(Carregando Índice)... (Carregando Índice)... |
Você está em:
Inicial  acp-medicine Hematologia
acp-medicine Hematologia
Última revisão: 31/07/2012
Comentários de assinantes: 1
Elizabeth A. Price, MD, MPH
Clinical Instructor, Department of Medicine, Division of Hematology, Stanford University School of Medicine, Stanford, CA
Stanley L. Schrier, MD
Professor of Medicine, Division of Hematology, Stanford University School of Medicine, and Staff Physician, Medicine-Hematology, Stanford Hospital and Clinics, Stanford, CA
Artigo original: Price EA, Schrier SL. Anemia: production defects. ACP Medicine. 2008;1-22.
[The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2011 Decker Intellectual Properties Inc. All Rights Reserved.]
Agradecimentos:
Figuras 1 e 6 – Talar Agasyan.
Figura 5 – Alan D. Iselin.
Figura 7 – Tom Moore.
Tradução: Soraya Imon de Oliveira
Revisão técnica: Dr. Euclides Furtado de Albuquerque Cavalcanti
Os defeitos de produção de hemácias causam uma anemia caracterizada por baixa contagem absoluta de reticulócitos. O exame das contagens do sangue periférico e da medula óssea ajuda a classificar estes distúrbios. A medula caracteristicamente apresenta um dos seguintes aspectos:
1. Proporção normal de células mieloides em relação às células eritroides (proporção M:E), celularidade normal e padrão normal de maturação eritroide.
2. A ausência total dos elementos medulares normais em decorrência da aplasia (ausência das células da medula) ou da substituição dos elementos normais da medula por fibrose, tumores sólidos, granulomas ou leucemia.
3. Hiperplasia eritroide com celularidade aumentada. Os defeitos da maturação eritroide resultam em eritropoiese inefetiva ou hemólise intramedular. Os precursores eritroides morrem na medula e poucas células chegam à periferia.
A anemia da inflamação crônica (também conhecida como anemia por doença crônica) é secundária a doenças neoplásicas, infecciosas e inflamatórias, além de outras doenças crônicas, como distúrbios hepáticos, insuficiência cardíaca congestiva e diabetes melito.1,2 Os valores de hematócrito geralmente variam de 27 a 35%, embora 20% dos pacientes apresentem hematócrito abaixo de 25%.2
A anemia por inflamação crônica usualmente resulta da combinação da sobrevida levemente diminuída das hemácias com o sequestro de ferro no sistema reticuloendotelial e níveis de eritropoietina abaixos do esperado para o grau de anemia.1,2 As hemácias usualmente apresentam aspecto morfológico normal, embora ocasionalmente possam estar levemente hipocrômicas e microcíticas. Os níveis séricos de ferro e transferrina são baixos, e a saturação do ferro frequentemente pode chegar a 15%,1,2 enquanto os níveis séricos de ferritina usualmente permanecem normais ou estão elevados.2,3 Todas estas alterações podem ser induzidas por citocinas inflamatórias (p. ex., interleucina [IL]-1; fator de necrose tumoral-alfa; interferons alfa, beta e gama; e talvez o fator transformador do crescimento tumoral-beta).4 Sob condições experimentais, estas citocinas diminuem a produção de eritropoietina, causam hipoferremia, aumentam os níveis séricos de ferritina, comprometem a eritropoiese e bloqueiam a liberação de ferro pelas células do sistema reticuloendotelial.5 A hepcidina, um mediador recém-descrito do metabolismo de ferro, emergiu como peça fundamental na patogênese da anemia por inflamação crônica.6 A hepcidina atua ligando-se ao único exportador de ferro conhecido localizado junto às células dos mamíferos – a ferroportina – com consequente internalização e degradação, que resulta na ablação da exportação do ferro.7 Isto, por sua vez, limita a absorção do ferro não-heme ao nível do intestino e também a liberação de ferro a partir dos macrófagos para as células progenitoras eritroides localizadas na medula óssea. A síntese de hepcidina é positivamente regulada pela inflamação (em particular, pela IL-6)8-10 e pela sobrecarga de ferro, sendo negativamente regulada em resposta à anemia e à hipóxia.11 Os níveis urinários de hepcidina estão aumentados em indivíduos com anemia por inflamação crônica.9 É interessante notar que camundongos transgênicos com expressão condicional de hepcidina recapitulam muitos dos aspectos da anemia por inflamação crônica em seres humanos, incluindo uma anemia de grau leve a moderado com eritropoiese ferro-restrita, acúmulo de ferro no baço e resposta embotada à eritropoietina endógena.12
Anemia branda, com níveis normais ou elevados de leucócitos e plaquetas, em paciente com doença crônica sugere o diagnóstico de anemia por inflamação crônica. O diagnóstico desta anemia normocítica ou hipocrômica e microcítica é facilmente confundido com anemia ferropriva, traço talassêmico ou anemia sideroblástica. Se após examinar cuidadosamente um esfregaço periférico o diagnóstico ainda for incerto, os testes mais úteis para estabelecer o diagnóstico são a medida dos níveis séricos de ferritina e, em raros casos, um exame de medula óssea que inclua uma coloração férrica [Tabela 1 e a Figura 1].
Tabela 1. Diagnóstico diferencial das anemias hipocrômicas
|
Variável Laboratorial |
Anemia por Inflamação Crônica (AIC) |
Anemia Ferropriva (AF) |
AIC e AF |
Traço Talassêmico |
Anemias Sideroblásticas |
|
Esfregaço |
Costuma ser normocrômica e normocítica, mas pode ser levemente hipocrômica e microcítica. |
Varia com o grau de anemia [ver Função da Hemácia e Distúrbios do Metabolismo do Ferro]. |
Varia com a doença e a extensão da anemia; usualmente normal. |
Hipocromia, células-alvo, micrócitos, pontilhado basofílico. |
As hemácias podem ser micro, normo ou macrocíticas. Entre portadores do sexo feminino, é possível observar uma população dimórfica. |
|
Nível sérico de ferro |
Baixo |
Baixo |
Baixo |
Normal |
Alto |
|
Capacidade de ligação de ferro |
Baixa/normal |
Alta |
Baixa/normal |
Normal |
Normal |
|
Percentual de saturação |
5 a 16 |
0 a 16 |
Baixo |
Normal (20 a 40) |
60 a 90 |
|
Níveis séricos de ferritina |
Normais ou altos |
Baixos |
Baixos/normais/ou um pouco elevados |
Normais |
Altos |
|
Receptor de transferrina solúvel (sTfR) |
Normal |
Alto |
Usualmente normal, mas pode estar aumentado diante de uma deficiência de ferro severa. |
Variável |
Variável |
|
sTfR/log ferritina |
Baixa |
Alta |
Normal ou alta |
Normal ou alta |
|
|
Ferro medular nas células reticuloendoteliais |
++ a +++ |
0 |
0 |
++ a +++ |
++++ |
|
Ferro medular nos sideroblastos |
0 |
0 |
0 |
++ a +++ |
++++ com sideroblastos anelares |
|
Precursores eritroides da medula |
Normais |
Geralmente normais; o citoplasma pode ser escasso. |
Normais |
Em geral, hiperplasia eritroide leve. |
Hiperplasia eritroide intensa, com diseritropoiese. |

Figura 1. Diagrama de fluxo mostrando as etapas do diagnóstico da anemia causada por defeitos de produção. Este tipo de anemia é sugerido por uma baixa contagem de reticulócitos corrigida ou pelo achado de anormalidades envolvendo leucócitos ou plaquetas no esfregaço de sangue periférico.
Em alguns casos, existe mais de uma causa de anemia e pode ser necessário realizar um exame abrangente do paciente para estabelecer a causa primária. Exemplificando, um paciente com anemia por inflamação crônica resultante de um carcinoma de cólon também pode apresentar deficiência de ferro decorrente de um sangramento intestinal. A anemia causada por inflamação combinada à ferropriva constitui um problema clínico bastante comum, que pode impor um desafio diagnóstico ao médico. O padrão-ouro, de uma forma geral, parece ser a avaliação do aspirado de medula óssea para determinação do conteúdo de ferro. Entretanto, trata-se de um procedimento invasivo raramente executado com esta indicação. A concentração de receptor de transferrina solúvel (RTS) dividida pelo logaritmo da concentração de ferritina pode ser particularmente útil em casos deste tipo. Os níveis de RTS estão elevados nos estados de deficiência de ferro e de eritropoiese aumentada,13 mas o mesmo não ocorre em condições inflamatórias.14 Um valor RTS/log(ferritina) maior do que 1,5 aponta deficiência de ferro em pacientes com anemia por inflamação crônica concomitante.15,16
A infecção pelo HIV produz efeitos hematológicos complexos, como a anemia hemolítica autoimune Coombs-positiva,17 mas também causa anemia por inflamação crônica na maioria dos pacientes com AIDS.
