(Carregando Índice)... (Carregando Índice)... |
Última revisão: 08/08/2017
Comentários de assinantes: 2
Saul Genuth, MD, FACP
Professor of Medicine, Division of Clinical and Molecular Endocrinology, Case Western Reserve University School of Medicine
Artigo original: Genuth S. Type 1
[The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2011 Decker Intellectual Properties Inc. All Rights Reserved.]
Tradução: André Islabão
Revisão Técnica: Dr. Euclides Furtado de Albuquerque Cavalcanti
O Diabetes melito tipo 1 é uma doença metabólica caracterizada pela destruição das células beta pancreáticas, resultando em deficiência da secreção de insulina e subsequente hiperglicemia.1 A destruição das células beta costuma resultar de um processo autoimune. As causas incomuns de diabetes tipo 1, nas quais não há evidência de atividade autoimune e para as quais não podem ser encontradas outras causas para a destruição das células beta pancreáticas (p. ex., fibrose cística) são classificadas como tipo 1 idiopáticas.1
A classificação atual do diabetes melito foi revisada por uma força-tarefa da American Diabetes Association (ADA) que incluiu representantes da Europa.1 As principais classes etiológicas da doença, junto com exemplos mais esotéricos, foram classificadas [Tabela 1]. O diabetes tipo 1 constitui 5 a 10% de todos os casos de diabetes; o tipo 2 é responsável pela maioria dos casos restantes.
Os diabetes tipos 1 e 2 eram conhecidos anteriormente como diabetes melito insulino-dependente (DMID) e diabetes melito não insulino-dependente (DMNID), respectivamente. Esta classificação foi abandonada em grande parte porque era difícil diferenciar pacientes com DMID de pacientes com DMNID que acabavam necessitando de tratamento com insulina para controle da hiperglicemia. A classificação atual, que se baseia na etiologia em vez do modo de tratamento, coloca maior ênfase na história e nas características dos pacientes para determinar a etiologia e o tipo prováveis.
Tabela 1. Classificação etiológica do diabetes.
|
Diabetes melito tipo 1* (destruição de células beta, geralmente causando deficiência absoluta de insulina) |
Imunomediado | |
|
Idiopático | ||
|
Diabetes melito tipo 2* (pode variar entre predomínio de resistência à insulina com deficiência relativa de insulina e predomínio de deficiência de secreção de insulina com resistência à insulina) | ||
|
Outros tipos específicos de diabetes |
Defeitos genéticos da função das células beta |
Cromossomo 12, HNF-1a (previamente MODY3) |
|
Cromossomo 7, glicoquinase (previamente MODY2) | ||
|
Cromossomo 20, HNF-4a (previamente MODY2) | ||
|
Defeitos de DNA mitocondriais | ||
|
Defeitos genéticos na ação de insulina |
Resistência à insulina do tipo A | |
|
Doença do pâncreas exócrino |
Pancreatite | |
|
Trauma/pancreatectomia | ||
|
Neoplasia | ||
|
Fibrose cística | ||
|
Hemocromatose | ||
|
Endocrinopatias |
Acromegalia | |
|
Síndrome de Cushing | ||
|
Glucagonoma | ||
|
Feocromocitoma | ||
|
Induzido por fármacos ou substâncias químicas |
Ácido nicotínico | |
|
Glicocorticosteroides | ||
|
Tiazídicos | ||
|
Pentamidina | ||
|
Interferon-alfa | ||
|
Infecções |
Rubéola congênita | |
|
Citomegalovírus | ||
|
Formas incomuns de diabetes imunomediado |
Rigidez muscular espasmódica (stiff-man syndrome) | |
|
Anticorpo antirreceptor de insulina | ||
|
Outras síndromes genéticas associadas com diabetes |
Síndrome de Down | |
|
Síndrome de Turner | ||
|
Ataxia de Friedreich | ||
|
Distrofia miotônica | ||
|
Diabetes melito gestacional (DMG) | ||
Nota: A lista de outros tipos específicos de diabetes não é completa. Existem muitas outras destas síndromes, em especial síndromes genéticas da função das células beta e síndromes genéticas associadas com diabetes.
*Os pacientes com qualquer forma de diabetes podem necessitar de tratamento com insulina em algum estágio da doença. Tal uso da insulina, por si só, não classifica o paciente.
Os estudos disponíveis sugerem que a prevalência de Diabetes melito tipo 1 nos Estados Unidos é de cerca de 1,8 por 1.000 em indivíduos com menos de 19 anos2 e 2,1 por 1.000 em adultos.3 Estima-se uma prevalência total de cerca de 500.000 casos. As estimativas atuais de incidência anual são de 18 por 100.000 pessoas na faixa de zero a 19 anos e de nove por 100.000 pessoas naqueles com mais de 20 anos.3 O pico de incidência ocorre com 11 anos de idade, mas casos novos podem, às vezes, aparecer em pacientes mais velhos. Estima-se que ocorram aproximadamente 30.000 novos casos de Diabetes melito tipo 1 por ano nos Estados Unidos, e isso é mais comum em brancos do que em afro-americanos. No mundo todo, a maior incidência anual de Diabetes melito tipo 1 é encontrada na Finlândia (45 casos por 100.000), e a menor é encontrada na Coreia (< 1 por 100.000). A incidência de diabetes tipo 1 está aumentando a uma taxa de cerca de 3% ao ano; em 2010, espera-se que a incidência anual em muitas populações exceda a 30 por 100.000.4
O diabetes envolve os aspectos mais fundamentais do metabolismo humano. As anormalidades hormonais do diabetes afetam todos os seguintes: a produção e o gasto de energia; a proporção usada de carboidratos, gorduras e proteínas como fonte de energia; o armazenamento de energia como carboidrato e gordura; e o equilíbrio entre síntese proteica (anabolismo) e degradação (catabolismo). Para entender a patogênese do diabetes melito, é útil começar com uma breve revisão do metabolismo normal.5
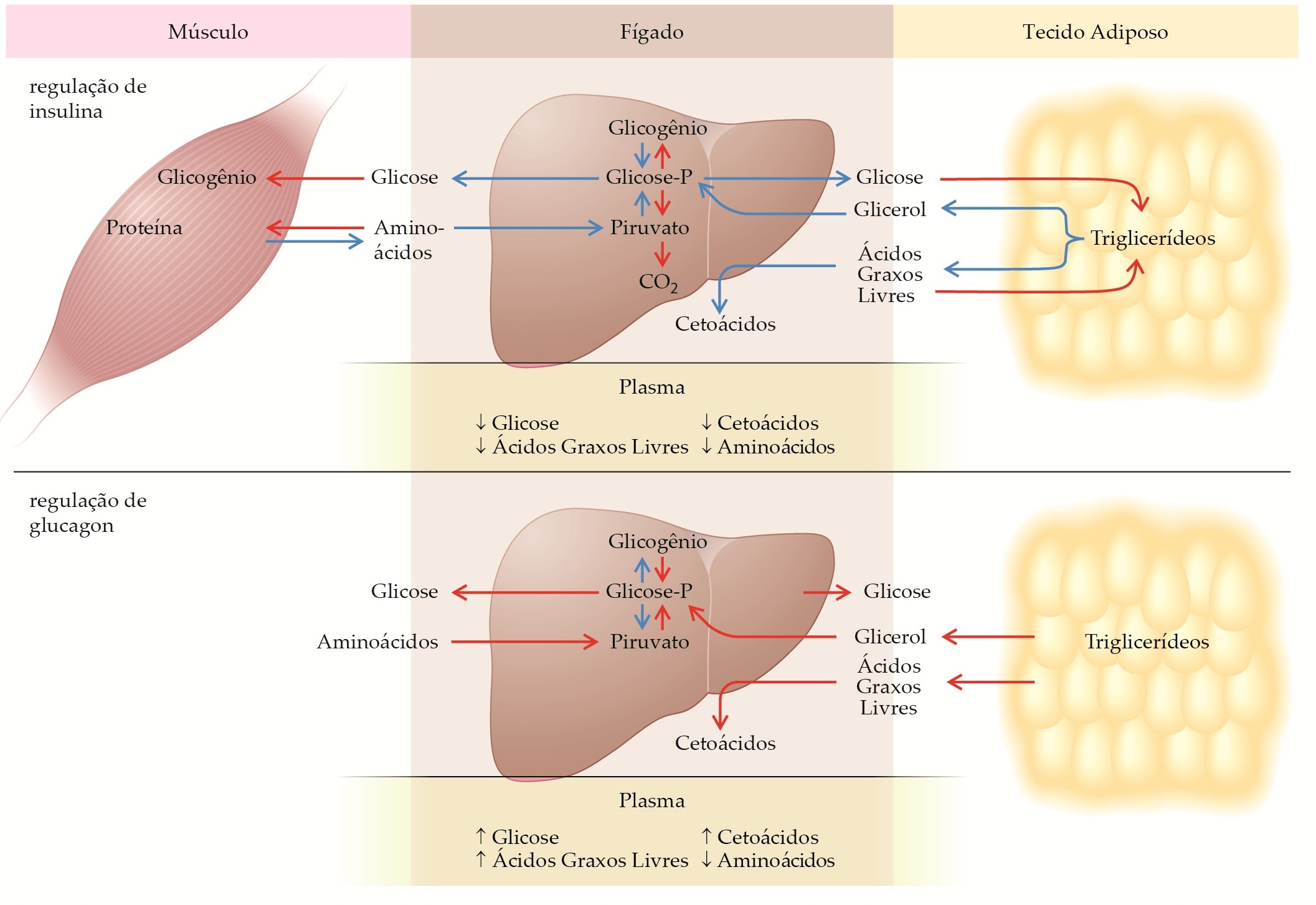
O equilíbrio adequado entre insulina e glucagon é um regulador hormonal crucial da homeostasia metabólica basal. A insulina primariamente facilita o armazenamento de glicose como glicogênio, de ácidos graxos livres como triglicerídeos e de aminoácidos como proteínas, e inibe a glicogenólise, a lipólise, a cetogênese, a proteólise e a gliconeogênese [Figura 1]. O glucagon estimula a mobilização de glicose, ácidos graxos livres e glicerol, bem como a captação hepática de aminoácidos e a conversão de seus esqueletos de carbono em glicose. O glucagon também estimula a cetogênese a partir de ácidos graxos livres [Figura 1]. Os níveis normais em estado de equilíbrio de insulina e glucagon ajudam a manter o nível de glicose plasmática durante o jejum noturno entre 60 e 110 mg/dL, o nível de ácidos graxos livres em menos de 0,7 mmol/L, de cetoácidos em menos de 0,2 mmol/L e cada aminoácido em seu próprio nível. Após uma refeição mista, a insulina plasmática aumenta rapidamente [Figura 2] e, com isso, a razão insulina/glucagon. Essa condição reverte todos os processos de mobilização de energia e síntese de glicose descritos anteriormente. O carboidrato da dieta é armazenado no glicogênio dos músculos e do fígado, os ácidos graxos livres são novamente esterificados e armazenados como triglicerídeos no tecido adiposo e o metabolismo das proteínas retorna em direção ao anabolismo. Quando todos os nutrientes tiverem sido assimilados e a glicose plasmática retornar ao nível pré-prandial basal, a insulina plasmática [Figura 2] e a razão insulina/glucagon retornam prontamente para os níveis basais, evitando um exagero de ação da insulina, o qual causaria hipoglicemia. Assim, uma elevação imediata, um pico precoce e uma queda rápida na secreção de insulina são requisitos do metabolismo pós-prandial normal [Figura 2].

Figura 1. As ações opostas da insulina e do glucagon, em especial dentro do fígado, sobre o fluxo e os níveis plasmáticos de substratos, são aqui mostradas. Os dois hormônios têm efeitos diretamente opostos sobre enzimas-chave, como glicogênio-sintase e fosforilase. Assim, os efeitos estimulatórios do glucagon sobre a produção de glicose e cetoácidos são ampliados quando há deficiência de insulina, como no Diabetes melito tipo 1. As setas vermelhas indicam estimulação. As setas azuis indicam inibição.
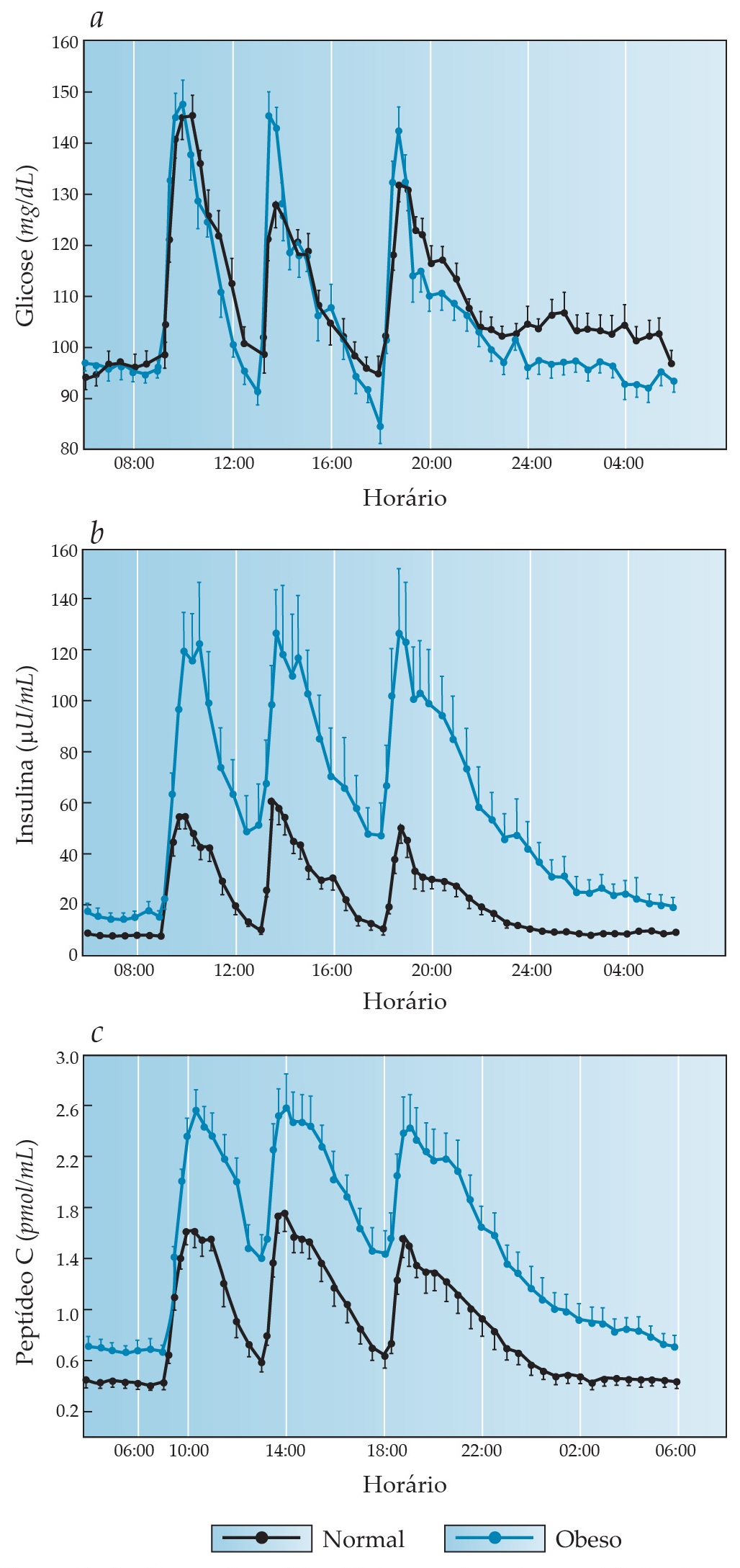
Figura 2. A glicose plasmática (a) é normalmente mantida dentro de uma faixa estreita ao longo do dia, em grande parte por causa da função das células beta. A insulina plasmática (b) e o peptídeo C plasmático (c) aumentam rapidamente a partir dos níveis basais em cada refeição e, após atingirem um pico, retornam prontamente aos níveis basais, que são mantidos ao longo da noite. Observe também que os níveis plasmáticos de insulina e de peptídeo C estão elevados em pessoas obesas, as quais são resistentes à insulina.
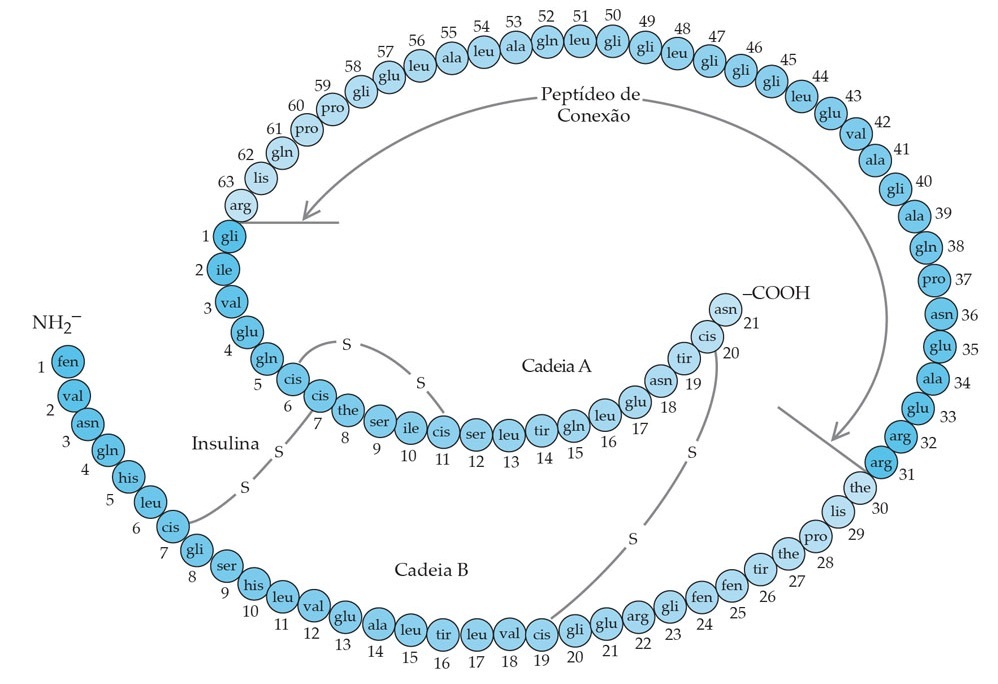
A insulina é sintetizada nas células beta das ilhotas pancreáticas a partir de uma molécula maior chamada pró-insulina, a qual é quebrada para gerar insulina, e um peptídeo de conexão intramolecular chamado peptídeo C [Figura 3]. As duas moléculas são armazenadas nos mesmos grânulos e são secretadas em uma razão equimolar quando a célula beta é estimulada. Assim, os níveis plasmáticos de peptídeo C são um marcador confiável da função das células beta [Figura 3].
