Doenças glomerulares – Roman GB Bonegio David J Salant
Roman G.B. Bonegio, MD, PHD
Assistant Professor of Medicine, Renal Section, Boston University School of Medicine, Boston, MA
David J. Salant, MD
Professor of Medicine, Boston University School of Medicine, Chief of Nephrology Section, Boston Medical Center, Boston, MA
Artigo original: Bonegio RGB, Salant DJ. Glomerular Diseases. ACP Medicine. 2011;1-27.
[The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2011 Decker Intellectual Properties Inc. All Rights Reserved.]
Agradecimentos: Figuras 1b, 10b – Christine Kenney; Figuras 2, 7, 10a, 10c, 16 – Seward Hung; Figuras 4, 9 – Cortesia do Dr. Vivette D’Agati, Columbia University; Figura 5 – Cortesia do Dr. Agnes Fogo, Vanderbilt University School of Medicine; Figura 11 – Cortesia do Dr. Dontscho Kerjaski, Viena, Austria.
Tradução: Soraya Imon de Oliveira
Revisão técnica: Dr. Euclides Furtado de Albuquerque Cavalcanti
O glomérulo é afetado por uma ampla variedade de processos patológicos. Alguns são agudos e autolimitados, enquanto outros levam à perda progressiva da função renal em decorrência da desorganização do aparelho de filtração normal por ação de processos inflamatórios, degenerativos ou infiltrantes, que culminam em esclerose glomerular e consequente atrofia tubular e fibrose intersticial. De fato, as doenças glomerulares são responsáveis por mais de 50% dos casos de doença renal crônica. Uma breve revisão sobre a estrutura e função glomerulares normais auxilia na avaliação das manifestações destes distúrbios complexos.
Anatomia e fisiologia do glomérulo
O glomérulo consiste em um arranjo em paralelo de alças capilares sustentado por células mesangiais e por uma matriz mesangial [Figura 1]. O sangue é seletivamente filtrado pelo glomérulo, à medida que se move da arteríola aferente através dos capilares glomerulares em direção à arteríola eferente drenante. Os componentes estruturais da parede do capilar glomerular isolam o sangue do espaço urinário e formam uma barreira à filtração, com seletividade para carga e tamanho. Esta barreira de filtração glomerular permite a passagem imediata de líquido e pequenas moléculas do sangue para o interior do espaço urinário, mas previne que as células sanguíneas e as proteínas plasmáticas sejam filtradas. A barreira de filtração é composta por 3 camadas [Figura 1b]: a camada de células endoteliais fenestradas, que reveste as alças capilares; a membrana basal glomerular (MBG); e a camada de células epiteliais glomerulares viscerais, frequentemente denominadas podócitos, por apresentarem processos podais interdigitantes. As cargas negativas presentes na superfície das células endoteliais e podócitos, bem como na MBG, conferem uma barreira carga-seletiva contra a passagem de moléculas aniônicas (p. ex., albumina). Contudo, a fenda do diafragma, que consiste em uma junção intercelular especializada situada entre os podócitos, forma a barreira tamanho-seletiva final contra a passagem das proteínas plasmáticas.

Figura 1. (a) Fotomicrografia ilustrando os diferentes componentes de um glomérulo normal, incluindo as células mesangiais (setas grossas), células endoteliais (setas finas), podócitos (seta pontilhada) e as células epiteliais parietais da cápsula de Bowman (pontas de seta) (coloração com ácido periódico de Schiff [PAS]). (b) Representação esquemática do glomérulo, mostrando os constituintes do glomérulo normal.
*Alças capilares.
B = espaço de Bowman; MBG = membrana basal glomerular; T = túbulos.
O glomérulo normal filtra cerca de 20% do plasma que o atravessa, a uma taxa de filtração glomerular (TFG) de 80 a 120 mL/min, ou seja, aproximadamente 120 a 150 L/dia. Como resultado da seletividade glomerular e de alguma reabsorção tubular, a excreção urinária de proteínas geralmente é mantida abaixo de 150 mg/dia, do quais menos de 20 mg/dia são de albumina. Os leucócitos e as hemácias raramente são encontrados no sedimento urinário normal (< 5 células/campo de maior aumento).
Classificação clínica e terminologia da doença glomerular
Em geral, o termo “glomerulonefrite” é utilizado para denotar as doenças glomerulares que resultam de um processo inflamatório. Estas doenças caracteristicamente possuem um componente imunológico. As doenças glomerulares não inflamatórias muitas vezes são denotadas pelo termo “nefropatia”. Existem numerosas doenças distintas capazes de lesar o glomérulo e produzir um ou mais dos aspectos cardinais de proteinúria, hematúria e azotemia, como resultado do comprometimento da depuração de solutos. Nós recomendamos primeiro estabelecer um diagnóstico sindrômico, que poderá estreitar de modo considerável o diagnóstico diferencial e, assim, permitir uma abordagem mais lógica de investigação e tratamento do paciente com doença glomerular.
Diagnóstico sindrômico da doença glomerular
A síndrome nefrítica é caracterizada pelo desenvolvimento de hematúria macro ou microscópica, com hemácias dismórficas ou cilindros de hemácias na urinálise e graus variáveis de azotemia, oligúria, hipertensão e edema. A nefrite ocorre subsequentemente à lesão ao endotélio, MBG ou mesângio; danifica as alças capilares glomerulares e permite que as células sanguíneas passem para a urina. O nível de proteinúria geralmente é menor do que aquele observado em pacientes com síndrome nefrótica (isto é, < 3,5 g/dia). A nefrite pode ser acompanhada de uma diminuição relativamente rápida da TFG. A doença pode estar limitada aos rins (primária) ou ser secundária a doenças autoimunes ou infecciosas [Tabela 1].
Tabela 1. Apresentação clínica e causas comuns de doença glomerular
|
|
Síndrome nefrítica |
GNRP |
Síndrome nefrótica |
|
Achados clínicos |
Hipertensão Edema TFG reduzida Hematúria com sedimento urinário ativo Proteinúria < 3,5 g/dia |
Síndrome nefrítica ou nefrítica-nefrótica, com deterioração rápida da função renal Hematúria com sedimento urinário ativo |
Edema Proteinúria > 3,5 g/dia ou proporção albumina:creatinina > 3 Hipoalbuminemia Hiperlipidemia Lipidúria |
|
Doenças glomerulares primárias |
NIgA* PHS Glomerulonefrite necrotizante idiopática GNMP primária*† DDD |
Doença anti-MBG |
Doença de lesões mínimas GESF Nefropatia membranosa |
|
Doenças glomerulares secundárias comuns |
Familiar Síndrome de Alport Farmacológica Vasculite ou lúpus fármaco-induzido† Infecciosa Glomerulonefrite pós-infecciosa† Crioglobulinemia associada à hepatite C*† Autoimune LES*† Vasculites ANCA-associadas |
Autoimune Vasculite ANCA-associada LES severo† Infecciosa Doença do imunocomplexo pós-infeccioso severa† |
Familiar Distúrbios hereditários de proteínas de podócitos Farmacológicas Fármacos AINH Toxinas Metais pesados (ouro/mercúrio) Metabólica Diabetes melito Infecciosa HIV Infecções parasitárias Associada à malignidade Amiloidose Doença da deposição de Ig monoclonal Paraneoplásica |
*Pode ocorrer na síndrome nefrítica-nefrótica.
†Baixo complemento.
AINH = anti-inflamatórios não hormonais; ANCA = anticorpo anticitoplasma de neutrófilo; DDD = doença de depósito denso; GESF = glomerulosclerose segmentar focal; GNMP = glomerulonefrite membranoproliferativa; GMRP = glomerulonefrite rapidamente progressiva; LES = lúpus eritematoso sistêmico; MBG = membrana basal glomerular; NIgA = nefropatia por IgA; PHS = púrpura de Henoch-Schönlein; TFG = taxa de filtração glomerular.
A síndrome nefrótica é definida pela presença de uma severa albuminúria (> 3,5 g/dia), edema e hipoalbuminemia, e geralmente acompanhada de lipidúria e hiperlipidemia. Além da perda de albumina pela urina, outras proteínas plasmáticas também podem atravessar o glomérulo na síndrome nefrótica severa. Os pacientes, portanto, podem apresentar hipercoagulabilidade decorrente da perda urinária de antitrombina e apresentam risco aumentado de desenvolvimento de trombose venosa e embolia pulmonar. Estes pacientes também são suscetíveis a infecções, devido à perda de complemento e de imunoglobulinas, bem como ao desenvolvimento de osteomalácia decorrente da perda de proteína ligadora de vitamina D. A síndrome nefrótica pode ocorrer no contexto de doenças hereditárias envolvendo os podócitos, doença sistêmica (p. ex., diabetes melito e amiloidose) ou doenças renais primárias que têm como alvo os podócitos [Tabela 1]. Além disso, as reações idiossincráticas a medicamentos como os AINH ou a intoxicação com metais pesados podem lesar os podócitos e produzir síndrome nefrótica.
A síndrome nefrítica-nefrótica constitui o termo utilizado para descrever os pacientes que apresentam aspectos de ambas as síndromes anteriormente descritas (isto é, > 3,5 g proteinúria/dia e sedimento urinário ativo contendo hemácias e cilindros de hemácias). Esta síndrome sugere a existência de uma inflamação extensiva junto ao tufo glomerular e geralmente está associada a doenças que produzem um padrão de lesão membranoproliferativa (glomerulonefrite membranoproliferativa [GNMP]) (ver adiante). O diagnóstico diferencial também inclui a nefrite lúpica e a nefropatia por IgA (NIgA), nefrite de Henoch-Schölein e certas formas de nefrite pós-infecciosa.
O termo “glomerulonefrite rapidamente progressiva” (GNRP) descreve um subconjunto de pacientes que apresentam lesão renal aguda e aspectos de glomerulonefrite (tipicamente, as síndromes nefrítica ou nefrítica-nefrótica). Observa-se uma perda rápida da TFG, que é indicada pela duplicação dos níveis séricos de creatinina em um período de 3 meses. É comum a GNPR estar associada à glomerulonefrite crescêntica [Figura 2] resultante de uma vasculite sistêmica, imunocomplexos circulantes ou anticorpos anti-MBG [Tabela 1]. A GNRP deve ser considerada uma emergência médica e requer encaminhamento imediato do paciente a um nefrologista, devido à necessidade urgente de diagnóstico e intervenção terapêutica para prevenção de perda irreversível da TFG, desenvolvimento de doença renal em estágio terminal (DRET) ou morte.
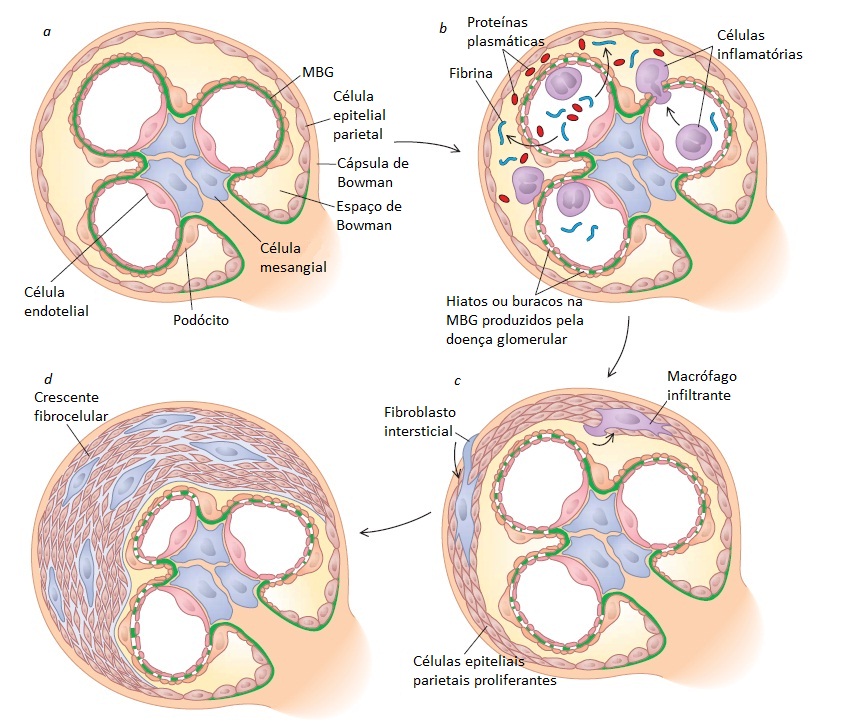
Figura 2. Ilustração esquemática dos eventos que levam à formação de crescentes. (a) Glomérulo normal. (b) A doença glomerular leva ao aparecimento de hiatos ou buracos na membrana basal glomerular (MBG), permitindo a infiltração de células inflamatórias, resultando na (c) proliferação das células epiteliais parietais e, por fim, levando à (d) formação do crescente fibrocelular.
Epidemiologia da doença glomerular
As doenças do glomérulo constituem as causas mais comuns de DRET em todo o mundo. Nos Estados Unidos, assim como nos países mais desenvolvidos, o diabetes melito predomina como causa de DRET (44%), enquanto a glomerulonefrite é responsável por cerca de 7% dos casos (http://www.usrds.org/). Nos países em desenvolvimento, onde a prevalência das doenças infecciosas é mais alta, a glomerulonefrite imunomediada constitui a principal causa de DRET. A verdadeira prevalência da doença glomerular pode estar subestimada, todavia, pois é estimado que apenas 10 a 20% dos pacientes com doença glomerular apresentem sintomas clínicos até manifestarem insuficiência renal crônica.
Patogênese da lesão glomerular
As doenças glomerulares podem ser hereditárias (p. ex., defeitos do colágeno da MBG observados na síndrome de Alport e proteínas podócito-associadas), imunomediadas ou causadas por estresse metabólico (p. ex., diabetes, síndrome metabólica) ou hemodinâmico (p. ex., hipertensão intraglomerular subsequente à diminuição da massa renal). A lesão mediada por anticorpos constitui o mecanismo mais comum e mais bem-descrito de glomerulonefrite imunomediada.
A lesão mediada por anticorpos é produzida por dois mecanismos distintos. Um mecanismo envolve autoanticorpos dirigidos contra antígenos-alvo específicos do glomérulo. Os principais exemplos são a síndrome de Goodpasture (doença anti-MBG), em que o paciente apresenta anticorpos contra o domínio não colágeno das cadeias de colágeno de tipo IV existentes na MBG, e a nefropatia membranosa idiopática, em que os anticorpos dirigidos contra o receptor da fosfolipase A2 (PLA2R – em inglês, phospholipase A2 receptor) de tipo M atacam os podócitos.1 Como alternativa, pode haver formação de imunocomplexos contendo anticorpos e antígeno na circulação ou durante o processo de filtração glomerular, com consequente deposição no glomérulo. Alguns exemplos deste tipo de doença por imunocomplexo são a glomerulonefrite pós-infecciosa, a nefrite lúpica e a GNMP atribuível à hepatite C. Em geral, os anticorpos e imunocomplexos que se depositam no glomérulo promovem lesão principalmente por ativarem a cascata do complemento. O sítio de deposição de anticorpos (mesangial, subendotelial ou subepitelial) é o principal determinante da inflamação glomerular, de quais células glomerulares são lesadas e, consequentemente, da síndrome clínica que se desenvolverá.
A imunidade celular também pode exercer algum papel em certas formas de glomerulonefrite que envolvem o influxo de células inflamatórias. As células T sensibilizadas por antígenos endógenos ou exógenos presentes nos glomérulos recrutam macrófagos e induzem uma reação de hipersensibilidade tardia. A imunidade celular também pode estar envolvida na doença de lesões mínimas, em que uma citocina putativa derivada de células T parece causar a lesão no podócito.
Diagnóstico
História e exame físico
Uma história detalhada fornece os indícios essenciais ao diagnóstico e à etiologia da doença glomerular, em cada paciente. Os sintomas sugestivos incluem um ganho de peso inexplicável, edema, episódios recentes ou passados de hematúria e urina espumosa. Devem ser feitas perguntas específicas sobre os sintomas extrarrenais de doenças sistêmicas, como artralgia, erupção prurídica ou sensível à luz solar, alopécia, doença crônica no trato respiratório superior e hemoptise. Uma história familiar de doença renal pode sugerir uma forma familiar de glomerulonefrite, como a síndrome de Alport, em especial se o indivíduo afetado apresentar perda auditiva. Também existem formas familiares de NIgA, glomerulosclerose segmentar e focal (GESF) e síndrome hemolítica-urêmica atípica. Além das formas mendelianas de doença renal, a história familiar pode ser importante como indicador de suscetibilidade à doença. A predisposição familiar à nefropatia diabética, nefrosclerose hipertensiva e nefropatia associada à infecção pelo HIV (NAHIV) está bem descrita, sobretudo entre pacientes de ascendência africana recente. Esta predisposição foi recentemente associada a um polimorfismo envolvendo o gene codificador da cadeia pesada 9 da miosina (MYH9), bem como ligada a variantes localizadas nas adjacências deste gene (ApoL1) que codificam uma forma tripanolítica da proteína ApoL1. A ApoL1 confere resistência à doença do sono africana.2-7
As células glomerulares também podem ser lesadas pela exposição a fármacos e toxinas específicos. Os podócitos, por exemplo, podem ser lesados pela exposição aos AINH ou ao interferon, causando uma doença de lesões mínimas. Contudo, a exposição destas células ao ouro, à penicilamina e ao mercúrio predispõe ao desenvolvimento de nefropatia membranosa. A disfunção endotelial causadora de microangiopatia trombótica ou síndrome hemolítica-urêmica foi associada ao uso de inibidores do fator de crescimento endotelial vascular (FCEV),8 ciclosporina, tacrolimo, mitomicina C e anticoncepcionais orais.
Durante o exame, é preciso procurar sinais de síndromes nefrítica ou nefrótica, incluindo hipertensão e edema, e usar o teste da vareta medidora de nível de urina para detectar hematúria e albuminúria. É notável que pacientes com anasarca decorrente da síndrome nefrótica não apresentem elevação da pressão venosa jugular nem congestão pulmonar. Estes achados devem alertar para a possível coexistência de uma doença cardíaca. Contudo, pacientes com nefrite aguda e hipertensão severa podem apresentar edema pulmonar e encefalopatia hipertensiva. É preciso procurar sinais da possível existência de doenças sistêmicas, incluindo um exame detalhado do trato respiratório superior, boca, articulações e pele. Deve ser feito um exame microscópico de urina, para determinar se o sedimento urinário é ativo e contém hemácias dismórficas e cilindros de hemácias – um sinal patognomônico de glomerulonefrite. A lipidúria (corpúsculos adiposos ovais, gotículas lipídicas e cilindros de gordura) é tipicamente observada na síndrome nefrótica.
Além de definir a síndrome apresentada pelo paciente, a revisão dos sistemas e o exame físico podem ser úteis para identificar infecções (faringite, endocardite, hepatite ou osteomielite) que possam estar associadas à glomerulonefrite. Algumas doenças em particular podem estar associadas a malignidades, incluindo: a nefropatia membranosa, que pode ocorrer em associação com tumores do pulmão, mama, intestino ou próstata; a doença de lesões mínimas, que pode estar associada à doença de Hodgkin; e a amiloidose, possivelmente associada ao mieloma múltiplo.
Exames laboratoriais e investigação inicial
Os exames de laboratório devem incluir as análises bioquímicas, que permitem o cálculo da TFG estimada e a identificação de distúrbios eletrolíticos capazes de complicar a insuficiência renal, tais como hipercalemia, hiperfosfatemia e hipocalcemia. Os níveis séricos de albumina, perfil lipídico de jejum e quantificação da excreção urinária de albumina, seja na urina de 24 horas ou via proporção proteína:creatinina em amostra isolada, são essenciais para o diagnóstico da síndrome nefrótica. Como já mencionado, é importante examinar um sedimento de urina fresco em busca de evidências de hemácias, cilindros de hemácias ou lipidúria.