Identificar e tratar a doença primária constituem as partes mais importantes do manejo da anemia por inflamação crônica. A terapia à base de ferro pode ser corretiva, em particular diante da suspeita ou confirmação de uma deficiência de ferro concomitante.18 Neste caso, as formulações de ferro endovenosas podem ser preferíveis, dada a possibilidade de os níveis de hepcidina aumentados limitarem a absorção oral de ferro.19 Os agentes estimuladores eritropoiéticos (AEEs), incluindo a eritropoietina recombinante e a formulação de ação prolongada da darbepoetina-alfa, constituem o tratamento padrão para pacientes com anemia por inflamação crônica. Para muitos pacientes, a administração de doses farmacológicas de eritropoietina corrige a anemia por inflamação crônica ao anular o defeito de produção de eritropoietina. É útil se obter uma medida basal dos níveis plasmáticos de eritropoietina, porque a ocorrência de uma resposta à eritropoietina é improvável em pacientes cujos níveis endógenos sejam superiores a 500 mU/mL. As respostas de eritropoietina foram descritas em pacientes com artrite reumatoide,20 AIDS,21 e enteropatia inflamatória.22 Para responder de forma satisfatória, o paciente deve ter boas reservas de ferro (p. ex., níveis de ferritina normais ou elevados, ou coloração férrica da medula adequada) [ver Abordagem de distúrbios hematológicos]. Anteriormente, a recomendação era iniciar o paciente em um curso de 100 a 150 U/kg por via subcutânea, 3 vezes por semana. Entretanto, a maioria dos médicos administra uma única dose subcutânea de 40.000 unidades de eritropoietina por semana.23 A darbepoetina pode ser administrada por via subcutânea, a doses de 100 mcg por semana ou doses de 200 mcg com intervalos de 1 semana. Se os níveis de hemoglobina não subirem após 12 semanas, a eritropoietina deve ser descontinuada.
Embora pacientes com câncer também tenham reagido positivamente,24 evidências crescentes sugerem que o uso de AEEs por pacientes com câncer ativo pode ser prejudicial, pois alguns estudos demonstraram tempos menores de sobrevida livre de progressão locorregional25 e de sobrevida geral.26 Um estudo concluído precocemente randomizou 989 pacientes com malignidades ativas, que não estavam recebendo quimioterapia concomitante, para o uso de darbepoetina ou placebo.27 Observou-se uma piora significativa da sobrevida geral no grupo de pacientes tratados com darbepoetina vs. o grupo tratado com placebo, com aumento da incidência de eventos tromboembólicos. Uma tarja preta foi adicionada às informações de prescrição da eritropoietina e da darbepoetina, indicando que os AEEs não são indicados para pacientes com malignidades hematológicas não mieloides ou sólidas que não estejam recebendo quimio ou radioterapia. Do mesmo modo, a Society of Hematology e a American Society of Clinical Oncology têm contraindicado o uso dos AEEs por esta população.27
A causa predominante da anemia na doença renal é a produção deficiente de eritropoietina pelos rins adoecidos. Se houver uma doença inflamatória renal subjacente, pode haver um componente da anemia por inflamação crônica.28 Anorexia e ingesta alimentar precária, extração frequente de amostras de sangue e perda de eritrócitos durante a hemodiálise podem acarretar deficiência de ferro. Deficiência de ácido fólico, hiperesplenismo e hiperparatireoidismo secundário com fibrose medular29 também podem promover anemia.
Em pacientes que realizam hemodiálise, a anemia também pode ser causada pela toxicidade do alumínio. Este tipo de anemia foi inicialmente identificado em pacientes que apresentam a conhecida demência da diálise. Níveis plasmáticos de alumínio bastante elevados provavelmente resultam da contaminação do líquido de diálise ou da absorção gastrintestinal dos géis ingeridos para quelar os fosfatos presentes na dieta. Experimentos in vitro mostraram que o alumínio inibe o crescimento dos precursores eritroides das unidades formadoras de colônias-eritroides (CFU-E, erythroid precursors colony-forming unit-erythroid) e das unidades formadoras de explosão-eritroides (BFU-E, burst-forming unit-erythroid).30
O esfregaço sanguíneo deve ser examinado levando-se em conta a existência de fragmentação eritrocitária ou equinocitose, a fim de excluir outras possíveis causas de anemia. A presença dos corpúsculos de Heinz sugere a ocorrência de hemólise, talvez causada por oxidantes presentes no líquido de hemodiálise.
A eritropoietina constitui o tratamento padrão para pacientes com doença renal e anemia de grau moderado a severo; é capaz de eliminar a necessidade de transfusão em pacientes que realizam hemodiálise e por aqueles com doença renal progressiva que ainda não necessitam de hemodiálise. Esta terapia promove uma melhora significativa da qualidade de vida destes pacientes.31 É costume iniciar a terapia com 50 U de eritropoietina/kg, 3 vezes por semana, seja por via endovenosa ou subcutânea, e aumentar a dosagem conforme o necessário para trazer os níveis de hemoglobina ao valor desejado. Dados recentes levantaram a questão sobre a supercorreção da anemia em pacientes com doença renal. Um estudo randomizou pacientes com taxa de filtração glomerular (TFG) entre 15 e 35 mL/min e níveis basais de hemoglobina de 11 a 12,5 g/dL para receber tratamento à base de epoetina-beta, com o objetivo de manter uma concentração de hemoglobina da ordem de 13 a 15 g/dL ou 10,5 a 11,4 g/dL.32 Decorridos 3 anos de seguimento, os pacientes do grupo de hemoglobina alta apresentaram melhora significativa da saúde geral e do funcionamento físico. Entretanto, os episódios hipertensivos, as dores de cabeça e as necessidades de diálise também foram mais prevalentes neste grupo. Em outro estudo envolvendo pacientes adultos com TFG de 15 a 50 mL/min e níveis basais de hemoglobina abaixo de 11 g/dL, 715 indivíduos foram randomizados para receber epoetina-alfa, com o objetivo de atingir uma concentração de hemoglobina igual a 13,5 g/dL, enquanto 717 pacientes foram randomizados para receber uma dose cuja meta era atingir níveis de hemoglobina de 11,3 g/dL.33 Após um período médio de seguimento de 16 meses, os indivíduos randomizados para o grupo de hemoglobina alta apresentaram um número significativamente maior de eventos adversos, incluindo morte, infarto do miocárdio, internação por insuficiência cardíaca congestiva e acidente vascular cerebral (AVC), levando o estudo a ser terminado precocemente. Assim, existe uma preocupação atual quanto ao uso dos AEEs por pacientes com doença renal crônica, em especial com correção agressiva da anemia para um nível de hemoglobina elevado. Em geral, uma concentração-alvo de 11 a 12 g/dL deve ser utilizada.34 Os pacientes devem ser monitorados de perto para garantir que seus níveis de hemoglobina não passem de 13 g/dL e também para prevenir o desenvolvimento de hipertensão e complicações tromboembólicas.
O status do ferro também pode ser monitorado via tratamento com AEEs, visto que a administração de suplementos de ferro por via parenteral melhora a resposta a estes agentes. O ImFed (uma forma de ferro dextrano) pode ser administrado por via intramuscular ou endovenosa, em doses que variam de 100 a 500 mg, com uma frequência antecipada de reações da ordem de 4,7%. O Ferrlecit (uma forma de gliconato férrico de sódio) pode ser infundido por via endovenosa (125 mg durante 1 hora), com eventual ocorrência de hipotensão, náusea e rubor.39 A sucrose de ferro (100 mg/sessão de hemodiálise ou 300 mg ao longo de 1,5 horas) está associada a uma menor incidência de reações de hipersensibilidade, em comparação a outras formulações de ferro parenterais,36 e pode permitir a diminuição da dose de AAE.37 Em um estudo envolvendo pacientes com anemia induzida pela toxicidade ao alumínio, o tratamento com deferoxamina IV (30 mg/kg, por via IV, ao final de cada sessão de diálise) produziu uma melhora significativa.38
A ingesta excessiva de bebidas alcoólicas – seja aguda ou crônica – exerce profundos efeitos hematológicos.39 A ingesta de cerca de 80 g de álcool (uma garrafa de vinho, cerca de 2,5 L de cerveja ou 1/3 de garrafa de uísque) diariamente pode acarretar macrocitose,40 estomatocitose,41 trombocitopenia,39 vacuolização de proeritroblastos, sideroblastos anelares,41 queda acentuada dos níveis séricos de ácido fólico e elevação dos níveis séricos de ferro. Também pode comprometer a resposta de reticulócitos ao ácido fólico administrado em pacientes com deficiência comprovada desta substância. O consumo agudo de álcool em si não produz anemia megaloblástica.39 Foi postulado que a toxicidade hematológica induzida pelo álcool é mediada pelo acetaldeído, principal metabólito do etanol, significativamente mais tóxico e reativo do que o próprio etanol. O mecanismo que provoca estas anormalidades álcool-induzidas pode ser a formação de anticorpos contra os adutos de acetaldeído-hemoglobina.41 Megaloblastos, macro-ovalócitos e neutrófilos polimorfonucleares (PMNs) hipersegmentados geralmente aparecem quando há uma deficiência de ácido fólico concomitante. O consumo abusivo crônico de álcool muitas vezes resulta numa deficiência simultânea de ácido fólico e ferro, doença hepática grave, sangramento gastrintestinal (GI), hiperesplenismo e anemia por inflamação crônica.
A inanição resultante da anorexia nervosa ou a deficiência proteica podem causar anemia e até pancitopenia. O paciente também pode apresentar hemólise [Figura 2]. O exame de biópsia de medula óssea revela a hipocelularidade, com um material gelatinoso de fundo característico, que consiste em mucopolissacarídeos ácidos. Pode haver anemia mesmo diante de níveis normais de ácido fólico e cobalamina (vitamina B12), que pode ser corrigida com uma dieta nutricional adequada.