Figura 3. A estrutura da pró-insulina humana, a molécula precursora da insulina. O peptídeo que conecta a amina terminal (NH2-) da cadeia A na carboxila terminal (-COOH) da cadeia B é chamado de peptídeo de conexão (peptídeo C). A pró-insulina é convertida em insulina e peptídeo C e essas duas moléculas são carregadas juntas nos grânulos secretores. Quando a célula beta é estimulada, o peptídeo C e a insulina são secretados em proporções equimolares. Assim, os níveis de peptídeo C refletem a capacidade funcional das células beta.
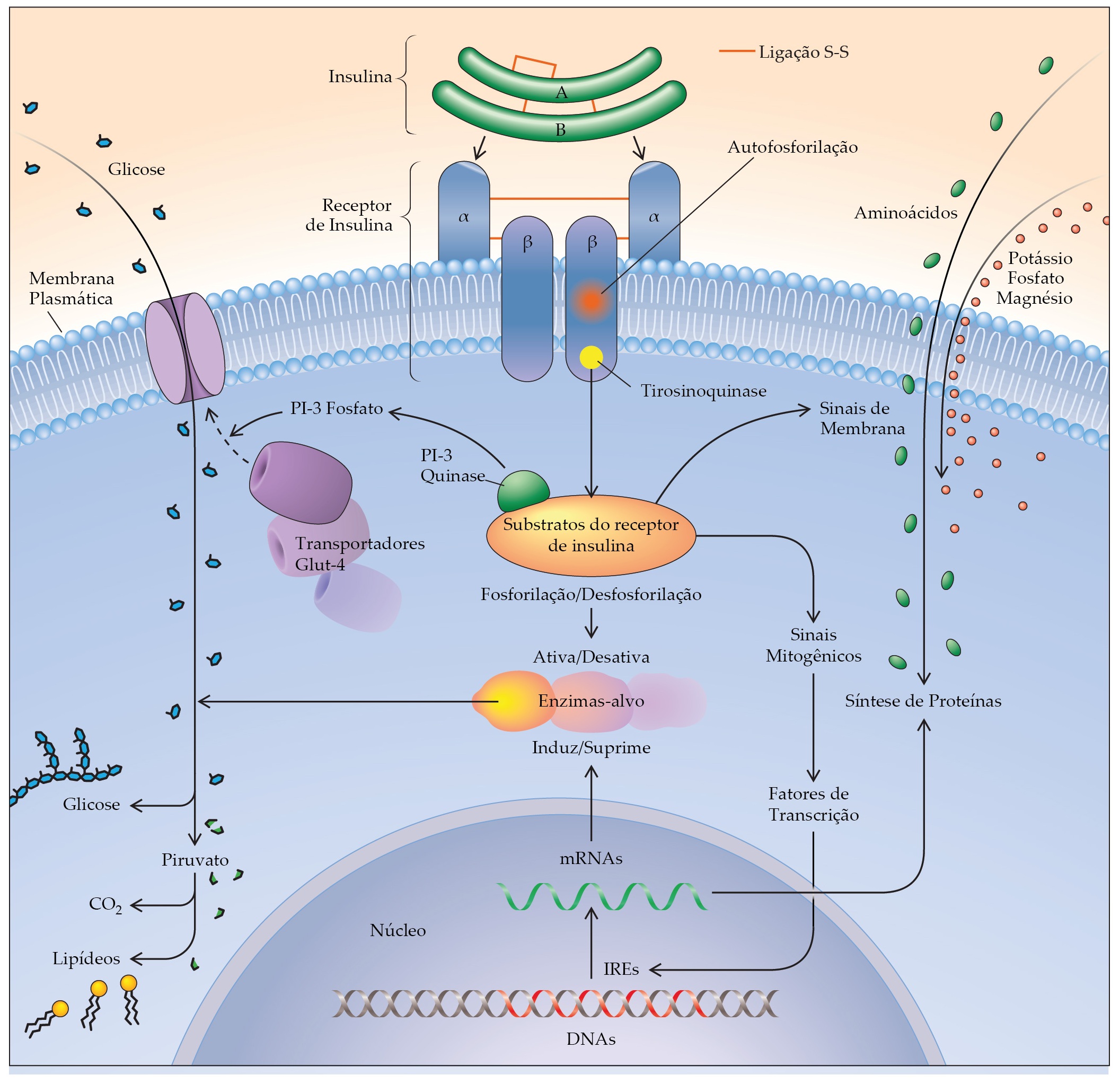
A insulina age por meio de um receptor de insulina plasmática que leva à geração de múltiplos mediadores dos numerosos efeitos intracelulares citoplasmáticos e nucleares da insulina [Figura 4]. A insulina regula as atividades e a síntese de enzimas-alvo. A sensibilidade de tecidos-alvo à insulina é outro principal determinante da ação da insulina. Existe uma alça de retroalimentação entre a responsividade à insulina em tecidos-alvo e a secreção de insulina nas células beta. Essa relação trabalha para aumentar a secreção de insulina em indivíduos relativamente resistentes à ação da insulina – por exemplo, pessoas obesas [Figura 2] – e para diminuir a liberação de insulina em indivíduos muito sensíveis à ação da insulina [Figura 5]. O resultado é um mecanismo fundamental para a manutenção dos níveis plasmáticos de jejum e pós-prandiais de glicose dentro de estreitos limites normais.
Figura 4. As ações celulares da insulina começam com a ligação ao seu receptor na membrana plasmática. Como resultado, certas moléculas de tirosina na porção intracelular do receptor transmembrana sofrem autofosforilação, criando atividade de tirosinoquinase no receptor. Vários substratos de receptores de insulina (IRSs) intracelulares são, então, fosforilados em tirosina pelo receptor. Os IRSs fosforilados reduzem e ativam ou inativam várias enzimas (p. ex., fosfatidilinositol-3-quinase [PI-3 quinase]) e outras moléculas mediadoras. Entre os principais efeitos dessa cascata estimulada pela insulina estão a translocação de transportadores da glicose (Glut-4) até a membrana plasmática, onde eles facilitam a difusão da glicose para dentro da célula; o desvio do metabolismo intracelular da glicose em direção ao armazenamento como glicogênio pela ativação da glicogênio-sintase; a estimulação da captação celular de aminoácidos, fosfato, potássio e magnésio; a estimulação da síntese proteica e a inibição da proteólise; e a regulação da expressão de genes por meio de elementos regulatórios de insulina (IREs) em moléculas-alvo de DNA. Diversos intermediários nessas vias, junto com as moléculas mencionadas anteriormente, são produtos de genes cuja mutação poderia produzir um estado de resistência à insulina característico do diabetes melito tipo 2. Os conectores vermelhos entre as cadeias A e B de insulina e entre as subunidades a e ß do receptor de insulina indicam ligações S-S. A cadeia A também tem uma ligação S-S intramolecular.
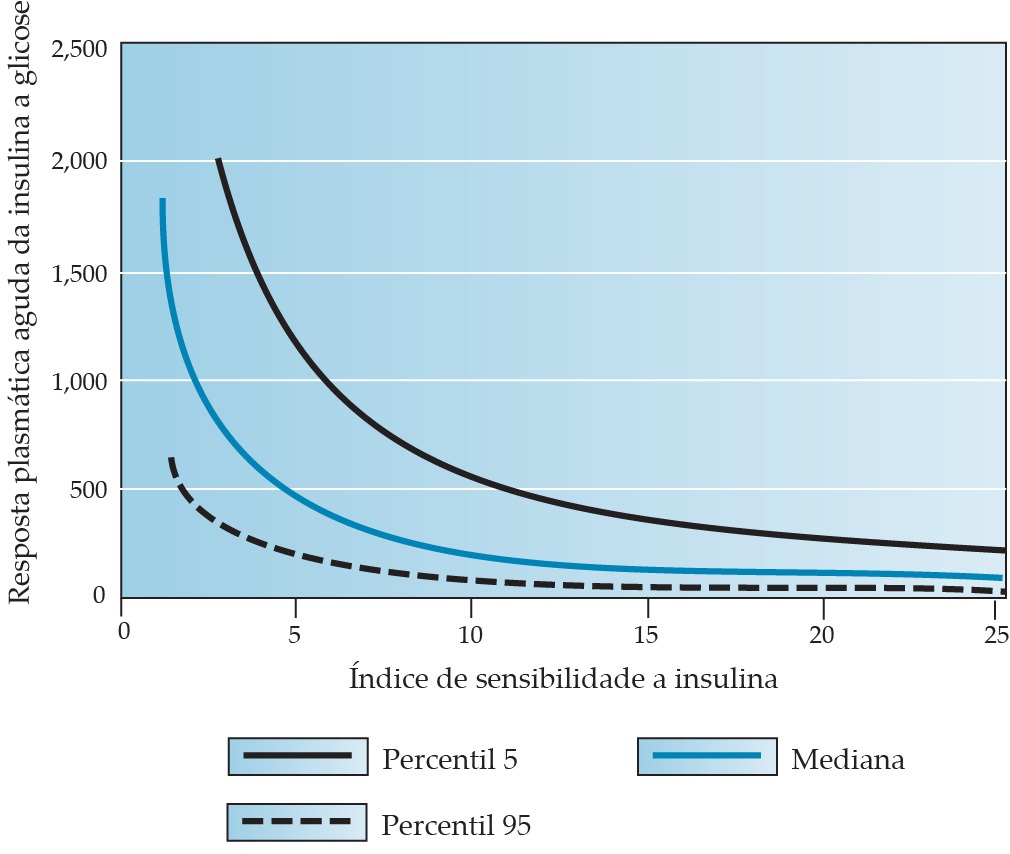
Figura 5. A medida dos níveis de insulina após a administração de glicose fornece uma indicação da sensibilidade (isto é, a captação de glicose pelos tecidos periféricos em resposta à insulina) e da responsividade das células beta pancreáticas. São mostrados a mediana e os percentis 5 e 95 da sensibilidade à insulina. Observe que a quantidade de insulina secretada em resposta à administração de glicose aumenta à medida que diminui a sensibilidade à insulina. Esse mecanismo de retroalimentação ajuda a manter os níveis de glicose plasmática na faixa fisiológica.
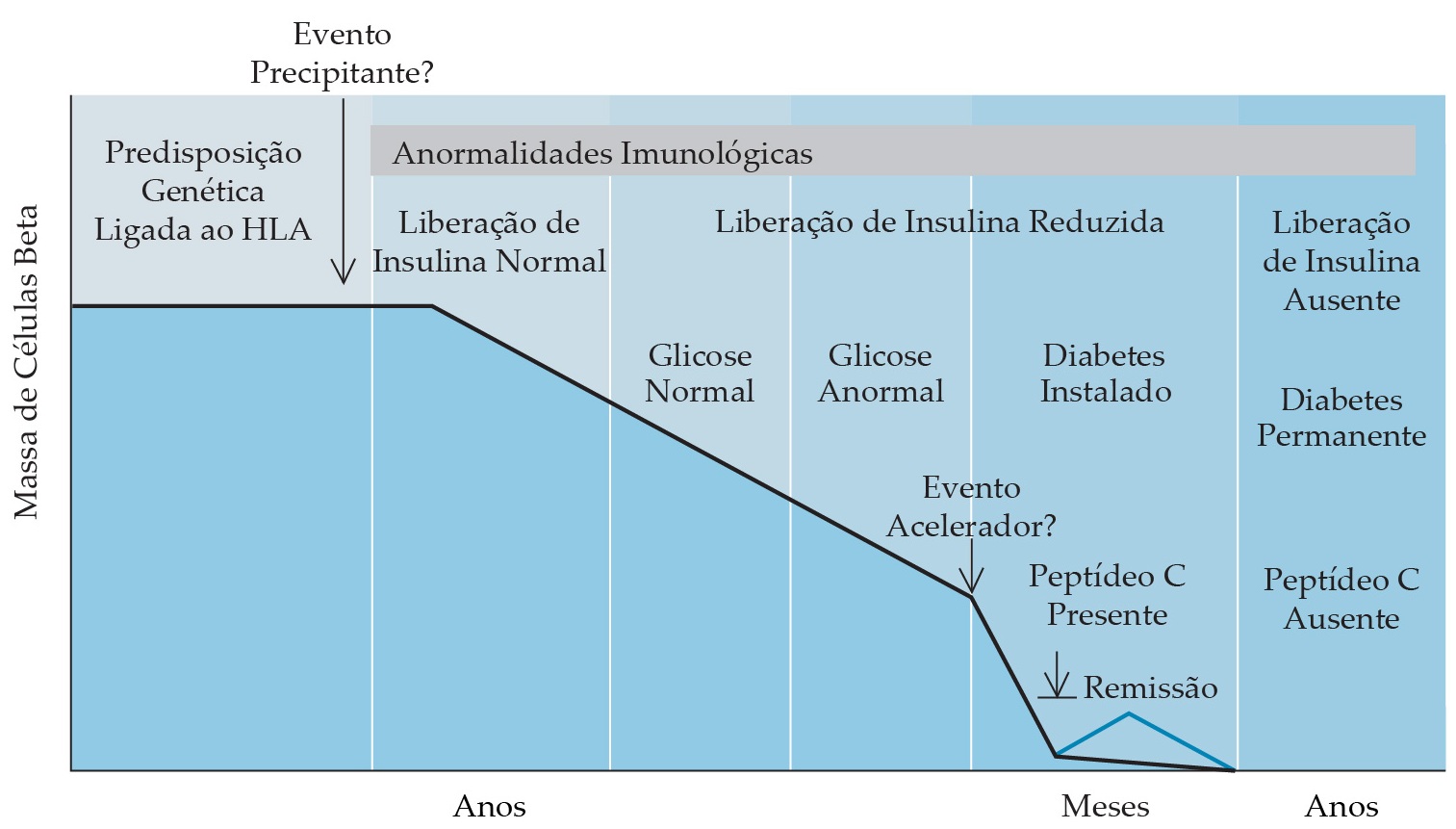
O diabetes tipo 1 se caracteriza por deficiência absoluta de insulina, tornando os pacientes dependentes da reposição de insulina exógena para a sobrevivência.1,6 A deficiência de insulina resulta da destruição ou desaparecimento das células beta produtoras de insulina que constituem 60 a 70% das ilhotas pancreáticas. O diabetes clínico ocorre quando 90% das células beta tiverem sido eliminadas [Figura 6].
Figura 6. Visão atual da patogênese do Diabetes melito tipo 1 autoimune. Em alguns indivíduos, genes ligados ao HLA iniciam um ataque autoimune contra as ilhotas pancreáticas, em especial as células beta. Em outros indivíduos, genes ligados ao HLA protegem contra a resposta destrutiva autoimune. Um evento inicial, como a exposição a um vírus com um epítopo antigênico que se parece com um antígeno das células beta ou a uma toxina, pode iniciar o processo de autodestruição. O desaparecimento das células beta pode ocorrer porque o antígeno viral acelera a taxa normal de apoptose (morte celular programada). À medida que o tempo passa, diminuem a produção e a secreção de insulina, apesar da hiperglicemia crescente. Quando a liberação de insulina cai para quantidades mínimas ou cessa por completo, ocorre a cetoacidose diabética. Outro evento externo pode desencadear essa catástrofe final das células beta. Algumas células beta podem sobreviver porque, após isso, pode ocorrer um breve período de remissão marcado pelo reaparecimento de peptídeo C no plasma se os níveis de glicose plasmática forem controlados de forma estrita com insulina exógena. Por fim, cessa toda a função das células beta, causando instabilidade metabólica.
Existem fortes evidências de que um processo autoimune mediado por células está envolvido na destruição de células beta na maioria dos casos de Diabetes melito tipo 1.7-9 Em vários casos em que a morte ocorreu por um acidente ou por uma doença que não a diabetes logo após o paciente ser diagnosticado com Diabetes melito tipo 1, foram encontrados infiltrados de linfócitos mononucleares nas ilhotas. A distribuição de células T em tais casos é marcada por um aumento nas células T supressoras-indutoras CD8+ e uma diminuição nas células T auxiliares indutoras CD4+.7 Uma resposta imunocelular semelhante tem sido encontrada em modelos animais de diabetes insulino-deficientes espontâneo.9 Em algumas situações, manipulações experimentais que evitam as respostas dos linfócitos T também evitam o desenvolvimento de diabetes. Além disso, também foi descrita a transferência de diabetes de animais afetados para animais não afetados por meio de linfócitos. Tem sido demonstrado que interleucinas e outras citocinas exibem efeitos tóxicos sobre as células beta e inibem a secreção de insulina.
Autoanticorpos contra uma série de antoantígenos de ilhotas e células beta estão presentes no soro de pacientes com Diabetes melito tipo 1 no momento do diagnóstico.10 Os autoantígenos incluem as enzimas descarboxilase do ácido glutâmico (DAG), carboxipeptidase H, uma proteína tirosinafosfatase rotulada ICA512 ou IA-2 e a própria insulina.10
Alguns estudos têm demonstrado que os autoanticorpos contra as ilhotas são capazes de inibir a secreção de insulina in vitro ou mesmo de causar a lise das células beta. Outras evidências sustentam a importância de fenômenos autoimunes na patogênese do Diabetes melito tipo 1. Por exemplo, estudos em gêmeos idênticos que eram discordantes para Diabetes melito tipo 1 mostraram que, na ausência de terapia imunossupressora, o gêmeo idêntico diabético irá rejeitar um transplante de pâncreas do gêmeo não diabético; presume-se que os autoanticorpos do receptor reconheçam antígenos nas ilhotas pancreáticas do gêmeo doador, os quais são idênticos aos do receptor. Se o tratamento do Diabetes melito tipo 1 com o agente imunossupressor ciclosporina for iniciado dentro de duas a seis semanas após o início clínico do diabete, a dependência da insulina pode ser eliminada ou a dose de insulina pode ser grandemente reduzida, mas apenas enquanto for mantido o tratamento imunossupressor.11,12 A toxicidade associada com a ciclosporina e outros agentes imunossupressores impede o uso dessa forma de terapia na prática clínica.
Atualmente, está claro que os fenômenos autoimunes iniciam muito antes do início da doença clínica. Autoanticorpos contra ilhotas ou células beta podem ser encontrados em 2 a 4% dos parentes em primeiro grau de pacientes com Diabetes melito tipo 1, o que é 10 a 20 vezes a prevalência em sujeitos-controle. Estudos longitudinais têm mostrado que o Diabetes melito tipo 1 tem muito mais chance de ocorrer em parentes não acometidos com altos títulos de autoanticorpos em relação a parentes sem tais anticorpos, e a doença irá se desenvolver em tais pacientes dentro de poucos anos.13,14 A resistência à insulina, avaliada com base no modelo de homeostasia, também é um fator de risco para a progressão do diabetes tipo 1 em indivíduos de risco alto e moderado.15 Testes longitudinais seriados das respostas plasmáticas da insulina à injeção de glicose intravenosa demonstram declínio progressivo na função das células beta em parentes com autoanticorpos positivos antes do início clínico do diabetes.15
Em parentes de pacientes com diabetes tipo 1, o risco de desenvolvimento da doença pode ser estratificado pela medida de autoanticorpos contra as ilhotas. O número de anticorpos presentes, bem como suas características (p. ex., título, subclasse e epítopos), indicam o grau de risco. As características dos autoanticorpos – e, assim, o grau de risco – podem mudar com o tempo, especialmente em pacientes mais jovens.16
Outras doenças autoimunes, como doença de Graves, doença de Addison, tireoidite linfocítica de Hashimoto e síndrome celíaca, ocorrem com frequência aumentada em pessoas com diabetes tipo 1.
