, Willett W, Curhan G. Alcohol intake and risk of incident gout in men: a prospective study. Lancet.2004;363(9417):1277-81.
Choi HK, Atkinson K, Karlson EW, Willett W, Curhan G. Purine-richfoods, dairy and protein intake, and the risk of gout in men. N Engl J Med.2004;350(11):1093-103.
Harris ED, Budd RC, Firestein GS, Genovese MC, Sergent JS, Ruddy S, et al., editors. Kelley’s textbook of rheumatology. 7th ed. Philadelphia: Saunders; 2007.
Hochberg MC, Silman AJ, Smolen JS, Weinblatt ME, Weisman MH, editors. Rheumatology. 4th ed. Edinburgh: Mosby; 2008.
Klippel JH, Stone JH, Crofford LeJ, White PJ, editors. Primer on the rheumatic disease, 13th ed. New York: Springer; 2008.
Moreira C, Castelar Pinheiro GR, Marques Neto JF, editores. Reumatologia essencial. Rio de Janeiro: Guanabara Koogan; 2009.
Skare TL. Reumatologia: princípios e prática. 2. ed. Rio de Janeiro: Guanabara Koogan; 2007.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.