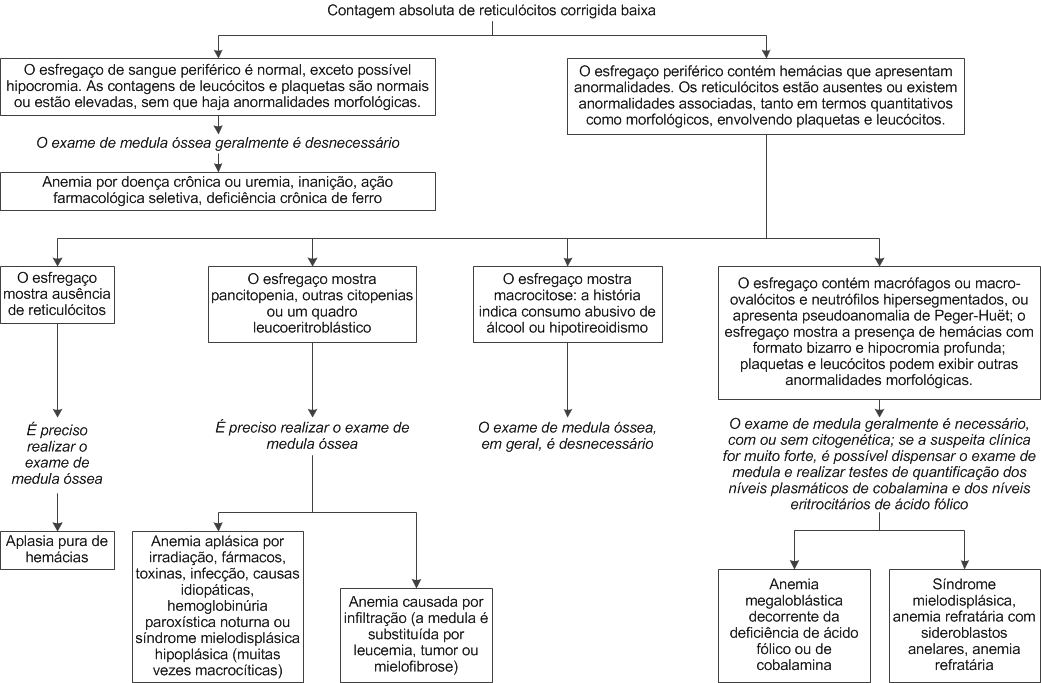
Figura 2. As alterações no esfregaço de sangue periférico observadas em casos de doença hepática severa ou inanição incluem (a) variação distinta do tamanho e formato das hemácias; células agudamente espiculadas (acantócitos) e eritrócitos recortados proeminentes. O esfregaço de sangue leucoeritroblástico (b) indica que a medula foi reposta com hematopoiese extramedular. Caracteriza-se por apresentar variação de tamanho e formato das hemácias, presença de hemácias nucleadas no sangue periférico, plaquetas gigantes e imaturidade das células da série mieloide. Na deficiência de ácido fólico ou de cobalamina (c), o esfregaço caracteriza-se pela variação do tamanho dos eritrócitos e por uma macrocitose distinta. Ocasionalmente, pode haver eritrócitos com “cauda de peixe”, além de neutrófilos hipersegmentados.
O hipotireoidismo compromete a produção de eritrócitos. Embora possa causar anemia macrocítica, o médico deve submeter o paciente a uma avaliação adicional para descobrir outras possíveis causas de anemia macrocítica, como a deficiência de ácido fólico ou de cobalamina.
A anemia branda associada ao pan-hipopituitarismo severo pode ser corrigida por meio da reposição dos hormônios da adrenal, tireoide e gônadas. O efeito intensificador dos andrógenos sobre a ação da eritropoietina é bem conhecido.
Os níveis de hemoglobina, índices de hemácias e contagens de leucócitos e plaquetas em indivíduos idosos sadios são similares aos de indivíduos adultos mais jovens. Este achado foi confirmado por um estudo envolvendo pacientes com idade mínima de 84 anos.42 Sendo assim, é necessário realizar uma avaliação quando estes indivíduos idosos desenvolvem anemia. Cerca de 10% dos homens e mulheres com mais de 65 anos de idade são anêmicos, sendo que 1/3 destes indivíduos apresentam um tipo de anemia inexplicável pelos mecanismos usuais.43 É importante notar que os afro-americanos podem apresentar níveis de hemoglobina fisiologicamente mais baixos em cerca de 1 g/dL, se comparados aos caucasianos,44,45 – diferença esta que ainda não foi totalmente explicada pela existência de deficiência de ferro ou alfa-talassemia.45 É fundamental avaliar e tratar idosos, pois a ocorrência de anemia (concentração de hemoglobina [Hgb] < 12 g/dL em mulheres e < 13 g/dL em homens) constitui um fator de risco independente para o declínio do funcionamento físico46,47 e da cognição,48 aumento das internações e mortalidade.49 Ainda é preciso investigar se a instituição da terapia para aumentar os níveis de hemoglobina em pacientes idosos com anemia inexplicável promove melhores resultados.
A combinação de anemia com neutropenia ou trombocitopenia, ou destas 3 anormalidades (p. ex., pancitopenia), usualmente aponta a ocorrência de dano na medula hematopoiética. Se a cavidade medular apresenta infiltração, porém as células-tronco pluripotentes continuam intactas, a hematopoiese extramedular muitas vezes se desenvolve nos órgãos onde ocorre hematopoiese fetal (p. ex., baço, fígado e ossos distais).
A pancitopenia pode ser congênita ou adquirida. O achado de citopenias combinadas ou de células imaturas no sangue (mielócitos, metamielócitos e eritroblastos) – ou seja, um esfregaço sanguíneo leucoeritroblástico – sugere a ocorrência de hematopoiese extramedular [Figura 2]. Estes achados constituem uma indicação para realização de exame de amostra de medula óssea obtida por aspiração e biópsia.
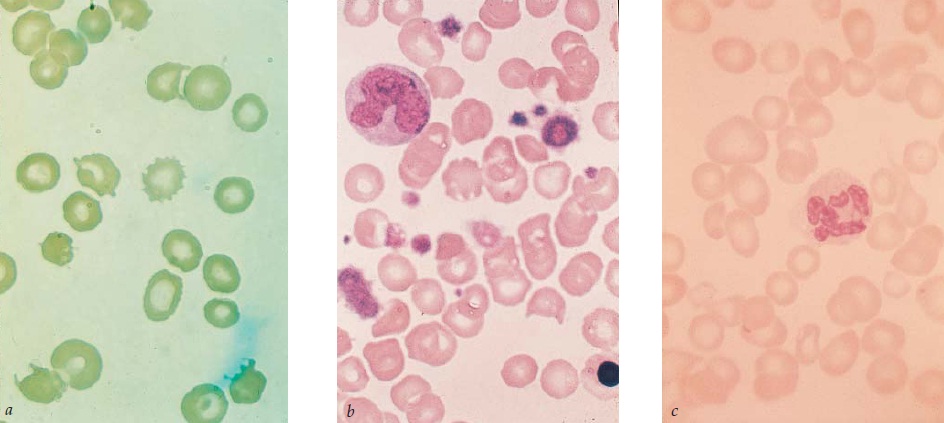
A pancitopenia (p. ex., anemia, neutropenia e trombocitopenia) e um exame de biópsia mostrando uma medula aplásica [Figura 3] estabelecem um diagnóstico funcional de anemia aplásica. A amostra de biópsia não deve ser obtida de um sítio medular que tenha sido irradiado. É essencial determinar o grau de severidade da doença. Uma anemia aplásica severa (AAS) é definida por: (1) uma medula com menos de 25% de celularidade normal ou uma medula com menos de 50% de celularidade normal na qual menos de 30% das células sejam hematopoiéticas; e (2) 2 a 3 valores sanguíneos periféricos anormais (contagem absoluta de reticulócitos < 40.000/mcL; contagem absoluta de neutrófilos [CAN] < 500/mcL, ou níveis de plaquetas < 20.000/mcL). Estes critérios foram considerados relativamente insensíveis. Alguns pesquisadores preferem identificar uma coorte de pacientes com anemia aplásica muito severa (AAMS), como sendo aqueles com CAN < 200/mcL.50
Figura 3. É possível observar em (a) uma biópsia normal de medula óssea e em (b) uma biópsia de medula de um paciente com anemia aplásica, apresentando uma aplasia quase total.
A anemia aplásica tem numerosas causas [Tabela 2], embora nem sempre sua causa exata possa ser determinada.
Tabela 2. Causas da anemia aplásica
|
Fármacos utilizados na radioterapia |
Agentes cujo uso regularmente causa mielossupressão |
|
Agentes alquilantes: melfalana, ciclofosfamida, clorambucil, bussulfano. | |
|
Antimetabólitos: azatioprina, 6-mercaptopurina, hidroxiurea, metotrexato. | |
|
Outros agentes antitumorais: daunorrubicina, doxorrubicina, carmustina, lomustina, amsacrina. | |
|
Agentes cujo uso ocasionalmente causa mielossupressão | |
|
Cloranfenicol, compostos de ouro, arsênico, sulfonamidas, mefenitoína, trimetadiona, fenilbutazona, quinacrina, indometacina, diclofenaco, felbamato. | |
|
Toxinas |
Benzeno, vapores de cola. |
|
Infecções |
Hepatite não A, não B e não C; mononucleose infecciosa; infecção por parvovírus (ataque de precursores eritroides); HIV. |
|
Distúrbios malignos |
Leucemia de células pilosas; leucemia linfocítica aguda; leucemia mieloide aguda (raramente); síndromes mielodisplásicas. |
|
Distúrbios clonais |
Hemoglobinúria paroxística noturna. |
|
Aplasia imuno-mediada |
Fasciíte eosinofílica. |
|
Distúrbios hereditários |
Anemia de Fanconi. |
|
Gravidez |
|
A radiação ionizante e os fármacos quimioterápicos empregados no tratamento de distúrbios malignos e imunológicos são capazes de destruir as células-tronco hematopoiéticas. Espera-se alcançar a recuperação com uma dosagem e um esquema cuidadosamente estabelecidos. Certos fármacos, como o cloranfenicol, produzem uma aplasia medular dose-independente. A terapia com ouro e a inalação de vapores de solventes orgânicos (p. ex., benzeno ou cola) também podem acarretar uma insuficiência medular fatal.