Como apenas 30 a 50% dos gêmeos idênticos não afetados de pacientes com Diabetes melito tipo 1 irão desenvolver a doença, provavelmente sera necessário um fator ambiental para desencadear o processo destrutivo autoimune.17 Além disso, indivíduos com genótipos que os colocam em alto risco para diabetes formam uma porcentagem menor da população diabética do que no passado – um fato que sugere uma causa ambiental para a incidência crescente de diabetes tipo 1.18
Vários candidatos virais foram propostos.17 A única associação confirmada é de que a prole de mães infectadas com rubéola durante a gestação tem risco aumentado de Diabetes melito tipo 1. Além disso, algumas evidências indiretas associam coxsackievírus B, enterovírus, rotavírus, parvovírus e citomegalovírus com Diabetes melito tipo 1.19,20 Toxinas no ambiente ou na dieta também podem iniciar a destruição de células beta geneticamente vulneráveis. Foi estabelecido um consórcio internacional para acompanhar pessoas com genótipos de antígenos leucocitários humanos (HLA) de alto risco [veja Fatores genéticos, adiante] a partir do nascimento até a adolescência, em um esforço para identificar agentes infecciosos, fatores dietéticos ou outros fatores ambientais que possam desencadear autoimunidade nas ilhotas nessa população com suscetibilidade genética.21
No momento do início clínico do Diabetes melito tipo 1, pelo menos um pequeno número de células beta ainda é potencialmente capaz de funcionar.22 Após várias semanas de tratamento com insulina exógena, em especial se for estabelecido um controle metabólico exemplar,23 a dependência de insulina exógena diminui ou cessa completamente por semanas ou meses em alguns pacientes. Essa remissão temporária (a chamada fase de “lua de mel”) é marcada por um aumento nos níveis séricos de peptídeo C, o que indica um aumento na secreção endógena de insulina [Figura 6]. No entanto, vários anos após o diagnóstico de Diabetes melito tipo 1, o peptídeo C costuma ser indetectável por exames de rotina no soro.24 Contudo, mesmo uma pequena quantidade de secreção residual de insulina endógena torna o controle da glicemia com insulina exógena menos variável.
O Diabetes melito tipo 1 não se desenvolve em todos os indivíduos com autoanticorpos positivos. Além disso, o período de latência entre o início da destruição das células beta e o aparecimento clínico do problema pode ser de muitos anos;25 a doença não aparece em alguns pacientes até muito mais tarde. Essa condição tem sido chamada de diabetes autoimune latente em adultos (LADA).26 A natureza gradual e indolente da doença nesses pacientes com autoanticorpos positivos também é sugerida pelo fato de que alguns podem ser tratados com fármacos estimulantes de células beta antes de necessitarem de insulina.27
Embora uma história familiar de Diabetes melito tipo 1 tenha mais chance de estar ausente do que presente em casos-índice, é realmente verdade que filhos e irmãos de pacientes com diabetes melito tipo1 têm risco aumentado para a doença.
Existe uma base genética para a suscetibilidade ao Diabetes melito tipo 1, mas nenhum genótipo específico leva de forma inevitável ao desenvolvimento da doença.28 A doença irá se desenvolver em 5 a 10% dos parentes em primeiro grau de pacientes com Diabetes melito tipo 1 e em 20% das pessoas com dois parentes de primeiro grau (p. ex., ambos os pais) com a doença. Estudos de associação e ligação têm incriminado vários genes envolvidos no risco de diabetes melito 1. O polimorfismo de genes HLA no lócus do complexo de histocompatibilidade maior (CHM) no cromossomo 6 é responsável por 50% do risco genético.28
DR3 e DR4 são alelos de suscetibilidade HLA que parecem operar de maneira sinérgica.29 Os indivíduos heterozigotos para DR3 e DR4 têm maior risco do que indivíduos homozigotos para DR3 ou DR4. O alelo DR2 diminui o risco de diabetes tipo 1 e domina o efeito de suscetibilidade de DR3 ou DR4 quando um deles está acompanhado do DR2. Os alelos HLA DR3,DQB1*0201 e DR4,DQB1*0302 estão associados com alto risco, enquanto o alelo DR1,DQB1*0501 está associado com risco moderado.30
Tem sido relatada uma associação não apenas com diabetes tipo 1, mas também com artrite reumatoide e lúpus eritematoso sistêmico em pessoas portadoras de polimorfismo em um único nucleotídeo do gene da proteína tirosinafosfatase N22 (PTPN22). A PTPN22 codifica uma proteína, a tirosinafosfatase Lyp, a qual está envolvida na regulação para baixo (downregulation) da sinalização de células T.31
Vários mecanismos têm sido sugeridos para explicar a maneira pela qual moléculas de HLA da classe II podem predispor ou proteger contra a doença.32 Apesar do acúmulo de conhecimentos consideráveis, o desenvolvimento de Diabetes melito tipo 1 ainda não pode ser previsto com certeza completa.33
O Diabetes melito tipo 1 está associado com pelo menos 15 lócus adicionais em nove outros cromossomos.33 De particular interesse é o fato de que um número variável de repetições em sequência na região promotora do gene da insulina tem sido associado à doença. No entanto, a própria molécula de insulina tem estrutura aparentemente normal em pacientes com Diabetes melito tipo 1. Com o genoma humano sequenciado e a tecnologia genética avançada se tornando custo-efetiva, é provável que os componentes genéticos do Diabetes melito tipo 1 sejam definidos de maneira que seja possível identificar pessoas suscetíveis que possam se beneficiar de terapias preventivas.
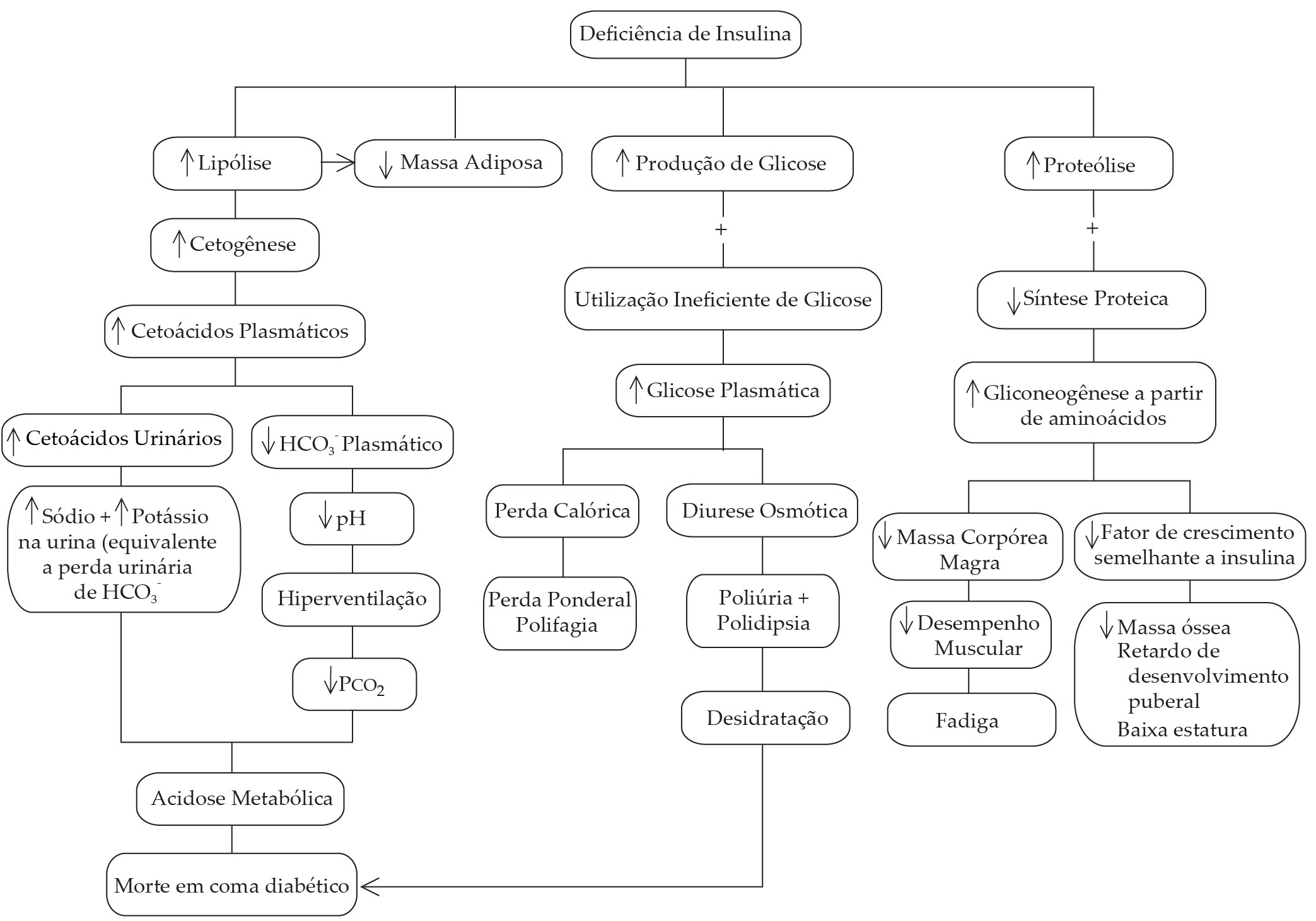
Todas as manifestações clínicas e bioquímicas do Diabetes melito tipo 1 podem ser consequência da deficiência de insulina [ver Figuras 1 e 7].34 A perda do efeito estimulante da insulina sobre a captação de glicose por músculos e tecido adiposo junto com a perda do efeito supressor da insulina sobre a liberação hepática de glicose levam à hiperglicemia severa. Os níveis da glicemia de jejum costumam aumentar para 300 a 400 mg/dL e os níveis de glicose pós-prandial aumentam para 500 a 600 mg/dL em pacientes antes do tratamento.34 Este aumento resulta na apresentação de uma elevada carga de glicose filtrada aos túbulos renais e, consequentemente, em diurese osmótica, manifestada por poliúria e polidipsia compensatória. A perda dos efeitos lipogênicos e antilipolíticos da insulina sobre o tecido adiposo leva a altos níveis plasmáticos e captação hepática aumentada de ácidos graxos livres. Essa condição aumenta a cetogênese e, por fim, altos níveis plasmáticos de cetoácidos causam acidose metabólica. A degradação proteica é favorecida na ausência das ações anticatabólicas e anabólicas da insulina. A proteólise de proteínas musculares fornece aminoácidos que mantêm taxas elevadas de gliconeogênese. Este processo resulta de perda de massa corpórea gorda e magra; a perda ponderal é agravada ainda mais por um aumento no gasto basal de energia.35 O balanço de nitrogênio negativo, acompanhado por perdas de potássio, magnésio e fosfato na urina, prejudica o crescimento e o desenvolvimento em crianças.
Figura 7. São mostrados os caminhos que levam da deficiência de insulina às principais manifestações clínicas do Diabetes melito tipo 1. Observe que a deficiência de insulina também resulta em uma diminuição no fator de crescimento semelhante à insulina e isso diminui a taxa de crescimento.
Em geral, as evidências sorológicas, bem como os marcadores genéticos, podem identificar pessoas com risco aumentado para o desenvolvimento de diabetes tipo 1. No momento do diagnóstico, cerca de 90% dos pacientes com Diabetes melito tipo 1 têm autoanticorpos contra GAD (Glutamic acid decarboxylase), IA-2 (insulinoma antigen 2) ou insulina, isoladamente ou em combinação. A presença de autoanticorpos contra células das ilhotas pancreáticas prediz o desenvolvimento de diabetes autoimune em parentes de primeiro grau, enquanto na população geral, a presença de dois ou mais autoanticorpos é altamente preditiva de diabetes no futuro; os anticorpos IA-2 indicam um risco muito alto.36 A existência de tais marcadores, bem como o longo período entre seu aparecimento e o desenvolvimento da doença, inspiraram pesquisas sobre a possibilidade de intervenções para evitar o diabetes clínico.37
Infelizmente, um ensaio financiado pelo National Institute of Diabetes and Digestive and Kidney Diseases não encontrou evidências de que o Diabetes melito tipo 1 possa ser evitado pela indução de tolerância imune à insulina humana exógena administrada por via subcutânea ou oral a parentes de pacientes com altos títulos de autoanticorpos contra as ilhotas. Contudo, uma análise sugeriu que os pacientes do estudo com níveis altos de autoanticorpos contra insulina e que receberam insulina oral podem ter tido um retardo e, talvez, uma redução na incidência de diabetes tipo 1.38 Essa hipótese está sendo testada em um estudo iniciado pelo Type 1 Diabetes TrialNet (www.diabetestrialnet.org), que é uma rede de ensaios clínicos criada pelo National Institutes of Health para a condução de estudos em pacientes com diabetes tipo 1 de início recente e em parentes em risco para a doença.
Estudos anteriores mostraram que o tratamento com ciclosporina oferecia um método efetivo para a supressão do diabetes tipo 1 [ver Patogênese, Fatores autoimunes, anteriormente]. Entretanto, a toxicidade da ciclosporina a longo prazo a torna insegura para ser usada com este propósito. Em estudos mais recentes, o uso de anticorpos monoclonais anti-CD3 com alvo em células T mostrou-se promissor para a proteção de células beta contra a destruição.39-41
Os estudos atuais do Type 1 Diabetes TrialNet estão investigando o tratamento de pacientes com diagnóstico recente de diabetes tipo 1 usando antagonistas adaptados e humanizados de moléculas em linfócitos que são importantes em sua ativação nos processos autoimunes. Isso inclui rituximabe, que está aprovado para o tratamento de linfomas selecionados e artrite reumatoide, e a combinação de micofenolato mofetil e daclizumabe (um inibidor do receptor de interleucina 2), que estão aprovados para a prevenção de rejeição após transplante de órgãos.
O diagnóstico de Diabetes melito tipo 1 ainda é quase sempre feito com base na história dos sintomas; o diagnóstico é confirmado pelo achado de um nível de glicemia plasmática maior do que 200 mg/dL, junto com glicosúria e, em geral, cetonúria. Os sintomas clássicos são poliúria, polidipsia, perda ponderal apesar de ingestão normal ou aumentada de alimentos, fadiga e visão borrada. Esses sintomas costumam se apresentar 4 a 12 semanas antes que o paciente busque ajuda médica. No futuro, porém, talvez seja possível fazer o diagnóstico antes do início clínico da doença por meio de métodos sorológicos, complementados por testes da função das células beta.
De todas as doenças crônicas, o diabetes é único porque o seu tratamento envolve o autocuidado diário pelo paciente e uma série de adaptações no estilo de vida. Para um controle metabólico ideal, os pacientes devem furar seus dedos pelo menos quatro vezes ao dia para testar o nível de glicemia, injetar insulina pelo menos três vezes ao dia, prestar atenção regularmente ao momento e ao conteúdo de suas refeições e tentar seguir um programa de exercícios. O paciente está realmente no centro de seus cuidados. O autocuidado pelo paciente exige educação intensiva com respeito às habilidades para a realização da injeção e do monitoramento da glicemia, do teste de cetonas na urina quando o paciente não se sentir bem, do planejamento das refeições, da detecção e tratamento da hipoglicemia e do manejo de doenças intercorrentes. Os membros da família e pessoas próximas do paciente devem ser incluídos, em especial com respeito ao reconhecimento e ao tratamento da hipoglicemia. Idealmente, o paciente deve compreender a patofisiologia do diabetes e suas complicações a longo prazo quase tão bem quanto os profissionais da saúde. Alguns aspectos dos cuidados exigem reforço educacional periódico, o qual costuma ser estimulado por algum problema terapêutico, como episódios preveníveis de hipoglicemia severa ou cetoacidose.
Os objetivos clínicos do tratamento incluem:
diminuição dos níveis de glicose plasmática e da excreção urinária de glicose para eliminar poliúria, polidipsia, polifagia, perda calórica e efeitos adversos, como visão borrada por edema do cristalino e suscetibilidade a infecções, em particular a vaginite em mulheres;
eliminação da cetoacidose;
indução de balanço de nitrogênio positivo para restauração da massa corpórea magra e da capacidade física e para manutenção do crescimento, desenvolvimento e funcionamento normais;
prevenção ou minimização de grande parte das complicações tardias do diabetes.
A introdução da insulina em 1922 permitiu que os pacientes com Diabetes melito tipo 1 vivessem o suficiente para experimentar as complicações microvasculares distintivas do diabetes – retinopatia e nefropatia – bem como neuropatia. O aparecimento dessas complicações na década de 1930 gerou um debate de 50 anos sobre se retinopatia, nefropatia e neuropatia diabéticas eram o resultado direto das anormalidades metabólicas, mais notavelmente da hiperglicemia, ou se eram uma consequência independente e paralela do diabetes que antes costumava não ser vista em função da morte por desequilíbrio metabólico extremo (isto é, coma diabético). O debate não era apenas acadêmico, pois ele se refletia em abordagens muito diferentes de tratamento. A crença de que as complicações eram causadas pela hiperglicemia impelia os médicos a trabalhar com meios inadequados para ajudar o paciente a alcançar a glicemia mais perto do normal possível. Por outro lado, a crença de que as complicações ocorriam independentemente da hiperglicemia encorajava uma abordagem um pouco mais despreocupada, na qual os médicos tentavam primariamente eliminar os sintomas imediatos, como poliúria, que ocorriam quando a glicose plasmática excedia o limiar renal (> 180 mg/dL). Além disso, o risco de hipoglicemia associado com a abordagem mais agressiva contra a hiperglicemia reforçava os argumentos dos profissionais conservadores. Por fim, foi acumulada uma grande quantidade de evidências para sustentar, mas não comprovar, a chamada hipótese da glicose (ou seja, a hipótese de que o tratamento que normalizava os níveis de glicose evitaria ou retardaria as complicações a longo prazo do diabetes melito). O Diabetes Control and Complications Trial (DCCT) encerrou este debate sobre o diabetes tipo 1.