As investigações subsequentes são realizadas com o objetivo de excluir causas secundárias de glomerulonefrite e devem ser adequadas à síndrome em questão. Os níveis de complemento são particularmente úteis para estreitar o diagnóstico diferencial do paciente que apresenta nefrite [Tabela 1]. Do mesmo modo, os títulos de anticorpos antinúcleo (Fator antinúcleo – FAN), anticorpos anticitoplasma de neutrófilo (ANCA), sorologia de hepatite e títulos de antiestreptolisina O podem ser úteis neste contexto. Contudo, os níveis de hemoglobina A1C (HbA1C), imunofixação ou eletroforese de proteínas séricas e urinárias, cadeias leves livres, testes para detecção de HIV e FAN podem ser mais úteis em casos de pacientes nefróticos.
A ultrassonografia renal geralmente é menos significativa, mas pode permitir a observação de rins pequenos e ecogênicos na doença avançada ou de rins aumentados na glomerulonefrite aguda, nefropatia diabética em estágio inicial e NAHIV.
Exame de biópsia renal
A ferramenta diagnóstica definitiva, em muitos casos de doença renal, especialmente em casos de doença renal primária, é a biópsia renal. As biópsias renais percutâneas costumam ser obtidas por um nefrologista ou radiologista que emprega uma técnica estéril e anestesia local. Guiada por ultrassom, uma agulha de biópsia disparada por um mecanismo de molas é utilizada na obtenção de um centro de tecido cortical de um dos rins. Havendo indicação, a biópsia deve ser realizada no momento adequado, uma vez que a rápida iniciação da terapia apropriada para a doença glomerular muitas vezes limita a lesão renal e preserva a função dos rins.
A obtenção de biópsias renais percutâneas orientada por ultrassom em geral constitui um procedimento seguro. Entretanto, pode haver complicações sérias, como o desenvolvimento de hematomas perinéfricos, hematúria grosseira, fístulas arteriovenosas, infecção e lesão de um órgão adjacente.9,10 Até 1/3 das complicações sérias não são reconhecidas durante as primeiras 8 horas subsequentes à conclusão do procedimento.9,10 Por isso, preferimos manter o paciente sob observação de um dia para o outro, após a obtenção da biópsia. A maioria das complicações, quando identificada prontamente, pode ser tratada de forma conservadora ou por radiologia intervencionista, necessitando de intervenção cirúrgica apenas em casos muito raros. A disfunção plaquetária no contexto da insuficiência renal em estágio avançado (creatinina > 5 mg/dL), ou atribuível a uma terapia concomitante com inibidores plaquetários, constitui o único fator preditor reconhecido de complicações de sangramento. O valor do exame de biópsia deve ser cuidadosamente ponderado contra o risco aumentado de sangramento para todos os pacientes, porém especialmente no caso daqueles com insuficiência renal em estágio avançado para os quais uma intervenção terapêutica pode não retardar a necessidade de diálise.
A biópsia renal estabelece um diagnóstico patológico, determina se a doença é mediada por anticorpos ou complemento e auxilia na determinação da terapia doença-específica. O exame de biópsia também fornece informação sobre o grau de cicatrização glomerular e intersticial. O grau de cicatrização intersticial é importante como indicador prognóstico, uma vez que a lesão renal fibrótica geralmente é considerada irreversível. Existem 4 situações em que o exame de biópsia renal deve ser obrigatoriamente considerado: (1) insuficiência renal de progressão rápida no contexto da doença glomerular (GNRP); (2) síndrome nefrótica (exceto no caso de crianças que podem ser consideradas com doença de alteração mínima e necessitam de biópsia somente quando a terapia à base de esteroides falha, bem como no caso de pacientes diabéticos que apresentam os aspectos clínicos clássicos da nefropatia diabética); (3) doença renal, no contexto de uma doença sistêmica (p. ex., lúpus eritematoso sistêmico [LES], mieloma, amiloide ou vasculite); e (4) disfunção inexplicável de um transplante renal. Alguns pacientes com proteinúria não nefrótica, hematúria ou doença renal crônica também podem ser beneficiados por um exame de biópsia renal para fins diagnósticos ou prognósticos. As biópsias são relativamente contraindicadas para pacientes com DRET, diátese hemorrágica ou que têm apenas um único rim.
O relatório do exame de biópsia renal inclui uma descrição de microscopia óptica, coloração de imunofluorescência e microscopia eletrônica.
Microscopia óptica
A microscopia óptica descreve a natureza da doença glomerular utilizando cortes de biópsia renal corados. O relatório do exame de biópsia deve conter uma descrição da celularidade glomerular, espessura da MBG e patência das alças capilares glomerulares, grau de cicatrização (esclerose) glomerular e presença ou ausência de crescentes glomerulares. Quando o glomérulo é hipercelular, a microscopia óptica é capaz de distinguir se o número aumentado de células glomerulares é atribuível à proliferação de células glomerulares residentes ou à infiltração do glomérulo por células inflamatórias, como os neutrófilos. A MBG pode ser espessada pela deposição de imunocomplexos, enquanto as alças capilares podem entrar em colapso ou ser preenchidas por material hialino. Os crescentes glomerulares consistem em camadas de células, incluindo células epiteliais, linfócitos e macrófagos, que são observadas no espaço urinário da cápsula de Bowman [Figura 2]. A presença de crescentes glomerulares aponta a existência de uma doença severa, geralmente associada à GNRP. A microscopia óptica também pode ser empregada para esclarecer se a doença glomerular é focal (isto é, < 50% dos glomérulos estão envolvidos) ou difusa (ou seja, > 50% dos glomérulos estão adoecidos). A doença é denominada segmentar quando tende a afetar uma pequena parte ou segmento do glomérulo. Em contraste, a doença que tende a envolver todo o tufo glomerular é denominada global. O relatório também deve incluir um comentário sobre o grau de atrofia tubular e fibrose intersticial, porque o grau de fibrose tubulointersticial é o melhor fator preditor do prognóstico. O uso de colorações especiais permite detectar, por microscopia óptica, a presença de maior concentração proteica na matriz (coloração com prata), fibrilas amiloides (corante vermelho Congo) ou corpúsculos de inclusão viral.
Imunofluorescência
A imunofluorescência determina a natureza do processo imune causativo. A coloração pode detectar a deposição de anticorpos específicos (p. ex., IgA ou IgG) e componentes do complemento (p. ex., C1q, C3, C4 ou C5b-9). A localização e o padrão de deposição dos anticorpos com frequência são diagnósticos. Os depósitos mesangiais de IgA, por exemplo, são típicos de NIgA ou púrpura de Henoch-Schönlein (PHS). Já os depósitos granulares de IgG e C3 junto à parede capilar são típicos dos imunocomplexos formados na nefropatia mesangial e na nefrite lúpica. A deposição linear de IgG na MBG é diagnóstica da doença anti-MBG. As colorações específicas para cadeias leves kappa e lambda permitem identificar as doenças de deposição de anticorpos monoclonais, como amiloidose AL e doença de deposição de cadeia leve.
Microscopia eletrônica
A microscopia eletrônica fornece informação sobre a ultraestrutura glomerular. É possível avaliar a localização dos imunodepósitos eletrondensos e o grau de lesão das células glomerulares. A integridade e a espessura da MBG também podem ser avaliadas. A extensão da obliteração do processo podal ajuda a distinguir as formas primárias de lesão ao podócito (como a doença de lesões mínimas ou a GESF, em que todos os podócitos dos processos podais são obliterados) da obliteração “adaptativa” do podócito (que é secundária à hipertensão glomerular e envolve redução do número de néfrons, com apenas uma parte dos podócitos afetados).
Princípios gerais no tratamento das doenças glomerulares
Tratamento das complicações agudas das glomerulopatias
O controle das complicações agudas da glomerulonefrite, entre as quais a anasarca, trombose venosa, infecção ou hipertensão acelerada, deve ser abordado com urgência. Como a sobrecarga de volume observada na síndrome nefrótica é causada pela retenção de sais e água, os pacientes respondem aos diuréticos de alça. A terapia diurética é especialmente importante quando um edema pulmonar, efusões pleurais, ascite severa ou edema periférico maciço acarretam uma morbidade significativa. A diálise deve ser prontamente iniciada em casos de pacientes oligúricos com edema pulmonar, azotemia severa ou sinais e sintomas de uremia. A trombose venosa e a embolia pulmonar precisam ser tratadas com heparina e administração subsequente de varfarina, até que a síndrome nefrótica seja controlada.
Tratamento geral das doenças glomerulares
A terapia destinada a baixar a pressão capilar intraglomerular e diminuir a proteinúria é oferecida à maioria dos pacientes com doença glomerular crônica, porque foi demonstrado que esta terapia melhora os sintomas de nefrose e retarda a progressão para insuficiência renal crônica. Os inibidores da enzima conversora de angiotensina (ECA) ou os bloqueadores do receptor de angiotensina II (BRA) diminuem a pressão intraglomerular ao promoverem dilatação da arteríola eferente, além de serem mais efetivos do que outros agentes anti-hipertensivos na redução das perdas urinárias de proteína e na prevenção da fibrose renal. Recomenda-se que as metas de pressão arterial sejam menores do que as metas adotadas para a população em geral (< 130/80 mmHg). O objetivo habitual da terapia consiste em diminuir o nível de proteinúria para menos de 1 g/dia ou reduzir a proporção proteína:creatinina para menos de 1. A administração de doses baixas de espironolactona e/ou de um inibidor de renina, o aliskiren, pode produzir um efeito redutor de proteínas adicional. É importante notar que a inibição do sistema da renina-angiotensina (SRA) é mais efetiva quando instituída no início do curso da doença. Além disso, a redução da pressão intraglomerular pode promover a diminuição aguda da TFG e precipitar insuficiência renal e hipercalemia em pacientes com doença renal crônica em estágios IV ou V. Por estes motivos, os riscos de bloqueio do SRA podem sobrepujar os benefícios proporcionados aos pacientes cujo estado se aproxima da DRET. A hipercolesterolemia severa e a hipertrigliceridemia ocorrem com frequência em pacientes nefróticos e requerem terapia com inibidores de HMG-CoA redutase, para diminuir os níveis lipoproteína de baixa densidade (LDL) para menos de 100 mg/dL.11 Os pacientes com síndrome nefrótica em geral, mas em particular aqueles com nefropatia membranosa, são extremamente hipercoaguláveis e correm risco de desenvolver trombose venosa. A terapia profilática com varfarina deve ser considerada para pacientes com síndrome nefrótica com história de trombose anterior ou que apresentam hipoalbuminemia severa persistente (albumina sérica < 2 g/dL). Os pacientes devem receber vacinação contra pneumococos, devido ao alto risco de infecção pneumocócica associada à síndrome nefrótica.
Tratamento das manifestações tardias da doença renal crônica
As doenças glomerulares frequentemente permanecem assintomáticas durante meses ou anos, antes de se manifestarem. Por isso, a 1ª manifestação apresentada por muitos pacientes é a insuficiência renal em estágio avançado. O tratamento destes pacientes é voltado para o controle das complicações da doença renal crônica.
As doenças glomerulares específicas e seu tratamento
Doenças associadas à síndrome nefrítica
Os aspectos clínicos da síndrome nefrítica são produzidos por uma lesão inflamatória severa e tipicamente aguda nos glomérulos, que leva à diminuição da função renal. As características clínicas incluem hematúria, proteinúria, hipertensão e aumento dos níveis de creatinina. Na síndrome nefrítica, a hematúria pode ser macro ou microscópica. Um sedimento urinário ativo contendo hemácias dismórficas e cilindros de hemácias é patognomônico para nefrite. A proteinúria observada na síndrome nefrítica geralmente é menor que 3,5 g/24 horas.
As principais complicações da síndrome nefrítica são a hipertensão, sobrecarga de volume, hipercalemia e acidose metabólica. Os pacientes podem apresentar um ou mais destes agravantes. A diminuição da TFG observada na síndrome nefrítica causa retenção acentuada de sais e água, com consequente desenvolvimento de hipertensão e edema. O paciente pode apresentar crise hipertensiva aguda acompanhada de insuficiência cardíaca congestiva, edema cerebral e edema pulmonar.
As formas mais comuns de glomerulonefrite manifestada como síndrome nefrítica podem ser classificadas com base nos respectivos mecanismos patológicos subjacentes, em 3 categorias: mediada por imunocomplexo (p. ex., glomerulonefrite pós-infecciosa, NIgA ou nefrite lúpica); induzida por anticorpo (p. ex., doença anti-MBG); e paucimune (p. ex., vasculites ANCA-associadas na ausência de imunodepósitos glomerulares detectados por imunofluorescência).
O curso clínico das doenças glomerulares individuais pode variar. Exemplificando, a glomerulonefrite pós-estreptocócica (GNPE) costuma ser aguda e autolimitada; a NIgA é intermitente e crônica; a doença anti-MBG e a vasculite ANCA-associada apresentam início rápido e são progressivas, levando ao desenvolvimento de insuficiência renal em estágio terminal dentro de um curto período, caso não sejam tratadas.
Síndrome nefrítica secundária a infecções
A glomerulonefrite pode resultar de infecções bacterianas (Streptococcus, Staphylococcus aureus, Salmonella, Escherichia coli, Legionella e Mycoplasma), infecções virais (hepatite B, hepatite C, HIV e citomegalovírus), infecções parasitárias (Schistosoma, Plasmodium, Trypanosoma) e infecções fúngicas (Aspergillus, Histoplasma, Candida).11 A doença renal associada à infecção costuma estar relacionada a imunocomplexos. Estes imunocomplexos são compostos de antígenos do agente infeccioso e de anticorpos do hospedeiro dirigidos contra os antígenos do patógeno. A doença glomerular clássica associada à infecção é a GNPE, que produz um quadro histológico glomerular distinto daquele observado na maioria das outras formas pós-infecciosas de glomerulonefrite.11 No momento, a hepatite C e a infecção pelo HIV constituem as principais causas de glomerulonefrite associada à infecção em países industrializados. Contudo, a GNPE continua sendo comum em outras partes do mundo.11 A glomerulonefrite associada à hepatite C e a GNPE tipicamente se manifestam como síndrome nefrítica,11 enquanto a infecção pelo HIV causa síndromes nefríticas ou nefróticas.12-14
Glomerulonefrite pós-estreptocócica (GNPE)
A GNPE tem como característica ser uma doença autolimitada, que se desenvolve após uma infecção de faringe ou de pele (p. ex., impetigo ou pioderma) pelas conhecidas cepas nefritogênicas de estreptococos do grupo A.11 Ocasionalmente, alguns casos foram associados aos estreptococos do grupo C. A GNPE afeta com mais frequência crianças com idade entre 2 e 10 anos, além de ser 2 vezes mais comum no sexo masculino do que no feminino. A doença é endêmica nas áreas densamente povoadas dos países em desenvolvimento, mas também ocorre sob a forma epidêmica em instituições e, às vezes, como casos esporádicos no mundo desenvolvimento.
Patogênese. A GNPE consiste em uma forma clássica de glomerulonefrite mediada por imunocomplexo. A lesão glomerular é induzida quando os imunocomplexos circulantes, compostos por antígeno estreptocócico e anticorpo dirigido contra este antígeno, depositam-se no mesângio e subendotélio do glomérulo. Os 2 antígenos estreptocócicos mais provavelmente envolvidos na patogênese são a gliceraldeído fosfato desidrogenase estreptocócica (GAPDH – em inglês, glyceraldehyde phosphate dehydrogenase; também conhecida como receptor de plasmina nefrite-associado) e a protease catiônica estreptocócica exotoxina B (SpeB).15 Ambos os antígenos ativam a via alternativa do complemento e foram detectados nos glomérulos de um significativo percentual de pacientes com GNPE. Os títulos de anticorpos anti-GAPDH e anti-SpeB estão correlacionados à doença clínica e raramente são detectados em pacientes com infecções estreptocócicas não causadoras de nefrite.16 A sensibilização aos antígenos estreptocócicos explica o típico período de latência de 1 a 3 semanas entre a infecção e o aparecimento da nefrite.
Patologia. À microscopia óptica, a GNPE caracteriza-se por uma hipercelularidade glomerular difusa resultante da proliferação de células mesangiais e endoteliais residentes, bem como da infiltração de neutrófilos [Figura 3]. Até 5% dos pacientes desenvolvem uma doença severa acompanhada de glomerulonefrite crescêntica. A coloração por imunofluorescência revela a presença de depósitos de IgG e C3 junto à parede capilar e ao mesângio, sem C1q nem C4, consistente com a via alternativa de ativação do complemento. Na GNPE, a microscopia eletrônica mostra a presença de múltiplos depósitos de imunocomplexos, grandes e em forma de cúpula, que são referidos como “corcundas” subepiteliais, por sua localização na base dos podócitos [Figura 4]. Estes depósitos do tipo “corcunda” diferem dos imunodepósitos subepiteliais, que são menores e mais frequentemente encontrados na nefropatia membranosa, na nefrite lúpica membranosa e em alguns casos de GNPE. As biópsias obtidas ainda no início do curso da GNPE podem apresentar depósitos de imunocomplexos junto ao mesângio e subendotélio.
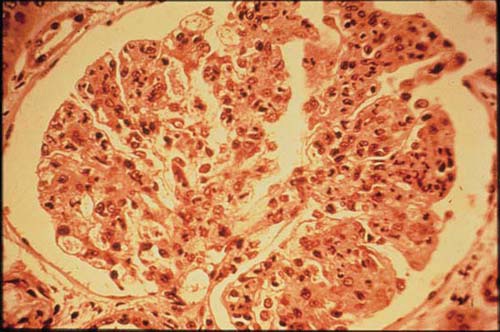
Figura 3. Fotomicrografia de glomerulonefrite pós-estreptocócica (GNPE) mostrando hipercelularidade glomerular.
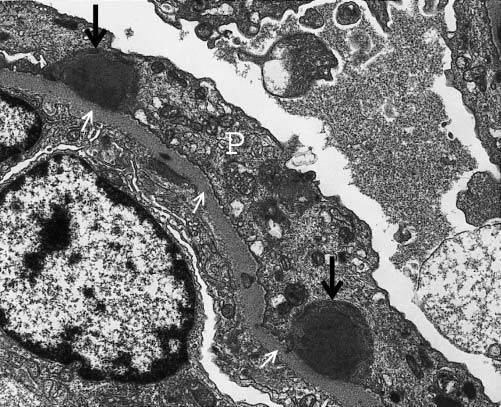
Figura 4. Micrografia eletrônica de transmissão mostrando os depósitos em forma de corcunda (setas pretas) caracteristicamente encontrados na glomerulonefrite pós-estreptocócica (GNPE), situados abaixo dos podócitos (P). A membrana basal glomerular (MBG) é indicada pelas setas brancas.
Achados clínicos e laboratoriais. Após um período de latência entre a faringite inicial (duração média de 10 dias) ou infecção cutânea (duração média de 21 dias), os pacientes desenvolvem um edema marcante que afeta sobretudo a face e a parte superior do corpo, além de produzirem pequenos volumes de urina escura ou turva contendo hemácias e cilindros de hemácias e de apresentarem uma hipertensão que pode ser severa.11,17 O níveis séricos de creatinina podem permanecer normais nos casos mais brandos, mas insuficiência renal aguda não é incomum. A proteinúria é tipicamente inferior a 500 mg/dia no curso da doença aguda. As culturas produzidas com material obtido da garganta e da pele geralmente resultam negativas, entretanto as determinações sorológicas de uma infecção estreptocócica recente, como a quantificação de antiestreptolisina O, resultam elevadas e permanecem aumentadas durante 3 a 6 meses. Os títulos de antiestreptolisina O podem ser embotados pelo uso recente de antibióticos e são menos marcantes após as infecções cutâneas. Como a GNPE é induzida por imunocomplexos pela via alternativa do complemento, os pacientes apresentam diminuição acentuada dos níveis de CH50 e C3, mas não de C1q, C2 nem C4, que são ativadas pela via clássica do complemento. A depressão persistente dos níveis de C3 deve sugerir a existência de outra causa, como GNMP ou doença de depósito denso (DDD) [ver adiante].