Em até 2 a 10% dos pacientes com hepatite, há desenvolvimento de uma severa aplasia dentro de 2 a 3 meses após a manifestação de um caso aparentemente típico de doença aguda, geralmente em homens jovens. Com frequência, a hepatite não tem causa evidente e os exames para hepatites A, B e C resultam negativos.51 Observa-se uma alta incidência de anemia aplásica após o transplante de fígado em pacientes com hepatite severa não A e não B.52
Diversas linhas de evidência sustentam a possibilidade de distúrbios imunológicos serem capazes de provocar aplasia. O tipo medular ocorre na doença do enxerto vs. hospedeiro (GVHD – do inglês graft versus host disease).53 O pré-condicionamento imunossupressor aumenta as chances de transplante de medula singênica bem-sucedido em pacientes com anemia aplásica,54 sendo que a terapia imunossupressora tem sido utilizada com sucesso no tratamento da anemia aplásica idiopática.53,54 O sangue de alguns pacientes com anemia aplásica parece conter células T supressoras que inibem o crescimento das células progenitoras comprometidas, conhecidas como unidades formadoras de colônias de granulócitos-macrófagos (CFU-GM, colony-forming unit-granulocyte-macrophage). As células T supressoras podem atuar produzindo interferon-gama.54 Foi demonstrado que os pacientes com anemia aplásica apresentam número reduzido de células T reguladoras, do mesmo modo como se observa em outros distúrbios autoimunes,55 e isto poderia levar a uma regulação deficiente das células T citotóxicas. O resultado destes complexos mecanismos imunes envolvendo células T supressoras é uma profunda diminuição da concentração de células hematopoiéticas, que pode ser medida pelo ensaio de células iniciadoras de cultura de longa duração e pela capacidade de formar colônias secundárias a partir de colônias que sobrevivem há 5 semanas em cultura de medula.56
Pacientes com mutações herdadas no mecanismo de reparo ou proteção dos telômeros parecem apresentar risco aumentado de desenvolvimento de anemia aplásica. Os telômeros são nucleoproteínas complexas importantes para a proteção, replicação e estabilização de genomas eucarióticos lineares.57 Nas células somáticas humanas, os telômeros consistem em repetições paralelas que são gradualmente perdidas com o envelhecimento celular. Quando um nível crítico de encurtamento é atingido, ocorre parada permanente do crescimento ou morte celular.57 O mecanismo de reparo de telômeros inclui um molde de RNA, codificado por TERC, e uma telomerase transcriptase reversa, codificada por TERT. Os telômeros são curtos em 1/3 dos pacientes com anemia aplásica aparentemente adquirida,58 sendo identificadas mutações em TERT59 e TERC.60
A aplasia também pode ser parte de um pródromo de leucemia de células pilosas [ver Leucemia linfoide crônica e distúrbios de plasmócitos], leucemia linfoblástica aguda [ver Leucemia aguda], em raros casos, leucemia mieloide aguda, ou pode ainda se desenvolver durante o curso de mielodisplasia [ver Leucemia aguda].
O paciente com anemia aplásica pode procurar atendimento médico devido à fadiga e falta de ar. A trombocitopenia concomitante pode causar petéquias, bolhas de sangue orais, sangramento gengival e hematúria, dependendo dos níveis de contagem plaquetária. Sem dúvida, o maior problema associado à anemia aplásica são as infecções bacterianas recorrentes causadas por uma profunda neutropenia. Sepse, pneumonia e infecções no trato urinário são comuns entre pacientes com anemia aplásica. As infecções fúngicas invasivas podem causar a morte do paciente, especialmente quando este apresenta neutropenia severa.
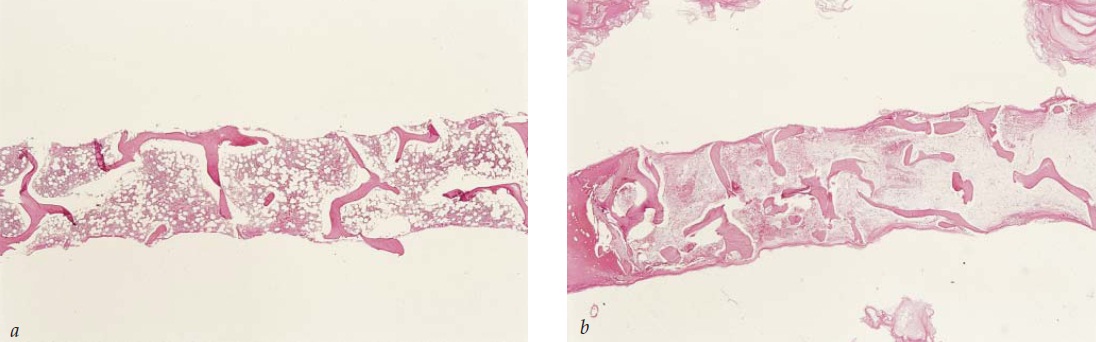
O diagnóstico de anemia aplásica requer o exame de amostras de aspirado e biópsia de medula [Figura 3], bem como a obtenção de uma história abrangente das exposições farmacológicas, infecções e em especial dos sintomas sugestivos de doença viral e resultados de exames sorológicos para hepatite, mononucleose infecciosa, HIV e parvovírus [Figura 4]. A medida da expressão de CD59 nas hemácias é útil para o diagnóstico de hemoglobinúria paroxística noturna.
Também é importante determinar o grau de severidade da anemia aplásica [ver Anemia Aplásica, Definição, anteriormente]. Os casos graves estão associados a uma taxa bastante baixa de remissão espontânea e a uma mortalidade de 70% dentro de 1 ano. Em contraste, 80% dos pacientes que apresentam formas mais brandas de anemia aplásica sobrevivem por 1 ano.50
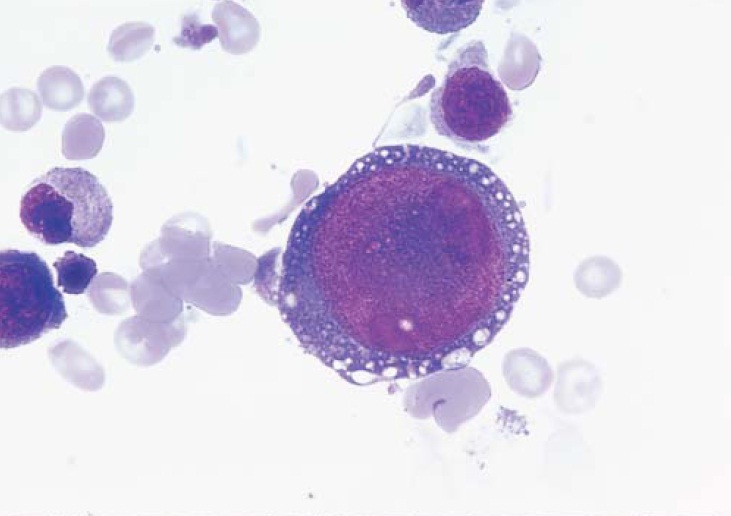
Figura 4. O promonoblasto gigante, evidente neste esfregaço de medula, sugere fortemente um diagnóstico de infecção por parvovírus.
O diagnóstico diferencial de pancitopenia inclui a leucemia linfocítica crônica, lúpus eritematoso sistêmico e esplenomegalia congestiva. Nestas doenças, contudo, a medula não está aplásica e, em vez disso, apresenta hiperplasia das linhagens celulares envolvidas. Outras condições que causam pancitopenia incluem a síndrome mielodisplásica hipoplásica, leucemia aguda, anemia megaloblástica e leucemia linfocítica granular de células grandes.61
O tratamento das formas mais brandas de anemia aplásica envolve a remoção do agente agressor e proporcionar uma terapia de suporte, principalmente a transfusão terapêutica, prevendo que as células-tronco pluripotentes restantes irão preencher novamente a medula.
Terapia de suporte. A trombocitopenia é um problema de saúde importante e frequente associado à anemia aplásica. Deve ser supervisionado com transfusão de plaquetas, conforme seja necessário para controlar ou prevenir o sangramento. Usualmente, um limiar de 10.000 plaquetas/mcL é utilizado para transfusão, ainda que o tratamento conservador seja melhor, e o mínimo possível de transfusões deve ser realizado. Uma extensiva substituição plaquetária pode resultar em alossensibilização às plaquetas, podendo complicar transplantes de medula óssea alogênicas futuras. As transfusões de hemácias são administradas conforme o necessário para controlar os sinais e sintomas de anemia.
O fator estimulador de colônias de granulócitos (G-CSF, granulocyte colony-stimulating factor) e o fator estimulador de colônias de granulócitos-macrófagos (GM-CSF, granulocyte-macrophage colony-stimulating factor) foram administrados aos pacientes para elevar a contagem de neutrófilos e ajudar a combater a infecção. Estes fatores geralmente são inefetivos quando utilizados de modo isolado, devido à severa deficiência de células precursoras que, por sua vez, constituem o alvo das ações do G-CSF e do GM-CSF.62 Geralmente, é preferível instituir um tratamento definitivo: a terapia imunossupressora ou, de preferência, o transplante de medula óssea alogênica (quando há disponibilidade de um irmão doador compatível) [ver Transplante de célula hematopoiética].63
Terapia definitiva. O transplante de um enxerto doado por um irmão compatível – após a instituição de um regime preparatório com altas doses de ciclofosfamida e globulinas antitimócitos (GAT), aliado ao uso de metotrexato e ciclosporina para profilaxia contra GVHD – constitui um regime bastante efetivo para pacientes com anemia aplásica.64 Os atuais resultados sugerem uma taxa de cura superior a 90% entre pacientes mais jovens.65 O uso de transplantes de sangue periférico pode diminuir a sobrevida, em comparação aos transplantes de medula, devido à maior incidência de GVHD.66 Os resultados obtidos com doadores incompatíveis ou compatíveis e sem parentesco não foram muito satisfatórios, embora tenham melhorado significativamente com as transplantações doadoras alternativas realizadas após o ano de 1997 vs. aquelas realizadas antes de 1997.67 Pacientes com anemia aplásica sem irmãos doadores compatíveis muitas vezes passam por um teste com terapia imunossupressora antes de serem submetidos ao transplante.