O DCCT foi um ensaio clínico randomizado que testou a hipótese da glicose em pacientes com Diabetes melito tipo 1.42 Metade dos pacientes com diabetes com um a cinco anos de duração participou em um ensaio de prevenção primária que excluía todos os pacientes com retinopatia ou microalbuminúria, e metade dos pacientes com diabetes com um a 15 anos de duração participava em um ensaio de intervenção secundária que incluía apenas pacientes que já tinham retinopatia diabética não proliferativa leve a moderada, mas com menos de 200 mg/dia de excreção urinária de albumina. Em ambos os ensaios DCCT, os pacientes foram designados de maneira aleatória para tratamento convencional (não mais do que duas injeções de insulina ao dia) ou para tratamento intensivo (três a quatro injeções de insulina por dia ou o uso de bomba de infusão de insulina subcutânea contínua [continuous subcutaneous insulin infusion - CSII]; automonitoramento da glicemia pelo menos quatro vezes ao dia; nível-alvo para a glicemia antes das refeições de 70 a 120 mg/dL; um objetivo de hemoglobina glicada [HbA1c] de menos de 6,05%; e contatos muito frequentes entre o paciente e a equipe de tratamento). Uma diferença na HbA1c de 1,8% (8,9% versus 7,1%), correspondendo a uma diferença média na glicemia de cerca de 75 mg/dL, foi mantida entre os dois grupos de tratamento.43
Após um período médio de acompanhamento de 6,5 anos, o tratamento intensivo produziu benefícios substanciais. O risco de desenvolvimento de retinopatia nova, ou seja, o aparecimento de microaneurismas, foi reduzido em 27%, enquanto o risco de progressão da retinopatia foi reduzido em 76%; o desenvolvimento de microalbuminúria foi reduzido em 35%; a macroalbuminúria (ou seja, proteinúria) foi reduzido em 56%; e o desenvolvimento de neuropatia clínica, confirmada por velocidades de condução nervosa ou testes de função do sistema autonômico anormais, foi reduzido em 60%.42 Os pacientes na coorte de prevenção primária, com uma duração média do diabetes de 2,5 anos, tiveram uma resposta global ao tratamento intensivo um pouco maior do que os pacientes na coorte de prevenção secundária, com uma duração média do diabetes de 8,5 anos.
O principal efeito adverso do tratamento intensivo foi um aumento de três vezes no risco de episódios hipoglicêmicos graves, definidos como episódios que necessitam da assistência de outras pessoas para tratamento e reversão.42,44 Pelo menos um evento destes por ano foi experimentado por 25% dos pacientes em tratamento intensivo, e 50% experimentaram mais de um destes episódios até o final do estudo; 14% experimentaram 10 ou mais episódios. Cerca de 25% de todos os episódios de hipoglicemia severa resultaram em coma ou convulsões. A taxa global de hipoglicemia severa foi de 62 eventos por 100 pacientes-ano para aqueles pacientes em tratamento intensivo, em comparação com 19 eventos por 100 pacientes-ano para os pacientes em tratamento convencional. Entretanto, análises adicionais dos dados não mostraram associação entre episódios múltiplos de hipoglicemia severa e déficits cognitivos nem efeitos do tratamento intensivo no desempenho neuropsicológico.45
Além dos episódios de hipoglicemia, o tratamento intensivo causou maior ganho de peso; um terço dos pacientes tinha mais de 120% do peso corpóreo ideal (índice de massa corpórea [IMC] aproximado de 27) no final do estudo.42 O tratamento intensivo também era mais caro do que o tratamento convencional,46 porém, o custo era parcialmente equilibrado por uma diminuição projetada nos custos associados com uma menor taxa de complicações.47 O custo estimado por ano de qualidade de vida ganha foi de US$ 28.661, o que representa um bom valor.
O ensaio clínico DCCT forneceu provas (grau A de evidências) de que as complicações microvasculares e neuropáticas podiam ser evitadas, ou pelo menos retardadas, de modo significativo com a manutenção de níveis de glicemia o mais perto do normal que as técnicas de tratamento permitissem, mas com segurança. O DCCT não conseguiu provar que a hiperglicemia causava complicações microvasculares, mas forneceu forte apoio para a hipótese. Foi demonstrado que o risco de retinopatia se relacionava com o nível médio de HbA1c de modo exponencial semelhante em cada grupo de tratamento do DCCT.48 O risco de retinopatia diminuiu 44% para cada diminuição proporcional de 10% na HbA1c (p. ex., uma diminuição na HbA1c de 9% para 8,1%). As relações entre níveis de HbA1c e riscos de desenvolvimento de microalbuminúria ou neuropatia foram semelhantes. Além disso, as análises do DCCT não indicaram qualquer limiar glicêmico abaixo do qual não haveria risco adicional de complicações microvasculares.49 Essa observação significa que a normoglicemia deve ser o objetivo ideal no tratamento do Diabetes melito tipo 1. Além disso, os benefícios do tratamento intensivo prévio (ou os efeitos adversos do tratamento convencional prévio) foram ainda demonstrados anos após o final do DCCT. O acompanhamento da coorte do DCCT, relatado no estudo Epidemiology of Diabetes Interventions and Complications (EDIC), mostrou que os níveis de HbA1c nos grupos de terapia intensiva e terapia convencional convergiam dentro de 2 anos após o final do DCCT. Contudo, em comparação com os participantes que receberam terapia convencional, aqueles que receberam terapia intensiva com insulina durante o DCCT tiveram menos progressão de retinopatia, nefropatia e neuropatia por 11 anos a partir de então.50-53 Além disso, para pacientes que receberam tratamento intensivo, o risco de eventos cardiovasculares foi reduzido ao longo da média de 17 anos de acompanhamento nos estudos DCCT e no EDIC,54 bem como o risco da progressão da aterosclerose (medida como o aumento na espessura íntima-média da artéria carótida55 ou a prevalência de calcificação coronariana).56
Assim, períodos sustentados de exposição glicêmica têm consequências no longo prazo. Um nível de hiperglicemia que seria atualmente considerado inaceitável continua a ter efeitos adversos mesmo após alguma melhora no controle metabólico, e uma redução marcada e prolongada na hiperglicemia com o tratamento intensivo no início do curso do diabetes tipo 1 continua a ter efeitos benéficos mesmo após alguma piora no controle metabólico. Deve-se notar ainda que o tratamento intensivo durante o DCCT não causou nenhuma piora na função cognitiva a longo prazo, conforme avaliado 10 anos após o final do DCCT.57
Os padrões de tratamento atuais da ADA refletem os resultados do DCCT [Tabela 2].58 Estes padrões incluem os seguintes objetivos:
manutenção de níveis pré-prandiais de glicemia capilar entre 90 e 130 mg/dL (5 a 7,2 mmol/L) e níveis pós-prandiais (1 a 2 horas após o início da refeição) de pico de glicemia capilar menores que de 180 mg/dL (< 10 mmol/L);
manutenção de um nível de HbA1c de menos de 7% (relativo a uma faixa de não diabéticos no DCCT de cerca de 4 a 6%).
De modo realista, as ferramentas terapêuticas atuais tornam difícil que se alcancem esses objetivos estritos em muitos pacientes com Diabetes melito tipo 1, em especial naqueles sem nenhuma secreção de insulina endógena. A relação exponencial entre o risco de complicações microvasculares e a HbA1c prediz que apenas níveis normais de HbA1c evitariam completamente essas complicações. No entanto, a manutenção de um nível de HbA1c abaixo de 7% removerá muito do risco absoluto para a maioria dos pacientes. Os esforços para se alcançar valores de HbA1c menores de 7% devem ser iniciados assim que seja possível fazê-lo de forma segura e devem ser continuados enquanto se puder minimizar a hipoglicemia.
Tabela 2. Padrões* da American Diabetes Association para o controle glicêmico em diabetes melito55
|
Medida |
Normal |
Objetivo |
|
Glicemia capilar pré-prandial |
< 100 mg/dL (< 5,5 mmol/L) |
90 a 130 mg/dL (5 a 7,2 mmol/L) |
|
Pico de glicemia capilar pós-prandial† |
< 140 mg/dL (< 7,7 mmol/L) |
< 180 mg/dL (< 10 mmol/L) |
|
Hemoglobina A1c (%) |
< 6 |
< 7 |
*Os valores mostrados nessa tabela são necessariamente generalizados para toda a população de pacientes com diabetes. Os objetivos devem ser individualizados: pacientes com comorbidades, muito jovens, adultos mais velhos e pacientes com condições ou circunstâncias incomuns podem precisar de objetivos de tratamento diferentes. Esses valores se referem a adultos não gestantes. Objetivos glicêmicos menos intensivos podem estar indicados em pacientes com hipoglicemias severas ou frequentes; objetivos glicêmicos mais estritos (ou seja, redução dos níveis de hemoglobina A1c para o normal [< 6%]) podem reduzir ainda mais as complicações, mas ao custo de risco aumentado de hipoglicemia. A hemoglobina A1c é o alvo primário para o controle glicêmico; a glicose pós-prandial pode ser o objetivo se o alvo de hemoglobina A1c não for alcançado, apesar de se alcançar o objetivo de glicemia pré-prandial. Ações adicionais sugeridas dependem de circunstâncias individuais dos pacientes. Tais ações podem incluir melhorias na educação para o autocuidado no diabetes, manejo combinado com uma equipe de tratamento, encaminhamento para um endocrinologista, alteração na terapia farmacológica, início ou aumento dos testes de automonitoramento da glicemia ou contatos mais frequentes com o paciente. A hemoglobina A1c tem como referência uma faixa não diabética de 4 a 6% (média de 5%; desvio padrão de 0,5%).
†As medidas de glicemia pós-prandial devem ser feitas 1 a 2 horas após o início da refeição.
Os medidores de glicose atuais são pequenos, usam pouco sangue, podem permitir o uso do antebraço além das pontas dos dedos como locais de punção, são menos vulneráveis à falta de acurácia por erros do paciente, e têm programas de memória que permitem que o paciente ou o cuidador avaliem o padrão do controle glicêmico nos 2 meses prévios, eliminando em grande parte o problema de transcrições de resultados incorretos ou fabricados pelo paciente.
Os sistemas de monitoramento contínuo de glicose (Continuous glucose monitoring systems - CGMS), que usam sensores subcutâneos para registrar os níveis de glicose médios a cada 5 minutos por até 72 horas, foram aprovados pelo Food and Drug Administration. Embora o perfil registrado possa fornecer apenas uma pequena janela dentro de uma vida de flutuações da glicemia, tal perfil pode guiar ajustes periódicos do regime pela identificação de instabilidade glicêmica (p. ex., hipoglicemia noturna ou hiperglicemia ou hiperglicemia pós-prandial excessiva) que podem escapar da detecção pela HbA1c ou por testes intermitentes com medidores de glicose. O teste da glicemia com CGMS em casa por período de 3 a 7 dias está entrando na prática clínica, permitindo que pacientes e médicos ajustem o tratamento com insulina de maneira mais precisa.59,60
O teste da glicemia antes de cada refeição ou lanche maior é essencial se o paciente quer ajustar cada dose da insulina de ação rápida ao nível da glicemia antes da refeição e à quantidade de carboidratos a ser ingerida. Os níveis glicêmicos também devem ser verificados de maneira periódica após refeições para assegurar que não esteja ocorrendo hiperglicemia pós-prandial indevida. Os pacientes também devem verificar os níveis glicêmicos antes ou depois de exercícios intensos para evitar ou abortar a hipoglicemia. Como a hipoglicemia severa pode ter efeitos adversos no julgamento e no tempo de reação de motoristas, os quais podem contribuir para acidentes, é muito importante verificar os níveis glicêmicos antes de dirigir.61
Leituras ocasionais da glicemia às 3:00 da manhã são úteis para monitorar hipoglicemias noturnas frequentes que passariam despercebidas. Mais importante ainda é o fato de que em doenças intercorrentes, em especial aquelas acompanhadas por náuseas, vômitos e redução na ingesta de líquidos ou calorias, os pacientes devem fazer testes sanguíneos com frequência para guiar a terapia com insulina. Além disso, o risco de cetoacidose sob estas circunstâncias demanda a testagem de urina ou sangue para cetoácidos. A presença de níveis significativos de cetoácidos é um sinal para contatar imediatamente o médico e estabelecer contatos frequentes para instruções com respeito a doses de insulina e ingesta de carboidratos.
A medida dos níveis de 1,5-anidroglucitol (1,5-AG) no soro pode ser usada para monitorar o controle glicêmico pós-prandial em pacientes com diabetes moderadamente bem controlado. A 1,5-AG é a forma 1-desóxi da glicose. Em pessoas normais, o equilíbrio entre uma pequena ingesta oral e a excreção urinária de 1,5-AG, bem como um grande conteúdo total de 1,5-AG, resulta em um nível sanguíneo constante. Quando os níveis glicêmicos excedem o limiar renal de 150 mg/dL, contudo, a glicose inibe de forma competitiva a reabsorção de 1,5-AG pelos túbulos renais. Como resultado, a excreção urinária de 1,5-AG aumenta e o nível sérico cai. A medida dos níveis de 1,5-AG tem sido usada clinicamente no Japão por mais de uma década; nos Estados Unidos, o FDA aprovou o uso de um teste sérico da 1,5-AG (GlycoMark) para medir os níveis glicêmicos pós-prandiais em 1 a 2 semanas.62
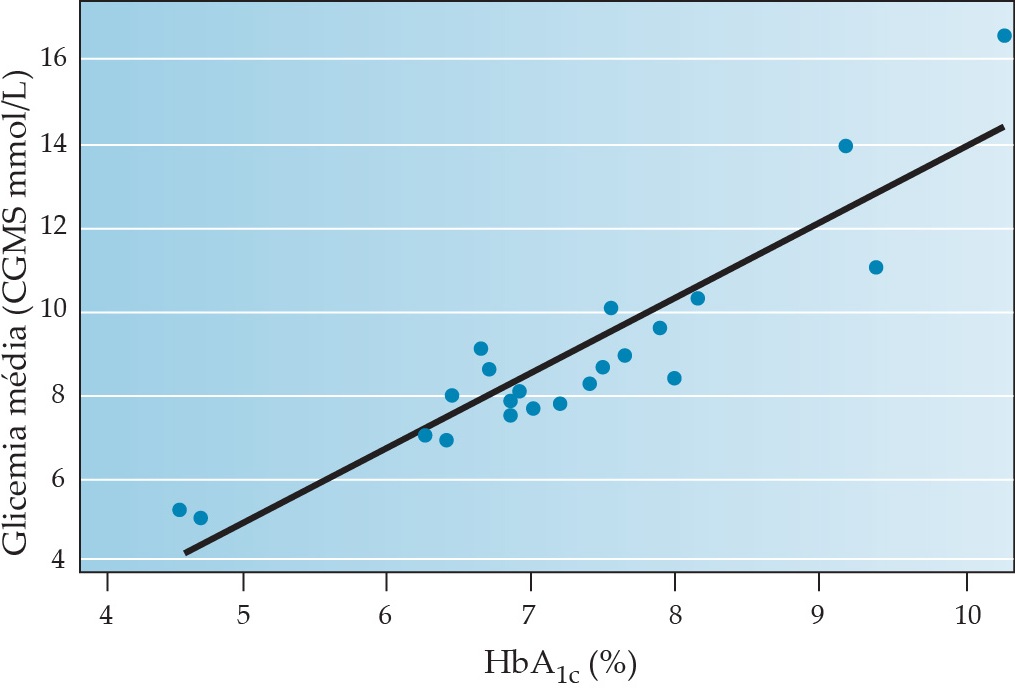
Para monitorar o controle glicêmico global e o risco de complicações tardias, a HbA1c é medida no consultório médico. Este produto da glicosilação não enzimática da hemoglobina fornece um excelente índice dos níveis glicêmicos médios 2 a 3 meses antes [Figura 8].63,64 O nível glicêmico médio medido por CGMS e HbA1c estão altamente correlacionados (r = 0,89-0,92).65 Em pelo menos um estudo, os pacientes cujo nível de HbA1c era medido de forma periódica tinham um melhor estado de saúde, níveis glicêmicos menores e menos hospitalizações do que um grupo selecionado de maneira aleatória com pacientes cujo nível de HbA1c permanecia desconhecido para o paciente e o médico.66
As recomendações atuais da ADA sugerem que se meça a HbA1c pelo menos duas vezes ao ano em pacientes que alcançam os objetivos do tratamento e têm controle glicêmico estável, e que se meça a HbA1c quatro vezes ao ano em pacientes que não alcançam os objetivos glicêmicos ou cuja terapia mudou; durante a gestação, os níveis deve ser medidos mensalmente.58 Como os métodos e resultados variam entre os laboratórios, foi estabelecido um programa nacional de padronização da hemoglobina glicada e a HbA1c deve ser medida em laboratórios certificados para o fornecimento de resultados equivalentes ao DCCT.64 O uso de testes de HbA1c de resultado rápido melhora a eficiência com que os cuidadores podem modificar o regime de tratamento dos pacientes nas consultas e pode melhorar os resultados do tratamento.67 A análise de outros produtos da glicosilação não enzimática, como a frutosamina e a albumina glicosilada, as quais refletem períodos mais curtos de glicemia crônica, são menos úteis no manejo de rotina do diabetes.64
Figura 8. Correlação entre a glicemia média calculada a partir de 3 meses de níveis intersticiais de glicose medidos com CGMS e o valor de HbA1c determinado no final dos 3 meses.65
A correção da deficiência de insulina é o componente mais importante no manejo do Diabetes melito tipo 1. Antes da disponibilidade de insulina, os pacientes com Diabetes melito tipo 1 e deficiência completa de insulina seguiam, de maneira inevitável, um curso previsível de piora clínica e morriam em coma diabético ou essencialmente de fome e inanição. A insulina extraída de pâncreas de bois e porcos e purificada até níveis cada vez maiores era o elemento fundamental da terapia até que a tecnologia de DNA recombinante tornasse possível a produção de insulina humana autêntica em grandes quantidades. Embora as insulinas animais sejam, sob o ponto de vista terapêutico, bioequivalentes à insulina humana, elas desapareceram do mercado à medida que os fabricantes passaram a produzir apenas insulina humana. Em raros casos de alergia local à insulina humana, pode-se substituir por insulina de ação rápida (p. ex., insulina lispro; ver adiante). Em situações de emergência, os pacientes com alergia sistêmica à insulina humana podem ser dessensibilizados pela administração de quantidades extremamente pequenas e por aumento gradual da dose ao longo de 6 a 24 horas até que o paciente seja tolerante e responsivo à insulina humana.
O princípio básico da reposição de insulina é fornecer um suprimento lento, contínuo e de longa duração que imite a secreção basal interprandial e noturna das células beta normais.68-70 Além disso, uma forma de ação rápida e relativamente curta de insulina administrada antes das refeições imita a rápida secreção normal de insulina estimulada pelas refeições [Figura 2]. Várias preparações de insulina para administração subcutânea estão atualmente disponíveis [Tabela 3]. É importante reconhecer que existe variabilidade considerável nas características farmacocinéticas dessas diferentes formas de insulina entre os pacientes e no mesmo paciente de um dia para o outro. As taxas de absorção de insulina a partir da pele variam conforme o local da injeção, a profundidade e a angulação da injeção, a temperatura ambiente e a realização de exercícios com o membro que recebeu a injeção. A injeção no tecido subcutâneo do abdome produz os resultados menos variáveis. A ação terapêutica esperada também pode ser afetada por flutuações na sensibilidade do paciente à insulina em momentos diferentes. Apesar da variabilidade dos resultados, podem ser esperados certos padrões médios com os regimes de múltiplas injeções diárias em uso comum [Figura 9]. CSII com o uso de uma bomba externa fornece uma liberação basal suave e aumentos agudos mais previsíveis na insulina plasmática para as refeições. Apenas insulina de ação rápida (isto é, insulina lispro, aspart ou glulisina) ou insulina regular devem ser usadas em tais bombas, o que é uma razão para a grande consistência de seus efeitos.