Tratamento, curso e prognóstico. O tratamento da GNPE é de suporte. O controle da hipertensão é fundamental; é aliado à diálise sempre que necessário, para os casos de insuficiência renal aguda oligúrica. A terapia antibiótica não tem utilidade na GNPE, uma vez que a infecção incitadora costuma ocorrer com antecedência de várias semanas. A maioria dos pacientes (> 95%) recupera a função renal normal, principalmente aqueles que apresentam as formas endêmica e epidêmica de GNPE. Embora a hematúria macroscópica e o edema em geral sejam resolvidos dentro de 2 semanas, a hematúria microscópica pode persistir durante vários anos. Vinte por cento dos pacientes desenvolvem uma proteinúria na faixa nefrótica, tipicamente durante a fase de recuperação da doença, que é resolvida espontaneamente. Contudo, a proteinúria na faixa não nefrótica pode durar até 2 anos. Os pacientes com a forma esporádica da GNPE muitas vezes têm idade mais avançada e podem apresentar uma disfunção renal persistente e/ou hipertensão, enquanto alguns podem progredir para insuficiência renal em estágio terminal. A GNPE recorrente é extremamente rara, talvez devido à imunidade de longa duração contra as cepas nefritogênicas.
Glomerulonefrite associada ao vírus da hepatite C (HCV)
A doença glomerular constitui uma manifestação extra-hepática pouco frequente da infecção crônica pelo vírus da hepatite C (HCV). Entretanto, considerando que mais de 170 milhões de pessoas em todo o mundo estão infectadas por este vírus, a nefrite associada ao HCV é bastante comum na prática clínica e pode se desenvolver decorridos vários anos da infecção viral inicial, na ausência de doença hepática evidente.18
Aspectos clínicos e laboratoriais. A maioria dos pacientes com glomerulonefrite associada ao HCV apresenta aspectos nefríticos e nefróticos mistos, tais como edema e proteinúria severa, hematúria microscópica com cilindros de hemácias, hipertensão e graus variáveis de disfunção renal. Cerca de 30% dos pacientes também apresentam aspectos sistêmicos de crioglobulinemia, manifestados sob a forma da tríade artralgia/vasculite cutânea com púrpura/úlceras penetrantes, além de fraqueza. Alguns pacientes apresentam vasculite severa e GNRP.18,19 O diagnóstico geralmente é estabelecido pela obtenção de resultados positivos em testes de detecção de anticorpos anti-HCV, bem como pela detecção de RNA de HCV e hipocomplementemia (níveis baixos de C3 e muito baixos de C4) em pacientes que apresentam aspectos clínicos característicos.
Patogênese e patologia. A glomerulonefrite associada ao HCV consiste em uma doença por imunocomplexos clássica e é a causa mais comum de GNMP de tipo I em países industrializados.20 A GNMP é caracterizada por uma hipercelularidade glomerular atribuível à infiltração de monócitos e proliferação celular glomerular, bem como à expansão mesangial e espessamento da parede capilar glomerular atribuível à deposição de imunocomplexos antígeno-anticorpo junto ao mesângio e subendotélio. No caso da GNMP associada ao HCV, os imunocomplexos são compostos pelo HCV e por anticorpo anti-HCV, que ativam a via clássica do complemento. A interposição das células inflamatórias sob o endotélio e a formação de novos materiais de membrana basal levam à característica reduplicação ou formação em “trilho de bonde” da MBG. A superestimulação crônica das células B pode resultar em expansão clonal e produção de IgM monoclonal com atividade reumatoide dirigida contra a IgG anti-HCV policlonal. Estes imunocomplexos são constituídos por IgM, IgG e HCV e constituem as crioglobulinas mistas causadoras da síndrome da crioglobulinemia mista, sendo visíveis por microscopia eletrônica junto ao subendotélio e capilares glomerulares, graças à subestrutura característica.18 Menos frequentemente, a infecção pelo HCV pode estar associada a NIgA, nefropatia membranosa, glomerulonefrite fibrilar, GESF (em afro-americanos) e microangiopatia trombótica (em receptores de transplante renal).21
Tratamento, curso e prognóstico. A nefrite associada ao HCV segue um curso tipicamente indolente e recorrente, com progressão gradual para DRET. Foi demonstrado que as medidas gerais (ver adiante) recomendadas para o tratamento dos pacientes com glomerulonefrite, incluindo o controle da pressão arterial, inibição do SRA e uso de agentes redutores de lipídios, retardam a progressão da doença renal em pacientes infectados pelo HCV.22 A base do tratamento da glomerulonefrite associada ao HCV está voltada para a eliminação do vírus e dos imunocomplexos. Atualmente, existem 2 formulações (alfa-2-a e alfa-2-b) de interferons conjugados ao polietilenoglicol (peguiladas) aprovadas para uso no tratamento da infecção por HCV em pacientes renais. O interferon peguilhado é mais efetivo quando combinado à ribavirina, que consiste em um metabólito inibidor da replicação do HCV. A ribavirina é depurada pelos rins, portanto, seu uso por pacientes renais precisa ser ajustado de acordo com a TFG, pois seu acúmulo pode causar uma hemólise prejudicial à vida.21,23 A ribavirina é contraindicada para pacientes com insuficiência renal em estágio avançado. Nos casos de crioglobulinemia com vasculite sistêmica severa ou GNRP, a plasmaférese, anticorpos anti-célula B monoclonais e terapia imunossupressora (corticosteroides e ciclofosfamida) são utilizados para promover uma diminuição aguda dos níveis de crioglobulina.18,21,24
Outras glomerulonefrites pós-infecciosas
A glomerulonefrite proliferativa focal e segmentar, que se manifesta como hematúria microscópica e hipocomplementemia, constitui uma ocorrência relativamente comum e um bom indício da existência de endocardite associada ao Streptococcus viridans. A endocardite estafilocócica, em especial em usuários de drogas endovenosas, pode causar uma nefrite mais severa com manifestações nefríticas-nefróticas mistas ou GNRP.25 Outras infecções bacterianas (p. ex., shunts infectados e abscessos viscerais),11 bem como certas infecções virais (p. ex., hepatite B)26 podem causar GNMP, que está associada a uma hipocomplementemia de via alternativa caracterizada por níveis baixos de C3 e níveis normais de C4. A terapia antibiótica elimina a infecção associada à endocardite e permite a resolução da resposta imune. Níveis de C3 persistentemente baixos sugerem que a infecção não foi eliminada.
Nefropatia por IgA (NIgA) e púrpura de Henoch-Schönlein (PHS)
A denominação NIgA deriva da deposição glomerular de IgA.27 Trata-se do padrão mais prevalente de glomerulonefrite em países onde o exame de biópsia renal é amplamente utilizado como ferramenta investigativa.28 É particularmente prevalente na Ásia, e isto sugere uma possível predisposição genética à doença.27 A NIgA afeta mais indivíduos do sexo masculino do que a população feminina (proporção 3:1) e é mais comum em adultos jovens. Os depósitos mesangiais de IgA também podem ser observados associados a certo número de doenças sistêmicas [Tabela 2]. Formas hereditárias também foram descritas.29
Tabela 2. Doenças associadas à deposição mesangial de IgA
|
Doenças dermatológicas |
Dermatite herpetiforme |
|
Psoríase | |
|
Síndrome de Reiter | |
|
Doenças gastrintestinais |
Doença celíaca |
|
Doença de Crohn | |
|
Colite ulcerativa | |
|
Doenças hepáticas |
Cirrose alcoólica |
|
Cirrose associada à hepatite | |
|
Doenças neoplásicas |
Carcinoma bronquial |
|
Gamopatia monoclonal de IgA | |
|
Micose fungoide | |
|
Síndrome de Sézary | |
|
Doenças reumáticas |
Espondilite anquilosante |
|
Artrite reumatoide | |
|
Doenças sem classificação |
Infecção pelo HIV |
|
Diabetes melito | |
|
Hemossiderose pulmonar idiopática | |
|
Deficiência de properdina | |
|
Fibrose retroperitoneal | |
|
Sarcoidose |
Aspectos clínicos e laboratoriais. É comum a NIgA se manifestar como hematúria macroscópica episódica, frequentemente acompanhada de dor no flanco, durante uma infecção no trato respiratório superior. Esta nefrite, referida como sinfaringítica, ocorre em cerca de 40 a 50% dos casos e contrasta com a nefrite de aparecimento tardio, que surge em 2 a 3 semanas após o desenvolvimento de faringite em indivíduos com GNPE. Estes pacientes tipicamente apresentam função renal normal, com hematúria microscópica persistente e pouca ou nenhuma proteinúria entre os episódios agudos de hematúria grosseira. Cerca de 30 a 40% dos pacientes apresentam uma proteinúria mais severa, e alguns desenvolvem síndrome nefrótica, além da hematúria. Poucos pacientes com NIgA apresentam insuficiência renal aguda atribuível à GNPR ou necrose tubular aguda.
A PHS é uma vasculite sistêmica caracterizada pela deposição tecidual de imunocomplexos contendo IgA, inclusive no mesângio glomerular. Portanto, esta condição compartilha muitos aspectos com a NIgA.30,31 A PHS manifesta-se clinicamente como uma tétrade clássica de erupção purpúrica (tipicamente, sobre os membros inferiores), artralgia (nos joelhos e tornozelos), dor abdominal e nefrite. Embora a PHS seja mais frequente em crianças com menos de 5 anos de idade, o envolvimento renal é mais comum em crianças maiores e adultos. Assim como a NIgA, a manifestação da PHS muitas vezes acompanha uma infecção no trato respiratório superior. O espectro das manifestações renais é quase igual ao da NIgA.
A urinálise, tanto na nefrite da NIgA como na nefrite da PHS, mostra uma hematúria microscópica e quantidades variáveis de proteinúria. Os cilindros hemáticos são encontrados com frequência durante as exacerbações agudas. Outras investigações laboratoriais geralmente não têm utilidade na NIgA, sendo que o diagnóstico definitivo é estabelecido pelo exame de biópsia renal. Os níveis séricos de complemento estão dentro da faixa normal, enquanto os níveis de IgA estão aumentados em cerca de 1/3 dos pacientes. Contudo, há relatos de que uma proporção IgA sérica:C3 elevada (> 3) tem utilidade diagnóstica e prognóstica para NIgA em casos de pacientes com hematúria e proteinúria.32 O exame de biópsia cutânea, em casos de PHS, tipicamente revela a existência de vasculite leucocitoclástica e imunocoloração para IgA junto à vasculatura.
O diagnóstico diferencial de hematúria glomerular microscópica isolada persistente inclui a glomerulonefrite hereditária (síndrome de Alport), doença da membrana basal fina, LES e glomerulonefrite pós-infecciosa.
Patogênese. As nefrites de NIgA e de PHS resultam da deposição mesangial de imunocomplexos contendo IgA, ativação da via alternativa do complemento e iniciação de uma resposta inflamatória local caracterizada pela proliferação mesangial [Figuras 5 e 6]. A tendência de as exacerbações de hematúria macro ou microscópica ocorrerem em 24 horas após uma infecção mucosa (p. ex., trato respiratório superior ou intestino) indica que a doença glomerular pode ser iniciada por uma resposta de IgA de mucosa anormal à infecção.28 Embora alguns estudos tenham sugerido que níveis séricos de IgA elevados podem contribuir para a doença,28 a IgA que se deposita nos glomérulos de pacientes com NIgA e PHS é incomum. A glicosilação aberrante da IgA1 na região da dobradiça da cadeia pesada modifica a estrutura da IgA e permite o desenvolvimento de anticorpos IgG anti-IgA. Assim, a teoria vigente sustenta que a IgA1 com glicosilação aberrante se deposita no mesângio, onde atua como um antígeno plantado para formação de imunocomplexos IgA-IgG, com consequente ativação local do complemento e lesão glomerular.33,34
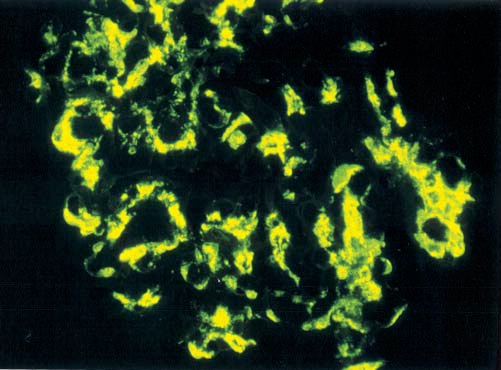
Figura 5. Imunocoloração de IgA mostrando a deposição de IgA com típica distribuição celular mesangial, na nefropatia por IgA (NIgA).
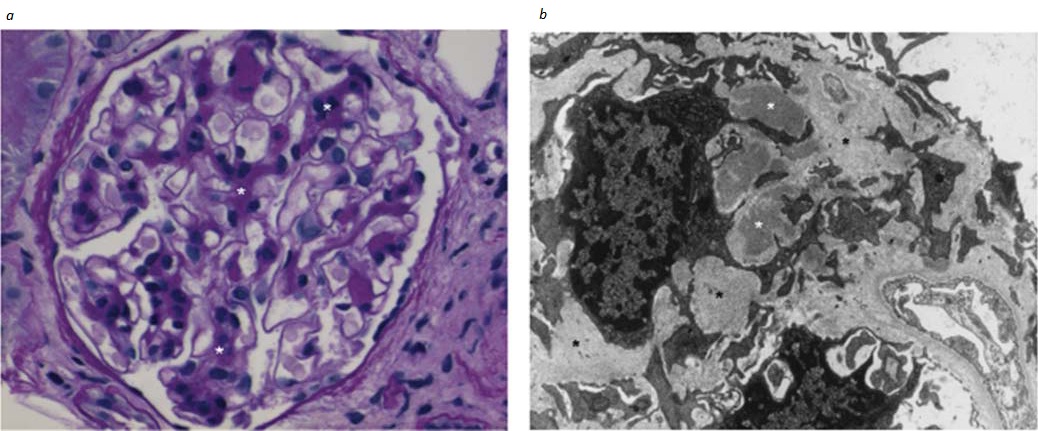
Figura 6. (a) Fotomicrografia obtida de um paciente com nefropatia por IgA (NIgA), mostrando a característica hipercelularidade mesangial e a expansão da matriz (*). (b) Micrografia eletrônica de transmissão do mesângio de um paciente com NIgA. Os imunodepósitos mesangiais (asterisco branco) e a matriz mesangial expandida (asterisco preto) são indicadas.
Patologia. As lesões glomerulares que se desenvolvem após a deposição glomerular de IgA são variáveis.30,35 A lesão característica consiste na expansão mesangial acompanhada de proliferação celular mesangial [Figura 6], que é visualizada à microscopia óptica como hipercelularidade de células mesangiais [Figura 7]. Ocasionalmente, há formação de crescentes glomerulares que costumam estar associados à GNRP. Nos casos severos que evoluem para insuficiência renal em estágio terminal, observa-se cicatrização mesangial e fibrose tubulointersticial. A coloração imunofluorescente é diagnóstica e revela a presença de IgA [Figura 5] no mesângio e, ocasionalmente, de IgG e IgM. O C3 com frequência é detectado sem C1q nem C4, de modo consistente com a ativação da via alternativa do complemento. À microscopia eletrônica, os imunodepósitos mesangiais são tipicamente visualizados como densidades eletrônicas.
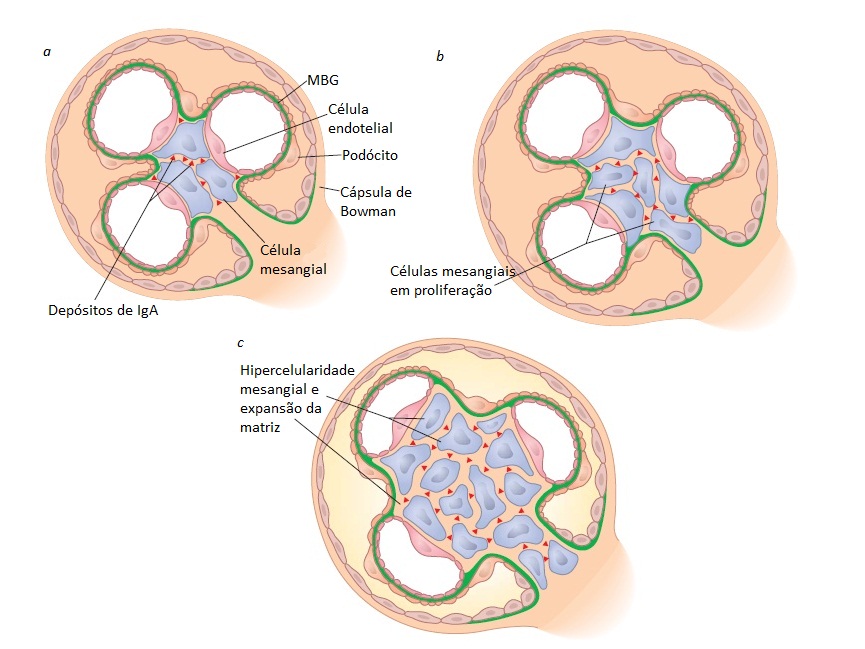
Figura 7. Representação esquemática destacando os eventos observados na nefropatia por IgA (NIgA) que levam ao desenvolvimento de hipercelularidade mesangial e expansão da matriz. (a) A deposição de IgA entre as células mesangiais é seguida de (b) proliferação de células mesangiais, que leva à (c) hipercelularidade mesangial e à expansão da matriz.
MBG = membrana basal glomerular.
Tratamento, curso e prognóstico. Na maioria dos pacientes, a NIgA segue um curso indolente. Os fatores de risco de progressão da doença incluem níveis elevados de creatinina sérica e pressão arterial aumentada no momento da manifestação da doença, bem como proteinúria persistente (> 1 g/dia), idade avançada e fibrose intersticial detectada no exame de biópsia renal.26,36 Por motivos ainda obscuros, a hematúria macroscópica recorrente está associada, em muitos casos, a um melhor prognóstico do que a proteinúria e a hematúria microscópica persistente. Vários estudos randomizados controlados, envolvendo pacientes com NIgA que apresentavam proteinúria, demonstraram que o uso de inibidores de ECA ou BRA diminui a proteinúria e retarda a progressão da doença.37-39 O uso combinado de inibidores de ECA e BRA pode intensificar o efeito antiproteinúrico, porém seu efeito renoprotetor ainda precisa ser demonstrado.40 Como é difícil prever exatamente quais pacientes com NIgA apresentarão evolução da doença, uma possibilidade seria considerar o tratamento de todos os pacientes com NIgA com um destes agentes. O consenso a que se chegou, após a realização de vários estudos, indica que a adição de um curso de 6 meses de prednisona a um tratamento com inibidor de ECA proporciona melhora da sobrevida renal em pacientes com NIgA que apresentam proteinúria acima de 1 g/dia.41,42 Resultados similares foram relatados por um pequeno estudo controlado sobre o uso de micofenolato de mofetil por um período de 6 meses.43 Entretanto, o papel deste agente na NIgA ainda precisa ser estabelecido.44 Como as evidências de eficácia são limitadas, sugerimos que a imunossupressão promovida por agentes citotóxicos45 seja restrita aos pacientes que mostram evidências de doença ativa e progressão mesmo com a inibição do SRA e a terapia à base de corticosteroides. No caso dos pacientes com GNRP, o tratamento deve ser iniciado com a terapia de pulsos endovenosos de metilprednisolona. O papel da tonsilectomia é controverso. Recomendamos reservar este procedimento para os casos de pacientes com tonsilite recorrente. Embora os estudos clínicos de avaliação da eficácia dos óleos de peixe no tratamento da NIgA tenham produzido resultados conflitantes,27,46 tais óleos não são tóxicos. Defendemos o uso de altas doses de ácidos graxos ômega-3 aliados a uma dieta pobre em ácidos graxos ômega-6, como adjunto dos inibidores de SRA.