Foi demonstrado que 3 formas de imunossupressão produzem remissão parcial da anemia aplásica.62,63,68 A GAT promoveu remissão sustentada em cerca de metade dos pacientes que participaram de um estudo randomizado.63 Altas doses de corticosteroide promoveram melhoras aproximadas de 40% nas contagens sanguíneas dos pacientes tratados, sendo que a ciclosporina também se mostrou benéfica.63 (Os andrógenos [p. ex., oximetolona] podem exercer algum papel no tratamento da AAS, mas não são administrados de maneira isolada.)62,68
Embora cada um destes agentes possa ser utilizado individualmente ou de forma consecutiva no tratamento da anemia aplásica, um estudo controlado sugeriu que os resultados obtidos são melhores quando os 3 agentes são usados ao mesmo tempo.62,63 A combinação da GAT com um corticosteroide e ciclosporina resultou numa sobrevida de 62% aos 36 meses. Os primeiros sinais de resposta surgiram em torno da quarta semana. A média do tempo decorrido até a remissão foi de 60 a 82 dias.63 Neste estudo, o resultado do paciente apresentou correlação com a qualidade da resposta hematológica. Um relato de seguimento de 11 anos confirmou a efetividade da combinação de GAT, corticosteroides e ciclosporina. A taxa de recidiva foi igual a 38% e doenças clonais ou malignas desenvolveram-se em 25% dos pacientes.69
Com base na usual disponibilidade de GAT de cavalo nos Estados Unidos,62,63 recomenda-se que seja administrada GAT de cavalo a uma dosagem de 40 mg/kg/dia, em 500 mL de salina e durante 4 dias, por um período de 4 a 5 horas, através de uma linha IV equipada com um filtro em linha microagregado. O efeito colateral tóxico da GAT é a doença do soro, que geralmente pode ser controlada com corticosteroides. A prednisona (60 a 100 mg/dia) é administrada por via oral, em doses divididas, ou adiciona-se metilprednisolona (40 mg) ao frasco de infusão, e a dose pode ser aumentada para 1 mg/kg/dia. A terapia à base de corticosteroide é ajustada para controlar a doença do soro, mas em geral pode ser diminuída gradativamente após 2 semanas e suspendida depois de 30 dias. Como a GAT pode abaixar as contagens plaquetárias, são realizadas transfusões de plaquetas de acordo com a necessidade, para manter a contagem de plaquetas acima de 20.000/mcL.
A ciclosporina (10 a 12 mg/kg/dia) é administrada por via oral em duas doses divididas, para que os níveis do fármaco no sangue total cheguem a 500 a 800 ng/mL ou os níveis séricos atinjam 100 a 200 ng/mL. Decorridos 29 dias, a dosagem de ciclosporina pode ser reduzida aos poucos para que os níveis de fármaco no sangue total sejam de 200 a 500 ng/mL.63,64 O curso de ciclosporina é mantido durante 6 meses, no mínimo. A ciclosporina pode causar hipertensão, toxicidade renal, hipomagnesemia, hipofosfatemia, vitiligo, tremores, hipertricose, suscetibilidade à pneumonia por Pneumocystis jiroveci (anteriormente denominada Pneumocystis carinii) e hiperplasia gengival.62,63 Em um estudo, 300 mg de pentamidina em aerossol foram administrados a cada 4 semanas, como medida profilática contra a pneumocistose.63
Em outro estudo, o G-CSF (5 mcg/kg/dia) foi administrado por via subcutânea durante os primeiros 90 dias, com metilprednisolona IV (2 mg/kg/dia nos dias 1 a 5, seguidas de 1 mg/kg/dia nos dias 6 a 10, e diminuída em 30 dias), promovendo resultados satisfatórios.70
Ao contrário dos pacientes submetidos ao transplante de medula óssea alogênico, os responsivos à terapia imunossupressora não se curam de fato. Muitos continuam apresentando citopenia moderada,71 sendo que 20 a 36% apresentam recidivas de anemia aplásica,62,63,71 ainda que o prolongamento da terapia com ciclosporina para mais de 6 meses possa diminuir o risco de recidiva.72 Até 20 a 36% dos pacientes eventualmente desenvolvem distúrbios clonais, como hemoglobinúria paroxística noturna, síndrome mielodisplásica e leucemia aguda.62,63 Os pacientes também apresentam risco aumentado de desenvolvimento de tumores sólidos após o tratamento da anemia aplásica, embora em um estudo tenha sido observado risco idêntico para pacientes submetidos à terapia imunossupressora e para aqueles que receberam transplante de medula óssea alogênica.73 Mais de 50% dos pacientes que apresentaram recidivas de anemia aplásica após a resposta inicial à terapia imunossupressora podem responder a um segundo curso terapêutico.62,63 No caso dos pacientes irresponsivos, uma triagem com GAT de coelho pode funcionar. A GAT de coelho (3,5 mg/kg/dia diluída em solução salina e infundida ao longo de 6 a 8 horas, durante 5 dias consecutivos)74 é administrada com ciclosporina (5 mg/kg/dia, PO, nos dias 1 a 180 e, então, reduzida) e G-CSF (5 mcg/kg/dia nos dias 1 a 90), embora em um dos estudos os pacientes tratados com GAT de coelho após terem falhado em responder à GAT de cavalo tenham apresentado uma taxa de resposta geral de apenas 30%.75 É provável que um terceiro curso de GAT seja benéfico apenas para aqueles que responderam previamente.76
Existe uma certa preocupação no sentido de que o uso de G-CFS pode aumentar o risco de desenvolvimento de síndrome mielodisplásica ou leucemia aguda,77 embora os resultados sejam variáveis.78,79 Em um estudo retrospectivo envolvendo 840 pacientes com AAS tratada com GAT e ciclosporina, 43% receberam G-CFS enquanto 57% não receberam este fator. Aqueles que receberam G-CSF apresentaram uma incidência acumulada de 10 anos de SMD e LMA igual a 10,9% vs. a incidência de 5,8% apresentada por aqueles que não receberam G-CSF (p = 0,07).80 O European Group for Blood and Marrow Transplantation (EBMT) está conduzindo um estudo randomizado prospectivo, no qual compara GAT e ciclosporina, com ou sem G-CSF, para responder a esta questão de forma mais definitiva.81
Há um relato intrigante sobre 10 pacientes com AAS submetidos ao tratamento com altas doses de ciclofosfamida IV (45 mg/kg/dia), durante 4 dias consecutivos.82 Alguns pacientes também receberam ciclosporina. Apenas um curso de ciclofosfamida IV foi administrado. Dentre os pacientes, 7 produziram uma resposta hematológica completa e 6 ainda estavam vivos após, em média, 10,8 anos de seguimento (variando de 7,3 a 17,8 anos). Em estudo utilizando uma dose elevada de ciclofosfamida (Cytoxan) isolada, como terapia de resgate para AAS, constatou-se que 9 dos 17 pacientes avaliados (53%) apresentaram remissão completa ou parcial da doença após um seguimento médio de 29 meses.83 No entanto, um estudo comparando o uso de uma dose alta de ciclofosfamida com GAT foi concluído precocemente devido à excessiva morbidade e mortalidade induzidas pela ciclofosfamida.84 Sendo assim, o papel da ciclofosfamida administrada em doses altas no tratamento da anemia aplásica ainda precisa ser extensivamente esclarecido.
O daclizumabe consiste num anticorpo monoclonal humanizado, que se liga ao receptor de IL-2 e bloqueia a produção de IL-2 por células T ativadas. Dentre os 16 pacientes elegíveis que apresentavam anemia aplásica moderada e foram tratados com daclizumabe durante 3 meses, 6 (38%) responderam ao tratamento.85
A escolha da terapia adequada para pacientes com AAS é influenciada pela idade e grau de severidade da doença. O EBMT relatou os resultados da terapia imunossupressora de 810 pacientes, que foram subdivididos em 3 grupos de faixas etárias: menos de 49 anos; 50 a 59 anos; e mais de 60 anos. As taxas de sobrevida de 5 anos para os indivíduos com AAS foram de 86%, 72% e 54%, respectivamente. Para aqueles com AAMS, as taxas comparáveis foram de 49%, 40% e 21%.86 Os pacientes de idade mais avançada desenvolveram sangramentos e infecções em maior quantidade.