Tabela 3. Preparações de insulina.
|
Tipo de insulina |
Início |
Duração (horas) |
Tempo até o pico (horas) |
|
Ação rápida |
|
|
|
|
Insulina regular |
30 a 60 min |
6 a 8 |
2 a 4 |
|
Insulina inalada |
15 a 30 min |
6 a 8 |
2 a 4 |
|
Ação muito rápida |
|
|
|
|
Lispro |
5 a 15 min |
4 a 6 |
1 a 2 |
|
Aspart |
5 a 15 min |
4 a 6 |
1 a 2 |
|
Glulisina |
5 a 15 min |
4 a 6 |
a 2 |
|
Ação intermediária |
|
|
|
|
Neutral protamina Hagedorn (NPH) |
1 a 2 horas |
10 a 14 |
4 a 8 |
|
Ação longa |
|
|
|
|
Detemir |
2 a 3 horas |
9 a 24 |
Sem pico |
|
Glargina |
1,5 a 3 horas |
20 a 24 |
Sem pico |
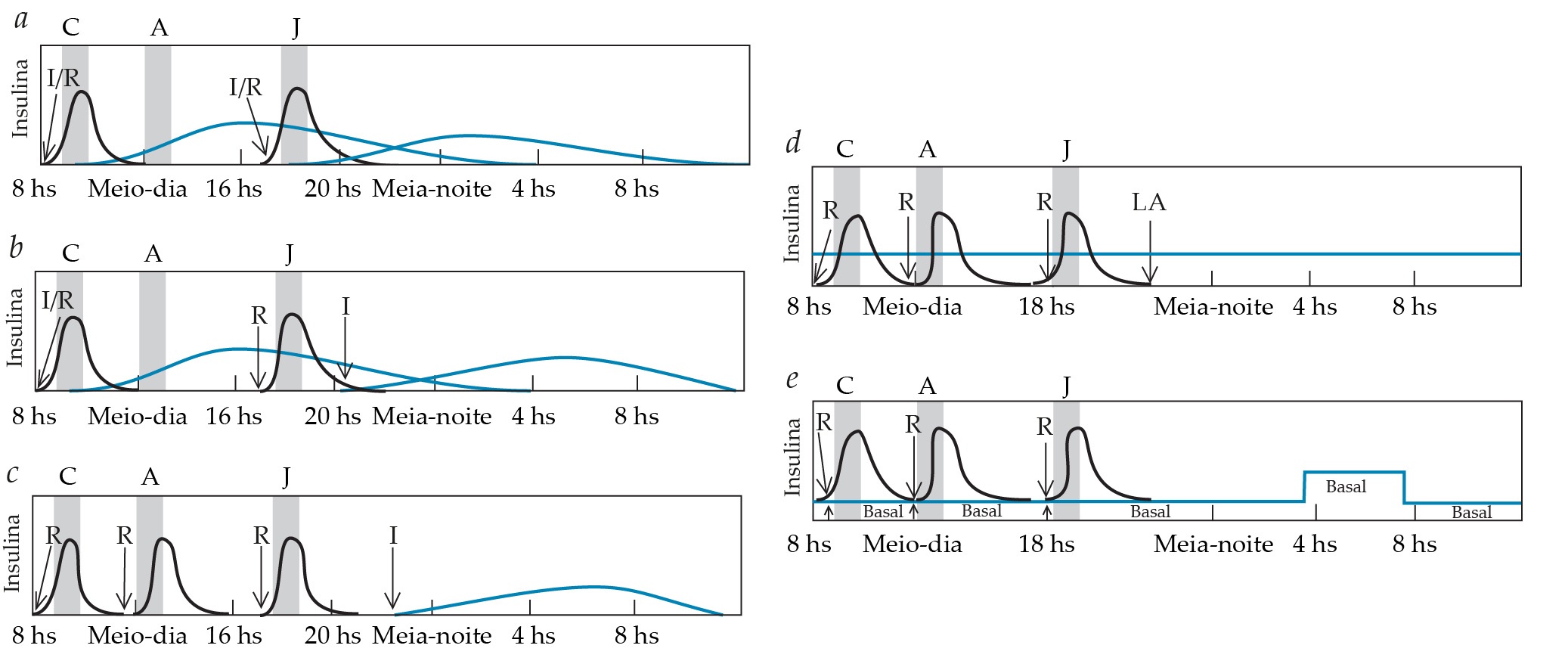
Figura 9. Combinações diferentes de várias preparações de insulina podem ser empregadas para estabelecer o controle glicêmico em pacientes com Diabetes melito tipo 1 (e naqueles pacientes com diabetes melito tipo 2 que alcançam um grau equivalente de deficiência de insulina). As setas indicam o momento da injeção. As curvas pretas representam insulina de ação rápida ou regular (R). As curvas azuis representam insulina NPH de ação intermediária (I) ou insulina de longa ação (LA) (p. ex., detemir ou glargina). (a) Uma injeção mista de insulina I e R é administrada antes do café da manhã e do jantar nesse regime não intensivo. Além do risco de hipoglicemia antes do almoço e no final da tarde, a administração de insulina I antes do jantar predispõe os pacientes à hipoglicemia entre 2 e 4 horas da manhã. (b) Neste regime mais seguro, o paciente recebe três injeções: uma injeção mista antes do café da manhã, insulina R antes do jantar e insulina I antes de deitar. O risco de hipoglicemia noturna é menor com esse regime e a glicemia de jejum tem um controle melhor. (c) Este regime intensivo combina três injeções pré-prandiais de insulina R com uma injeção de insulina I antes de deitar. As doses pré-prandiais de insulina R são ajustadas conforme a glicemia e o tamanho da refeição. (d) Este regime intensivo utiliza insulina de longa ação ao deitar para repor a secreção basal de insulina. Em alguns pacientes, injeções de insulina de longa ação devem ser administradas para melhorar os níveis glicêmicos antes do almoço e do jantar. As doses pré-prandiais de insulina R são ajustadas conforme os níveis glicêmicos e o conteúdo previsto de carboidratos na refeição. (e) Este regime intensivo fornece apenas insulina R como insulina de ação rápida ou regular. Uma infusão subcutânea contínua de insulina R mantida por uma bomba repõe a secreção basal de insulina. As taxas basais podem ser alteradas durante diferentes momentos do dia ou conforme as atividades. Por exemplo, a taxa basal pode ser diminuída ou mesmo suspensa durante períodos de exercício aeróbico intenso. A taxa basal noturna pode ser aumentada em 1,5 a 2 vezes entre 3 ou 4 horas da manhã e o café para acomodar o aumento nas necessidades de insulina do início da manhã conhecido como fenômeno do alvorecer (dawn). As doses em bolo pré-prandiais são individualmente ajustadas e rapidamente bombeadas, conforme os níveis glicêmicos e o conteúdo de carboidratos previsto na refeição. (C = café da manhã, A = almoço, J = jantar).
Insulina de ação rápida. Vários análogos da insulina foram criados para gerar uma farmacocinética que se aproximasse melhor da secreção e das necessidades da insulina fisiológica.71 Uma das características da insulina humana natural (ou sintética) é que seis moléculas se associam com uma molécula de zinco para formar hexâmeros. Os hexâmeros de insulina devem se dissociar em monômeros antes que possam ser absorvidos dos locais de injeção subcutânea. Essa necessidade é a principal razão pela qual a insulina cristalina com zinco (insulina regular) tem um pico de ação duas a quatro horas após a injeção e deve ser administrada 30 a 60 minutos antes da refeição para ter qualquer chance de limitar a hiperglicemia pós-prandial.
As companhias farmacêuticas desenvolveram uma variedade de análogos da insulina para evitar a formação de hexâmeros e criar um monômero que fosse rapidamente absorvido a partir do local de injeção. Nesses análogos, diferentes aminoácidos substituem aminoácidos na cadeia B da molécula de insulina. Com a insulina lispro, o primeiro análogo da insulina de ação rápida a ser desenvolvido, lisina e prolina são trocados nas posições 28 e 29 da cadeia B [ver Figura 3]. Com a insulina aspart, o ácido aspártico substitui a prolina na posição 28.72 Na insulina glulisina, a lisina toma o lugar da asparagina na posição 3 e o ácido glutâmico substitui a lisina na posição 29.73 O início de ação desses análogos da insulina começa dentro de 15 minutos, o pico do efeito ocorre em uma a duas horas e a duração da ação é de apenas quatro a seis horas. Dessa forma, a insulina de ação rápida injetada logo antes de uma refeição fornece um perfil de insulina plasmática pós-prandial semelhante àquele da secreção de insulina humana normal [ver Figura 2]. Os principais benefícios do uso de insulinas de ação rápida são a redução de picos glicêmicos pós-prandiais e alguma diminuição na hipoglicemia que pode resultar da elevação tardia da ação da insulina regular.74,75 No entanto, a perda daquela ação tardia pode causar hiperglicemia recorrente antes da próxima refeição. Assim, os pacientes que mudam da insulina regular para uma insulina de ação rápida podem não ter redução na HbA1c a menos que aumentem a dose da insulina basal (neutral protamine Hagedorn [NPH], insulina glargina ou insulina detemir ou a velocidade de infusão basal na CSII).76 Pode ser útil combinar uma insulina de ação rápida com a insulina regular em uma única injeção para otimizar o controle pós-prandial.
Insulina inalada. A insulina inalada, produzida por uma empresa farmacêutica, foi aprovada pelo FDA em janeiro de 2006. As propriedades farmacocinéticas da insulina inalada lembram aquelas das insulinas de ação rápida, de modo que ela é adequada para uso pré-prandial, mas deve ser administrada em conjunto com uma insulina basal injetada. As doses necessárias são muito maiores em relação à insulina injetada e o custo é mais elevado. O uso da insulina inalada tem sido associado com melhora na satisfação do paciente e na qualidade de vida, presumivelmente por redução do número de injeções,77 mas, em ensaios clínicos, os níveis de HbA1c alcançados com a insulina inalada não foram menores do que aqueles alcançados com insulina injetada.
Em ensaios clínicos, ocorreram diminuições na função pulmonar nas primeiras semanas do tratamento com insulina inalada; por essa razão, o uso da insulina inalada não era recomendado para pacientes com doença pulmonar subjacente ou que são tabagistas, e todos os pacientes candidatos ao tratamento com insulina inalada devem ser submetidos à espirometria e, talvez, à medida da capacidade de difusão de monóxido de carbono.78 As diminuições na função pulmonar com a insulina inalada são pequenas e não progressivas durante até dois anos de tratamento.79 Contudo, os testes de função pulmonar devem ser repetidos após os primeiros seis meses de tratamento com insulina inalada e anualmente depois disso, mesmo que o paciente não tenha sintomas pulmonares.
Por causa do decepcionante nível de adoção por médicos e pacientes da única formulação de insulina inalada e seu dispositivo de administração aprovados pelo FDA, o fabricante os retirou do mercado em 2007. Entretanto, pelo menos outras duas formulações de insulina inalada e seus sistemas de administração estão em desenvolvimento.
Insulina de longa ação. Dois análogos da insulina de longa ação, glargina e detemir, foram sintetizados para uso como insulina basal.80-82 Esses análogos não têm um pico de ação discernível e têm uma duração de ação maior do que a insulina ultralenta, a qual foi retirada do mercado em 2005. A glargina tem duas argininas adicionais na carboxila terminal da cadeia B da insulina, B31 e B32, e tem uma glicina no lugar da arginina na posição A21; a detemir foi criada com a omissão da treonina na posição B30 e a adição de uma cadeia de ácido graxo C14 na posição B29 [ver Figura 3]. A detemir tem uma duração de ação longa porque o componente de ácido graxo se liga de maneira reversível à albumina plasmática, o que diminui a sua velocidade de saída do plasma e diminui o acesso às células-alvo. A detemir tem uma menor afinidade pelos receptores de insulina em relação à insulina humana, a qual necessita do uso de doses maiores. A redução do peso corpóreo é uma vantagem adicional da detemir.
A glargina e a detemir costumam ser administradas como uma dose única ao deitar para fornecer insulina basal por 24 horas com menos hipoglicemia noturna.73,74 Algumas vezes, são necessárias injeções pela manhã e à noite para o controle glicêmico ideal.
Os regimes de tratamento intensivo são a forma preferida de tratamento para pacientes com diabetes tipo 1, incluindo crianças, embora os objetivos glicêmicos devam ser mais altos em crianças para evitar hipoglicemia severa. Combinações diferentes de preparações de insulina podem ser usadas para se aproximar (mas nunca reproduzir de forma confiável) os perfis normais de insulina plasmática [Figura 9]. O Diabetes melito tipo 1 quase nunca pode ser controlado de forma satisfatória com menos de duas injeções por dia de insulina de ação intermediária ou uma a duas injeções ao dia de insulina de longa ação combinada com insulina de ação rápida antes das refeições. Um controle metabólico satisfatório só pode ser obtido por um tempo com uma única injeção diária de insulina em pacientes com uma remissão tipo “lua de mel” ou em pacientes com Diabetes melito tipo 1 de início tardio. Tal sucesso só é possível pela presença de alguma secreção normalmente regulada de insulina endógena.
A dose diária total média de insulina é de 0,6 a 0,7 unidades por quilo de peso.43 Essa média aumenta para 1 U/kg durante a adolescência, quando aumenta temporariamente a resistência à insulina. Como regra geral, a injeção de insulina basal e a injeção de insulina nas refeições constituem, cada uma, cerca de 50% da dose diária total média. A dose da insulina de ação rápida ou regular antes de cada refeição é escolhida pelo paciente com base no nível glicêmico, na quantidade estimada de carboidratos a ser ingerida ou ambos. Um regime típico necessitaria de 1 a 2 unidades extra de insulina para cada 50 mg/dL de aumento na glicemia acima do alvo pré-prandial de 80 a 120 mg/dL, ou 1 U para cada 15 g de carboidratos extra a ser ingeridos acima da quantidade habitual de carboidratos prescrita no plano nutricional. Pacientes muito sofisticados podem combinar ambas as diretrizes.
Misturas de insulinas com doses fixas não são muito adequadas sob o ponto de vista patofisiológico para pacientes com Diabetes melito tipo 1. Porém, para pacientes que só podem ou vão implementar tal tratamento convencional, um regime típico seria uma dose total diária de 0,6 a 0,7 U/kg. Entre dois terços e três quartos da dose devem ser administrados antes do café da manhã e o restante antes do jantar, a razão entre insulina de ação intermediária e insulina de ação rápida estaria entre 2:1 e 4:1 antes do café da manhã e 1:1 antes do jantar. Como a administração de insulina NPH ou de longa ação antes do jantar aumenta o risco de hipoglicemia entre 2 e 4 horas da manhã, os pacientes em tratamento convencional devem ser solicitados a trocar para um regime de três injeções, usando insulina intermediária ou de longa ação ao deitar para evitar hipoglicemia noturna e para um melhor controle do nível de glicemia antes do café da manhã. As insulinas glargina ou detemir também podem ser úteis para minimizar a hipoglicemia noturna.81,82
As necessidades de insulina são aumentadas por uma maior ingesta calórica, em especial de carboidratos, por ganho de peso de massa magra ou gorda, pelo início da puberdade, por infecções e outros estresses clínicos ou cirúrgicos, pela administração de glicocorticoides e, algumas vezes, pelas alterações fisiológicas que precedem o início da menstruação. Durante doenças agudas, os pacientes necessitarão de doses adicionais de insulina de ação rápida quando aumentar a hiperglicemia e, em especial, se ocorrer cetose. Os contatos telefônicos frequentes com os cuidadores permitem uma orientação profissional adequada para as doses adicionais de insulina, a ingestão de nutrientes para evitar hipoglicemia e a ingesta de líquidos para evitar desidratação. Os análogos da insulina de ação rápida são especialmente úteis nessas circunstâncias, pois o efeito de uma dose excessiva é de curta duração e é menos provável que ocorra hipoglicemia.
As mulheres com diabetes tipo 1 em idade reprodutiva devem ser instruídas a informar os seus médicos quando decidirem engravidar. A concepção com o diabetes controlado de forma inadequada aumenta muito o risco de anormalidades congênitas importantes no feto. Este risco pode ser reduzido até uma taxa semelhante à de não diabéticas quando o controle glicêmico é excelente.83,84 Assim, o nível de HbA1c da paciente deve ser mantido o mais próximo possível do normal antes da concepção com tratamento intensivo, devendo ser mantido assim ao longo da gestação. O valor médio para os testes glicêmicos pré-prandiais e pós-prandiais em casa deve ser de menos de 126 mg/dL.84
Ao longo da gestação, é necessária uma normoglicemia (relativa ao estado gestacional normal) para evitar morte intrauterina e morbidade e mortalidade perinatais. Os alvos glicêmicos pré-prandiais durante a gestação são de 60 a 90 mg/dL; os alvos pós-prandiais são de 120 a 140 mg/dL.85,86 As necessidades de insulina podem diminuir no primeiro trimestre, mas depois aumentam para doses acima do nível pré-gestacional no terceiro trimestre, quando aumenta de maneira marcada a resistência à insulina. A insulina não é necessária durante o trabalho de parto; na verdade, pode ser necessária uma infusão de glicose durante o trabalho de parto ativo para evitar a hipoglicemia materna.87 Da mesma forma, durante as primeiras 48 horas após o parto as necessidades de insulina podem ser muito menores do que aquelas do terceiro trimestre, mas elas logo retornam às quantidades habituais da paciente.
O CSII melhorou de forma considerável desde a sua introdução na década de 1970. As modernas bombas de infusão de insulina permitem que se programem múltiplas taxas basais, conseguindo-se uma flexibilidade durante o dia, bem como um ajuste automático das doses ao dormir. Com frequência, a taxa basal deve ser menor na primeira metade da noite para depois aumentar e acomodar o chamado fenômeno do alvorecer [Figura 9]. Este último é uma lenta elevação no nível glicêmico antes do paciente acordar; ele é demonstrável em pessoas normais, mas é exagerado em pessoas com Diabetes melito tipo 1 que não conseguem limitá-lo com o aumento da secreção endógena de insulina. Por outro lado, a interrupção da administração de insulina pela bomba por um período de apenas oito horas pode resultar em hiperglicemia extrema, cetoacidose diabética (CAD) e hipercalemia. A interrupção pode ser o resultado de deslocamento da agulha, o qual pode ser imperceptível para o paciente. Em consequência disso, recomenda-se o monitoramento da glicemia pelo menos quatro vezes ao dia em pacientes que utilizam a bomba de insulina. No DCCT, os pacientes que usavam uma bomba de insulina tinham uma taxa de eventos para CAD que era um pouco maior, mas essa diferença era significativa (1,8 por 100 pacientes-ano) em relação aos pacientes em regimes de múltiplas injeções diárias (0,8 por 100 pacientes-ano).43 Não houve diferença no risco de hipoglicemia severa entre pacientes tratados com bombas de insulina e pacientes tratados com múltiplas injeções diárias, embora episódios resultando em coma ou convulsões fossem mais comuns nos pacientes tratados com CSII.43 A taxa de infecção no local do cateter foi mantida muito baixa por meio de frequentes mudanças de cateter e do uso preemptivo de antibióticos ao surgirem os primeiros sinais de infecção. O uso da bomba cresceu de maneira exponencial nos últimos 10 anos; estima-se que a bomba seja atualmente usada por 200.000 pacientes ou mais. As bombas podem ser usadas de forma adequada em adolescentes,88 um grupo cujo diabetes é especialmente difícil de ser controlado por razões fisiológicas, comportamentais e sociais.