Apesar da imunossupressão mais satisfatória e da terapia de suporte com inibição de ECA ou uso de BRA, a NIgA continua sendo uma causa significativa de DRET, levando até 40% dos pacientes afetados a necessitarem de terapia de substituição renal dentro de um período de 20 anos após a manifestação da condição.27,36 Pode haver recidiva de NIgA subsequentemente ao transplante renal, mas são raros os casos de falha de transplante, uma vez que a progressão da doença geralmente é lenta.
Nefrite lúpica
A nefrite lúpica abrange um espectro de doenças glomerulares, incluindo a deposição mesangial de imunocomplexos ou a hipercelularidade (classes I e II), glomerulonefrite proliferativa focal ou difusa (classes III e IV), nefrite membranosa (classe V) e esclerose em estágio avançado (classe VI).47,48 No lúpus, a doença glomerular é causada por uma combinação de doença por imunocomplexos (que está associada à hipocomplementemia), formação de imunocomplexos in situ e microangiopatia trombótica. Dada a gama de patologias possíveis na nefrite lúpica, não deve surpreender que esta condição possa ser observada na síndrome nefrítica com ou sem GNRP, na síndrome nefrótica ou diante de um quadro nefrítico-nefrótico misto.
A nefrite lúpica proliferativa (classes III ou IV) constitui uma indicação para a terapia imunossupressora intensiva. Até pouco tempo atrás, a terapia-padrão para a doença de classes III e IV era a administração endovenosa de ciclofosfamida mensal durante 6 meses e, subsequentemente, a cada 3 meses por até 2 anos, combinada a prednisona em desmame progressivo.49 Múltiplos estudos clínicos recentes demonstraram que o micofenolato de mofetil pode ser utilizado em substituição à ciclofosfamida, seja como terapia de indução50-55 ou para manutenção da remissão induzida por regimes de ciclofosfamida de curta duração.56 O micofenolato de mofetil se mostrou equivalente à ciclofosfamida na indução da remissão da nefrite lúpica de classes III ou IV, apresentando um perfil de efeitos colaterais similar ou melhor que o da ciclofosfamida. Além disso, um estudo recente sugeriu que o micofenolato de mofetil também pode ser útil para o tratamento da nefrite lúpica membranosa (classe V).57
Doença anti-membrana basal glomerular (anti-MBG)
A doença anti-MBG (também conhecida como anti-GBM – do inglês glomerular basement membrane) pode ocorrer como uma forma de GNRP limitada aos rins ou pode ser acompanhada de hemorragia pulmonar prejudicial à vida (síndrome de Goodpasture). Esta doença contribui para cerca de 20% dos casos de GNRP, mas continua sendo uma condição rara, cuja incidência anual foi estimada em 0,5 a 0,9 casos por milhão na população branca, enquanto a incidência nas outras raças é ainda menor. Observa-se uma distribuição bimodal de idades com picos na 3ª e na 6ª décadas de vida. A maioria dos pacientes é do sexo masculino, porém a doença foi descrita em indivíduos de ambos os sexos, bem como em todas as faixas etárias.
Aspectos clínicos e laboratoriais. A doença anti-MBG manifesta-se tipicamente com um declínio rápido da função renal (isto é, GNRP), sedimento urinário ativo contendo hemácias e cilindros de hemácias, e proteinúria leve. Cerca de metade dos pacientes apresentam glomerulonefrite isolada, enquanto o restante também apresenta sintomas indicativos de hemorragia pulmonar, incluindo dispneia, tosse e hemoptise.58 Este último grupo de pacientes pode apresentar predisposição ao desenvolvimento de doença pulmonar em decorrência de uma infecção viral recente, exposição a toxinas pulmonares (p. ex., fumaça de cigarro59 ou solventes contendo hidrocarboneto60). A existência de anemia ferropriva é indicativa de perda de sangue por hemorragia pulmonar subclínica. A hipertensão constitui um aspecto tardio e resulta da retenção de sais e água atribuível a um severo comprometimento renal.
Os anticorpos anti-MBG circulantes constituem a principal característica desta doença. Entretanto, o diagnóstico é frequentemente estabelecido com base no exame de biópsia renal (ver adiante), que, por sua vez, deve ser realizado em caráter de urgência, uma vez que o atraso no início do tratamento resulta na perda irreversível da função renal. Resultados positivos nos testes de detecção de ANCA são obtidos em 20 a 30% dos pacientes com doença anti-MBG. Contudo, a vasculite sistêmica raramente é observada nestes casos, e a doença comporta-se de modo análogo a uma nefrite anti-MBG severa.61
Patogênese. A doença anti-MBG é causada por autoanticorpos patogênicos dirigidos contra um epítopo conformacional presente nos domínios não colagenosos das cadeias alfa-3 e alfa-5 do colágeno de tipo IV,62-65 que é um dos principais componentes das membranas basais glomerular (MBG) e alveolar. Os anticorpos induzem uma resposta inflamatória local que danifica a membrana basal, com consequente formação de crescentes glomerulares [Figuras 2 e 8] e produção de hemorragia pulmonar.66 Uma doença glomerular idêntica acomete uma pequena proporção dos pacientes com nefrite hereditária de Alport ligada ao X, após o transplante renal de um rim normal. Isto resulta do desenvolvimento de aloanticorpos dirigidos contra o domínio não colagenoso da cadeia alfa-5 do colágeno de tipo IV existente no rim transplantado, contra o qual estes anticorpos são naive em decorrência da deficiência hereditária.65
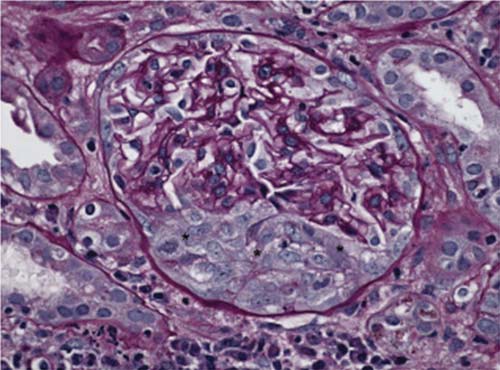
Figura 8. Um amplo crescente em múltiplas camadas junto ao espaço de Bowman, típico da glomerulonefrite crescêntica observada na doença anti-membrana basal glomerular (anti-MBG).
Patologia. O achado característico encontrado na biópsia renal é uma glomerulonefrite crescêntica e necrotizante difusa. Apesar de a lesão inicial ocorrer junto ao tufo glomerular, as interrupções existentes na MBG permitem que produtos plasmáticos extravasem dos capilares para dentro do espaço de Bowman, levando à proliferação extracapilar do epitélio residente, infiltração do espaço de Bowman por células inflamatórias e formação de crescente [Figuras 2 e 8].67 Tipicamente, na doença anti-MBG severa, os crescentes são observados em mais de 50% dos glomérulos do paciente [Figura 8].68 Os crescentes celulares iniciais e fibrocelulares tardios comprimem o tufo glomerular e podem obstruir a origem do túbulo proximal [Figura 8]. Deste modo, a severidade da doença está relacionada em parte à extensão da formação de crescentes.69-71 O achado patognomônico fornecido pela biópsia renal consiste na deposição linear de IgG e C3 ao longo da MBG [Figura 9].
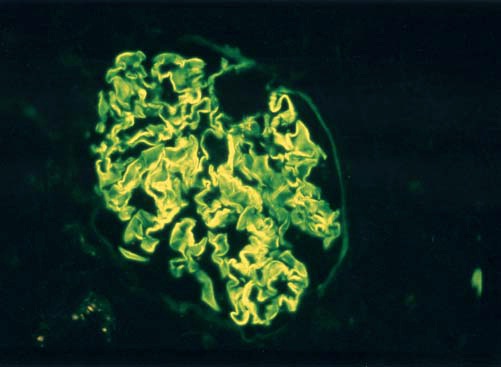
Figura 9. Padrão linear de imunocoloração de IgG ao longo da membrana basal glomerular (MBG), em um paciente com doença anti-membrana basal glomerular (anti-MBG).
Tratamento, curso e prognóstico. Sem terapia, a doença anti-MBG tem como consequência a rápida perda da função renal e, se houver hemorragia pulmonar, geralmente é fatal. O tratamento de escolha consiste em plasmaférese, prednisona e ciclofosfamida.69 A plasmaférese é utilizada para remover os anticorpos anti-MBG e mediadores inflamatórios, enquanto os agentes imunossupressores minimizam a formação de anticorpos adicionais. Muitos dos pacientes com doença anti-MBG que recebem tratamento imediato e adequado têm a função renal suficientemente preservada para evitar a necessidade de diálise. A insuficiência renal aguda oligúrica com níveis séricos de creatinina acima de 6 mg/dL está associada a um prognóstico precário em termos de recuperação renal, sendo que aqueles que necessitam de diálise no momento da manifestação da doença raramente se recuperam, mesmo que recebam tratamento. Contudo, todos os pacientes com hemorragia pulmonar decorrente da doença anti-MBG devem ser tratados, conforme descrito anteriormente, independentemente da função renal.58 A doença recorrente é extremamente rara e, em contraste com os pacientes que apresentam vasculite, a imunossupressão prolongada costuma ser desnecessária. Os pacientes que desenvolvem DRET são candidatos adequados ao transplante renal, sendo que a recidiva da doença no rim transplantado raramente ocorre se os anticorpos anti-MBG forem indetectáveis no momento do transplante.
Vasculite renal e sistêmica
O envolvimento renal ocorre com frequência na vasculite sistêmica. Alguns exemplos incluem: poliangiite microscópica, granulomatose de Wegener, síndrome de Churg-Strauss e glomerulonefrite crescêntica paucimune isolada. Estas doenças são discutidas em detalhes em outra seção. Em resumo, é preciso mencionar que as síndromes de vasculite podem ser limitadas aos rins ou apresentar aspectos clínicos de vasculite sistêmica (púrpura palpável, hemorragia pulmonar, artralgias e outras doenças em órgãos-alvo). A vasculite renal afeta cerca de 30% dos pacientes que apresentam aspectos de GNRP, incluindo a rápida perda de função renal, um sedimento urinário ativo e hipertensão. A proteinúria é um achado comum, mas raramente está na faixa nefrótica. O diagnóstico geralmente é estabelecido pela detecção de uma glomerulonefrite crescêntica necrotizante sem imunodeposição (glomerulonefrite paucimune) ao exame de biópsia renal, sendo sustentado por uma sorologia positiva para anticorpos dirigidos contra a mieloperoxidase (MPO-ANCA) ou proteinase 3 (PR3-ANCA) em mais de 80% dos casos.
Há indicação para instituição de uma terapia de indução agressiva, à base de glicocorticoides combinados com ciclofosfamida oral (1,5 a 2 mg/kg/dia) ou pulsos de ciclofosfamida endovenosa (15 mg/kg/dia, a cada 2 a 3 semanas). Esta indicação baseia-se na observação de que a taxa de mortalidade associada à vasculite sistêmica não tratada é de aproximadamente 90% em 2 anos, em geral secundária ao desenvolvimento de insuficiência pulmonar ou renal.72 Além disso, estudos recentes envolvendo pacientes com doença severa sob tratamento com diálise ou apresentando disfunção renal severa (níveis séricos de creatinina > 5,8 mg/dL) comprovaram os benefícios proporcionados pela plasmaférese.73,74 A maioria dos pacientes (> 90%) tratados com agentes imunossupressores entra em remissão durante os primeiros 3 a 6 meses, de modo que a terapia de indução geralmente é mantida por mais 2 a 3 meses depois que o paciente entra em remissão.75 Devido ao risco de recidiva de 10 a 20%, os pacientes que atingem a remissão devem ser mantidos sob regime com azatioprina (2 mg/kg/dia) por no mínimo 18 meses.75
Doenças associadas à síndrome nefrótica
A síndrome nefrótica é caracterizada por proteinúria intensa (> 3,5 g/24 horas), hipoalbuminemia (< 3 g/dL), edema periférico, hiperlipidemia (níveis elevados de colesterol total e LDL colesterol) e lipidúria. A síndrome nefrótica resulta de um aumento marcante da permeabilidade glomerular à albumina, entre outras macromoléculas. A função renal geralmente está normal no início do curso da doença, mas é comum haver uma elevação dos níveis séricos de creatinina nos estágios mais avançados da doença. O sedimento urinário tipicamente não contém hemácias, leucócitos nem cilindros celulares, mas com frequência apresenta gotículas de gordura, cilindros de colesterol e corpúsculos adiposos ovais.
A principal característica da síndrome nefrótica é a intensa proteinúria (> 3,5 g/dia). É bastante incomum que uma proteinúria desta magnitude seja causada por outras condições que não sejam a doença glomerular. Na falta de uma amostra de urina de 24 horas, uma amostra isolada de urina pode ser utilizada para estimar o grau de proteinúria.76 Uma proporção proteína:creatinina (mg/mg) urinária normal é menor que 0,15 e sugere uma taxa de excreção proteica aproximada de 150 mg/dia. Contudo, uma proporção maior que 3,5 sugere a existência de uma proteinúria na faixa nefrótica. Os testes de urina com vareta medidora de nível são úteis para avaliação de albuminúria, mas não detectam a presença das cadeias leves de imunoglobulina (proteína de Bence-Jones) em casos de mieloma múltiplo. O teste com vareta medidora de nível mede a concentração de albumina e, portanto, deve ser interpretado tendo como referência a concentração urinária determinada pela gravidade específica. A microalbuminúria refere-se a níveis urinários de albumina (> 30 mg/g de creatinina) abaixo da faixa de variação detectável pelo teste da vareta medidora de nível, sendo tipicamente encontrada no início do curso da nefropatia diabética. Baixos níveis de proteinúria, às vezes naturalmente transitórios, podem ser encontrados em numerosas condições, incluindo insuficiência cardíaca, exercícios extenuantes, febre, doença vascular renal, hipertensão severa e distúrbios tubulointersticiais. A possibilidade de proteinúria ortostática – uma condição benigna em que há desenvolvimento de proteinúria de baixo grau apenas quando o paciente se posiciona na vertical – deve ser excluída em casos de pacientes com menos de 30 anos de idade, antes de iniciar uma investigação mais extensiva.
A etiologia da síndrome nefrótica é dividida em causas primárias e secundárias. As doenças glomerulares primárias mais comuns causadoras de síndrome nefrótica em adultos são a GESF, a nefropatia membranosa e a doença de lesões mínimas [Tabela 1]. Embora estas entidades patológicas geralmente sejam primárias ou idiopáticas, cada uma delas também pode ser secundária a uma infecção subjacente, doença sistêmica ou exposição à toxina.
Glomerulosclerose segmentar e focal (GESF)
A GESF constitui a principal causa de síndrome nefrótica em adultos, sendo responsável por 25% de todos os casos e por mais de 50% dos casos de indivíduos afro-americanos afetados.
Aspectos clínicos e laboratoriais. Cerca de 90% das crianças e 70% dos adultos com GESF apresentam achados clínicos e laboratoriais de síndrome nefrótica. A incidência e severidade da GESF são maiores entre os afro-americanos, em parte devido à ligação aos polimorfismos associados à doença envolvendo os genes MYH9 e ApoL1 nesta população.6,7 Aproximadamente 50% dos pacientes adultos apresentam hipertensão, enquanto 30% apresentam insuficiência renal.77 Pode haver hematúria microscópica acompanhada de lipidúria, porém os cilindros celulares não são encontrados na urinálise. Não existem achados laboratoriais específicos para o diagnóstico da GESF idiopática. Exames específicos, como aqueles utilizados na detecção da infecção pelo HIV, podem ser úteis para excluir possíveis causas secundárias (ver adiante). Embora estudos tenham demonstrado que um fator de permeabilidade circulante está presente em alguns pacientes, atualmente não há ensaios comercializados para detecção deste fator.78
Patogênese e patologia. A denominação GESF deriva dos achados histológicos. No início do processo patológico, menos de 50% dos glomérulos estão envolvidos (focal) e, dentre os glomérulos afetados, apenas alguns segmentos apresentam cicatrização (esclerótica) [Figuras 10 e 11]. A GESF pode ser primária, hereditária ou secundária ao número reduzido de néfrons [Tabela 3]. Embora a esclerose seja visível apenas em alguns segmentos de uma minoria dos glomérulos de pacientes com GESF primária durante a manifestação inicial, todos os glomérulos apresentam extravasamento de proteínas e retração dos processos podais dos podócitos. Em contraste com a GESF primária, os pacientes com as formas hereditária e secundária da GESF apresentam processos podais retraídos somente em alguns segmentos glomerulares.79 A imunofluorescência resulta negativa nos glomérulos não escleróticos, mas pode apresentar aprisionamento inespecífico de IgM e C3 em áreas escleróticas. Os patologistas são capazes de distinguir os vários subtipos de GESF primária.80 Exceto pela variável colapsante (ver adiante), que segue um curso de progressão rápida, e pela variante de extremidade, que tende a ser mais responsiva ao tratamento, os aspectos clínicos, curso e resposta à terapia são bastante similares.
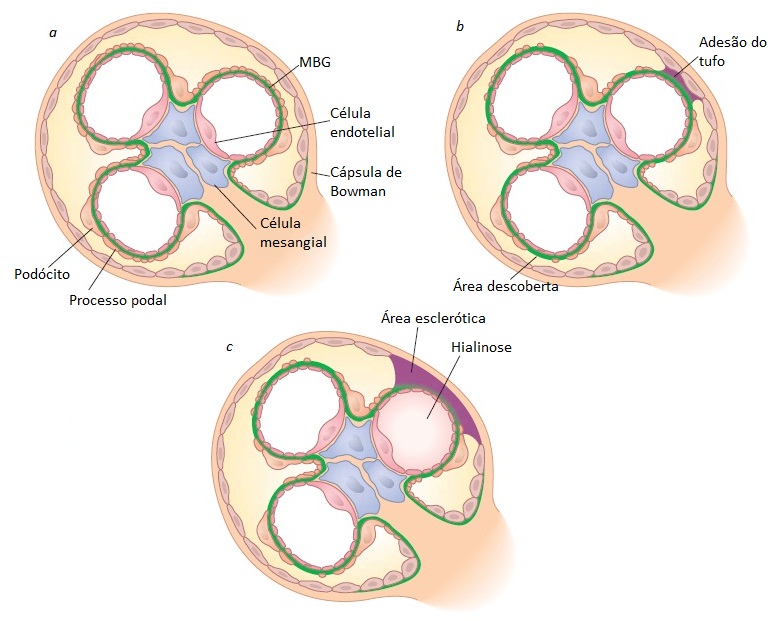
Figura 10. Ilustração esquemática da patogênese da glomerulosclerose segmentar e focal (GESF). (a) Glomérulo normal. (b) A perda ou lesão de podócitos resulta em áreas de membrana basal glomerular (MBG) descoberta, permitindo a adesão do tufo à cápsula de Bowman. (c) A adesão do tufo leva à formação de áreas escleróticas, que constitui a 1ª etapa do desenvolvimento de glomerulosclerose e hialinose, capaz de obliterar as alças capilares adjacentes.
