Glomerulonefrite Difusa Aguda ou Glomerulonefrite Pós-estreptocócica
INTRODUÇÃO
A glomerulonefrite difusa aguda (GNDA) ou glomerulonefrite pós-estreptocócica (GNPE) caracteriza-se pelo processo inflamatório de origem imunológica que acomete todos os glomérulos de ambos os rins. Juntamente com a febre reumática, é considerada uma sequela tardia, e não supurativa, de uma infecção estreptocócica prévia.
Esta doença é o exemplo clássico da síndrome nefrítica aguda. A glomerulonefrite aguda ou síndrome nefrítica aguda manifesta-se geralmente de forma abrupta e é caracterizada clinicamente pela tríade clássica: edema, hipertensão e hematúria. O quadro clínico completo inclui oligúria, queda do ritmo de filtração glomerular (RFG) e proteinúria.
EPIDEMIOLOGIA
A GNPE pode ocorrer de forma esporádica ou epidêmica. Acomete predominantemente crianças em idade pré-escolar e escolar, com pico de incidência ao redor dos 7 anos de idade, sendo raro o acometimento de crianças menores de 2 anos de idade (5%) e adultos acima de 40 anos (5 a 10%).
O sexo masculino geralmente é 2 vezes mais acometido, embora alguns autores relatem que quando o processo é secundário à estreptococcia cutânea, não há predomínio de sexo.
A faringite estreptocócica ocorre, predominantemente, em escolares entre 5 e 15 anos de idade, sendo mais comum nos meses de inverno e início da primavera.
ETIOLOGIA
A glomerulonefrite pós-estreptocócica (GNPE) geralmente é precedida por uma infecção da orofaringe ou da pele por certas cepas “nefritogênicas” dos estreptococos beta-hemolíticos dos grupos A, C e G. As cepas ditas nefritogênicas mais relacionadas a infecções respiratórias são as dos sorotipos 1, 4, 12 e 25, e a infecção de pele está mais relacionada aos sorotipos 2, 42, 49, 56 e 60.
Os fatores que permitem que as cepas sejam nefritogênicas permanecem obscuros. Entretanto, quadro clínico semelhante já foi descrito em associação a outros agentes infecciosos, como bactérias, vírus e parasitas.
PATOGENIA
Os estudos morfológicos e a redução sérica do complemento (principalmente do C3) sugerem que a GNDA seja doença de base imunológica, mediada por imunocomplexos, embora este mecanismo ainda esteja não bem determinado.
Dentre os vários mecanismos patogenéticos propostos, os mais aceitos são:
• deposição glomerular de imunocomplexos de antígeno estreptococo-anticorpo ou de produtos produzidos pelo estreptococo (como a estreptoquinase);
• mecanismos autoimunes, pelos quais certos antígenos estreptocócicos simulariam anticorpos que apresentam reação cruzada com antígenos glomerulares renais (glicoproteínas da membrana basal glomerular);
• complexos terminais do complemento, formados a partir da ativação da cascata do complemento, que estimulam a produção de prostanoides pelas células mesangiais glomerulares e a elaboração de interleucinas, estando, assim, implicados na injúria glomerular aguda e, particularmente, como um estímulo para a proliferação mesangial.
A patogenia do antígeno autólogo baseia-se na ação de substância produzida pelo estreptococo, a neuraminidase, que atuaria sobre a imunoglobulina G (IgG) tornando-a antigênica.
A patogênese do complexo imunológico é aceita, mas o(s) antígeno(s) causador(es) ainda é/são controvertido(s). Nos últimos anos, dois antígenos estreptocócicos – a exotoxina cisteína-proteinase catiônica B (SPE B) e o receptor de plasmina, um gliceraldeído fosfato desidrogenase (Pir, GAPDH) –, atraíram a atenção porque foram localizados nos glomérulos de pacientes com glomerulonefrite pós-estreptocócica aguda (GNPEA) e porque o anticorpo sérico para esses antígenos foi associado às infecções nefritogênicas estreptocócicas.