Bombas implantáveis, que liberam insulina na cavidade peritoneal e resultam em uma primeira passagem de insulina mais fisiológica através do fígado, têm fornecido níveis aceitáveis de HbA1c com uma frequência de hipoglicemia severa mais baixa.89 Há relatos de dificuldades com obstrução da liberação de insulina e de infecção. As bombas implantáveis ainda não estão aprovadas para uso comercial.
Os modelos atuais de bombas externas se aproximam, mas não alcançam, o ideal de liberação de insulina completamente em alça fechada, com os níveis glicêmicos monitorados de forma contínua e os resultados ajustando de maneira automática as taxas de infusão. Algumas bombas têm um sensor de glicose separado que pode ser usado por até 3 dias por vez e que se comunica sem fio com a bomba, fornecendo leituras contínuas e avisando sobre níveis altos ou baixos.
Os pacientes podem utilizar estes dados para fazer os ajustes necessários na infusão de insulina.
A insulina não é o único hormônio que regula os níveis glicêmicos. A amilina, que é produzida pelas células beta pancreáticas, contribui para a redução pós-prandial dos níveis de glicose; como a insulina, ela está ausente ou deficiente em pessoas com diabetes melito.
A pranlintide, um análogo sintético da amilina, foi aprovada pelo FDA em 2005 como tratamento adjunto para pacientes com diabetes melito que não conseguem alcançar o controle glicêmico desejado apesar da terapia ideal com insulina nas refeições. Quando acrescentada a um regime preexistente de insulina, a pranlintide reduziu os níveis de HbA1c em 0,5 a 0,7% em relação ao basal.90 A pranlintide é injetada por via subcutânea logo antes das principais refeições, em uma seringa diferente daquela usada para injeções de insulina. Ao iniciar a pranlintide, os pacientes devem inicialmente reduzir em 50% suas doses pré-prandiais de insulina de ação rápida ou regular. Para minimizar as náuseas e vômitos, a pranlintide é iniciada em uma dose baixa (15 µg) e aumentada gradualmente conforme a necessidade, em intervalos de pelo menos 3 dias, até uma dose de manutenção de 30 a 60 µg.91
Até 2004, tinham sido realizados mais de 17.000 transplantes de pâncreas nos Estados Unidos e a taxa chegou a 1.400 casos por ano.92 A grande maioria dos transplantes de pâncreas ainda é realizada como uma opção, em conjunto com um transplante de rim necessário. Transplantes solitários de pâncreas necessitando de imunossupressão e transplantes de pâncreas realizados algum tempo depois do transplante de rim em pacientes já imunossuprimidos completam o restante e estão crescendo lentamente em número. Ao longo do período de 2000 a 2004, as taxas de sobrevida do enxerto em um ano eram de cerca de 80% e as taxas de sobrevida dos pacientes eram de 95%.92 Quando o procedimento é bem-sucedido, os transplantes de pâncreas fornecem níveis de HbA1c como de pessoas não diabéticas, liberando os pacientes do rigor da dieta, dos testes de glicose e das injeções de insulina, e eles virtualmente eliminam os episódios de hipoglicemia.93 Ocorre algum grau de resistência à insulina, em especial quando é usada a prednisona para imunossupressão. Nas técnicas mais modernas, o ducto pancreático drena para dentro do trato intestinal e a drenagem venosa pancreática é dirigida à veia porta. A qualidade de vida costuma melhorar. Por outro lado, o paciente corre o risco de mortalidade e morbidade operatórias e deve permanecer em tratamento imunossupressor, com os riscos de infecção e de doença maligna. A permanência hospitalar, as taxas de readmissão, a morbidade e o número de episódios de rejeições agudas são maiores para pacientes submetidos a transplante de pâncreas em comparação com aqueles submetidos a transplante de rim.
Um estudo concluiu que, embora o transplante de pâncreas seja capaz de restaurar a secreção endógena de insulina por 10 anos ou mais em alguns pacientes, o controle glicêmico piorava, mesmo quando os valores de glicose permaneciam dentro da faixa normal.94
O transplante isolado de ilhotas, que ainda é objeto de pesquisa, pode ser feito sem necessidade de uma grande cirurgia. Além disso, a capacidade de modular imunologicamente ilhotas isoladas em laboratório (mascarando ou removendo antígenos de superfícies celulares) pode, algum dia, permitir o transplante com pouca ou nenhuma imunossupressão. De outro modo, as ilhotas podem ser colocadas em tubos ocos semipermeáveis que permitem que a glicose entre e a insulina saia, mas que protegem as ilhotas de reações inflamatórias a um corpo estranho. O sucesso no transplante de ilhotas, quando a independência do tratamento com insulina dura pelo menos 1 ano, tem sido alcançado em 58% de 116 tentativas na América do Norte.95
Relatos iniciais de transplante de ilhotas usando um protocolo desenvolvido em Edmonton, Canadá, sugeriram uma alta taxa de sucesso.96 No protocolo de Edmonton, ilhotas purificadas de pâncreas de até cinco doadores cadavéricos são infundidas na veia porta do receptor. O protocolo não utiliza glicocorticoides para a imunossupressão; em vez disso, a terapia de indução com interleucina 2 é seguida por imunossupressão com sirolimo ou dose baixa de tacrolimo.
No entanto, um estudo de acompanhamento de cinco anos em 65 pacientes submetidos a transplante de ilhotas com o protocolo Edmonton mostrou que, embora 80% tivessem mantido a secreção de peptídeo C, apenas cerca de 10% ainda estavam independentes da insulina.97 As complicações incluíram sangramento ou oclusão da veia porta e toxicidade por terapia imunossupressora, bem como necessidades aumentadas de múltiplas medicações anti-hipertensivas e de estatinas. Embora muitos pacientes não consigam manter a independência da insulina, mesmo um funcionamento parcial das ilhotas fornece benefícios pela melhora do controle glicêmico;98 o benefício mais importante é a virtual cessação completa de eventos severos de hipoglicemia. Assim, os pacientes que sofrem dessa complicação grave e, algumas vezes, fatal da terapia com insulina exógena, são os melhores candidatos para este procedimento de pesquisa.
Uma modificação no protocolo Edmonton usando ilhotas de um único doador cadavérico e indução de imunossupressão com globulina antitimócitos, daclizumabe e etanercepte tem sido estudada.99 Cinco de oito pacientes mantiveram a independência da insulina por mais de 1 ano. É claro que permanecem desafios a serem vencidos antes que o transplante de ilhotas possa liberar insulina de maneira fisiológica por períodos prolongados em pacientes com deficiência de insulina.100
O tratamento com insulina não produzirá resultados satisfatórios a menos que esteja relacionado com a ingesta de nutrientes. Para facilitar a combinação entre as doses de insulina e as refeições e para evitar a hipoglicemia, os pacientes com Diabetes melito tipo 1 devem comer refeições consistentes e regulares com cerca de 50% das calorias sendo carboidratos. Embora a dose de insulina pré-refeição seja determinada em grande parte pelo conteúdo absoluto de carboidratos da refeição, os pacientes também devem prestar atenção na ingesta total de energia a partir de proteínas e lipídeos. Entre 60 e 70% da ingesta de energia deve estar na forma de carboidratos e gordura monoinsaturada. Menos de 10% da ingesta de energia deve estar na forma de gorduras saturadas e a ingesta de colesterol deve ser de menos de 300 mg ao dia.101 Em pacientes com níveis de colesterol LDL de 100 mg/dL ou mais, pode ser benéfica a redução dos níveis de gordura saturada para menos de 7% e de colesterol para menos de 200 mg/dia. Têm sido utilizados vários métodos para ensinar os pacientes a avaliar as quantidades de alimentos e seu conteúdo de nutrientes e calorias. Esses métodos incluem listas de equivalências que colocam os alimentos em seis categorias; cada categoria tem aproximadamente a mesma quantidade de carboidratos, proteínas e calorias por porção. Essas categorias equivalentes são pão, carne, leite, frutas, gorduras e vegetais. Outra abordagem é focar apenas no conteúdo de carboidrato dos alimentos, pois os carboidratos causam a maior parte da hiperglicemia pós-prandial. Como diferentes carboidratos são digeridos e absorvidos em velocidades diferentes e, dessa forma, têm efeitos diferentes nos níveis plasmáticos de glicose, foram desenvolvidos índices glicêmicos para os alimentos comuns, levando em conta seus efeitos diferentes.102 Numerosos estudos desfizeram o mito de que a sucrose aumenta a glicemia mais do que quantidades equivalentes de outros carboidratos.101 Para uma instrução ideal e reforço da terapia nutricional deve haver um nutricionista na equipe de cuidados do paciente diabético.
Os exercícios são outro componente importante do cuidado com o diabetes, pois ajudam a manter o condicionamento cardiovascular, a sensibilidade à insulina e o bem-estar geral. Contudo, os pacientes devem aprender a forma como seus níveis glicêmicos respondem a diferentes formas de exercícios, e devem ser instruídos sobre como ajustar suas refeições, a dose e o momento da aplicação da insulina ou ambos, para evitar hipoglicemia durante, imediatamente depois ou mesmo seis a 12 horas após os exercícios, à medida que são repostos os depósitos de glicogênio nos músculos a partir da glicose plasmática.103
Em geral, os pacientes devem evitar atividades físicas se seus níveis glicêmicos em jejum estiverem em 250 mg/dL ou mais e tiverem cetose, e eles devem ter cuidado se os níveis glicêmicos estiverem acima de 300 mg/dL e não houver cetose.104 Eles devem ingerir carboidratos adicionais antes dos exercícios se os níveis glicêmicos estiverem abaixo de 100 mg/dL. Os pacientes devem monitorar a glicemia antes e depois de atividade física moderada a intensa, devendo ter algum alimento à base de carboidratos prontamente disponível durante e após a atividade.
Esportes de alto impacto estão contraindicados para pacientes com retinopatia avançada, os quais estão em risco para hemorragia vítrea, bem como para pacientes com neuropatia periférica ou doença vascular, que estão sob risco de sofrerem traumas nos pés.
A CAD é o resultado final da deficiência de insulina104 [ver Figura 6]; ela é agravada por elevações induzidas pelo estresse nos níveis de glucagon, cortisol, hormônio do crescimento, epinefrina e norepinefrina, os quais acrescentam um componente de resistência à insulina.105 A cada ano, a CAD ocorre em 2 a 5% dos pacientes com Diabetes melito tipo 1. Nos pacientes acompanhados de perto no DCCT, as taxas globais de eventos foram de 2 por 100 pacientes-ano no grupo de tratamento intensivo e de 1,8 por 100 pacientes-ano no grupo de tratamento convencional.44 A mortalidade relatada varia no mundo todo de 0 a 10%. A maioria dos casos ocorre em pacientes já com diagnóstico de Diabetes melito tipo 1, mas a CAD ainda pode ser a primeira manifestação do diabetes, em especial em crianças. O automonitoramento da glicemia e das cetonas na urina e o contato próximo com a equipe de cuidados em diabete devem facilitar o reconhecimento e o abortamento da CAD em evolução por meio de tratamento agressivo e precoce em casa com quantidades adicionais de insulina e líquidos. Cerca de metade dos casos de CAD são precipitados por infecção. Sepse, infarto do miocárdio e outras comorbidades importantes costumam ser mais frequentemente a causa da morte do que a causa do desequilíbrio metabólico. Em crianças, o edema cerebral é uma complicação rara e grave. Ele costuma aparecer seis a 12 horas após o início do tratamento quando é notada uma melhora bioquímica; ainda assim, ele costuma ser fatal.
Achados de apresentação. Os pacientes com CAD apresentam sinais e sintomas de desidratação secundária a diurese osmótica e vômitos e, algumas vezes, à diarreia causada por uma gastrenterite concomitante; de hiperventilação compensatória para eliminar o CO2; e de vários graus de alterações mentais e diminuição do nível de consciência. As convulsões não costumam ser resultado da CAD. Um quadro de coma completo quase com certeza indica um longo período de CAD antes de o paciente receber atenção médica. A CAD apresenta vários achados laboratoriais característicos [ver Tabela 4]. A acidose metabólica representa um ânion gap secundário a níveis elevados de acetoacetato e beta-hidroxibutirato, com pequenas contribuições de lactato e ácidos graxos livres. Embora os níveis séricos de potássio e fosfato costumem estar normais ou até elevados no início do quadro, este achado mascara a depleção corpórea total desses eletrólitos, bem como a depleção de magnésio. Se os níveis séricos estiverem baixos na admissão, a depleção é profunda. Os desvios em relação aos padrões comuns criam armadilhas diagnósticas. Como os testes atuais detectam apenas acetoacetato ou acetona, as cetonas podem estar ausentes no soro se o potencial redox do paciente for muito alto e o equilíbrio da cetoacidose estiver desviado em direção ao beta-hidroxibutirato (como pode ocorrer na intoxicação alcoólica). Testes sanguíneos rápidos para o beta-hidroxibutirato estão atualmente disponíveis em muitos laboratórios hospitalares para confirmação do diagnóstico de CAD. Os níveis séricos de bicarbonato podem estar normais se houver acidose respiratória concomitante. O pH sanguíneo arterial pode estar normal se também houver alcalose metabólica causada por ingestão de diuréticos ou vômitos perniciosos. Em algumas ocasiões, os níveis glicêmicos estão abaixo de 250 mg/dL em função de jejum,106 grande ingesta de álcool, inanição profunda ou gestação.
Tratamento. O tratamento da CAD exige a monitoração cuidadosa do paciente [ver Tabela 4].104,107,108 A reposição de volume é tão importante quanto a terapia com insulina.109 Deve-se iniciar com solução salina a 0,9% intravenosa mesmo antes de se estabelecer o diagnóstico. Após a administração inicial de um litro em 30 a 60 minutos, a terapia com fluidos deve continuar de forma agressiva até que o volume circulante esteja reposto, conforme indicado por um aumento na pressão arterial para níveis normais e uma redução na taquicardia compensatória. A reposição subsequente do volume total é feita de forma mais lenta a uma taxa de 150 a 500 mL/hora com solução salina a 0,45%, mudando-se para soluções contendo glicose a 5% quando a glicemia tiver diminuído para 250 mg/dL. Os déficits de fluidos típicos variam de 50 a 100 mEq/kg. A média do déficit de sódio é de 7 mEq/kg e, mais importante ainda, os déficits de potássio podem ser de até 7 mEq/kg. A neutralização dos fortes ácidos orgânicos acetoacetato e beta-hidroxibutirato em seus sais de sódio e potássio e a perda desses sais na urina causam depleção efetiva do bicarbonato corpóreo total e resultam em acidose metabólica hiperclorêmica durante o período de recuperação.
Tabela 4. Achados laboratoriais típicos e monitoração na cetoacidose diabética.
|
Teste |
Média |
Variação |
|
Glicose plasmática |
600 mg/dL (33 mmol/L) |
200 a 2.000 mg/dL (11 a 110 mmol/L) |
|
Cetonas plasmáticas (positivo) |
1:16 |
1:2-1:64 |
|
Beta-hidroxibutirato (mmol/L) |
– |
3 a 25 |
|
HCO3 plasmático (mEq/L) |
10 |
4 a 15 |
|
pH sanguíneo |
7,15 |
6,80 a 7,30 |
|
PCO2 (mm Hg) |
20 |
14 a 30 |
|
Ânion gap plasmático (Na+ - [Cl + HCO3]) (mEq/L) |
23 |
16 a 30 |
Realizar hemograma completo, medida de ureia, creatinina, exame comum de urina, culturas apropriadas e radiografia de tórax.
1. Peso na admissão e a cada 12 horas.
2. Registrar a ingesta e débitos cumulativos a cada 1 a 2 horas (cateter de Foley, se houver incontinência).
3. Verificar pressão arterial, pulso, respiração e estado mental a cada 1 a 2 horas e a temperatura a cada 8 horas.
4. Verificar a glicose no sangue (ponta de dedo) ou plasma (laboratório) a cada 1 a 2 horas.
5. Verificar o potássio sérico a cada 2 a 4 horas; verificar outros eletrólitos e níveis séricos de cetonas ou ß-hidroxibutirato a cada 4 horas.
6. Verificar pH e gasometria arterial na admissão (em crianças, pode ser usado o pH venoso, acrescentando 0,1 ao resultado). Se o pH for < 7,0 na admissão, verificar novamente conforme a necessidade até que esteja > 7,1.
7. Verificar níveis séricos de fosfato, magnésio e cálcio na admissão. Se estiverem baixos, repetir a cada 4 horas; caso contrário, a cada 8 a 12 horas.
8. Avaliar amostras de urina para cetonas e glicose.
9. Realizar um ECG na admissão; repetir se o acompanhamento do nível de potássio sérico for anormal ou não estiver disponível.
Nota: 1-9 devem ser realizados até que o paciente esteja estável, com níveis de glicose estáveis em 250 mg/dL e com a acidose em grande parte resolvida (HCO3 plasmático > 15 a 18, ânion gap plasmático < 16). É preferido o tratamento em unidade de terapia intensiva.
A reposição de potássio (10 a 40 mEq/hora) deve iniciar imediatamente após a administração de insulina e assim que se tiver descartado hipercalemia e oligúria ou anúria. Caso contrário, haverá hipocalemia grave à medida que a insulina estimular a entrada de potássio nas células [Figura 4]. Se o nível sérico de potássio estiver abaixo de 4 mEq/L na admissão, existe um déficit muito grande e a reposição deve ser feita a uma velocidade maior para manter níveis não menores do que 3,5 a 4 mEq/L. Deve-se esperar para iniciar a administração de insulina em tais situações até que o nível sérico de potássio alcance 4 mEq/L. A hipocalemia é a causa de morte mais trágica como resultado de um julgamento terapêutico errado.
A injeção subcutânea de um análogo da insulina de ação rápida é uma rota simples e efetiva para a administração de insulina no tratamento da CAD.110 Essa importante nova abordagem pode ser empregada em um ambiente médico geral. Neste regime, é administrada uma insulina de ação rápida (p. ex., insulina lispro) em uma dose de 0,15 U/kg a cada duas horas.
Uma abordagem mais antiga é a administração intravenosa de insulina regular ou de ação rápida, iniciando com um bolo de 10 U ou 0,1 U/kg, seguido pela mesma dose por hora em infusão preferivelmente em bomba de infusão e por meio de acesso venoso exclusivo. Essa abordagem exige a admissão em uma unidade de tratamento intensivo, onde há mais segurança para a monitoração cuidadosa e respostas rápidas a mudanças nas circunstâncias clínicas e resultados laboratoriais. Nos pacientes mais graves, deve-se preferir esse tipo de tratamento.