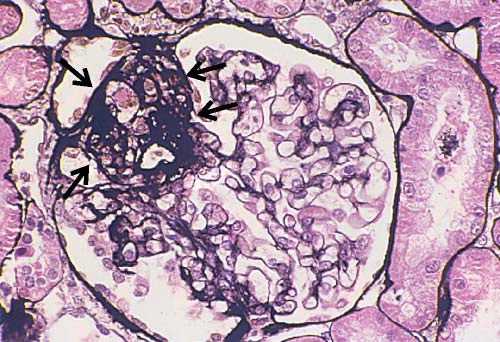
Figura 11. Fotomicrografia mostrando os aspectos característicos da glomerulosclerose segmentar e focal (GESF) com obstrução das alças capilares, hialinose e adesão do tufo (setas).
Tabela 3. Causas de GESF
|
GESF primária (idiopática) |
Lesão de extremidade glomerular |
|
Lesão hilar | |
|
Variante celular de GESF | |
|
Glomerulopatia colapsante idiopática | |
|
GESF secundária à massa renal reduzida e glomerulomegalia |
Hipoplasia/displasia renal |
|
Nefropatia de refluxo | |
|
Estágios finais de outra doença glomerular: NIgA; Nefrite lúpica; GNPE; Nefropatia membranosa | |
|
Idade avançada | |
|
Anemia falciforme | |
|
Obesidade mórbida | |
|
Falha de transplante renal | |
|
GESF secundária a infecções e toxinas |
Glomerulopatia colapsante vírus-associada (HIV, poliomavírus e outros) |
|
GESF associada ao uso de bisfosfonato (glomerulopatia colapsante) | |
|
Formas familiares de GESF |
Mutações envolvendo proteínas da fenda diafragmática (p. ex., podocina, CD2AP) |
|
Mutações envolvendo proteínas sinalizadoras (p. ex., PLCE1, TRYPC6) | |
|
Mutações que afetam o citoesqueleto de actina (p. ex., alfa-actinina 4, INF2) | |
|
Outras citopatias (mitocondriais, doença de Charcot-Marie-Tooth, trissomia do 21) | |
|
Genes de suscetibilidade (p. ex., MYH9, ApoL1) |
GESF = glomerulosclerose segmentar e focal; GNPE = glomerulonefrite pós-estreptocócica; NIgA = nefropatia por IgA.
A glomerulopatia colapsante é considerada uma entidade clínica distinta. Costuma ser observada em pacientes de ascendência africana que foram infectados pelo HIV, mas também ocorre com o tratamento à base de altas doses de bisfosfonato e na ausência de uma causa identificável.80,81 Em contraste com a GESF clássica, a GESF colapsante está associada à proliferação de podócitos e ao colapso do tufo glomerular.
A patofisiologia da GESF varia de acordo com a causa subjacente. Na GESF primária ou idiopática, há evidências de que, em alguns casos, um fator de permeabilidade glomerular circulante provoca lesão nos podócitos, contudo a natureza exata deste fator e seu papel na patogênese da doença ainda precisam ser determinados.78,82 A GESF hereditária resulta de mutações em genes codificadores de proteínas de podócitos [Tabela 3] e deve ser suspeitada em casos de pacientes com familiares que apresentam doença renal proteinúrica. A herança pode ser autossômica dominante ou recessiva. As proteínas mutantes formam um complexo que é essencial à integridade da fenda diafragmática de filtração, que atua como ponte junto ao espaço existente entre processos podais adjacentes e forma a barreira final à permeabilidade proteica, bem como à estabilidade do citoesqueleto do podócito.
A GESF também pode ocorrer como consequência de alterações mal adaptativas dos podócitos induzidas pela exposição prolongada a uma pressão intraglomerular aumentada – denominada GESF secundária. Entre as causas de GESF secundária listadas na Tabela 3, considera-se que a obesidade apresenta proeminência aumentada, embora a proteinúria geralmente esteja fora da faixa nefrótica. Seja qual for a causa, o mecanismo subjacente do desenvolvimento das lesões escleróticas é o dano e a perda dos podócitos com consequente desnudamento da MBG subjacente [Figura 10].83 Isto promove a formação de adesões ou fixações sinequiais entre o tufo glomerular e a cápsula de Bowman. O avanço do processo esclerótico leva à obsolescência dos glomérulos afetados, atrofia dos túbulos associados e fibrose intersticial.
Tratamento, curso e prognóstico. O tratamento da GESF é dirigido às doenças subjacentes e complicações da síndrome da nefrótica. A etapa inicial consiste em reconhecer a diferença existente entre as formas primária e secundária de GESF. Todos os pacientes com GESF devem ser tratados com um inibidor de ECA ou BRA, a fim de diminuir a pressão arterial para menos de 125/80 mmHg e reduzir a proteinúria. No entanto, estes agentes são especialmente efetivos para pacientes com GESF secundária. Os pacientes com GESF primária e proteinúria moderada (< 2 g/dia), cujos níveis séricos de creatinina estão estáveis, podem ser seguidos sob terapia com inibidor de SRA combinado a agentes redutores de lipídios e ao uso judicioso de diuréticos de alça, quando necessário.84 Aqueles com proteinúria na faixa nefrótica ou síndrome nefrótica franca podem ser beneficiados por um tratamento adicional com prednisona [Figura 12]. Cerca de 50 a 60% dos pacientes tratados por até 6 meses alcançam a remissão parcial ou completa da proteinúria, que, por sua vez, está associada a um melhor prognóstico.85 No caso dos pacientes que apresentam recidiva após a retirada da prednisona, é possível introduzir a ciclosporina para manter a remissão, conforme a prednisona é desmamada.86 A ciclosporina também oferece a perspectiva de induzir remissão em certa proporção dos pacientes resistentes aos esteroides. A terapia com ciclosporina com duração mínima de 12 meses é necessária em todos os casos, para manter a remissão, enquanto o monitoramento dos níveis séricos deste fármaco se faz necessário para limitar a nefrotoxicidade.86 A ciclofosfamida constitui a base do tratamento da síndrome nefrótica em crianças, seja dependente ou resistente aos esteroides, mas raramente é utilizada por mais tempo em pacientes adultos. A experiência com o uso de micofenolato de mofetil no tratamento da GESF é limitada. Entretanto, a ausência de nefrotoxicidade torna este agente potencialmente atraente como alternativa à ciclosporina para manutenção da remissão em casos de recidiva subsequente à retirada do esteroide.
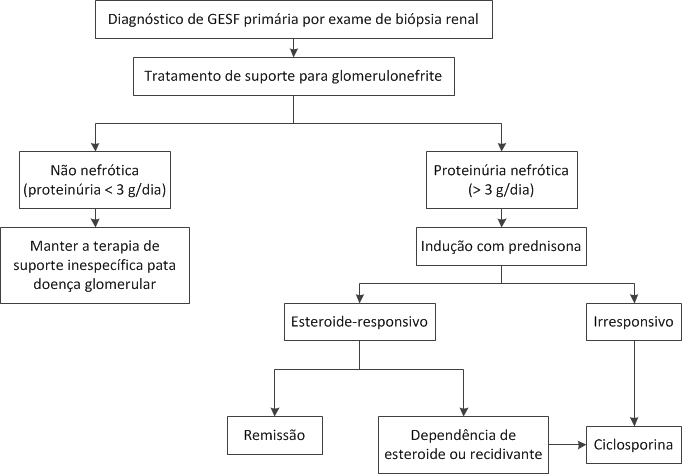
Figura 12. Algoritmo para tratamento da glomerulosclerose segmentar e focal (GESF) primária.
De uma forma geral, o prognóstico da glomerulopatia colapsante é precário, com o paciente apresentando proteinúria incessante e perda bastante rápida da função renal, independentemente do tratamento.81 Os inibidores de SRA e os diuréticos são indicados para diminuir a proteinúria e controlar o edema. Foi demonstrado que a terapia antirretroviral altamente ativa (HAART – em inglês, highly active antiretroviral therapy) estabiliza a função renal nos casos em que a glomerulopatia colapsante é atribuível à NAHIV.12,87 A rápida identificação da glomerulopatia colapsante induzida por bisfosfonato e a interrupção do curso do fármaco também podem impedir a progressão da condição.
Níveis séricos de creatinina aumentados no momento da manifestação, proteinúria na faixa nefrótica, hipercelularidade mesangial difusa e fibrose intersticial são fatores indicadores de prognóstico ruim na GESF. Qualquer resposta à terapia (uma remissão parcial ou total) confere um prognóstico favorável.84,86 Notavelmente, o número de glomérulos com cicatrização não constitui um fator prognóstico. A evolução para insuficiência renal, na GESF, não está correlacionada à idade, sexo nem presença de hematúria ou hipertensão no momento da apresentação.85 Os pacientes com insuficiência renal em estágio terminal decorrente de GESF são considerados candidatos excelentes ao transplante. Entretanto, a recidiva da doença no rim transplantado ocorre subsequentemente em 20 a 30% dos pacientes, sobretudo em pacientes jovens que apresentam proteinúria maciça e um intervalo pequeno entre apresentação e desenvolvimento de GESF. A plasmaférese tem se mostrado útil em alguns casos de pacientes com GESF recorrente após o transplante, em que provavelmente existe um fator de permeabilidade circulante causando a doença.78
Nefropatia membranosa
A nefropatia membranosa constitui a causa mais comum de síndrome nefrótica entre pacientes adultos brancos, após a nefropatia diabética; também é a principal causa de síndrome nefrótica em indivíduos com mais de 60 anos de idade.88,89 A nefropatia membranosa é assim denominada em decorrência do espessamento da MBG caracteristicamente observado em casos avançados, por microscopia óptica. Entretanto, é importante notar que o sítio primário de lesão na nefropatia membranosa é o podócito. Em adultos, 80% dos casos são primários ou idiopáticos, enquanto os 20% restantes são casos secundários à doença sistêmica, fármacos ou toxinas ambientais [Tabela 4]. A nefropatia membranosa idiopática afeta indivíduos de ambos os sexos, em todas as faixas etárias e de qualquer raça. A proporção homens:mulheres é de 2:1 a 3:1, sendo observada uma distribuição etária bifásica de 30 a 40 anos e 50 a 60 anos.90 A nefropatia membranosa é relativamente rara em crianças, sendo que em mulheres jovens o diagnóstico deve levar à suspeita de nefrite lúpica.
Tabela 4. Condições associadas à nefropatia membranosa
|
Tumores (p. ex., intestino, mama, pulmão, próstata) |
|
Infecções (p. ex., hepatite B, hepatite C, sífilis, filariose, doença hidática, esquistossomose, malária, lepra) |
|
Fármacos e toxinas (p. ex., ouro, penicilinamina, fármacos AINH, mercúrio) |
|
Doenças imunes (p. ex., LES, artrite reumatoide, doença de Hashimoto, doença de Graves, doença mista do tecido conectivo, síndrome de Sjögren, cirrose biliar primária, penfigoide bolhoso, síndrome da enteropatia de intestino delgado, dermatite herpetiforme, espondilite anquilosante, doença do enxerto vs. hospedeiro, síndrome de Guillain-Barré) |
|
Condições diversas (p. ex., sarcoidose, doença de Kimura [hiperplasia angiolinfoide com eosinofilia], hiperplasia de linfonodo angiofolicular) |
AINH = anti-inflamatórios não hormonais; LES = lúpus eritematoso sistêmico.
Aspectos clínicos e laboratoriais. Em contraste com a doença de lesões mínimas ou a GESF primária, que podem surgir de modo abrupto, a maioria dos pacientes com nefropatia membranosa (cerca de 80%) apresenta ganho de peso progressivo e formação de edema no decorrer de várias semanas ou meses, associados a uma proteinúria severa (> 3,5 g/dia). A proteinúria geralmente está na faixa de 5 a 15 g/dia, contudo até 20% dos pacientes com nefropatia membranosa apresentam níveis subnefróticos de proteinúria e, portanto, podem ser assintomáticos. A hematúria microscópica é observada em 50% dos pacientes, porém os cilindros celulares e leucócitos estão ausentes.91 O declínio da TFG ocorre de forma lenta na nefropatia membranosa, de modo que a função renal se encontra normal em 80% dos pacientes no momento da apresentação. A hipertensão está presente em 30% dos casos.
Além dos achados laboratoriais típicos da síndrome nefrótica (isto é, hipoalbuminemia, hipercolesterolemia e proteinúria), 75 a 80% dos pacientes com nefropatia membranosa idiopática (e não a forma secundária) apresentam anticorpos circulantes dirigidos contra o PLA2R de tipo M (anti-PLA2R). Os níveis séricos de complemento total e de componentes individuais de complemento encontram-se normais na nefropatia membranosa idiopática. A detecção de níveis baixos de complemento sugere o diagnóstico de LES, hepatite B ou hepatite C, os quais devem ser excluídos em todos os casos por meio da obtenção de uma sorologia adequada. Embora a hipercoagulabilidade possa complicar qualquer forma de síndrome nefrótica, a trombose em veia profunda e veia renal, como também a embolia pulmonar ocorrem mais frequentemente na nefropatia membranosa. A possibilidade de malignidade oculta deve ser excluída, em especial no caso de pacientes com mais de 65 anos de idade. Relatos apontam que 5 a 20% dos pacientes apresentam tumor maligno, e este pode não ser diagnosticado no momento da apresentação. As associações mais frequentes são com tumores sólidos de pulmão, mama, rim, intestino e próstata.
Patogênese e patologia. A nefropatia membranosa consiste em uma doença autoimune órgão-específica. A condição é caracterizada pela deposição subepitelial de imunocomplexos contendo IgG e complemento, expansão da MBG e obliteração dos processos podais do podócito.91 No início da doença, os glomérulos podem parecer totalmente normais à microscopia óptica, mas em certos casos a MBG sofre espessamento. A coloração da MBG com prata revela um padrão de picos característico, que resulta da deposição de uma nova membrana basal entre e ao redor dos imunodepósitos por ação dos podócitos lesados [Figura 13]. Como ocorre na maioria das formas de doença glomerular crônica, a atrofia tubular e a fibrose intersticial são encontradas nos casos em que há perda progressiva da função renal. Na nefropatia membranosa idiopática, a imunofluorescência resulta positiva para IgG, especialmente IgG4, bem como para C3 e C5b-9 [Figura 13]. Entretanto, o C1q tipicamente está ausente. A microscopia eletrônica revela a presença de numerosos depósitos eletrondensos subepiteliais, obliterações difusas de processos podais de podócitos sobrejacentes e rompimento das fendas diafragmáticas de filtração [Figura 14]. Em contraste, nos casos de nefropatia membranosa secundária, como a nefrite lúpica de classe V, a imunofluorescência muitas vezes mostra a presença de C1q e a predominância de outras subclasses de IgG, enquanto a microscopia eletrônica pode revelar a presença de depósitos mesangiais e subendoteliais além daqueles encontrados no espaço subepitelial.
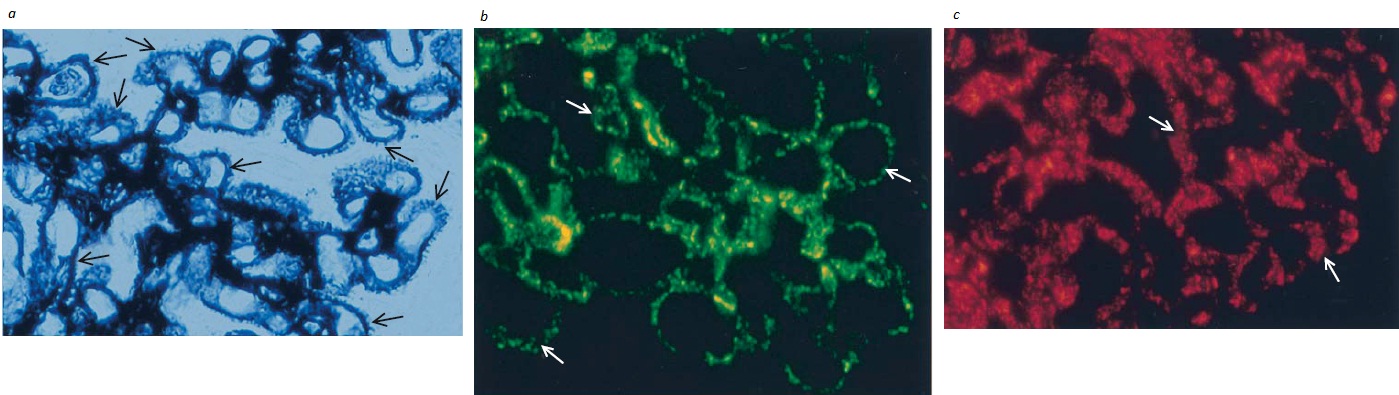
Figura 13. (a) Coloração com prata, mostrando a membrana basal glomerular (MBG) espessa com aparência de picos (setas). Estes picos representam pontos de membrana basal nova que são depositados em torno dos imunodepósitos. (b) Coloração imunofluorescente, mostrando o padrão granular típico da IgG observado na nefropatia membranosa. (c) Imunocoloração de complemento em padrão similar ao da IgG.
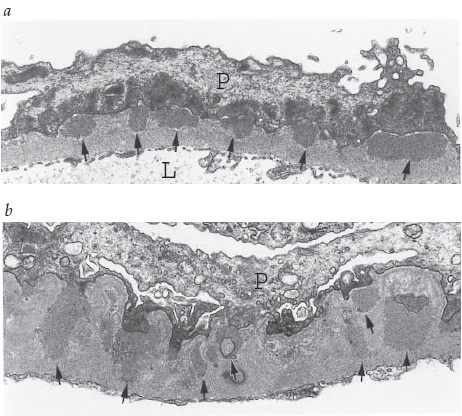
Figura 14. Micrografia eletrônica de transmissão, mostrando os imunodepósitos subepiteliais característicos (setas) das fases (a) inicial e (b) tardia da nefropatia membranosa. Os podócitos (P) e o lúmen capilar (L) são indicados.