Acredita-se atualmente que os imunocomplexos responsáveis pelo dano glomerular nesta nefropatia são formados primariamente in situ e depositam-se no lado subendotelial da parede capilar. A partir daí, ocorre:
• ativação do sistema complemento (tanto pela via clássica quanto pela via alternada, na maioria dos casos);
• liberação de C5a e C5b, que possuem atividades quimiotáxicas;
• ativação de neutrófilos que, por sua vez, secretarão proteases;
• ativação de substâncias oxidantes que irão determinar alterações na membrana basal glomerular (MBG), principalmente no que se refere à proteinúria.
Verifica-se a passagem do imunocomplexo para o lado epitelial, que constitui o chamado humps, que representa o imunocomplexo, podendo ser mais bem visibilizado pela microscopia eletrônica (ME) e pela técnica de imunofluorescência (IF).
FISIOPATOLOGIA
A fisiopatologia da GNDA inclui a redução no ritmo de filtração glomerular determinada pela infiltração de células inflamatórias e pela queda da permeabilidade basal glomerular. Durante a fase aguda, há importante processo inflamatório de natureza imunológica e alterações dos capilares glomerulares com redução de seus lúmens por tumefação de suas paredes ou por obstrução causada por coagulação intravascular. Este processo leva a uma queda da filtração glomerular com aumento dos níveis séricos, de ureia e creatinina.
As lesões dos capilares glomerulares permitem a passagem de hemácias da luz capilar para o espaço de Bowman, causando hematúria.
A própria reação inflamatória nos glomérulos altera as condições de permeabilidade da membrana glomerular às proteínas, condicionando proteinúria de pequena intensidade.
A baixa aguda do ritmo de filtração glomerular leva à retenção de sódio pelas células tubulares, principalmente distais. O sódio plasmático, em geral, fica diminuído, porém a massa total de sódio, em razão da expansão do espaço extracelular, fica aumentada. A reabsorção do sódio no nível dos túbulos está preservada, principalmente nos túbulos distais.
Este fato resultará na expansão do volume extracelular e na consequente supressão do sistema renina-angiotensina-aldosterona (SRAA).
São discutíveis as condições que induzem à hipertensão na GNDA. Atribui-se seu aparecimento à existência de vasoespasmo generalizado, associado à hipervolemia por retenção de sódio e água, levando à expansão do volume intravascular, visto que o SRAA não está ativado nesta patologia.
ANATOMIA PATOLÓGICA
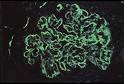
À microscopia óptica (MO) na síndrome nefrítica aguda, o padrão histológico mais comumente encontrado é o da glomerulonefrite proliferativa pura. A proliferação endocapilar existente nesse tipo de nefropatia se faz à custa das células mesangiais e endoteliais. Os tufos capilares tornam-se tumefeitos, com luzes vasculares colapsadas em razão do aumento da celularidade e da expansão da matriz mesangial. A imunofluorescência (IF) tem padrão bem característico. Os humps apresentam-se como depósitos glomerulares grosseiros, morfologicamente semelhantes entre si, dispostos ao acaso sob as paredes dos capilares glomerulares. Fixam-se intensamente os antissoros específicos para fator C3 do complemento e IgG.
Figura 1: Glomerulonefrite pós-estreptocócica: glomérulo volumoso, hipercelular, com infiltrado neutrofílico (MO, aumento original = 400 vezes).
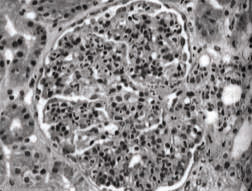
Figura 2: Imunofluorescência positiva para fatores do complemento.
MANIFESTAÇÕES CLÍNICAS
A GNPE é uma doença predominantemente infantil. A história típica é precedida de infecção estreptocócica e o intervalo entre a ocorrência da estreptococcia e a glomerulonefrite é geralmente de
O espectro clínico dos sinais e sintomas na GNDA é bastante diverso, podendo incluir casos assintomáticos com evidência subclínica (edema subclínico, hipertensão e complemento diminuído) ou laboratorial de envolvimento renal (hematúria microscópica) até quadros mais graves como insuficiência cardíaca congestiva, encefalopatia hipertensiva e insuficiência renal aguda.