Não foi demonstrado que a adição rotineira de bicarbonato de sódio ou fosfato de potássio acelere a recuperação em casos ordinários de CAD.104 Possíveis indicações para a administração de bicarbonato de sódio (50 a 200 mEq) incluem pH arterial abaixo de 7,0, alterações eletrocardiográficas indicativas de hipercalemia, hipotensão que não responde à infusão rápida de soro fisiológico e insuficiência cardíaca esquerda. Se for administrada a terapia com bicarbonato, deve-se monitorar a cada uma hora o potássio sérico e o pH arterial e administrar potássio adicional para se evitar a hipocalemia. A hipofosfatemia grave (< 1,5 mg/dL) pode causar rabdomiólise, hemólise e deterioração do sistema nervoso central; essas condições exigem a administração intravenosa de fosfato de potássio, 60 mmol (aproximadamente 2 g de fósforo) em seis horas. Quando o ânion gap estiver reduzido para perto do normal, o nível de beta-hidroxibutirato reduzido para menos de 3 mmol/L e o nível de bicarbonato aumentado para 15 a 18 mEq/L, a taxa de infusão de insulina pode ser diminuída para 2 U/hora. Em geral, é melhor manter a infusão de insulina em uma taxa de 1 a 2 U/hora em conjunto com infusão de glicose a 5 ou 10%, com o objetivo de manter os níveis de glicose plasmática ao redor de 150 mg/dL até a manhã seguinte, quando se pode iniciar um regime de insulina mista subcutânea ou reiniciar a dieta.
Vômitos persistentes exigem a sondagem gástrica, e a via aérea de um paciente torporoso deve ser protegida para prevenção de aspiração. Qualquer suspeita de sepse demanda tratamento com antibióticos de amplo espectro, um agente antifúngico, ou ambos, quando apropriado.
A hipoglicemia é uma emergência mais comum do que a CAD e é potencialmente perigosa. A hipoglicemia clínica pode variar de sintomas desagradáveis acompanhando um nível glicêmico baixo (< 50 a 60 mg/dL) até confusão, convulsões ou coma. Qualquer episódio que exija a intervenção de outra pessoa para sua reversão é classificado como hipoglicemia severa. A hipoglicemia severa pode ter consequências desastrosas, em especial se o paciente estiver dirigindo qualquer tipo de veículo, trabalhando em lugares altos ou operando maquinário potencialmente perigoso.
As causas mais comuns de hipoglicemia são a falta de refeições e lanches,111 erros na dose de insulina, exercícios, álcool e fármacos como betabloqueadores. Durante o DCCT, 55% dos episódios de hipoglicemia ocorreram durante o sono.101 Tais episódios costumam não ser detectados.112
Glucagon e epinefrina são os principais hormônios contrarregulatórios secretados em resposta à hipoglicemia.113 Ambos restauram os níveis de glicose por aumentarem o débito hepático de glicose, sendo que a epinefrina ainda diminui a sensibilidade dos músculos à insulina. Além disso, a secreção de catecolaminas alerta o paciente para tratar o episódio, pois ela produz os sintomas simpático-adrenais observados adiante. O cortisol e o hormônio do crescimento também são secretados em resposta à hipoglicemia e desempenham um papel na manutenção dos níveis de glicose, mas não na rápida recuperação da hipoglicemia.
Achados de apresentação. Os sintomas mais comuns da hipoglicemia leve inicial são adrenérgicos e incluem palpitações (por taquicardia), tremores, ansiedade e sudorese.114 A sudorese necessita da ativação simpática dos nervos colinérgicos que inervam as glândulas sudoríparas. Também pode ocorrer visão borrada e tontura.
Fatores que afetam a gravidade dos episódios hipoglicêmicos. O desenvolvimento de insuficiência adrenal primária ou secundária, hipopituitarismo e hipotireoidismo podem aumentar o risco de hipoglicemia por aumentar a sensibilidade à insulina, diminuir o apetite ou ambos. Estresse, exercícios ou uso de álcool ou drogas ilícitas podem mascarar ou impedir o reconhecimento da hipoglicemia. Os pacientes que reconhecem a hipoglicemia incipiente, mas que de forma consciente não respondem rápido (p. ex., podem esperar uma refeição em um restaurante ou continuar a dirigir após o aparecimento dos sintomas) também têm risco aumentado de hipoglicemia grave. Além disso, alguns fatores de risco para a hipoglicemia têm múltiplos efeitos que podem precipitar, prolongar ou piorar a gravidade da hipoglicemia. O álcool, por exemplo, prejudica o julgamento e inibe a gliconeogênese e o débito hepático de glicose, retardando, assim, a recuperação. Quando a hipoglicemia não é tratada de forma adequada, ela costuma evoluir para hipoglicemia mais grave.
Por fim, como o glucagon e a epinefrina são os principais hormônios de defesa contra a hipoglicemia prolongada, a sua ausência promove episódios mais prolongados e graves por meio de dois mecanismos: (1) o débito hepático compensatório de glicose é diminuído quando não é estimulado por glucagon ou epinefrina e (2) os sintomas adrenérgicos familiares podem cessar na ausência de epinefrina, resultando em incapacidade de reconhecer o episódio. A resposta do glucagon à hipoglicemia costuma diminuir em pacientes após alguns anos de diabetes tipo 1. Na ausência do glucagon, a secreção de epinefrina ainda fornece defesa contrarregulatória adequada; porém, a resposta da epinefrina também pode ser perdida, algumas vezes em associação com outras neuropatias autonômicas e, por vezes, de maneira seletiva. Muitos pacientes perdem a capacidade de contrarregular de maneira efetiva a hipoglicemia durante os primeiros 10 anos de Diabetes melito tipo 1.
Dada a importância dos regimes intensivos para evitar complicações microvasculares pela hiperglicemia, é uma pena que se observe um limiar mais baixo de glicemia para a liberação de glucagon e epinefrina em resposta à hipoglicemia, sobretudo em pacientes sob terapia intensiva com insulina.115 O nível de glicose mais baixo necessário para estimular a contrarregulação diminui a margem de segurança do tratamento. Por exemplo, o primeiro sintoma de hipoglicemia pode ocorrer apenas com níveis de glicose tão baixos como 35 mg/dL (diferente de 55 a 60 mg/dL) e podem consistir de confusão ou perda da capacidade de julgamento, interferindo com o autotratamento. Algumas evidências sugerem que a falta de percepção da hipoglicemia é autogerada, pois cada episódio pode diminuir o limiar no qual inicia a contrarregulação autonômica em episódios subsequentes.116 O inverso disso é o fato de que um período livre de hipoglicemia, produzido por contato terapêutico diário com os cuidadores, pode restaurar a percepção da hipoglicemia, 116,117 embora não possa restaurar as respostas contrarregulatórias normais.116 O aumento da captação de glicose pelo cérebro na presença de hipoglicemia118,119 é uma explicação provável para a relativa infrequência de catástrofes hipoglicêmicas clínicas.
Tratamento. Os pacientes reconhecem rapidamente a maioria dos episódios de hipoglicemia e podem tratar-se de maneira efetiva com carboidratos orais capazes de serem absorvidos de forma imediata. Cerca de 15 g de carboidratos costumam ser suficientes para restaurar níveis glicêmicos normais. Essa quantidade é fornecida por aproximadamente 180 mL de suco de laranja, 120 mL de refrigerante tipo cola (normal, e não dietético), 3 a 4 colheres de chá de açúcar de mesa, cinco balas açucaradas grandes ou três tabletes de glicose (cada um contendo 5 g de glicose). O uso de carboidratos complexos e alimentos com elevado conteúdo de gordura, como o chocolate, pode retardar a digestão e a absorção da glicose; esses alimentos não são a primeira escolha para o tratamento da hipoglicemia. Se o paciente não puder engolir ou estiver incapacitado para o autotratamento, pode-se administrar um gel contendo glicose e carboidratos simples pela boca; tal gel é aplicado entre as gengivas e a bochecha, de onde percorre um caminho lento e geralmente seguro até o estômago. O glucagon (1 mg administrado por via subcutânea ou intramuscular) também irá aumentar os níveis glicêmicos de maneira suficiente dentro de 15 a 30 minutos, quando o paciente poderá, então, ingerir carboidratos orais. O glucagon é fornecido em kits de emergência e deve sempre estar disponível para pacientes com história de episódios hipoglicêmicos graves. O glucagon pode causar náuseas, vômitos e cefaleia, em especial no caso de crianças. Quando todo o restante falhar, o pessoal de atendimento de emergência deve administrar glicose intravenosa ou o paciente deve ser levado a uma emergência (o que for mais rápido). Quando o momento de um episódio sugere que tenha sido causado por insulina de ação longa ou intermediária ou por exercícios prévios, o nível glicêmico pode cair de novo para níveis de hipoglicemia, sendo necessário um novo tratamento. Assim, um paciente que tenha necessitado do auxílio de outros para a reversão da hipoglicemia deve ser mantido sob vigilância por algum tempo.
Os pacientes com hipoglicemia severa costumam responder de forma rápida ao tratamento, embora os pacientes em estado pós-ictal ou em coma prolongado possam necessitar de dias para recuperar o estado mental e a função cognitiva normais. Muitas vezes os pacientes têm amnésia com episódios prolongados, incluindo um período anterior ao início da hipoglicemia. Em casos raros, os déficits neurológicos podem ser permanentes. Em geral, porém, as consequências da hipoglicemia a longo prazo não foram detectadas em adultos.45,120,121 Em crianças com até cinco anos de idade, episódios repetidos de hipoglicemia podem estar associados com déficits cognitivos tardios na infância.122,123 Em vista das potenciais consequências dos episódios prolongados, a hipoglicemia deve ser sempre tratada de forma imediata.
Prevenção. Os pacientes devem ser instruídos a tratarem-se como se tivessem hipoglicemia sempre que suspeitarem disso, mesmo se não puderem fazer um teste confirmatório no momento. O limiar para os sintomas de hipoglicemia varia entre as pessoas e mesmo em ocasiões diferentes na mesma pessoa. Assim, sempre que possível, um teste glicêmico confirmatório deve ser realizado para ajudar a diferenciar sintomas inespecíficos de hipoglicemia verdadeira. Os pacientes com risco aumentado de hipoglicemia severa devem monitorar seus níveis glicêmicos com maior frequência.
As complicações do Diabetes melito tipo 1 são discutidas em detalhes em outro capítulo.
O autor não tem relações comerciais com fabricantes de produtos ou fornecedores de serviços discutidos nesse capítulo.
1. American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2007 Jan;30 Suppl 1:S42-7.
2. SEARCH for Diabetes in Youth Study Group, Liese AD, D'Agostino RB Jr, Hamman RF, Kilgo PD, Lawrence JM, et al. The burden of diabetes mellitus among US youth: prevalence estimates from the SEARCH for Diabetes in Youth Study. Pediatrics. 2006 Oct;118(4):1510-8.
3. Harris MI, Flegal KM, Cowie CC, Eberhardt MS, Goldstein DE, Little RR, et al. Prevalence of diabetes, impaired fasting glucose, and impaired glucose tolerance in U.S. adults. The Third National Health and Nutrition Examination Survey, 1988-1994. Diabetes Care. 1998 Apr;21(4):518-24.
4. DIAMOND Project Group. Incidence and trends of childhood Type 1 diabetes worldwide 1990-1999. Diabet Med. 2006 Aug;23(8):857-66.
5. Genuth S. Hormones of the pancreatic islets. Physiology. 4th ed. St. Louis: Mosby; 1998.
6. Haller MJ, Atkinson MA, Schatz D. Type 1 diabetes mellitus: etiology, presentation, and management. Pediatr Clin North Am. 2005 Dec;52(6):1553-78.
7. Eisenbarth GS. Type I diabetes mellitus. A chronic autoimmune disease. N Engl J Med. 1986 May 22;314(21):1360-8.
8. Foulis AK, McGill M, Farquharson MA. Insulitis in type 1 (insulin-dependent) diabetes mellitus in man--macrophages, lymphocytes, and interferon-gamma containing cells. J Pathol. 1991 Oct;165(2):97-103.
9. Rossini AA, Greiner DL, Friedman HP. Immunopathogenesis of diabetes mellitus. Diabetes Reviews. 1993;1:43.
10. Wasserfall CH, Atkinson MA. Autoantibody markers for the diagnosis and prediction of type 1 diabetes. Autoimmun Rev. 2006 Jul;5(6):424-8. Epub 2005 Dec 29.
11. Feutren G, Papoz L, Assan R, Vialettes B, Karsenty G, Vexiau P, et al. Cyclosporin increases the rate and length of remissions in insulin-dependent diabetes of recent onset. Results of a multicentre double-blind trial. Lancet. 1986 Jul 19;2(8499):119-24.
12. Bougnères PF, Landais P, Boisson C, Carel JC, Frament N, Boitard C, et al. Limited duration of remission of insulin dependency in children with recent overt type I diabetes treated with low-dose cyclosporin. Diabetes. 1990 Oct;39(10):1264-72.
13. Riley WJ, Maclaren NK, Krischer J, Spillar RP, Silverstein JH, Schatz DA, et al. A prospective study of the development of diabetes in relatives of patients with insulin-dependent diabetes. N Engl J Med. 1990 Oct 25;323(17):1167-72.
14. Achenbach P, Bonifacio E, Ziegler AG. Predicting type 1 diabetes. Curr Diab Rep. 2005 Apr;5(2):98-103.
15. Srikanta S, Ganda OP, Gleason RE, Jackson RA, Soeldner JS, Eisenbarth GS. Pre-type I diabetes. Linear loss of beta cell response to intravenous glucose. Diabetes. 1984 Aug;33(8):717-20.
16. Achenbach P, Warncke K, Reiter J, Williams AJ, Ziegler AG, Bingley PJ, et al. Type 1 diabetes risk assessment: improvement by follow-up measurements in young islet autoantibody-positive relatives. Diabetologia. 2006 Dec;49(12):2969-76. Epub 2006 Sep 26.
17. Peng H, Hagopian W. Environmental factors in the development of Type 1 diabetes. Rev Endocr Metab Disord. 2006 Sep;7(3):149-62.
18. Gillespie KM, Bain SC, Barnett AH, Bingley PJ, Christie MR, Gill GV, et al. The rising incidence of childhood type 1 diabetes and reduced contribution of high-risk HLA haplotypes. Lancet. 2004 Nov 6-12;364(9446):1699-700.
19. Jones DB, Armstrong NW. Coxsackie virus and diabetes revisited. Nat Med. 1995 Apr;1(4):284.
20. van der Werf N, Kroese FG, Rozing J, Hillebrands JL. Viral infections as potential triggers of type 1 diabetes. Diabetes Metab Res Rev. 2007 Mar;23(3):169-83.
21. The Environmental Determinants of Diabetes in the Young (TEDDY) Consortium. National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland, 2006. Disponível em: www.niddk.nih.gov/patient/TEDDY/TEDDY.htm
22. Agner T, Damm P, Binder C. Remission in IDDM: prospective study of basal C-peptide and insulin dose in 268 consecutive patients. Diabetes Care. 1987 Mar-Apr;10(2):164-9.
23. Shah SC, Malone JI, Simpson NE. A randomized trial of intensive insulin therapy in newly diagnosed insulin-dependent diabetes mellitus. N Engl J Med. 1989 Mar 2;320(9):550-4.
24. Effects of age, duration and treatment of insulin-dependent diabetes mellitus on residual beta-cell function: observations during eligibility testing for the Diabetes Control and Complications Trial (DCCT). The DCCT Research Group. J Clin Endocrinol Metab. 1987 Jul;65(1):30-6.
25. Gorsuch AN, Spencer KM, Lister J, McNally JM, Dean BM, Bottazzo GF, et al. Evidence for a long prediabetic period in type I (insulin-dependent) diabetes mellitus. Lancet. 1981 Dec 19-26;2(8260-61):1363-5.
26. Groop LC, Bottazzo GF, Doniach D. Islet cell antibodies identify latent type I diabetes in patients aged 35-75 years at diagnosis. Diabetes. 1986 Feb;35(2):237-41.
27. Karjalainen J, Salmela P, Ilonen J, Surcel HM, Knip M. A comparison of childhood and adult type I diabetes mellitus. N Engl J Med. 1989 Apr 6;320(14):881-6.
28. Pugliese A. Unraveling the genetics of insulin-dependent type 1A diabetes: the search must go on. Diabetes Reviews. 1999;7:39.
29. Baisch JM, Weeks T, Giles R, Hoover M, Stastny P, Capra JD. Analysis of HLA-DQ genotypes and susceptibility in insulin-dependent diabetes mellitus. N Engl J Med. 1990 Jun 28;322(26):1836-41.
30. Skyler JS. Cellular therapy for type 1 diabetes: has the time come? JAMA. 2007 Apr 11;297(14):1599-600.
31. Ladner MB, Bottini N, Valdes AM, Noble JA. Association of the single nucleotide polymorphism C1858T of the PTPN22 gene with type 1 diabetes. Hum Immunol. 2005 Jan;66(1):60-4.
32. Nepom GT. Immunogenetics and IDDM. Diabetes Reviews. 1993;1:93.
33. Palmer JP. Predicting IDDM: use of humoral immune markers. Diabetes Reviews. 1993;1:104.
34. Genuth SM. Plasma insulin and glucose profiles in normal, obese, and diabetic persons. Ann Intern Med. 1973 Dec;79(6):812-22.
35. Nair KS, Halliday D, Garrow JS. Increased energy expenditure in poorly controlled Type 1 (insulin-dependent) diabetic patients. Diabetologia. 1984 Jul;27(1):13-6.
36. Gillespie KM. Type 1 diabetes: pathogenesis and prevention. CMAJ. 2006 Jul 18;175(2):165-70.
37. Skyler JS. Prediction and prevention of type 1 diabetes: progress, problems, and prospects. Clin Pharmacol Ther. 2007 May;81(5):768-71. Epub 2007 Mar 28.
38. Skyler JS, Krischer JP, Wolfsdorf J, Cowie C, Palmer JP, Greenbaum C, et al. Effects of oral insulin in relatives of patients with type 1 diabetes: The Diabetes Prevention Trial--Type 1. Diabetes Care. 2005 May;28(5):1068-76.
39. Herold KC, Gitelman SE, Masharani U, Hagopian W, Bisikirska B, Donaldson D, et al. A single course of anti-CD3 monoclonal antibody hOKT3gamma1(Ala-Ala) results in improvement in C-peptide responses and clinical parameters for at least 2 years after onset of type 1 diabetes. Diabetes. 2005 Jun;54(6):1763-9.
40. Keymeulen B, Vandemeulebroucke E, Ziegler AG, Mathieu C, Kaufman L, Hale G, et al. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med. 2005 Jun 23;352(25):2598-608.