Pacientes com menos de 20 anos. O transplante alogênico de medula óssea deve ser realizado em casos de pacientes com menos de 20 anos de idade, quando há disponibilidade de um irmão compatível. Apesar dos riscos existentes, incluindo a GVHD crônica e a disfunção orgânica induzida pelo programa de condicionamento,62 é possível curar 50 a 80% dos pacientes, e a incidência de distúrbios clonais tardios é bastante baixa.68 O transplante alogênico de medula óssea, aliado aos programas de condicionamento que consistem na administração de ciclofosfamida e GAT, produziu uma taxa de sobrevida de 69% após 15 anos.68 Nos casos de pacientes com menos de 20 anos que não contam com um irmão compatível como doador, deve ser considerada um transplante com um doador compatível sem parentesco com o recebedor. O transplante alogênico com doador compatível sem parentesco inicialmente produziu uma taxa de sobrevida de 2 anos de apenas 29%, devido à ocorrência de GVHD severa .62 Um estudo envolvendo 15 pacientes que receberam transplante de doadores sem parentesco revelou que todos os pacientes haviam sobrevivido após 2 a 86 meses (média do tempo de seguimento igual a 51 meses) da realização do procedimento. Apenas um paciente desenvolveu uma GVHD extensa, enquanto 5 (33%) desenvolveram GVHD moderada. Estes resultados sugerem que os regimes de condicionamento contendo GAT e ciclofosfamida estão melhorando os resultados do tratamento em casos de transplante com doador sem parentesco, neste grupo de pacientes.87
Pacientes com 20 a 45 anos de idade. Os pacientes incluídos nesta faixa etária, cujo estado de saúde seja excelente e que disponham de um irmão totalmente compatível como doador, podem ser capazes de tolerar uma GVHD e, assim, serem beneficiados pelo potencial curativo do transplante alogênico de medula óssea. Alguns especialistas propõem que o transplante deva ser considerado em casos de pacientes que estejam dentro desta faixa etária,68 particularmente porque os programas de condicionamento mais modernos parecem ser capazes de diminuir a severidade da GVHD.62,88
Pacientes com mais de 45 anos. Antigamente, acreditava-se que o impacto da GVHD era severo demais para pacientes com mais de 45 anos de idade. Então, foi sugerido que estes pacientes fossem submetidos à terapia imunossupressora.62,68 No entanto, os programas de condicionamento contendo GAT e ciclofosfamida pareceram ser mais toleráveis, visto que mesmo os pacientes com até 59 anos de idade expostos ao tratamento intensivo apresentaram um resultado satisfatório após transplante de medula alogênica.64
Em adultos, a aplasia pura de células vermelhas (APCV) constitui um distúrbio adquirido. A anemia é severa (o hematócrito usualmente está abaixo de 20%), a reticulocitopenia é profunda (frequentemente a 0%), a contagem absoluta de reticulócitos costuma ser inferior a 10.000/mcL e os precursores eritroides medulares estão praticamente ausentes. No entanto, os elementos medulares mieloides e megacariocíticos são preservados e as contagens de plaquetas e de leucócitos no sangue periférico também permanecem normais.
Na APCV, considera-se que a eritropoiese seja primariamente inibida por mecanismos imunes, entre os quais a supressão dos progenitores eritroides mediada por autoanticorpos e por células T, em geral no estágio subsequente ao estágio de CFU-E da diferenciação eritroide e antes da formação dos proeritroblastos. As células T, em particular o linfócito grande granular (T-LGL, large granular lymphocyte), podem estar envolvidas na supressão da eritropoiese, sendo que em alguns casos há evidências de que esta supressão seja causada por células T clonais.89 A inibição da eritropoietina mediada por autoanticorpo também foi descrita.90 Contudo, a APCV mediada por anticorpos associada à terapia com agentes estimuladores eritropoiéticos (AEEs) é mais comum. Os anticorpos formados em resposta aos AEEs inibem o crescimento das colônias de células progenitoras eritroides in vitro91 e apresentam reação cruzada com a eritropoietina endógena, bem como com todos os AEEs atualmente disponíveis. Existem outros 2 mecanismos que provavelmente causam APCV: (1) um ataque específico do parvovírus B19 sobre os precursores eritroides (um relato indicou que 14% dos casos eram causados por este vírus)92; e (2) uma anormalidade clonal hematopoiética subjacente, que pode ser um pródromo da síndrome mielodisplásica.93
A APCV pode ser causada por uma variedade de processos, incluindo neoplasias, distúrbios autoimunes, fármacos e infecções [Tabela 3].
Tabela 3. Causas da aplasia pura de hemácias adquirida
|
Primárias |
|
Associada ao timoma em 10 a 15% dos casos.14 |
|
Causas idiopáticas. |
|
Secundárias |
|
Neoplasia: leucemia linfocítica crônica, leucemia mieloide crônica, linfomas de Hodgkin e não Hodgkin; distúrbios proliferativos linfocíticos grandes granulares; pródromo de síndromes mielodisplásicas.44 |
|
Lúpus eritematoso sistêmico ou artrite reumatoide. |
|
Associada à gravidez. |
|
Associada à anemia hemolítica autoimune. |
|
Fármacos: aqueles mais comumente associados são a fenitoína, clorpromazida, zidovudina (AZT),49 trimetroprima-sulfametoxazol, isoniazida.44 |
|
Insuficiência glandular endócrina múltipla. |
|
Amiloidose primária. |
|
Infecções: mononucleose infecciosa, hepatite viral, infecção por parvovírus, HIV.44 |
|
Transplante de medula óssea com incompatibilidade ABO. |
A associação da APCV com proliferação de LGL e leucemia tem sido cada vez mais reconhecida.62 O uso rotineiro de estudos sobre rearranjo do gene codificador do receptor de célula T em uma série mostrou que 9 em 14 pacientes apresentavam um distúrbio clonal de LGL.95 Provavelmente, estas células LGL fazem a mediação direta da inibição da eritropoiese.92,94 Em até 20% dos casos, talvez a APCV possa ser um pródromo das síndromes mielodisplásicas ou da leucemia mieloide aguda.92,95
A eritroblastopenia também ocorre em um pequeno percentual de pacientes com anemia hemolítica autoimune [ver Hemoglobinopatias e anemias hemolíticas] e pode ser causada por um ataque de autoanticorpos aos normoblastos em maturação.
O tratamento da infecção pelo HIV com zidovudina (AZT) produz, em quase todos os pacientes, uma anemia usualmente marcada por uma significativa macrocitose.96 Uma hipoplasia eritroide moderada constitui a causa habitual desta anemia, que pode progredir para APCV.
A infecção pelo parvovírus causa crises aplásicas transientes que ocorrem em pacientes com distúrbios hemolíticos severos. A medula dos pacientes com estes distúrbios deve compensar a hemólise periférica aumentando sua produção em até 7 vezes e, por isso, tipicamente mostra uma intensa hiperplasia eritroide. Embora o parvovírus possa afetar todas as células precursoras, os precursores das hemácias são as células mais profundamente afetadas.92
A APCV pode complicar um transplante de medula óssea alogênica ABO-incompatível. O soro do receptor continua a expressar iso-hemaglutininas anti-A ou anti-B contra o antígeno A ou B do doador, expresso na superfície das células progenitoras eritroides.95 Na APCV da gravidez, os anticorpos contra BFU-E costumam desaparecer após o parto e este fato coincide com a remissão clínica da condição.97
A APCV mediada por anticorpos e associada à terapia com AEEs apresentou um pico pronunciado de incidência em 2002, atribuído às lixívias oriundas dos pistões de borracha descobertos das seringas,98 que desde então foram substituídos por tampões de Teflon®.99 Entretanto, casos esporádicos continuam a ocorrer.
O paciente com APCV apresenta sintomas característicos de anemia como fraqueza, fadiga e falta de ar. As contagens de leucócitos e de plaquetas permanecem normais em termos de morfologia e função. Uma contagem de reticulócitos muito baixa – seja um valor relativo de reticulócitos abaixo de 0,2% ou uma contagem absoluta de reticulócitos significativamente baixa de menos de 10.000/mcL – deve levar o médico a solicitar um aspirado de medula óssea. Tipicamente, a saturação da transferrina está alta, uma vez que o ferro não utilizado é deslocado para fora da medula. Em um pacientes com APCV, um aspirado e biópsia de medula óssea caracteristicamente mostram uma mielopoiese, linfopoiese e megacariocitopoiese normais, enquanto a eritropoiese é quase inexistente. Na ausência de alguma causa evidente de APCV, existem 4 condições possíveis a serem consideradas: APCV idiopática, timoma, síndromes mielodisplásicas hipoplásicas e leucemia LGL. A avaliação para estabelecer o diagnóstico de APCV usualmente inclui um exame de tomografia computadorizada do tórax para avaliar a possibilidade de timoma; análise imunofenotípica dos linfócitos presentes no sangue circulante ou na medula para identificar a proliferação de LGL; citogenética de medula para avaliar a possibilidade de SMD; e testes de anticorpos contra o parvovírus.92 Uma característica diagnóstica marcante da infecção pelo parvovírus consiste no aparecimento de pronormoblastos gigantes na medula [Figura 4]. Pode ser difícil estabelecer a diferença entre a APCV associada às síndromes mielodisplásicas e a leucemia mieloide aguda no momento do diagnóstico, a menos que seja detectada uma típica anormalidade citogenética mielodisplásica durante o exame de medula óssea.
Se houver suspeita de APCV mediada por anticorpos, o paciente deve ser submetido a testes para detecção de anticorpos antieritropoietina neutralizantes.100 Outro indício do diagnóstico de APCV mediada por anticorpo é um baixo nível sérico de eritropoietina,91 em contraste com os elevados níveis tipicamente detectados em outras formas de APCV.