Na maioria das vezes, o paciente encontra-se em bom estado geral, com queixas vagas como indisposição, inapetência, cefaleia e edema periorbital. Outros sintomas menos frequentes podem acompanhar o quadro, como mal-estar, letargia, cólicas abdominais ou dor nos flancos, hipertermia e vômitos.
O quadro clínico clássico constitui-se de:
• edema com intensidade variável, geralmente leve, frio, mole e gravitacional, sendo mais evidente em região periorbitária e no período matutino, mas pode atingir as extremidades inferiores e as regiões lombar ou genital;
• hipertensão arterial presente em cerca de 90% dos casos;
• hematúria macroscópica presente em aproximadamente 50% dos casos e microscópica observada em praticamente todos os casos na forma persistente ou intermitente.
Pode ser observada também congestão circulatória associada à retenção de sal e água (20%) e oligúria e/ou anúria transitórias, presentes em até 25% dos casos, sendo rara a forma de oligoanúria mantida.
Na regressão destes sintomas, constata-se inicialmente o desaparecimento do edema, em média de
DIAGNÓSTICO
Diante de um paciente com síndrome nefrítica, a primeira ação diagnóstica deve ser tentar encontrar, pela anamnese e exame físico, a presença de manifestações extrarrenais que possam indicar uma etiologia específica (p.ex., rash malar e artrite para o lúpus eritematoso). Caso a síndrome nefrítica seja a única condição do paciente, o médico deve perguntar a ele sobre faringite ou piodermite prévias recentes, verificar se o período entre as infecções estreptocócicas prévias e o início dos sintomas da GNDA é compatível, documentar laboratorialmente a infecção estreptocócica (ASLO, anti-DNAse B, anti-NADase, anti-hialuronidase) e avaliar queda transitória típica de complemento (C3 e CH50), com um retorno a níveis normais entre 2 e 8 semanas a contar dos primeiros sinais de nefropatia.
DIAGNÓSTICO LABORATORIAL
Urina I
Na maioria dos casos, mostra sinais de inflamação glomerular ativa, com hemácias dismórficas, presença de cilindros hemáticos, granulosos, hialinos e leucocitários, osmolaridade elevada e proteinúria positiva (raramente maciça).
Função Renal
• Níveis séricos de ureia e creatinina podem estar elevados;
• a função tubular costuma estar preservada;
• hiponatremia, acidose metabólica e hipercalemia podem ocorrer quando a queda no ritmo de filtração glomerular for importante, causando insuficiência renal aguda.
Hemograma e Perfil de Coagulação
• Anemia decorrente da expansão do volume, que gera leve diluição da concentração de hemoglobina;
• plaquetopenia transitória pela diminuição da meia-vida plaquetária;
• fatores de coagulação possivelmente alterados, com diminuição do fator XIII e alfamacroglobulina, diminuição do nível e da atividade da antitrombina III e aumento discreto da alfa-1-antitripsina.
Aspectos Imunológicos
A resposta imunológica à estreptococcia de orofaringe é diferente daquela encontrada na forma cutânea. A infecção de orofaringe leva à elevação dos títulos de antiestreptolisina O (ASLO), anti-hialuronidase e anti-DNAse. Todos esses anticorpos fazem parte do streptozyme test, muito utilizado para o diagnóstico de estreptococcia prévia recente.
Na infecção cutânea, porém, títulos de ASLO não se elevam, devendo-se utilizar a anti-DNAse para o diagnóstico (detectado em 60 a 70% dos casos), seguido pelo anti-hialuronidase. Na GNDA pós-faringoamidalite, o anticorpo mais encontrado é o ASLO (em 80 a 90% dos casos), seguido pelo anti-DNAse B (em 75% dos casos). Os níveis de ASLO geralmente se elevam de
O complemento total e algumas de suas frações encontram-se diminuídos em aproximadamente 90% dos casos na fase aguda da GNPE. C3, C5 e properdina estão frequentemente diminuídos; C4 está em níveis normais. A hipocomplementemia resolve-se em, aproximadamente, 8 semanas após o início do quadro; 90% dos pacientes apresentam hipergamaglobulinemia, com elevação de IgM e IgG; 75% dos casos apresentam crioglobulinemia e 50% apresentam positividade para o fator reumatoide.