41. Bisikirska B, Colgan J, Luban J, Bluestone JA, Herold KC. TCR stimulation with modified anti-CD3 mAb expands CD8+ T cell population and induces CD8+CD25+ Tregs. J Clin Invest. 2005 Oct;115(10):2904-13. Epub 2005 Sep 15.
42. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med. 1993 Sep 30;329(14):977-86.
43. Implementation of treatment protocols in the Diabetes Control and Complications Trial. Diabetes Care. 1995 Mar;18(3):361-76.
44. Adverse events and their association with treatment regimens in the diabetes control and complications trial. Diabetes Care. 1995 Nov;18(11):1415-27.
45. Effects of intensive diabetes therapy on neuropsychological function in adults in the Diabetes Control and Complications Trial. Ann Intern Med. 1996 Feb 15;124(4):379-88.
46. Resource utilization and costs of care in the diabetes control and complications trial. Diabetes Care. 1995 Nov;18(11):1468-78.
47. Lifetime benefits and costs of intensive therapy as practiced in the diabetes control and complications trial. The Diabetes Control and Complications Trial Research Group. JAMA. 1996 Nov 6;276(17):1409-15.
48. The relationship of glycemic exposure (HbA1c) to the risk of development and progression of retinopathy in the diabetes control and complications trial. Diabetes. 1995 Aug;44(8):968-83.
49. The absence of a glycemic threshold for the development of long-term complications: the perspective of the Diabetes Control and Complications Trial. Diabetes. 1996 Oct;45(10):1289-98.
50. Martin CL, Albers J, Herman WH, Cleary P, Waberski B, Greene DA, et al. Neuropathy among the diabetes control and complications trial cohort 8 years after trial completion. Diabetes Care. 2006 Feb;29(2):340-4.
51. Writing Team for the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group. Sustained effect of intensive treatment of type 1 diabetes mellitus on development and progression of diabetic nephropathy: the Epidemiology of Diabetes Interventions and Complications (EDIC) study. JAMA. 2003 Oct 22;290(16):2159-67.
52. Genuth S. Insights from the diabetes control and complications trial/epidemiology of diabetes interventions and complications study on the use of intensive glycemic treatment to reduce the risk of complications of type 1 diabetes. Endocr Pract. 2006 Jan-Feb;12 Suppl 1:34-41.
53. Writing Team for the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group. Effect of intensive therapy on the microvascular complications of type 1 diabetes mellitus. JAMA. 2002 May 15;287(19):2563-9.
54. Nathan DM, Cleary PA, Backlund JY, Genuth SM, Lachin JM, Orchard TJ, et al. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med. 2005 Dec 22;353(25):2643-53.
55. The effect of intensive diabetes therapy on carotid atherosclerosis in type 1 diabetes mellitus. DCCT Research Group and EDIC Research Group. N Engl J Med. 2003;348:2294.
56. Cleary PA, Orchard TJ, Genuth S, Wong ND, Detrano R, Backlund JY, et al. The effect of intensive glycemic treatment on coronary artery calcification in type 1 diabetic participants of the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Study. Diabetes. 2006 Dec;55(12):3556-65.
57. Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Study Research Group, Jacobson AM, Musen G, Ryan CM, Silvers N, Cleary P, et al. Long-term effect of diabetes and its treatment on cognitive function. N Engl J Med. 2007 May 3;356(18):1842-52.
58. American Diabetes Association. Standards of medical care in diabetes--2007. Diabetes Care. 2007 Jan;30 Suppl 1:S4-S41.
59. Diabetes Research in Children Network (DirecNet) Study Group, Buckingham B, Beck RW, Tamborlane WV, Xing D, Kollman C, et al. Continuous glucose monitoring in children with type 1 diabetes. J Pediatr. 2007 Oct;151(4):388-93, 393.e1-2. Epub 2007 Aug 24.
60. Deiss D, Bolinder J, Riveline JP, Battelino T, Bosi E, Tubiana-Rufi N, et al. Improved glycemic control in poorly controlled patients with type 1 diabetes using real-time continuous glucose monitoring. Diabetes Care. 2006 Dec;29(12):2730-2.
61. Stork AD, van Haeften TW, Veneman TF. Diabetes and driving: Desired data, research methods and their pitfalls, current knowledge, and future research. Diabetes Care. 2006 Aug;29(8):1942-9.
62. Dungan KM, Buse JB, Largay J, Kelly MM, Button EA, Kato S, et al. 1,5-anhydroglucitol and postprandial hyperglycemia as measured by continuous glucose monitoring system in moderately controlled patients with diabetes. Diabetes Care. 2006 Jun;29(6):1214-9.
63. Rohlfing CL, Wiedmeyer HM, Little RR, England JD, Tennill A, Goldstein DE. Defining the relationship between plasma glucose and HbA(1c): analysis of glucose profiles and HbA(1c) in the Diabetes Control and Complications Trial. Diabetes Care. 2002 Feb;25(2):275-8.
64. Goldstein DE, Little RR, Lorenz RA, Malone JI, Nathan D, Peterson CM, et al. Tests of glycemia in diabetes. Diabetes Care. 2004 Jul;27(7):1761-73.
65. Nathan DM, Turgeon H, Regan S. Relationship between glycated haemoglobin levels and mean glucose levels over time. Diabetologia. 2007 Nov;50(11):2239-44. Epub 2007 Sep 13.
66. Larsen ML, Hørder M, Mogensen EF. Effect of long-term monitoring of glycosylated hemoglobin levels in insulin-dependent diabetes mellitus. N Engl J Med. 1990 Oct 11;323(15):1021-5.
67. Cagliero E, Levina EV, Nathan DM. Immediate feedback of HbA1c levels improves glycemic control in type 1 and insulin-treated type 2 diabetic patients. Diabetes Care. 1999 Nov;22(11):1785-9.
68. Skyler JS. Insulin treatment. Therapy for Diabetes Mellitus and Related Disorders. 3rd ed. American Diabetes Association; 1998.
69. Mooradian AD, Bernbaum M, Albert SG. Narrative review: a rational approach to starting insulin therapy. Ann Intern Med. 2006 Jul 18;145(2):125-34.
70. Hirsch IB. Type 1 diabetes mellitus and the use of flexible insulin regimens. Am Fam Physician. 1999 Nov 15;60(8):2343-52, 2355-6.
71. Hirsch IB. Insulin analogues. N Engl J Med. 2005 Jan 13;352(2):174-83.
72. Raskin P, Guthrie RA, Leiter L, Riis A, Jovanovic L. Use of insulin aspart, a fast-acting insulin analog, as the mealtime insulin in the management of patients with type 1 diabetes. Diabetes Care. 2000 May;23(5):583-8.
73. Robinson DM, Wellington K. Insulin glulisine. Drugs. 2006;66(6):861-9.
74. Garg SK, Carmain JA, Braddy KC, Anderson JH, Vignati L, Jennings MK, et al. Pre-meal insulin analogue insulin lispro vs Humulin R insulin treatment in young subjects with type 1 diabetes. Diabet Med. 1996 Jan;13(1):47-52.
75. Anderson JH Jr, Brunelle RL, Koivisto VA, Pfützner A, Trautmann ME, Vignati L, et al. Reduction of postprandial hyperglycemia and frequency of hypoglycemia in IDDM patients on insulin-analog treatment. Multicenter Insulin Lispro Study Group. Diabetes. 1997 Feb;46(2):265-70.
76. Del Sindaco P, Ciofetta M, Lalli C, Perriello G, Pampanelli S, Torlone E, et al. Use of the short-acting insulin analogue lispro in intensive treatment of type 1 diabetes mellitus: importance of appropriate replacement of basal insulin and time-interval injection-meal. Diabet Med. 1998 Jul;15(7):592-600.
77. Royle P, Waugh N, McAuley L, McIntyre L, Thomas S. Inhaled insulin in diabetes mellitus. Cochrane Database Syst Rev. 2004;(3):CD003890.
78. Dunn C, Curran MP. Inhaled human insulin (Exubera): a review of its use in adult patients with diabetes mellitus. Drugs. 2006;66(7):1013-32.
79. Skyler JS, Jovanovic L, Klioze S, Reis J, Duggan W; Inhaled Human Insulin Type 1 Diabetes Study Group. Two-year safety and efficacy of inhaled human insulin (Exubera) in adult patients with type 1 diabetes. Diabetes Care. 2007 Mar;30(3):579-85.
80. Heinemann L, Linkeschova R, Rave K, Hompesch B, Sedlak M, Heise T. Time-action profile of the long-acting insulin analog insulin glargine (HOE901) in comparison with those of NPH insulin and placebo. Diabetes Care. 2000 May;23(5):644-9.
81. Rosenstock J, Park G, Zimmerman J; U.S. Insulin Glargine (HOE 901) Type 1 Diabetes Investigator Group. Basal insulin glargine (HOE 901) versus NPH insulin in patients with type 1 diabetes on multiple daily insulin regimens. U.S. Insulin Glargine (HOE 901) Type 1 Diabetes Investigator Group. Diabetes Care. 2000 Aug;23(8):1137-42.
82. Insulin detemir (levemir), a new long-acting insulin. Med Lett Drugs Ther. 2006 Jul 3;48(1238):54-5.
83. Jovanovic L, Nakai Y. Successful pregnancy in women with type 1 diabetes: from preconception through postpartum care. Endocrinol Metab Clin North Am. 2006 Mar;35(1):79-97, vi.
84. Langer O. Is normoglycemia the correct threshold to prevent complications in the pregnant diabetic patient? Diabetes Reviews. 1996;4:2.
85. de Veciana M, Major CA, Morgan MA, Asrat T, Toohey JS, Lien JM, et al. Postprandial versus preprandial blood glucose monitoring in women with gestational diabetes mellitus requiring insulin therapy. N Engl J Med. 1995 Nov 9;333(19):1237-41.
86. Kjos SL, Buchanan TA. Gestational diabetes mellitus. N Engl J Med. 1999 Dec 2;341(23):1749-56.
87. Jovanovic L. Glucose and insulin requirements during labor and delivery: the case for normoglycemia in pregnancies complicated by diabetes. Endocr Pract. 2004 Mar-Apr;10 Suppl 2:40-5.
88. Boland EA, Grey M, Oesterle A, Fredrickson L, Tamborlane WV. Continuous subcutaneous insulin infusion. A new way to lower risk of severe hypoglycemia, improve metabolic control, and enhance coping in adolescents with type 1 diabetes. Diabetes Care. 1999 Nov;22(11):1779-84.
89. Dunn FL, Nathan DM, Scavini M, Selam JL, Wingrove TG. Long-term therapy of IDDM with an implantable insulin pump. The Implantable Insulin Pump Trial Study Group. Diabetes Care. 1997 Jan;20(1):59-63.
90. Whitehouse F, Kruger DF, Fineman M, Shen L, Ruggles JA, Maggs DG, et al. A randomized study and open-label extension evaluating the long-term efficacy of pramlintide as an adjunct to insulin therapy in type 1 diabetes. Diabetes Care. 2002 Apr;25(4):724-30.
91. Lebovitz HE. Therapeutic options in development for management of diabetes: pharmacologic agents and new technologies. Endocr Pract. 2006 Jan-Feb;12 Suppl 1:142-7.
92. Gruessner AC, Sutherland DE. Pancreas transplant outcomes for United States (US) and non-US cases as reported to the United Network for Organ Sharing (UNOS) and the International Pancreas Transplant Registry (IPTR) as of June 2004. Clin Transplant. 2005 Aug;19(4):433-55.
93. Robertson RP, Davis C, Larsen J, Stratta R, Sutherland DE. Pancreas and islet transplantation for patients with diabetes. Diabetes Care. 2000 Jan;23(1):112-6.
94. Dieterle CD, Arbogast H, Illner WD, Schmauss S, Landgraf R. Metabolic follow-up after long-term pancreas graft survival. Eur J Endocrinol. 2007 May;156(5):603-10.
95. Close N, Alejandro R, Hering B, Appel M; CITR Investigators. Second annual analysis of the collaborative islet transplant registry. Transplant Proc. 2007 Jan-Feb;39(1):179-82.
96. Shapiro AM, Lakey JR, Ryan EA, Korbutt GS, Toth E, Warnock GL, et al. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N Engl J Med. 2000 Jul 27;343(4):230-8.
97. Ryan EA, Paty BW, Senior PA, Bigam D, Alfadhli E, Kneteman NM, et al. Five-year follow-up after clinical islet transplantation. Diabetes. 2005 Jul;54(7):2060-9.
98. Shapiro AM, Ricordi C, Hering BJ, Auchincloss H, Lindblad R, Robertson RP, et al. International trial of the Edmonton protocol for islet transplantation. N Engl J Med. 2006 Sep 28;355(13):1318-30.
99. Hering BJ, Kandaswamy R, Ansite JD, Eckman PM, Nakano M, Sawada T, et al. Single-donor, marginal-dose islet transplantation in patients with type 1 diabetes. JAMA. 2005 Feb 16;293(7):830-5.
100. Rother KI, Harlan DM. Challenges facing islet transplantation for the treatment of type 1 diabetes mellitus. J Clin Invest. 2004 Oct;114(7):877-83.
101. Franz MJ, Bantle JP, Beebe CA, Brunzell JD, Chiasson JL, Garg A, et al. Nutrition principles and recommendations in diabetes. Diabetes Care. 2004 Jan;27 Suppl 1:S36-46.
102. Jenkins DJ, Wolever TM, Taylor RH, Barker H, Fielden H, Baldwin JM, et al. Glycemic index of foods: a physiological basis for carbohydrate exchange. Am J Clin Nutr. 1981 Mar;34(3):362-6.
103. Zinman B, Ruderman N, Campaigne BN, Devlin JT, Schneider SH; American Diabetes Association. Physical activity/exercise and diabetes. Diabetes Care. 2004 Jan;27 Suppl 1:S58-62.
104. Umpierrez GE, Kitabchi AE. Diabetic ketoacidosis: risk factors and management strategies. Treat Endocrinol. 2003;2(2):95-108.
105. Barrett EJ, DeFronzo RA, Bevilacqua S, Ferrannini E. Insulin resistance in diabetic ketoacidosis. Diabetes. 1982 Oct;31(10):923-8.
106. Burge MR, Hardy KJ, Schade DS. Short-term fasting is a mechanism for the development of euglycemic ketoacidosis during periods of insulin deficiency. J Clin Endocrinol Metab. 1993 May;76(5):1192-8.
107. DeFronzo RA, Matsuda M, Barrett EJ. Diabetic ketoacidosis: a combined metabolic-nephrologic approach to therapy. Diabetes Reviews. 1994;2:209.
108. Trachtenbarg DE. Diabetic ketoacidosis. Am Fam Physician. 2005 May 1;71(9):1705-14.
109. Waldhäusl W, Kleinberger G, Korn A, Dudczak R, Bratusch-Marrain P, Nowotny P. Severe hyperglycemia: effects of rehydration on endocrine derangements and blood glucose concentration. Diabetes. 1979 Jun;28(6):577-84.
110. Della Manna T, Steinmetz L, Campos PR, Farhat SC, Schvartsman C, Kuperman H, et al. Subcutaneous use of a fast-acting insulin analog: an alternative treatment for pediatric patients with diabetic ketoacidosis. Diabetes Care. 2005 Aug;28(8):1856-61.
111. Epidemiology of severe hypoglycemia in the diabetes control and complications trial. The DCCT Research Group. Am J Med. 1991 Apr;90(4):450-9.
112. Gale EA, Tattersall RB. Unrecognised nocturnal hypoglycaemia in insulin-treated diabetics. Lancet. 1979 May 19;1(8125):1049-52.
113. Cryer PE, Davis SN, Shamoon H. Hypoglycemia in diabetes. Diabetes Care. 2003 Jun;26(6):1902-12.
114. Heller SR, Macdonald IA, Herbert M, Tattersall RB. Influence of sympathetic nervous system on hypoglycaemic warning symptoms. Lancet. 1987 Aug 15;2(8555):359-63.
115. Mokan M, Mitrakou A, Veneman T, Ryan C, Korytkowski M, Cryer P, et al. Hypoglycemia unawareness in IDDM. Diabetes Care. 1994 Dec;17(12):1397-403.
116. Dagogo-Jack S. Hypoglycemia in type 1 diabetes mellitus: pathophysiology and prevention. Treat Endocrinol. 2004;3(2):91-103.
117. Fanelli C, Pampanelli S, Epifano L, Rambotti AM, Di Vincenzo A, Modarelli F, et al. Long-term recovery from unawareness, deficient counterregulation and lack of cognitive dysfunction during hypoglycaemia, following institution of rational, intensive insulin therapy in IDDM. Diabetologia. 1994 Dec;37(12):1265-76.
118. Boyle PJ, Kempers SF, O'Connor AM, Nagy RJ. Brain glucose uptake and unawareness of hypoglycemia in patients with insulin-dependent diabetes mellitus. N Engl J Med. 1995 Dec 28;333(26):1726-31.
119. Criego AB, Tkac I, Kumar A, Thomas W, Gruetter R, Seaquist ER. Brain glucose concentrations in healthy humans subjected to recurrent hypoglycemia. J Neurosci Res. 2005 Nov 15;82(4):525-30.
120. Effects of intensive diabetes therapy on neuropsychological function in adults in the Diabetes Control and Complications Trial. Ann Intern Med. 1996 Feb 15;124(4):379-88.
121. Kramer L, Fasching P, Madl C, Schneider B, Damjancic P, Waldhäusl W, et al. Previous episodes of hypoglycemic coma are not associated with permanent cognitive brain dysfunction in IDDM patients on intensive insulin treatment. Diabetes. 1998 Dec;47(12):1909-14.
122. Ryan C, Vega A, Drash A. Cognitive deficits in adolescents who developed diabetes early in life. Pediatrics. 1985 May;75(5):921-7.
123. Bjørgaas M, Gimse R, Vik T, Sand T. Cognitive function in type 1 diabetic children with and without episodes of severe hypoglycaemia. Acta Paediatr. 1997 Feb;86(2):148-53.
Figuras 1 e 4 Seward Hung.
"Caro Marcos, Notamos que há interesse em aprofundar o assunto. De imediato, podemos sugerir livros que são específicos da área de Endocrinologia e Diabetes, conforme os links a seguir: Fundamentos em diabetes: http://www.grupoa.com.br/livros/clinica-medica-e-semiologia/fundamentos-em-diabetes/9788536320687 Diabetes melito: http://www.grupoa.com.br/livros/clinica-medica-e-semiologia/joslin-diabetes-melito/9788536317151 Endocrinologia baseada em evidências: http://www.grupoa.com.br/livros/clinica-medica-e-semiologia/endocrinologia-2ed/9788536313764 Boa leitura! Atenciosamente, Os Editores"
"Parabéns pela iniciativa. Minha esposa tem DM I ha 40 anos e usa bomba de insulina ha 6 anos. Tem um bom controle renal e da retina; entretanto está desenvolvendo lipodistrofia importante nos braços abdômen e coxas.Alguém poderia me ajudar com sugestões ou artigos de referências? Ficarei muito agradecido.Obrigado. Dr Mar4cos Oliveira da Silva CRMPe 7253"
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.