Os princípios gerais de tratamento para os pacientes com APCV são as transfusões para tratamento de anemia sintomática e a suspensão do uso de fármacos possivelmente agressores. Nenhuma terapia específica é indicada para as formas de APCV que sejam autolimitadas, como gravidez, transplante de medula óssea ABO-incompatível e alguns casos de infecção por parvovírus.95,97 O tratamento da APCV depende da causa identificada. Se houver um timoma, este deve ser removido cirurgicamente. Este procedimento promoveu a melhora dos pacientes em cerca de 1/3 dos casos de uma série,101 embora possa ser necessário instituir um tratamento adicional.102 Na impossibilidade de cirurgia, é preciso considerar a administração de um curso combinado de prednisona com octreotide, um análogo da somatostatina que se liga aos timomas, podendo inibir a função das células imunes tímicas.103
O tratamento de outras causas de APCV baseia-se na suposição de que o ataque seja imunomediado e, portanto, responsivo à terapia imunossupressora. O tratamento pode começar com a administração de 60 mg de prednisona oral, diariamente, em doses divididas. Este regime deve ser continuado por 1 a 3 meses.92 Se o paciente falhar em responder, conforme indicado por uma elevação da contagem de reticulócitos, devem ser adicionadas ciclofosfamida ou azatioprina na dose de 2 a 3 mg/kg/dia, por via oral. Pacientes que apresentam anormalidades citogenéticas medulares sugestivas de síndrome mielodisplásica são pouco responsivos.92,94 Alguns pacientes refratários a outras formas de terapia têm respondido bem à administração IV de IgG (0,4 g/kg/dia, durante 5 dias).104
Pacientes nos quais a proliferação de LGL constitui a causa subjacente são bem responsivos à ciclofosfamida.94,105 Usualmente, baixas doses de ciclofosfamida (50 a 100 mg/dia, PO) administradas durante 3 a 6 meses são o suficiente para promover a remissão que, às vezes, está associada ao desaparecimento da proliferação de LGL.94,105 Os pacientes pouco responsivos geralmente respondem à ciclosporina administrada por via oral.94,106 Foi demonstrado que a ciclosporina (12 mg/kg/dia) produz respostas em cerca de 65% dos casos, mesmo quando se trata de pacientes que não responderam à terapia com corticosteroides, plasmaférese, ciclofosfamida ou azatioprina.95,106
Para os pacientes nos quais a infecção por parvovírus constitui a causa da APCV, a administração IV de IgG funciona bem. A dosagem padrão é 0,4 g/kg/dia, durante 5 dias.92 Para pacientes com AIDS que apresentam infecção por parvovírus e APCV, a administração IV de IgG talvez tenha que ser continuada.107 A recuperação das crises de infecção por parvovírus ocorre espontaneamente, em 1 a 2 semanas após o aparecimento da infecção.
A terapia à base de AEEs para pacientes com APCV refratária é similar àquela instituída para pacientes com anemia aplásica (40 mg/kg/dia, IV, durante 4 dias).98 Outros fármacos utilizados em casos refratários são a azatioprina (2 a 3 mg/kg/dia), globulina antilinfócito e anticorpos monoclonais anti-CD20.62 Em casos bastante refratários, o transplante de medula óssea pode ser efetivo.108
No caso da APCV associada aos AEEs, o AEE em uso deve ser suspendido imediatamente e um tratamento imunossupressor deve ser iniciado.109 Em um estudo, 57% dos pacientes que receberam imunossupressão se recuperaram, em comparação aos 2% dos pacientes não submetidos à terapia imunossupressora.110 Pacientes novamente tratados com AEEs após terem desenvolvido APCV são mais propensos a responderem se o anticorpo neutralizador já não for mais detectável,110 embora haja relatos de recidiva subsequente.111 A Hematide (Affymax Inc., Palo Alto, California, USA) consiste em um antagonista de receptor de eritropoietina112 que não parece apresentar reação cruzada e foi utilizado com sucesso em um modelo experimental de APCV anticorpo-mediada em ratos.113 Um estudo clínico está em andamento para testar a eficácia e segurança da Hematide em pacientes com APCV mediada por anticorpo.100
Pode haver anemia com baixa contagem de reticulócitos, mesmo com uma intensa hiperplasia eritroide medular. Esta situação paradoxal constitui a principal característica da eritropoiese inefetiva ou da hemólise intramedular. É possível que haja comprometimento eritroide generalizado ou envolvimento de subpopulações específicas de precursores eritroides. Algumas destas subpopulações escapam da morte na medula, porém sua produção é tão severamente danificada que acabam sendo rapidamente removidas da circulação e, assim, conferem o quadro de hemólise periférica. Outros sinais de eritropoiese inefetiva incluem a icterícia, níveis séricos muito altos de desidrogenase lática, e 75 a 90% de saturação da capacidade de ligação do ferro sérico. O clássico quadro ferrocinético mostra uma rápida depuração do ferro plasmático, que indica a existência de uma intensa atividade precursora eritroide. A distribuição das hemácias marcadas para a circulação periférica, todavia, é dramaticamente reduzida, sugerindo que os precursores estão sendo destruídos por uma hemólise intramedular.
O diagnóstico diferencial inclui as anemias megaloblásticas e sideroblásticas, talassemias [ver Hemoglobinopatias e Anemias Hemolíticas], síndromes mielodisplásicas [ver Leucemia Aguda] e metaplasia mieloide agnogênica [ver Leucemia Mielógena Crônica e Outros Distúrbios Mieloproliferativos].
As anemias megaloblásticas são causadas pela deficiência de cobalamina (vitamina B12) ou de ácido fólico; por fármacos que interferem na síntese de DNA ou na absorção ou metabolismo da cobalamina; bem como por distúrbios genéticos que interferem no metabolismo do DNA ou na absorção ou distribuição da cobalamina.
A eritropoiese megaloblástica é caracterizada por uma síntese de DNA defeituosa e pela parada do ciclo na fase G2, com comprometimento da maturação e acúmulo de células que não sintetizam DNA, além de conterem DNA anômalo. Esta condição acarreta uma maturação assincrônica entre o núcleo e o citoplasma.114 A produção de RNA e síntese proteica continuam. Desta forma, células maiores (ou megaloblastos) são produzidas. Como consequência, a eritropoiese é inefetiva, havendo discordância quanto à ocorrência de um aumento da apoptose.115,116 Presume-se que defeitos similares de síntese de DNA caracterizem as anormalidades de mucosa ao nível do estômago e da língua. Na linhagem de células granulocíticas, a presença de metamielócitos gigantes representa a ocorrência de uma granulopoiese inefetiva.114
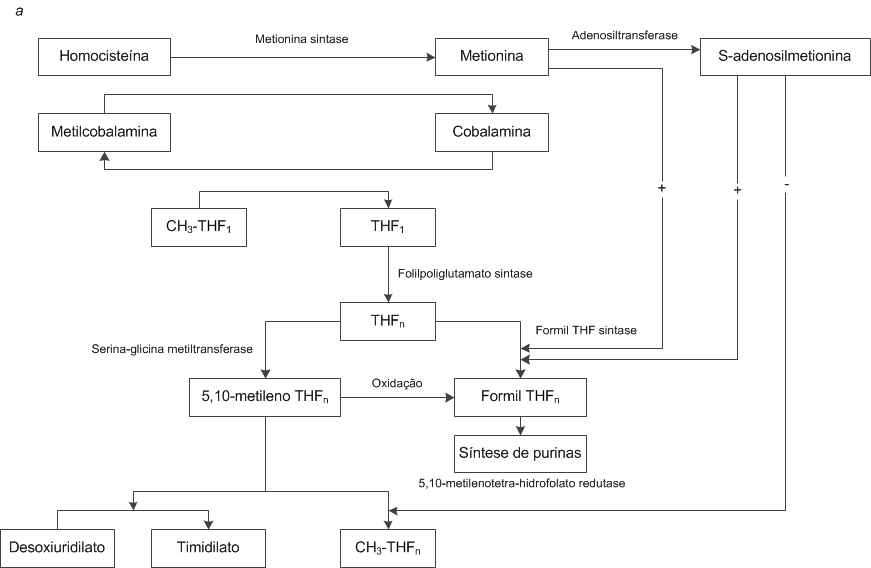
Papel do ácido fólico e da cobalamina. As interações entre o ácido fólico e a cobalamina são fundamentais para o metabolismo das unidades de carbono individuais, principalmente dos análogos de formil e metileno, que exercem papel central na síntese de DNA e purinas [Figura 5a].117 A cobalamina e o ácido fólico [Figura 6] se combinam na reação catalisada pela metionina sintase [Figura 5a], em que o grupo metil do CH3-THF1 é transferido para a cobalamina para formar metilcobalamina. Esta, por sua vez, transfere seu grupo metil para a homocisteína e forma metionina. O THF1 monoglutamatado que se forma nesta reação é poliglutamatado por ação da enzima folilpoliglutamato sintase, e um grupo metileno é adicionado pela serina-glicina metiltransferase para formar 5,10-metileno THFn. O 5,10-metileno THFn doa seu grupo metileno para converter a desoxiuridilato em timidilato, numa etapa essencial da síntese de DNA. O 5,10-metileno THFn também pode ser diretamente convertido a CH3-THF1 pela enzima 5,10-metilenotetra-hidrofolato redutase, disponibilizando assim o seu grupo metil.
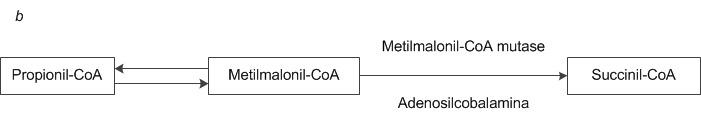
Figura 5. (a) A atividade de cofator interdependente intracelular da cobalamina e do ácido fólico é essencial na síntese e metabolismo de DNA.62 (b) A adenosilcobalamina atua como cofator na síntese de succinil-coenzima A a partir da metilmalonil-coenzima A.62 CoA = coenzima A.
O formil THFn (também denominado leucovorina, ácido folínico ou fator citrovorum) exerce papel importante na síntese de purinas e no metabolismo de DNA. Pode ser gerado pela oxidação do 5,10-metileno THFn por ação da enzima formil THF sintase, com a metionina fornecendo o grupo formato [Figura 5a].117 Quando há deficiência de cobalamina, o CH3-THF1 não consegue transferir seu grupo metil para a cobalamina. Dessa forma, o THF1 não fica livre para ser poliglutamatado pela folilpoliglutamato sintase [Figura 5a]. A forma poliglutamatada é necessária para a síntese do 5,10-metileno THFn ou do formil THFn. Sendo assim, as sínteses de DNA e de purinas são bloqueadas. Esta hipótese, denominada hipótese da captura de metilfolato, é sustentada pelo achado de níveis aumentados de CH3-THF1 no plasma de pacientes com deficiência de cobalamina. Uma explicação alternativa seria a hipótese da inanição de formato, segundo a qual a deficiência de cobalamina compromete a geração de metionina que, consequentemente, não pode fornecer os grupos metil necessários para que a enzima formil THFn sintase produza THFn.