Os estudos sobre nefropatia membranosa experimental que utilizaram um modelo com ratos denominado nefrite de Heymann forneceram indícios de que o autoantígeno-alvo consiste em um componente de superfície celular do podócito e os imunodepósitos subepiteliais são formados in situ.92 Estes achados levaram à descoberta recente de que o principal antígeno envolvido na nefropatia membranosa humana é o PLA2R e uma alta proporção dos pacientes com nefropatia idiopática, mas não aqueles com nefropatia membranosa secundária, apresentam autoanticorpos anti-PLA2R circulantes.1 O PLA2R é expresso nos podócitos humanos e serve de alvo para a formação in situ de imunocomplexos. Os anticorpos dirigidos contra a aldose redutase e a manganês superóxido dismutase também foram detectados em alguns casos de nefropatia membranosa idiopática e podem participar do processo de lesão.93 Algumas evidências apontam que o e-antígeno do vírus da hepatite B (HBV) pode atuar como antígeno plantado na nefropatia membranosa associada à infecção pelo HBV, porém os alvos encontrados na nefrite lúpica membranosa e na nefropatia membranosa fármaco-associada e tumor-associada ainda são indefinidos. Os aloanticorpos dirigidos contra a endopeptidase neutra (EPN), outro antígeno do podócito, são responsáveis por uma forma inusitada de nefropatia membranosa antenatal que ocorre em bebês nascidos de mães com deficiência de EPN.94,95
Tratamento, curso e prognóstico. O tratamento da nefropatia membranosa inclui o tratamento da doença subjacente nas formas secundárias de nefropatia membranosa, bem como a prevenção ou redução das complicações da síndrome nefrótica. Para os casos não tratados de nefropatia membranosa idiopática, a sobrevida renal é de 65 a 75% aos 10 anos e de 60% aos 15 anos. Em geral, cerca de 1/3 dos pacientes apresentam remissão completa e espontânea dentro de um período de 3 a 5 anos, com resolução da proteinúria e normalização da TFG. A remissão parcial é alcançada por 1/3 dos pacientes, que também apresentam proteinúria persistente de menos de 2 g/dia e, todavia, TFG normal. Por fim, a progressão para DRET ocorre no restante (1/3) dos casos.96,97 Alguns estudos mostraram que aproximadamente 50% dos pacientes evoluem para o estágio terminal. Nestes pacientes, a TFG cai pela metade no decorrer de 3 a 4 anos. Entre os marcadores de prognóstico ruim, estão: sexo masculino, idade acima de 60 anos, hipertensão, proteinúria persistente na faixa nefrótica ou diminuição da TFG no momento da apresentação.96 Em contraste, o prognóstico é excelente para os pacientes que apresentam proteinúria subnefrótica (< 3,5 g/dia), remissão espontânea ou induzida por tratamento, declínio de proteinúria superior a 50% durante um período qualquer de 6 meses de seguimento ou, ainda, aqueles que respondem bem à terapia com inibidor de ECA ou BRA.90,97
Todos os pacientes com nefropatia membranosa devem receber inibidores de SRA para abaixar a pressão arterial para 125/80 mmHg ou menos, bem como estatinas para controlar a hiperlipidemia. Estas medidas devem ser tomadas em paralelo ao uso judicioso dos diuréticos, a fim de controlar o edema e também devido ao efeito sinergístico destes últimos com os inibidores de SRA sobre a redução da proteinúria. Algumas autoridades defendem o uso da anticoagulação enquanto os pacientes com nefropatia membranosa permanecerem nefróticos. Nós reservamos a anticoagulação para os casos de síndrome nefrótica severa incessante e para os pacientes que apresentam evidências de trombose venosa. Os pacientes sujeitos a um alto risco de desenvolvimento de insuficiência renal progressiva ou que permanecem nefróticos após um período de avaliação de 6 meses sob terapia conservativa são tratados com um dos vários regimes que incluem um curso de 6 meses de ciclofosfamida e corticosteroides ou ciclosporina.86,98,99 Quando introduzidas antes da produção de um dano estrutural considerável, ambas as formas de terapia induzem remissão total ou parcial em cerca de 75% dos casos. Entretanto, as remissões tendem a ser mais duradouras após o tratamento com ciclofosfamida.99 Este fármaco pode ser administrado diariamente por via oral e com prednisona, ou de acordo com um regime que envolva 3 ciclos mensalmente alternados de ciclofosfamida oral e corticosteroides. Quando limitados a um curso de 6 meses, os tratamentos são bem tolerados. Entretanto, os pacientes do sexo masculino que pretendem ter filhos são aconselhados a guardar seus espermatozoides em um banco de espermas, enquanto as mulheres devem ser esclarecidas quanto aos riscos de infertilidade. As terapias alternativa e experimental incluem o uso de hormônio adrenocorticotrópico (ACTH)100,101 e de rituximabe.102,103 Atualmente, as informações disponíveis sobre a eficácia do micofenolato de mofetil na nefropatia membranosa idiopática são insuficientes para levar a quaisquer conclusões. O transplante renal para pacientes com insuficiência renal em estágio terminal decorrente de nefropatia membranosa é incentivado. Apesar dos relatos de taxas de recidiva subsequente de até 40 a 50% no rim transplantado, o rituximabe parece ser efetivo na indução da remissão em uma elevada proporção dos casos.104,105
Doença de lesões mínimas
A doença de lesões mínimas constitui a causa mais comum de síndrome nefrótica entre crianças e é a 3ª forma mais frequente de doença glomerular primária causadora de síndrome nefrótica em adultos.106 Não há predileção sexual entre os pacientes adultos, todavia a condição afeta 2 vezes mais meninos do que meninas. Embora a maioria das formas de doença de lesões mínimas seja idiopática, é preciso excluir as causas secundárias [Tabela 5]. Uma combinação de síndrome nefrótica e insuficiência renal deve alertar o clínico para a possibilidade de doença de lesões mínimas induzida por fármacos AINH agravada por uma nefrite intersticial.107,108
Tabela 5. Doenças associadas à doença de lesões mínimas
|
Fármacos (p. ex., fármacos AINH, interferon-alfa, lítio, ouro) |
|
Alergias (p. ex., pólen, poeira doméstica, picadas de insetos, imunizações) |
|
Malignidades (p. ex., linfoma de Hodgkin) |
AINH = anti-inflamatórios não hormonais.
Aspectos clínicos e laboratoriais. Os pacientes com doença de lesões mínimas tipicamente apresentam manifestação abrupta de edema severo e proteinúria maciça (> 6 g/dia). As anormalidades laboratoriais refletem a severidade da síndrome nefrótica com hipoalbuminemia severa e hiperlipidemia. Hipertensão e declínio da função renal são observados em menos de 20% dos adultos afetados. A urinálise mostra apenas a ocorrência de albuminúria e lipidúria. É raro haver hematúria.
Patogênese e patologia. Os glomérulos parecem normais ou apresentam uma expansão mesangial leve à microscopia óptica, em casos de doença de lesões mínimas. A imunofluorescência resulta negativa para imunodeposição, e o achado clássico de microscopia eletrônica consiste na extensiva obstrução dos processos podais do podócito. A causa da doença de lesões mínimas é desconhecida. O papel da imunidade celular é sugerido por sua associação ocasional à doença de Hodgkin, responsividade extraordinária aos corticosteroides e estudos realizados in vitro, os quais demonstraram a secreção de um fator de permeabilidade pelas células T de pacientes com a doença.109
Tratamento, curso e prognóstico. A maioria das crianças e adultos com doença de lesões mínimas responde ao tratamento com corticosteroides, apresentando remissão completa da proteinúria.106,107,110 Entretanto, mais de 60% dos pacientes sofrem recidiva durante ou após a retirada dos corticosteroides, sendo que alguns se tornam dependentes de esteroides. Os poucos pacientes resistentes aos esteroides podem subsequentemente apresentar GESF. A progressão para DRET é bastante incomum entre os pacientes responsivos aos esteroides. Devido à alta frequência da doença de lesões mínimas entre crianças pequenas com síndrome nefrótica, estas são tratadas em geral de forma empírica com prednisona, sem que se recorra ao exame de biópsia renal para o estabelecimento do diagnóstico. Embora os protocolos de tratamento sejam variáveis para os pacientes adultos,111 começamos o tratamento com 1 dose diária de prednisona e, então, mudamos para um regime de dias intercalados, quando os pacientes entram em remissão (geralmente, dentro de 8 semanas). Em seguida, o fármaco é gradualmente desmamado no decorrer de 6 a 8 semanas. Tratamos a 1ª recidiva deste mesmo modo, porém os pacientes que sofrem recidivas frequentes são mais bem-tratados com outro agente que mantenha a remissão e evite a toxicidade produzida pelo uso prolongado de esteroides [Figura 15]. Favorecemos o uso da ciclosporina em doses baixas, ajustadas para atingir níveis da ordem de 100 a 150 ng/mL, que são relativamente seguros e podem ser utilizados durante 1 ano ou mais para manter a remissão, desde que esta tenha sido induzida pela prednisona.110,112-114 Os pacientes são seguidos atentamente para detecção de evidências de nefrotoxicidade e hipertensão. No entanto, mesmo que a ciclosporina controle a proteinúria na maioria dos pacientes com recidivas frequentes e doença esteroide-dependente, os casos de cura são raros, e as recidivas ocorrem com frequência diante da descontinuação da ciclosporina. Embora existam menos publicações sobre o uso de micofenolato de mofetil no tratamento da doença recidivante,115-117 constatamos que este fármaco é tão efetivo quanto a ciclosporina e constitui uma alternativa satisfatória e menos nefrotóxica para os pacientes que conseguem tolerar seus efeitos gastrintestinais. Há muitos anos, a ciclofosfamida tem sido a base do tratamento da doença de lesões mínimas polirrecidivante para crianças pré-adolescentes. Esta terapia também não é curativa, mas prolonga os períodos livres de doença entre as recidivas. O levamisol, um imunoestimulante, também tem sido utilizado com algum sucesso por crianças com síndrome nefrótica dependente de esteroides.118
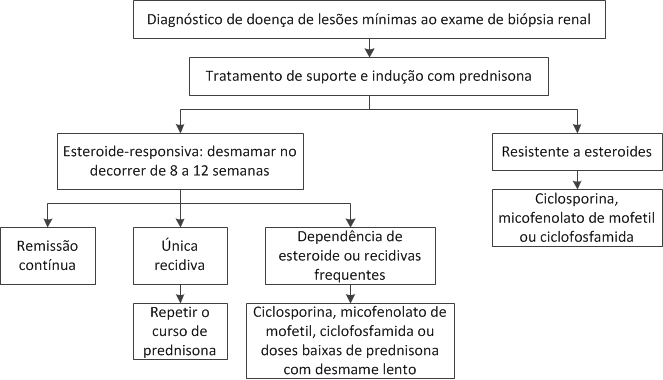
Figura 15. Algoritmo para tratamento da doença de lesões mínimas.
Síndrome nefrótica secundária a doenças sistêmicas comuns
Nefropatia diabética
A doença renal diabética constitui a causa mais comum de proteinúria nos países industrializados. O risco de 20 anos de desenvolvimento de microalbuminúria (> 30 mg/dia de albumina ou > 30 mg/g de creatinina), que representa a 1ª evidência de nefropatia diabética na maioria dos pacientes, tem diminuído atualmente, graças à moderna terapia anti-hipertensiva e antidiabética. Em pacientes com diabetes diagnosticado entre as décadas de 1960 e 1980, o risco de 20 anos de microalbuminúria variou de 20 a 50%. Contudo, estudos mais recentes, conduzidos após a introdução da inibição de ECA e do controle glicêmico mais rigoroso, relataram a ocorrência de microalbuminúria em cerca de 10% dos pacientes diabéticos decorridos 20 anos do estabelecimento do diagnóstico.119-121 Nem todos os pacientes com microalbuminúria evoluem ao ponto de desenvolver uma nefropatia diabética manifesta. De fato, estudos recentes sugerem que a maioria dos pacientes submetidos a um bom tratamento contra hipertensão e diabetes não progridem, sendo que alguns podem apresentar até mesmo regressão para um estado de normoalbuminúria. No United Kingdom Prospective Diabetes Study (Estudo prospectivo sobre o diabetes do Reino Unido), o maior estudo disponível sobre diabetes de tipo 2, o risco de progressão da microalbuminúria para macroalbuminúria (> 300 mg/dia de albumina ou > 300 mg/g de creatinina) foi de aproximadamente 2% ao ano, enquanto a progressão subsequente da macroalbuminúria para diminuição da TFG ou DRET foi observada a uma taxa de 2 a 3% dos pacientes/ano.122 Entretanto, apesar destas melhoras em termos de resultados alcançados pelos pacientes individuais, a incidência da nefropatia diabética continua aumentando, sobretudo em certos grupos populacionais, devido aos números epidêmicos de pacientes com diabetes de tipo 2. Como resultado, 44% dos pacientes que iniciam a diálise desenvolvem DRET atribuível à nefropatia diabética (United States Renal Disease System, www.usrds.org/). Os pacientes de maior risco são aqueles com controle glicêmico precário, diabetes de longa duração, hipertensão122,123 e suscetibilidade genética (evidenciada por uma história familiar de DRET), além de naturalidade africana ou ascendência afro-americana.124
A maioria dos pacientes com nefropatia diabética estabelecida apresenta a tríade clássica de hipertensão, proteinúria intensa e retinopatia no momento da apresentação. Os níveis de cretinina sérica em geral começam a aumentar decorridos 1 a 2 anos. O diagnóstico de nefropatia diabética estabelecido por exame de biópsia nem sempre é necessário. Quando o quadro clínico está totalmente de acordo com o diagnóstico de nefropatia diabética, no caso de um paciente com diabetes de longa duração mal controlado e acompanhado de retinopatia,125 muitos médicos estabelecem um diagnóstico provável e instituem o tratamento da nefropatia diagnóstica. A histologia deve ser realizada se os pacientes apresentarem: (1) síndrome nefrótica de aparecimento rápido ou deterioração acelerada da função renal (que são ambas condições incomuns em casos de nefropatia diabética);122 (2) hematúria substancial (que é incomum no diabetes); ou (3) evidências de doença sistêmica, como hepatite C, infecção pelo HIV, gamopatia monoclonal ou doença autoimune (que também causa proteinúria e requer terapia doença-específica). Quando são realizados exames de biópsia em pacientes com diabetes, a nefropatia diabética é diagnosticada na maioria dos casos.
A lesão glomerular típica do diabetes inclui a expansão mesangial, a esclerose glomerular e o espessamento da MBG. A esclerose pode conferir ao glomérulo uma aparência nodular (rim de Kimmelstiel-Wilson). A fibrose intersticial e a doença vascular constituem achados de prognóstico precário.
O tratamento da nefropatia diabética foi revolucionado pelo uso dos inibidores de ECA e BRA, que são ambos empregados para controlar a pressão arterial sistêmica, mantendo-a abaixo de 130/80 mmHg.126-128 Além disso, também foi demonstrado que um controle glicêmico rigoroso (HbA1C < 7) e um tratamento eficaz para dislipidemia com estatinas são capazes de retardar a progressão da microalbuminúria para a macroalbuminúria e nefropatia estabelecida.121
Nefropatia associada ao HIV (NAHIV)
A infecção pelo HIV pode causar várias lesões glomerulares distintas.1,14,87 A doença mais comum é a NAHIV, que se manifesta como síndrome nefrótica e é causada por uma glomerulopatia colapsante (ver anteriormente).1,129 A NAHIV ocorre em casos avançados de infecção por HIV/AIDS (CD4 < 200 células/mcL) e afeta de modo predominante indivíduos afro-americanos que apresentam variantes específicas do gene codificador da cadeia pesada de miosina 9 (MYH9).130 A infecção pelo HIV também pode levar ao desenvolvimento da síndrome hemolítica-urêmica e de púrpura trombocitopênica trombótica, NIgA, glomerulonefrite proliferativa mesangial e GNMP.88 Deste modo, é recomendável que seja feito o exame de biópsia renal para confirmação do diagnóstico, em especial no caso de pacientes com contagens de CD4 acima de 400 células/mcL. Na era da HAART,14 houve uma melhora da sobrevida dos pacientes. A maioria dos pacientes com NAHIV é iniciada na HAART em decorrência da infecção pelo HIV em estágio avançado. Segundo alguns estudos de coortes, a HAART, a inibição da ECA ou o uso de esteroides podem melhorar ou prevenir a NAHIV.12,13 Entretanto, nos estudos randomizados, nenhuma terapia foi capaz de retardar a progressão da NAHIV já estabelecida; ademais, muitos pacientes evoluem rapidamente para DRET e diálise.
Amiloidose
O termo “amiloidose” é utilizado para descrever um grupo de doenças causadas pela deposição extracelular de fibrilas insolúveis junto aos tecidos. A doença renal constitui uma complicação frequente da amiloidose e uma fonte importante de mortalidade para os pacientes. A amiloidose renal não tratada progride frequentemente para insuficiência renal em estágio terminal. Foram descritas 25 proteínas plasmáticas distintas na composição das fibrilas amiloides. Entretanto, a maioria dos casos de amiloidose renal resulta da deposição de cadeias leves monoclonais amiloidogênicas (amiloidose AL) ou de amiloide A sérico (amiloidose AA secundária).111 A amiloidose AL pode ser um distúrbio primário ou secundário ao mieloma múltiplo. As formas familiares de amiloidose raramente afetam os rins. A amiloidose AL é produzida por um clone de plasmócitos produtores de cadeias leves anômalas, enquanto a amiloidose AA é atribuível à deposição do fragmento N-terminal do amiloide A sérico (AAS), que é induzida por doenças inflamatórias crônicas, como artrite reumatoide, febre do Mediterrâneo familiar, enteropatia inflamatória e infecções crônicas.
Aspectos clínicos e laboratoriais. A suspeita diagnóstica é levantada quando o paciente apresenta proteinúria, que pode variar de subnefrótica a mais de 20 g/dia. Muitos pacientes exibem os aspectos típicos da síndrome nefrótica com ou sem disfunção renal. Os pacientes também podem apresentar síndrome de Fanconi ou diabetes insípido nefrogênico atribuível aos depósitos tubulares de fibrilas amiloides. Os rins, que são infiltrados por proteínas amiloides, muitas vezes são de tamanho aumentado ao serem vistos no exame de ultrassonografia. A amiloidose renal pode ser a forma de manifestação da doença amiloide ou pode ocorrer em pacientes que apresentam evidências clínicas de deposição amiloide em outros órgãos. Utilizando um ensaio específico e sensível, é possível detectar níveis séricos elevados de cadeias leves kappa ou lambda.
Patologia. Todas as formas de deposição amiloide são positivamente marcadas com corante vermelho Congo, que produz uma birrefringência em tom maçã-verde característica sob luz polarizada. A biópsia do coxim adiposo abdominal (menos invasiva do que a biópsia renal) é diagnóstica em 80 a 90% dos casos de pacientes com amiloidose AL e em 65 a 75% dos casos de amiloidose AA; por isso, deve ser realizada primeiro. Entretanto, um resultado negativo de exame de biópsia de coxim adiposo não exclui o diagnóstico e deve ser seguido do exame de biópsia renal. O achado histológico renal característico consiste na detecção de material amorfo junto ao mesângio glomerular e alças capilares. Nos casos severos, a microscopia óptica permite observar nódulos de material amiloide semelhantes à nefropatia diabética. Contudo, em contraste com os nódulos corados com ácido periódico de Schiff (PAS) na nefropatia diabética, os nódulos associados à amiloidose são fracamente corados pelo PAS. Isto se deve à composição destes nódulos por fibrilas amiloides, em vez de proteínas de matriz. O corante vermelho Congo é empregado para confirmar o diagnóstico, sendo que a birrefringência muitas vezes pode ser observada junto aos glomérulos, túbulos e vasculatura. A coloração imunofluorescente resulta negativa para IgG e complemento, porém é positiva para cadeias leves kappa ou lambda na amiloidose AL. Os anticorpos anti-AA podem ser utilizados para corar depósitos amiloides e confirmar o diagnóstico de amiloidose AA. Por fim, a microscopia eletrônica revela a presença de fibrilas não ramificantes, medindo 8 a 10 nm de diâmetro, que infiltram o mesângio, a parede capilar glomerular e a vasculatura renal.
Tratamento, curso e prognóstico. O prognóstico da amiloidose renal costuma ser precário no caso de pacientes não tratados, a maioria dos quais progride para insuficiência renal dentro de 1 a 2 anos.131 O controle das complicações da síndrome nefrótica, como edema e sobrecarga de volume, pode ser uma tarefa desafiadora, uma vez que muitos pacientes com amiloidose renal também apresentam doença cardíaca e doença do sistema nervoso autônomo. A meta da terapia da amiloidose AL consiste em erradicar o clone de plasmócitos produtores de cadeia leve amiloidogênica, por meio da utilização de altas doses de agentes quimioterápicos e transplante autólogo da medula óssea.111 O tratamento deve ser realizado em um centro especializado, por uma equipe multidisciplinar de nefrologistas, oncologistas e cardiologistas. Os regimes menos tóxicos, como a combinação de melfalano a altas doses de dexametasona,132 bortezomibe ou lenalidomida, são utilizados em casos de pacientes com doença cardíaca severa, que não apresentam boa tolerância ao transplante de células-tronco. A abordagem da amiloidose AA consiste em erradicar a fonte de inflamação crônica, sempre que possível. Entretanto, há relatos de resultados promissores obtidos com o uso de eprodisato, um análogo de glicosaminoglicana projetado para inibidor a deposição amiloide AA.133
Doenças comumente observadas na síndrome nefrítica-nefrótica mista
Glomerulonefrite membranoproliferativa (GNMP)
A GNMP constitui uma descrição patológica e não é uma doença específica. Este padrão de lesão pode ser observado em diversas doenças por imunocomplexo (incluindo uma condição idiopática denominada GNMP de tipo I), em microangiopatias trombóticas (p. ex., síndrome do anticorpo antifosfolipídio), na nefropatia crônica do transplante renal e na DDD (conhecida no passado como GNMP de tipo II) [Tabela 6].