DIAGNÓSTICO DIFERENCIAL
• Glomerulonefrite membranoproliferativa (GNMP);
• outras glomerulonefrites pós-infecciosas (p.ex., endocardite bacteriana aguda);
• glomerulonefrite lúpica;
• púrpura de Henoch-Schönlein (PHS);
• doença de Berger.
Todas essas condições podem ser facilmente excluídas por critérios clínicos e laboratoriais, com exceção da GNMP, que pode ocorrer após infecções estreptocócicas em crianças e ainda apresentar um padrão semelhante de ativação da via alternada do complemento. A GNMP pode ser suspeitada caso haja proteinúria nefrótica, ou caso a hipocomplementemia persista por mais de 8 semanas.
TRATAMENTO
O tratamento da GNDA é geralmente sintomático e ambulatorial. A hospitalização do paciente nem sempre é obrigatória, tornando-se necessária em casos de complicações: congestão cardiocirculatória, insuficiência renal aguda ou encefalopatia hipertensiva. É indicada a avaliação diária do paciente, visando reconhecer a evolução do edema e o peso, com controle da pressão arterial e do débito urinário.
Repouso
O repouso deve ser limitado pelo próprio paciente. É recomendado enquanto persistirem a hematúria macroscópica, a hipertensão e o edema.
Medidas Dietéticas
A dieta deve ser restrita em sal (limitada à fase de oligúria, edema e hipertensão). Superada a fase aguda, a dieta é gradativamente transicionada para a dieta comum e é um erro manter a dieta assódica por muito tempo.
A restrição hídrica está indicada, sendo especialmente importante na fase inicial, quando há oligúria e hipervolemia. O volume hídrico prescrito depende da gravidade do quadro clínico, oferecendo-se inicialmente 400 mL/m2/dia (ou 20 mL/kg/dia) para cobrir as perdas insensíveis. Adiciona-se a esse volume inicial uma fração gradualmente maior de volume, dependendo do débito urinário de 24 horas e da melhora do edema e da hipertensão arterial.
A restrição proteica está indicada quando a filtração glomerular permanecer muito diminuída. Devem ser prescritas dietas com baixo teor proteico (0,5 g/kg/dia) e a restrição de potássio deve ser iniciada apenas em presença de oligúria importante (diurese < 240 mL/m2/dia), isto é, nos primeiros 2 a 3 dias de doença.
Diuréticos
Nos casos de edema mais intenso, com hipertensão arterial e/ou sinais de congestão cardiocirculatória, indica-se o uso de diuréticos como a furosemida (1 a 2 até 6 mg/kg/dia). Nas hipertensões mais graves, deve-se administrar drogas hipotensoras, como os bloqueadores de canais de cálcio (nifedipina 0,15 até 1,5 mg/kg/dia a cada 6 horas).
A insuficiência renal na GNPE, em geral, é transitória e de curta duração. No entanto, quando severa e duradoura, podem ocorrer hipercalemia, hipocalcemia, hiperfosfatemia e acidose metabólica. A hipercalemia leve, sem alterações ecocardiográficas, pode ser conduzida com restrição dietética de potássio e uso de furosemida. Podem ser utilizadas também a resina trocadora de cálcio pelo potássio (Sorcal 0,5 a 1 mg/kg até 4 a 6 horas), a glicose hipertônica a 25% (1 a 3 mL/kg/h), junto com a insulina (3 U/3 a 5 g de glicose), e o bicarbonato de sódio a 3% (2 mEq/kg IV em 10 a 15 minutos). Na presença de hipercalemia severa, com alterações eletrocardiográficas graves, como ausência de onda P, alargamento de complexo QRS e arritmias, deve-se acrescentar o gluconato de cálcio a 10% (0,5 a 1 mL/kg IV, 5 a 10 minutos, com monitoração eletrocardiográfica) e indicar a diálise.