Tabela 6. Causas da DDD
|
Idiopática |
|
Fator nefrítico C3 |
|
Defeito de fator H |
|
Associação com a lipodistrofia parcial |
DDD = doença de depósito denso.
Aspectos clínicos e laboratoriais. Além do compartilhamento de um padrão histológico, a maioria dos casos de GNMP envolve uma mistura de aspectos nefríticos e nefróticos. Todos apresentam hematúria microscópica frequentemente associada à presença de cilindros de hemácias, bem como proteinúria muitas vezes na faixa nefrótica (> 3,5 g/dia). Cerca de 35% dos pacientes apresentam hematúria microscópica e proteinúria não nefrótica. Em torno de 35% dos pacientes apresentam síndrome nefrótica e uma leve diminuição da função renal; 20% dos pacientes apresentam glomerulonefrite progressiva crônica; e cerca de 10% dos pacientes apresentam GNPR. A hipertensão constitui um achado bastante comum, observado em 50 a 80% dos pacientes.
Em paciente adultos, a GNMP de tipo I está mais frequentemente associada a uma ou outra das causas secundárias listadas na Tabela 7.134 Entre estas causas, a infecção pelo HCV, com ou sem crioglobulinemia mista,135 é sem dúvida a principal causa [ver Glomerulonefrite associada ao vírus da hepatite C (HCV), anteriormente]. Entretanto, esta condição também pode ocorrer diante de outras infecções bacterianas crônicas, tais como abscessos, osteomielite e endocardite infecciosa, bem como associada à leucemia e ao linfoma de células B.134 Uma manifestação nefrítica-nefrótica mista e uma lesão histológica de GNMP são igualmente observadas em pacientes com nefrite lúpica. Em contraste, este tipo de manifestação é mais comumente idiopática em crianças com GNMP.
Tabela 7. Causas de GNMP de tipos I/III
|
Idiopática |
|
Doenças vasculares colágenas: LES, artrite reumatoide, síndrome de Sjögren |
|
Deficiência do complemento: adquirida (p. ex., fator nefrítico C4), hereditária (p. ex., C1q, C2, C3, C4) |
|
Crioglobulinemia (tipos 2 e 3) |
|
Infecções: viral (p. ex., hepatite C, hepatite B crônica, infecção por HIV); bacteriana (p. ex., endocardite infecciosa, abscesso visceral, desvio infectado); outras (p. ex., Plasmodium malariae, Schistosoma, Mycoplasma) |
|
Malignidades (p. ex., leucemia linfocítica crônica, linfoma, carcinoma de células renais) |
|
Outras (p. ex., deficiência de alfa-1-antitripsina, outras doenças hepáticas crônicas, vasculite com urticária hipocomplementêmica) |
GNMP = glomerulonefrite membranoproliferativa; LES = lúpus eritematoso sistêmico.
Os achados laboratoriais refletem a severidade da síndrome nefrótica e o grau de disfunção renal. A diminuição dos níveis séricos de C3 e C4 em decorrência da ativação da via clássica é tipicamente observada com os achados patológicos distintivos descritos adiante. Esta combinação deve levar à investigação por meio de uma sorologia adequada, culturas e outros exames que permitam descobrir a causa subjacente, antes de se declarar que a doença é a GNMP de tipo I, sobretudo no caso de pacientes adultos.
Patologia e patogênese. Em todas as variantes da GNMP de tipo I, incluindo a forma idiopática, ocorre deposição de imunocomplexos ativadores do complemento no mesângio e abaixo do endotélio glomerular, em que uma intensa reação inflamatória é desencadeada [Figura 16]. A lesão patológica resultante apresenta as características anteriormente descritas [ver Glomerulonefrite associada ao vírus da hepatite C (HCV), anteriormente]. Algumas autoridades reconhecem uma 3ª lesão, denominada GNMP de tipo III, em que também há imunodeposição subepitelial. Porém, é questionável se esta lesão difere da GNMP de tipo I.
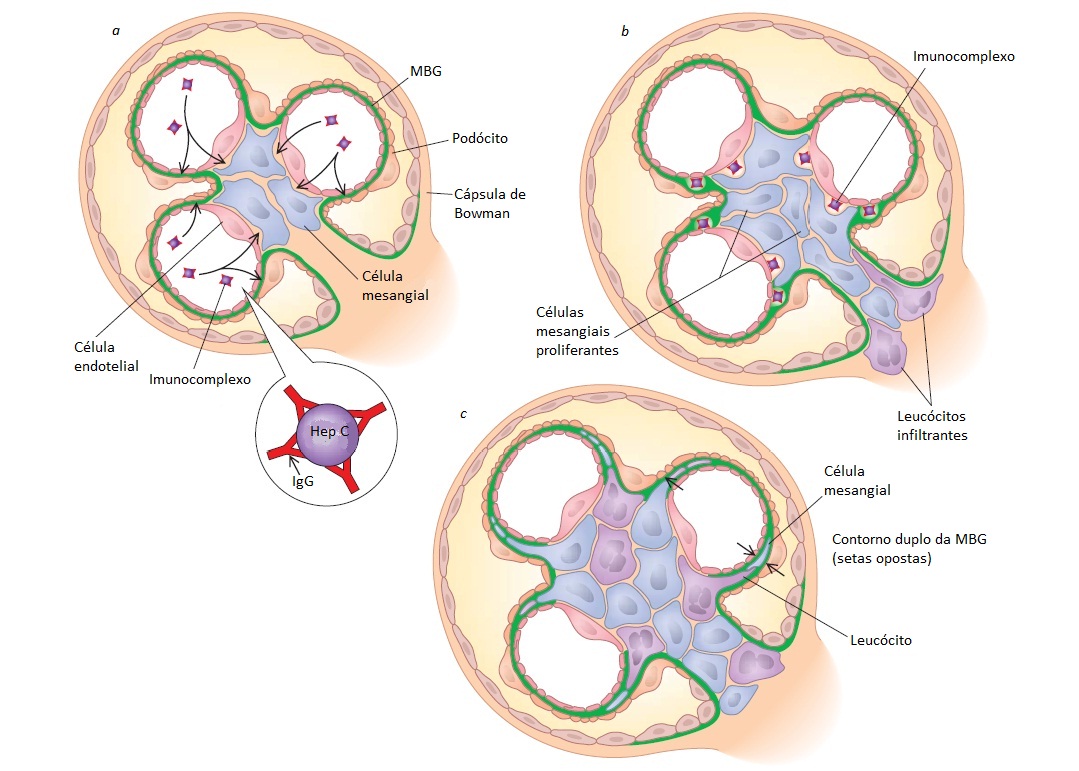
Figura 16. A patogênese da glomerulonefrite membranaproliferativa (GNMP) associada à infecção pelo vírus da hepatite C (HCV). (a) Imunocomplexos circulantes contendo antígenos virais e anticorpos anti-HCV entram no endotélio. Como resultado, há (b) proliferação de células mesangiais e infiltração de leucócitos. (c) A interposição de células mesangiais e leucócitos leva à formação de um contorno duplo da membrana basal glomerular (MBG).
Tratamento, curso e prognóstico. A melhor chance de controlar a GNMP de tipo I e preservar a função renal está na identificação e tratamento da causa. A GNMP de tipo I idiopática apresenta um prognóstico relativamente desfavorável, sendo que aproximadamente 40% dos pacientes evoluem para insuficiência renal em um período de 10 anos.136 A detecção de níveis séricos elevados de creatinina no momento do diagnóstico, proteinúria na faixa nefrótica e evidências de fibrose tubulointersticial ao exame de biópsia constituem fatores indicativos de prognóstico ruim.137
A terapia ideal para a GNMP idiopática ainda não foi definida. O tratamento da hipertensão com inibidores de SRA, uso judicioso de diuréticos e controle da hiperlipidemia são ações fundamentais. Estudos não controlados demonstraram que a terapia com prednisona em dias alternados e de longa duração preserva a função renal de crianças com GNMP de tipos I e III.138 Contudo, o uso de esteroides por pacientes adultos continua sendo controverso.137 Com base em estudos pediátricos, pode-se indicar a administração de doses baixas de prednisona para pacientes adultos com proteinúria na faixa nefrótica, mas esta terapia não deve ser prolongada diante da piora da hipertensão ou da perda da função renal. No caso da GNMP associada à GNRP, a terapia com administração endovenosa de metilprednisolona, com ou sem agentes citotóxicos, pode retardar o declínio acelerado da função renal.137 Os estudos iniciais, envolvendo pacientes adultos com GNMP idiopática, mostraram os potenciais benefícios proporcionados pela terapia antiplaquetária (p. ex., aspirina e dipiridamol) e pelo uso da varfarina.137 No entanto, estas terapias caíram em desuso. Estudos controlados, utilizando agentes alquilantes, falharam em mostrar os benefícios alcançados a longo prazo, enquanto o benefício terapêutico da ciclosporina ainda precisa ser devidamente comprovado. Rituximabe, micofenolato de mofetil e inibidores de complemento ainda não foram estudados. O transplante renal é indicado para os casos de GNMP de tipo I que evoluem para DRET, apesar de 20 a 50% dos casos apresentarem recaída.
Doença por depósitos densos (DDD – antiga GNMP de tipo II)
A DDD é uma doença rara que ocorre principalmente em crianças e adultos jovens.139,140 Embora também se manifeste tipicamente com uma apresentação nefrítica-nefrótica mista e seus vários padrões histológicos incluam a GNMP, sua patogênese difere da patogênese da GNMP de tipo I. A DDD é causada pela ativação inapropriada da via alternativa do complemento e está associada a baixos níveis de C3 e CH50, bem como a níveis normais de C1q e C4. O achado patognomônico consiste na presença de material eletrondenso em forma de faixas junto à MBG e na retina, que se coram positivamente para C3 e não contém IgG, C1q nem C4.139,140 As anormalidades que afetam proteínas regulatórias do complemento causam DDD [Tabela 6]. Um autoanticorpo circulante, conhecido como fator nefrítico C3 (C3Nef),140 é detectado em cerca de 80% dos casos de DDD. A C3Nef estabiliza a C3 convertase (C3bBb), permitindo a ativação descontrolada da via alternativa.136 Os defeitos hereditários ou as disfunções adquiridas de outras proteínas regulatórias do complemento, incluindo o fator H e a proteína cofator de membrana, também foram observados em um pequeno número de casos de DDD. Um quarto dos pacientes com DDD atribuível ao C3Nef também apresentam lipodistrofia parcial adquirida. A perda de gordura subcutânea na face e na parte superior do corpo, que confere ao paciente uma aparência abatida, pode ser um indício clínico importante para o diagnóstico.
O prognóstico da DDD é desfavorável, e 75% dos pacientes desenvolvem doença persistente, em muitos dos quais há evolução para insuficiência renal em estágio terminal.140 Não existem estratégias terapêuticas baseadas em evidências. Contudo, foi relatado que a troca de plasma remove C3Nef e estabiliza a função renal em alguns pacientes, podendo ser indicada para indivíduos com deficiências hereditárias de proteínas regulatórias do complemento. A troca de plasma, assim como outras terapias em potencial, são revistas por Appel et al. e Nasr et al.136,139 A recorrência da DDD após o transplante renal é quase universal.
Resumo
As doenças glomerulares abrangem um espectro por vezes mistificadamente amplo de manifestações clínicas, anormalidades patológicas e etiologias associadas a uma variedade de cursos, prognósticos e respostas ao tratamento. Por meio da definição da síndrome manifestada – nefrótica, nefrítica, nefrítica-nefrótica mista e de progressão rápida –, torna-se possível limitar o diagnóstico diferencial, desenvolver um plano lógico de investigação e determinar se há necessidade de pronto encaminhamento para tratamento. Por meio da identificação da presença ou ausência de quaisquer doenças sistêmicas ou hereditárias associadas, via obtenção de uma história detalhada, realização de exames e investigação laboratorial seletiva, é possível estreitar o diagnóstico diferencial e estabelecer a necessidade de obtenção de uma biópsia renal para definir a patologia subjacente. Uma vez estabelecido o diagnóstico, as ações terapêuticas gerais destinam-se a (1) controlar as complicações das síndromes nefrótica ou nefrítica, por meio da utilização de agentes anti-hipertensivos, estatinas e diuréticos; e (2) limitar a proteinúria e a perda de função renal, promovendo a diminuição da pressão intraglomerular com inibidores de SRA. Embora existam terapias altamente específicas para eliminação da causa disponibilizadas apenas para algumas doenças glomerulares, como a GNMP associada à hepatite C, os tratamentos imunossupressores, quando empregados de forma judiciosa, podem tanto preservar os rins como salvar a vida de pacientes para cujos casos não há terapia específica.
Os autores não possuem relações comerciais com os fabricantes de produtos ou prestadores de serviços mencionados neste capítulo.
Referências
1. Beck LH Jr, Bonegio RG, Lambeau G, et al. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med 2009;361:11–21.
2. Divers J, Freedman BI. Susceptibility genes in common complex kidney disease. Curr Opin Nephrol Hypertens 2010;19:79–84.
3. Freedman BI, Hicks PJ, Bostrom MA, et al. Non-muscle myosin heavy chain 9 gene MYH9 associations in African Americans with clinically diagnosed type 2 diabetes mellitus-associated ESRD. Nephrol Dial Transplant 2009; 24:3366–71.
4. Freedman BI, Hicks PJ, Bostrom MA, et al. Polymorphisms in the non-muscle myosin heavy chain 9 gene (MYH9) are strongly associated with end-stage renal disease historically attributed to hypertension in African Americans. Kidney Int 2009;75:736–45.
5. Kao WH, Klag MJ, Meoni LA, et al. MYH9 is associated with nondiabetic end-stage renal disease in African Americans. Nat Genet 2008;40:1185–92.
6. Kopp JB, Smith MW, Nelson GW, et al. MYH9 is a major-effect risk gene for focal segmental glomerulosclerosis. Nat Genet 2008;40:1175–84.
7. Genovese G, Friedman DJ, Ross MD, et al. Association of trypanolytic ApoL1 variants with kidney disease in African-Americans. Science 2010;329:841–5. Epub 2010 July 15.
8. Eremina V, Jefferson JA, Kowalewska J, et al. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med 2008;358:1129–36.
9. Stratta P, Canavese C, Marengo M, et al. Risk management of renal biopsy: 1387 cases over 30 years in a single centre. Eur J Clin Invest 2007;37:954–63.
10. Whittier WL, Korbet SM. Renal biopsy: update. Curr Opin Nephrol Hypertens 2004;13:661–5.
11. Kanjanabuch T, Kittikowit W, Eiam-Ong S. An update on acute postinfectious glomerulonephritis worldwide. Nat Rev Nephrol 2009;5:259–69.
12. Atta MG, Lucas GM, Fine DM. HIV-associated nephropathy: epidemiology, pathogenesis, diagnosis and management. Expert Rev Anti Infect Ther 2008;6:365–71.
13. Fine DM, Atta MG. Kidney disease in the HIV-infected patient. AIDS Patient Care STDS 2007;21:813–24.
14. Haas M, Kaul S, Eustace JA. HIV-associated immune complex glomerulonephritis with “lupus-like” features: a clinicopathologic study of 14 cases. Kidney Int 2005;67: 1381–90.
15. Yoshizawa N. Acute glomerulonephritis. Intern Med 2000;39:687–94.
16. Yoshizawa N, Yamakami K, Fujino M, et al. Nephritis-associated plasmin receptor and acute poststreptococcal glomerulonephritis: characterization of the antigen and associated immune response. J Am Soc Nephrol 2004;15: 1785–93.
17. Lewy JE, Salinas-Madrigal L, Herdson PB, et al. Clinico-pathologic correlations in acute poststreptococcal glomerulonephritis. A correlation between renal functions, morphologic damage and clinical course of 46 children with acute poststreptococcal glomerulonephritis. Medicine (Baltimore) 1971;50:453–501.
18. Perico N, Cattaneo D, Bikbov B, Remuzzi G. Hepatitis C infection and chronic renal diseases. Clin J Am Soc Nephrol 2009;4:207–20.
19. Monti G, Galli M, Invernizzi F, et al. Cryoglobulinaemias: a multi-centre study of the early clinical and laboratory manifestations of primary and secondary disease. GISC. Italian Group for the Study of Cryoglobulinaemias. QJM 1995;88:115–26.
20. D’Amico G, Fornasieri A. Cryoglobulinemic glomerulonephritis: a membranoproliferative glomerulonephritis induced by hepatitis C virus. Am J Kidney Dis 1995;25:361–9.
21. Meyers CM, Seeff LB, Stehman-Breen CO, Hoofnagle JH. Hepatitis C and renal disease: an update. Am J Kidney Dis 2003;42:631–57.
22. Chadban SJ, Atkins RC. Glomerulonephritis. Lancet 2005;365:1797–806.
23. Bruchfeld A, Lindahl K, Schvarcz R, Stahle L. Dosage of ribavirin in patients with hepatitis C should be based on renal function: a population pharmacokinetic analysis. Ther Drug Monit 2002;24:701–8.
24. Saadoun D, Resche-Rigon M, Sene D, et al. Rituximab combined with Peg-interferon-ribavirin in refractory hepatitis C virus-associated cryoglobulinaemia vasculitis. Ann Rheum Dis 2008;67:1431–6.
25. Neugarten J, Baldwin DS. Glomerulonephritis in bacterial endocarditis. Am J Med 1984;77:297–304.
26. Cacoub P, Terrier B. Hepatitis B-related autoimmune manifestations. Rheum Dis Clin North Am 2009;35: 125–37.
27. Donadio JV, Grande JP. IgA nephropathy. N Engl J Med 2002;347:738–48.
28. Barratt J, Smith AC, Molyneux K, Feehally J. Immunopathogenesis of IgAN. Semin Immunopathol 2007;29: 427–43.
29. Kiryluk K, Julian BA, Wyatt RJ, et al. Genetic studies of IgA nephropathy: past, present, and future. Pediatr Nephrol 2010;25:2257–68. Epub 2010 Apr 13.
30. Coppo R, Troyanov S, Camilla R, et al. The Oxford IgA nephropathy clinicopathological classifi cation is valid for children as well as adults. Kidney Int 2010;77:921–7.
31. Haas M. Histologic subclassifi cation of IgA nephropathy: a clinicopathologic study of 244 cases. Am J Kidney Dis 1997;29:829–42.
32. Nakayama K, Ohsawa I, Maeda-Ohtani A, et al. Prediction of diagnosis of immunoglobulin A nephropathy prior to renal biopsy and correlation with urinary sediment findings and prognostic grading. J Clin Lab Anal 2008;22: 114–8.
33. Novak J, Moldoveanu Z, Renfrow MB, et al. IgA nephropathy and Henoch-Schoenlein purpura nephritis: aberrant glycosylation of IgA1, formation of IgA1-containing immune complexes, and activation of mesangial cells. Contrib Nephrol 2007;157:134–8.
34. Suzuki H, Fan R, Zhang Z, et al. Aberrantly glycosylated IgA1 in IgA nephropathy patients is recognized by IgG antibodies with restricted heterogeneity. J Clin Invest 2009; 119:1668–77. Epub 2009 May 26.