A diálise peritoneal remove efetivamente o potássio corpóreo e está indicada nas seguintes situações clínicas: anúria com duração de 48 horas, sobrecarga de volume (resultando em insuficiência cardíaca congestiva), acidose metabólica intratável, hipercalemia refratária ao tratamento, uremia sintomática, pericardite urêmica e hiponatremia grave.
O tratamento medicamentoso compreende a erradicação da estreptococcia. Utiliza-se penicilina benzatina nas doses de 600.000 UI para crianças menores de
COMPLICAÇÕES
As complicações são:
• congestão cardiocirculatória;
• encefalopatia hipertensiva;
• insuficiência renal aguda.
Salienta-se que, em certas circunstâncias, as 3 complicações podem aparecer simultaneamente.
PREVENÇÃO
A antibioticoterapia sistêmica precoce das infecções orofaríngeas e cutâneas estreptocócicas não elimina o risco de glomerulonefrite. Os familiares de pacientes com glomerulonefrite pós-estreptocócica aguda devem realizar exame de cultura quanto à procura de estreptococos beta-hemolíticos do grupo A e tratados, se a cultura for positiva. Após o início da antibioticoterapia adequada, o doente deve ser mantido em isolamento respiratório por 24 horas, para evitar a disseminação das cepas nefritogênicas dos estreptococos.
PROGNÓSTICO
A recuperação completa ocorre em mais de 95% das crianças com GNPE aguda. Em torno de 1 semana após o quadro inicial, há a normalização da pressão arterial, aumento da diurese e queda dos níveis de ureia e creatinina séricas.
A resolução da hematúria macroscópica ocorre em torno de
A hipocomplementemia retorna em níveis normais em
INDICAÇÕES DE BIÓPSIA RENAL
• Oligoanúria com duração maior que 48 a 72 horas;
• oligúria e/ou azotemia persistente por mais de 4 semanas;
• hipertensão arterial persistente por mais de 4 semanas;
• hematúria macroscópica por mais de 4 semanas;
• complemento total e frações persistentemente baixas por mais de 8 semanas;
• proteinúria nefrótica (> 50 mg/kg/dia) presente por mais de 4 semanas.
Pacientes com história anterior sugestiva de nefropatia ou antecedentes familiares sugestivos de afecções renais hereditárias devem ser observados com atenção e eventualmente biopsiados, se apresentarem evolução atípica.
BIBLIOGRAFIA
1. Wells WC. Transactions of a society for the improvement medical and chirurgical knowledge. Vol 3. Society for the improvement medical and chirurgical knowledge. London, 1812. p.3.
2. Mosquera JA, Rodriguez-Iturbe B. Extracellular neuraminidase production of streptococci with acute nephritis. Clin Nephrol 1984; 21:21.
3. Marin C, Mosquera JA, Rodriguez-Iturbe B. Neuraminidase promotes neutrophil, lymphocyte and macrophage infiltration in the normal kidney. Kidney Int 1995; 47:88.
4. Michael AF, McLean RH, Roy LP et al. Immunologic aspects of the nephritic syndrome. Kidney Int 1973; 3:105.
5. Lange K, Seligton G, Cronin W. Evidence for the in situ origin of postestreptococcal GN: glomerular localization of endoestreptosin and the clinical significance of the subsequent antibody response. Clin Nephrol 1983; 19:3.
6. Heptinstall RH. Pathology of the kidney. 3.ed. Boston: Little, Brown; 1983.
7. Brun C, Gormsen H, Hilden T, Iversen P, Raaschou F. Kidney biopsy in acute glomerulonephritis. Acta Med Scand 1958; 160:155.
8. Rodriguez-Iturbe B, Batsford SR et al. O antígeno nefritogênico na glomerulonefrite pós-estreptocócica é a exotoxina pirogênica B (SPE B) ou a GAPDH? Kidney Int 2006; 1:101-10.
9. Toporovski J, Mello VR et al. Nefrologia pediátrica. 2.ed. 2006.