35. Yamamoto R, Imai E. A novel classifi cation for IgA nephropathy. Kidney Int 2009;76:477–80.
36. Cattran DC, Coppo R, Cook HT, et al. The Oxford classification of IgA nephropathy: rationale, clinicopathological correlations, and classifi cation. Kidney Int 2009;76:534–45.
37. Coppo R, Peruzzi L, Amore A, et al. IgACE: a placebo-controlled, randomized trial of angiotensin-converting enzyme inhibitors in children and young people with IgA nephropathy and moderate proteinuria. J Am Soc Nephrol 2007;18:1880–8.
38. Praga M, Gutierrez E, Gonzalez E, et al. Treatment of IgA nephropathy with ACE inhibitors: a randomized and controlled trial. J Am Soc Nephrol 2003;14:1578–83.
39. Li PK, Leung CB, Chow KM, et al. Hong Kong study using valsartan in IgA nephropathy (HKVIN): a double-blind, randomized, placebo-controlled study. Am J Kidney Dis 2006;47:751–60.
40. Russo D, Minutolo R, Pisani A, et al. Coadministration of losartan and enalapril exerts additive antiproteinuric effect in IgA nephropathy. Am J Kidney Dis 2001;38:18–25.
41. Pozzi C, Andrulli S, Del Vecchio L, et al. Corticosteroid effectiveness in IgA nephropathy: long-term results of a randomized, controlled trial. J Am Soc Nephrol 2004;15:157–63.
42. Manno C, Torres DD, Rossini M, et al. Randomized controlled clinical trial of corticosteroids plus ACE-inhibitors with long-term follow-up in proteinuric IgA nephropathy. Nephrol Dial Transplant 2009;24:3694–701.
43. Tang SC, Tang AW, Wong SS, et al. Long-term study of mycophenolate mofetil treatment in IgA nephropathy. Kidney Int 2010;77:543–9.
44. Appel AS, Appel GB. An update on the use of mycophenolate mofetil in lupus nephritis and other primary glomerular diseases. Nat Clin Pract Nephrol 2009;5: 132–42.
45. Ballardie FW. Quantitative appraisal of treatment options for IgA nephropathy. J Am Soc Nephrol 2007;18:2806–9.
46. Hogg RJ, Lee J, Nardelli N, et al. Clinical trial to evaluate omega-3 fatty acids and alternate day prednisone in patients with IgA nephropathy: report from the Southwest Pediatric Nephrology Study Group. Clin J Am Soc Nephrol 2006;1:467–74.
47. Weening JJ, D’Agati VD, Schwartz MM, et al. The classifi cation of glomerulonephritis in systemic lupus erythematosus revisited. J Am Soc Nephrol 2004;15: 241–50.
48. Weening JJ, D’Agati VD, Schwartz MM, et al. The classifi cation of glomerulonephritis in systemic lupus erythematosus revisited. Kidney Int 2004;65:521–30.
49. Balow JE, Austin HA 3rd. Treatment of proliferative lupus nephritis. Am J Kidney Dis 2004;43:383–5.
50. Ginzler EM, Dooley MA, Aranow C, et al. Mycophenolate mofetil or intravenous cyclophosphamide for lupus nephritis. N Engl J Med 2005;353:2219–28.
51. Appel GB, Contreras G, Dooley MA, et al. Mycophenolate mofetil versus cyclophosphamide for induction treatment of lupus nephritis. J Am Soc Nephrol 2009;20:1103–12.
52. Chan TM, Li FK, Tang CS, et al. Effi cacy of mycophenolate mofetil in patients with diffuse proliferative lupus nephritis. Hong Kong-Guangzhou Nephrology Study Group. N Engl J Med 2000;343:1156–62.
53. Chan TM, Tse KC, Tang CS, et al. Long-term study of mycophenolate mofetil as continuous induction and maintenance treatment for diffuse proliferative lupus nephritis. J Am Soc Nephrol 2005;16:1076–84.
54. Hu W, Liu Z, Chen H, et al. Mycophenolate mofetil vs cyclophosphamide therapy for patients with diffuse proliferative lupus nephritis. Chin Med J (Engl) 2002;115: 705–9.
55. Ong LM, Hooi LS, Lim TO, et al. Randomized controlled trial of pulse intravenous cyclophosphamide versus mycophenolate mofetil in the induction therapy of proliferative lupus nephritis. Nephrology (Carlton) 2005;10:504–10.
56. Contreras G, Pardo V, Leclercq B, et al. Sequential therapies for proliferative lupus nephritis. N Engl J Med 2004; 350:971–80.
57. Radhakrishnan J, Moutzouris DA, Ginzler EM, et al. Mycophenolate mofetil and intravenous cyclophosphamide are similar as induction therapy for class V lupus nephritis. Kidney Int 2010;77:152–60.
58. Pusey CD. Anti-glomerular basement membrane disease. Kidney Int 2003;64:1535–50.
59. Donaghy M, Rees AJ. Cigarette smoking and lung haemorrhage in glomerulonephritis caused by autoantibodies to glomerular basement membrane. Lancet 1983;2:1390–3.
60. Beirne GJ. Goodpasture’s syndrome and exposure to solvents. JAMA 1972;222:1555.
61. Levy JB, Hammad T, Coulthart A, et al. Clinical features and outcome of patients with both ANCA and anti-GBM antibodies. Kidney Int 2004;66:1535–40.
62. Saus J, Wieslander J, Langeveld JP, et al. Identifi cation of the Goodpasture antigen as the alpha 3(IV) chain of collagen IV. J Biol Chem 1988;263:13374–80.
63. Turner N, Mason PJ, Brown R, et al. Molecular cloning of the human Goodpasture antigen demonstrates it to be the alpha 3 chain of type IV collagen. J Clin Invest 1992;89: 592–601.
64. Pedchenko V, Bondar O, Fogo AB, et al. Molecular architecture of the Goodpasture autoantigen in anti-GBM nephritis. N Engl J Med 2010;363:343–54.
65. Salant DJ. Goodpasture’s disease—new secrets revealed. N Engl J Med 2010;363:388–91.
66. Salant DJ. Immunopathogenesis of crescentic glomerulonephritis and lung purpura. Kidney Int 1987;32:408–25.
67. Moeller MJ, Soofi A, Hartmann I, et al. Podocytes populate cellular crescents in a murine model of infl ammatory glomerulonephritis. J Am Soc Nephrol 2004;15:61–7.
68. Holdsworth S, Boyce N, Thomson NM, Atkins RC. The clinical spectrum of acute glomerulonephritis and lung haemorrhage (Goodpasture’s syndrome). Q J Med 1985;55: 75–86.
69. Levy JB, Turner AN, Rees AJ, Pusey CD. Long-term outcome of anti-glomerular basement membrane antibody disease treated with plasma exchange and immunosuppression. Ann Intern Med 2001;134:1033–42.
70. Kriz W, Hahnel B, Hosser H, et al. Pathways to recovery and loss of nephrons in anti-Thy-1 nephritis. J Am Soc Nephrol 2003;14:1904–26.
71. Le Hir M, Besse-Eschmann V. A novel mechanism of nephron loss in a murine model of crescentic glomerulonephritis. Kidney Int 2003;63:591–9.
72. de Groot K, Harper L, Jayne DR, et al. Pulse versus daily oral cyclophosphamide for induction of remission in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized trial. Ann Intern Med 2009;150:670–80.
73. de Lind van Wijngaarden RA, Hauer HA, Wolterbeek R, et al. Chances of renal recovery for dialysis-dependent ANCA-associated glomerulonephritis. J Am Soc Nephrol 2007;18:2189–97.
74. Jayne DR, Gaskin G, Rasmussen N, et al. Randomized trial of plasma exchange or high-dosage methylprednisolone as adjunctive therapy for severe renal vasculitis. J Am Soc Nephrol 2007;18:2180–8.
75. Jayne D, Rasmussen N, Andrassy K, et al. A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. N Engl J Med 2003;349:36–44.
76. Schwab SJ, Christensen RL, Dougherty K, Klahr S. Quantitation of proteinuria by the use of protein-to-creatinine ratios in single urine samples. Arch Intern Med 1987; 147:943–4.
77. Beaufi ls H, Alphonse JC, Guedon J, Legrain M. Focal glomerulosclerosis: natural history and treatment. A report of 70 cases. Nephron 1978;21:75–85.
78. Savin VJ, McCarthy ET, Sharma M. Permeability factors in focal segmental glomerulosclerosis. Semin Nephrol 2003; 23:147–60.
79. Henderson JM, Alexander MP, Pollak MR. Patients with ACTN4 mutations demonstrate distinctive features of glomerular injury. J Am Soc Nephrol 2009;20:961–8.
80. D’Agati VD. The spectrum of focal segmental glomerulosclerosis: new insights. Curr Opin Nephrol Hypertens 2008;17:271–81.
81. Schwimmer JA, Markowitz GS, Valeri A, Appel GB. Collapsing glomerulopathy. Semin Nephrol 2003;23: 209–18.
82. Meyrier AY. Treatment of focal segmental glomerulosclerosis with immunophilin modulation: when did we stop thinking about pathogenesis? Kidney Int 2009;76:487–91.
83. Kriz W. Progressive renal failure—inability of podocytes to replicate and the consequences for development of glomerulosclerosis. Nephrol Dial Transplant 1996;11: 1738–42.
84. Cameron JS. Focal segmental glomerulosclerosis in adults. Nephrol Dial Transplant 2003;18 Suppl 6:vi45–51.
85. Troyanov S, Wall CA, Miller JA, et al. Focal and segmental glomerulosclerosis: defi nition and relevance of a partial remission. J Am Soc Nephrol 2005;16:1061–8.
86. Cattran DC, Alexopoulos E, Heering P, et al. Cyclosporin in idiopathic glomerular disease associated with the nephrotic syndrome: workshop recommendations. Kidney Int 2007;72:1429–47.
87. Fine DM, Fogo AB, Alpers CE. Thrombotic microangiopathy and other glomerular disorders in the HIV-infected patient. Semin Nephrol 2008;28:545–55.
88. Korbet SM, Genchi RM, Borok RZ, Schwartz MM. The racial prevalence of glomerular lesions in nephrotic adults. Am J Kidney Dis 1996;27:647–51.
89. Haas M, Meehan SM, Karrison TG, Spargo BH. Changing etiologies of unexplained adult nephrotic syndrome: a comparison of renal biopsy findings from 1976-1979 and 1995-1997. Am J Kidney Dis 1997;30:621–31.
90. Cattran DC. Idiopathic membranous glomerulonephritis. Kidney Int 2001;59:1983–94.
91. Wasserstein AG. Membranous glomerulonephritis. J Am Soc Nephrol 1997;8:664–74.
92. Cybulsky AV, Quigg RJ, Salant DJ. Experimental membranous nephropathy redux. Am J Physiol Renal Physiol 2005;289:F660–71.
93. Prunotto M, Carnevali ML, Candiano G, et al. Autoimmunity in membranous nephropathy targets aldose reductase and SOD2. J Am Soc Nephrol 2010;21:507–19.
94. Debiec H, Guigonis V, Mougenot B, et al. Antenatal membranous glomerulonephritis due to anti-neutral endopeptidase antibodies. N Engl J Med 2002;346:2053– 60.
95. Debiec H, Nauta J, Coulet F, et al. Role of truncating mutations in MME gene in fetomaternal alloimmunisation and antenatal glomerulopathies. Lancet 2004;364:1252–9.
96. Donadio JV Jr, Torres VE, Velosa JA, et al. Idiopathic membranous nephropathy: the natural history of untreated patients. Kidney Int 1988;33:708–15.
97. Polanco N, Gutierrez E, Covarsi A, et al. Spontaneous remission of nephrotic syndrome in idiopathic membranous nephropathy. J Am Soc Nephrol 2010;21:697–704.
98. Ponticelli C, Passerini P. Treatment of membranous nephropathy. Nephrol Dial Transplant 2001;16 Suppl 5:8– 10.
99. Jha V, Ganguli A, Saha TK, et al. A randomized, controlled trial of steroids and cyclophosphamide in adults with nephrotic syndrome caused by idiopathic membranous nephropathy. J Am Soc Nephrol 2007;18:1899–904.
100. Berg AL, Nilsson-Ehle P, Arnadottir M. Benefi cial effects of ACTH on the serum lipoprotein profi le and glomerular function in patients with membranous nephropathy. Kidney Int 1999;56:1534–43.
101. Rauen T, Michaelis A, Floege J, Mertens PR. Case series of idiopathic membranous nephropathy with long-term benefi cial effects of ACTH peptide 1-24. Clin Nephrol 2009;71:637–42.
102. Ruggenenti P, Chiurchiu C, Brusegan V, et al. Rituximab in idiopathic membranous nephropathy: a one-year prospective study. J Am Soc Nephrol 2003;14:1851–7.
103. Fervenza FC, Sethi S, Specks U. Idiopathic membranous nephropathy: diagnosis and treatment. Clin J Am Soc Nephrol 2008;3:905–19.
104. El-Zoghby ZM, Grande JP, Fraile MG, et al. Recurrent idiopathic membranous nephropathy: early diagnosis by protocol biopsies and treatment with anti-CD20 monoclonal antibodies. Am J Transplant 2009;9:2800–7.
105. Sprangers B, Lefkowitz GI, Cohen SD, et al. Benefi cial effect of rituximab in the treatment of recurrent idiopathic membranous nephropathy after kidney transplantation. Clin J Am Soc Nephrol 2010;5:790–7.
106. Nolasco F, Cameron JS, Heywood EF, et al. Adult-onset minimal change nephrotic syndrome: a long-term follow-up. Kidney Int 1986;29:1215–23.
107. Bargman JM. Management of minimal lesion glomerulonephritis: evidence-based recommendations. Kidney Int Suppl 1999;70:S3–16.
108. Lowenstein J, Schacht RG, Baldwin DS. Renal failure in minimal change nephrotic syndrome. Am J Med 1981;70: 227–33.
109. Koyama A, Fujisaki M, Kobayashi M, et al. A glomerular permeability factor produced by human T cell hybridomas. Kidney Int 1991;40:453–60.
110. Dember LM, Salant DJ. Minimal change disease. In: Wilcox CS, editor. Therapy in nephrology and hypertension. 3rd ed. Philadelphia: W. B. Saunders; 2008. p. 205.
111. Dember LM. Amyloidosis-associated kidney disease. J Am Soc Nephrol 2006;17:3458–71.
112. Ponticelli C, Edefonti A, Ghio L, et al. Cyclosporin versus cyclophosphamide for patients with steroid-dependent and frequently relapsing idiopathic nephrotic syndrome: a multicentre randomized controlled trial. Nephrol Dial Transplant 1993;8:1326–32.
113. Meyrier A, Noel LH, Auriche P, Callard P. Long-term renal tolerance of cyclosporin A treatment in adult idiopathic nephrotic syndrome. Collaborative Group of the Societe de Nephrologie. Kidney Int 1994;45:1446–56.
114. Ittel TH, Clasen W, Fuhs M, et al. Long-term cyclosporine A treatment in adults with minimal change nephrotic syndrome or focal segmental glomerulosclerosis. Clin Nephrol 1995;44:156–62.
115. Dorresteijn EM, Kist-van Holthe JE, Levtchenko EN, et al. Mycophenolate mofetil versus cyclosporine for remission maintenance in nephrotic syndrome. Pediatr Nephrol 2008;23:2013–20.
116. Mendizabal S, Zamora I, Berbel O, et al. Mycophenolate mofetil in steroid/cyclosporine-dependent/resistant nephrotic syndrome. Pediatr Nephrol 2005;20:914–9.
117. Pesavento TE, Bay WH, Agarwal G, et al. Mycophenolate therapy in frequently relapsing minimal change disease that has failed cyclophosphamide therapy. Am J Kidney Dis 2004;43:e3–6.
118. Alshaya HO, Kari JA. Levamisole treatment in steroid sensitive nephrotic syndrome. Saudi Med J 2002;23: 1101–4.
119. Bojestig M, Arnqvist HJ, Hermansson G, et al. Declining incidence of nephropathy in insulin-dependent diabetes mellitus. N Engl J Med 1994;330:15–8.
120. Bojestig M, Karlberg BE, Lindstrom T, Nystrom FH. Reduction of ACE activity is insuffi cient to decrease microalbuminuria in normotensive patients with type 1 diabetes. Diabetes Care 2001;24:919–24.
121. Nordwall M, Bojestig M, Arnqvist HJ, Ludvigsson J. Declining incidence of severe retinopathy and persisting decrease of nephropathy in an unselected population of Type 1 diabetes—the Linkoping Diabetes Complications Study. Diabetologia 2004;47:1266–72.
122. Adler AI, Stevens RJ, Manley SE, et al. Development and progression of nephropathy in type 2 diabetes: the United Kingdom Prospective Diabetes Study (UKPDS 64). Kidney Int 2003;63:225–32.
123. Retinopathy and nephropathy in patients with type 1 diabetes four years after a trial of intensive therapy. The Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group. N Engl J Med 2000;342:381–9.
124. Freedman BI, Bostrom M, Daeihagh P, Bowden DW. Genetic factors in diabetic nephropathy. Clin J Am Soc Nephrol 2007;2:1306–16.
125. Wolf G, Muller N, Mandecka A, Muller UA. Association of diabetic retinopathy and renal function in patients with types 1 and 2 diabetes mellitus. Clin Nephrol 2007; 68:81–6.
126. Brenner BM, Cooper ME, de Zeeuw D, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 2001;345:861–9.
127. Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med 1993;329:1456–62.
128. Lewis EJ, Hunsicker LG, Clarke WR, et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med 2001;345:851–60.
129. Laurinavicius A, Hurwitz S, Rennke HG. Collapsing glomerulopathy in HIV and non-HIV patients: a clinicopathological and follow-up study. Kidney Int 1999;56: 2203–13.
130. Nunez M, Saran AM, Freedman BI. Gene-gene and gene-environment interactions in HIV-associated nephropathy: a focus on the MYH9 nephropathy susceptibility gene. Adv Chronic Kidney Dis 2010;17:44–51.
131. Gertz MA, Kyle RA, O’Fallon WM. Dialysis support of patients with primary systemic amyloidosis. A study of 211 patients. Arch Intern Med 1992;152:2245–50.
132. Jaccard A, Moreau P, Leblond V, et al. High-dose melphalan versus melphalan plus dexamethasone for AL amyloidosis. N Engl J Med 2007;357:1083–93.
133. Dember LM, Hawkins PN, Hazenberg BP, et al. Eprodisate for the treatment of renal disease in AA amyloidosis. N Engl J Med 2007;356:2349–60.
134. Rennke HG. Secondary membranoproliferative glomerulonephritis. Kidney Int 1995;47:643–56.
135. Johnson RJ, Gretch DR, Yamabe H, et al. Membranoproliferative glomerulonephritis associated with hepatitis C virus infection. N Engl J Med 1993;328:465–70.
136. Appel GB, Cook HT, Hageman G, et al. Membranoproliferative glomerulonephritis type II (dense deposit disease): an update. J Am Soc Nephrol 2005;16:1392–403.
137. Levin A. Management of membranoproliferative glomerulonephritis: evidence-based recommendations. Kidney Int Suppl 1999;70:S41–6.
138. McEnery PT. Membranoproliferative glomerulonephritis: the Cincinnati experience—cumulative renal survival from 1957 to 1989. J Pediatr 1990;116(5):S109–14.
139. Nasr SH, Valeri AM, Appel GB, et al. Dense deposit disease: clinicopathologic study of 32 pediatric and adult patients. Clin J Am Soc Nephrol 2009;4:22–32.
140. Smith RJ, Alexander J, Barlow PN, et al. New approaches to the treatment of dense deposit disease. J Am Soc Nephrol 2007;18:2447–56.