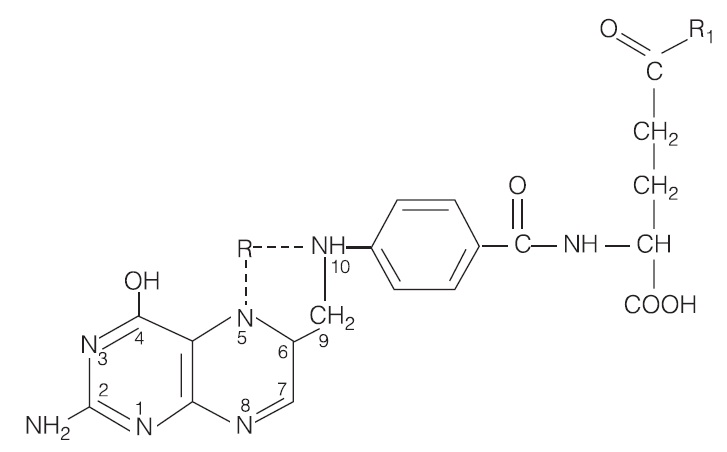
Figura 6. O ácido fólico atua como uma coenzima nas reações de transferência de carbono único. Permanece fisiologicamente inativa até ser reduzida nas posições 5, 6, 7 e 8 a tetra-hidrofolato. Grupos com carbono único (R), como os análogos de metil e formato, são adicionados na posição 5 ou 10, ou podem atuar como pontes para as posições 5 a 10, conforme mostra a ilustração. Vários glutamatos podem ser fixos em sequência (R1), convertendo o monoglutamato em poliglutamato. As enzimas da mucosa intestinal partem novamente os poliglutamatos em monoglutamato, enquanto as enzimas hepáticas adicionam glutamato ao tetra-hidrofolato ou a qualquer molécula de ácido fólico reduzido.
Outros aspectos do metabolismo do ácido fólico e da cobalamina. O ácido fólico e a cobalamina não são produzidos pelo corpo humano em quantidades adequadas. Ambos devem ser absorvidos a partir dos alimentos. A cobalamina, em particular, é derivada de fontes microbianas e ingerida por meio de carnes e ovos.
A maior parte do ácido fólico oriundo da dieta encontra-se na forma de poliglutamato e é absorvido pela mucosa intestinal. Sua absorção com marcação radioativa corresponde a aproximadamente 80% de uma dose de 200 mcg114,117 e os níveis séricos parecem ser mantidos pelo ácido fólico absorvido a partir dos alimentos. Observou-se a existência de uma circulação entero-hepática de ácido fólico: ele passa para a bile e intestino delgado e é quantitativamente reabsorvido. Em um modelo animal, a administração de etanol bloqueia a entrada do ácido fólico na bile. Este efeito poderia ser responsável, em parte, pela queda aguda dos níveis séricos de ácido fólico observada em 8 horas após o consumo de álcool. Uma queda semelhante dos níveis séricos de ácido fólico ocorre subsequentemente à ingesta de fenitoína. A necessidade diária de cobalamina é de aproximadamente 1 mcg. No entanto, a ingesta diária recomendada é de 2,4 mcg para adultos, a fim de garantir a quantidade de 1 mcg que o corpo precisa. Uma dieta ocidental típica que seja rica em produtos de origem animal geralmente fornece cerca de 5 a 15 mcg.118
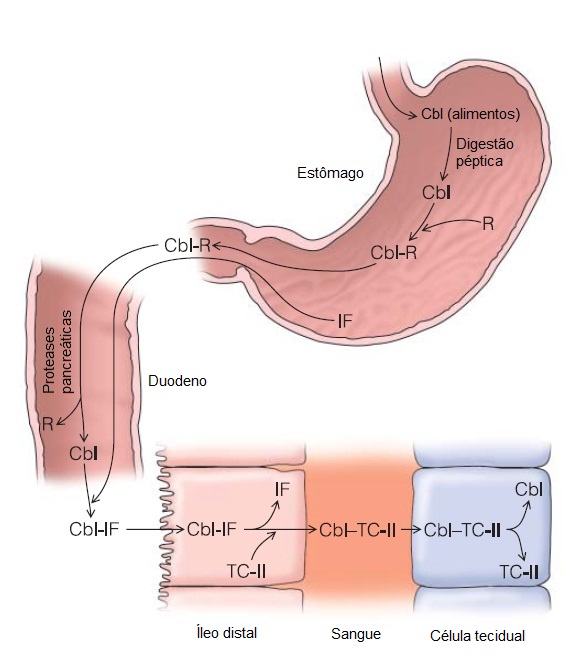
As proteínas R constituem uma classe de glicoproteínas ligadoras de cobalamina, encontradas na saliva e no suco gástrico. São produzidas por granulócitos e outros tecidos. O fator intrínseco (FI) consiste em uma glicoproteína de 45 kDa secretada pelas células parietais gástricas, altamente específica para a cobalamina inalterada. Após a ingesta, a cobalamina chega ao estômago ligada a proteínas animais e é liberada pela ação da pepsina e do ácido hidroclorídrico. A cobalamina livre, então, liga-se às proteínas R. O complexo proteína R/cobalamina não se liga aos receptores existentes no íleo e, assim, não é absorvido. No estômago, a cobalamina liga-se preferencialmente às proteínas R, em vez do FI.114,117,118 Portanto, o complexo proteína R/cobalamina fisiologicamente inativo é descarregado no duodeno. No duodeno e intestino delgado, todavia, as proteases pancreáticas degradam as proteínas R, liberando a cobalamina e permitindo sua ligação ao FI. O complexo FI/cobalamina, na presença de Ca2+ e em pH > 5,4, liga-se especificamente a um número limitado de sítios existentes nas microvilosidades das células mucosas localizadas junto à porção terminal do íleo, onde ocorre a absorção [Figura 7].117,188
Figura 7. Assimilação da cobalamina. A cobalamina oriunda da dieta (Cbl) entra no estômago e se liga às proteínas R. O complexo formado fisiologicamente inativo entra no duodeno. No intestino delgado, enzimas pancreáticas e pepsina digerem a proteína R e a Cbl se liga ao fator intrínseco (FI). O complexo FI/Cbl atravessa o intestino até atingir os receptores existentes nos microvilos das células mucosas localizadas no íleo distal. A Cbl, então, é transferida para a transcobalamina II (TC-II), que circula no sangue até se ligar aos receptores existentes nas células do corpo e ser internalizada.
No plasma, a maior parte da cobalamina encontra-se ligada à transcobalamina I (TC-I), que fica saturada em aproximadamente 70% com a cobalamina.119 A proteína de transporte fisiologicamente importante é a transcobalamina II (TC-II), que apresenta uma especificidade considerável pela cobalamina, sendo saturada por esta em apenas 5 a 10%. Os receptores para o complexo TC-II/cobalamina estão presentes em muitas membranas celulares. A TC-II liga cerca de 90% de uma dose de cobalamina recém-inoculada e o complexo formado é rapidamente depurado, com uma meia-vida de 6 a 9 minutos.120,121 Em indivíduos com deficiência congênita de TC-II, que resulta em uma severa anemia megaloblástica, há comprometimento tanto do transporte plasmático da cobalamina como de sua absorção. A absorção comprometida da cobalamina implica num papel para a TC-II junto ao enterócito ileal, onde a cobalamina é transferida do FI para a TC-II.
A elevação dos níveis de cobalamina observada em pacientes com leucemia granulocítica crônica ou granulocitose significativa é causada pelo aumento da concentração de TC-I, que é produzida nos granulócitos.
A etiologia da deficiência de cobalamina pode ser classificada como decorrente de uma deficiência dietética, anemia perniciosa (falta de FI), má absorção da cobalamina contida nos alimentos (incapacidade de liberar a cobalamina dos alimentos e disponibilizá-la para ligação ao FI), doença envolvendo o intestino delgado, insuficiência pancreática (embora seja discutido se a insuficiência pancreática de fato é capaz de causar deficiência de cobalamina) e distúrbios congênitos raros. Enquanto a má absorção da cobalamina contida nos alimentos é comum entre pacientes com deficiência subclínica, indivíduos com manifestações clínicas de deficiência de cobalamina são mais propensos a terem anemia perniciosa.122 A deficiência de cobalamina na anemia perniciosa é considerada resultante de uma gastrite autoimune e de um ataque autoimune ao FI gástrico. Existem 2 tipos de anticorpos anti-FI: um que bloqueia a fixação da cobalamina ao FI e outro que bloqueia a fixação do complexo FI/cobalamina aos receptores ileais.120 Clinicamente, anticorpos com alta especificidade para FI são encontrados em cerca de 70% dos pacientes com anemia perniciosa. Esta condição também pode ser causada por uma gastrite atrófica crônica, com consequente perda da produção de FI. A gastrite atrófica crônica na anemia perniciosa também está associada a um risco aumentado de desenvolvimento de câncer gástrico do tipo intestinal e de tumores carcinoides gástricos.121 A anemia perniciosa ocorre em associação com outros distúrbios autoimunes. Em um estudo, distúrbios autoimunes tireoidianos foram observados em 24% dos 162 pacientes com anemia perniciosa.123
Manifestações clínicas. Além da anemia megaloblástica e macrocítica, o paciente com deficiência de cobalamina pode apresentar fraqueza, letargia, icterícia e demência, bem como atrofia das papilas linguais e glossite. Uma neuropatia é o aspecto manifestado por cerca de 12% dos pacientes com deficiência de cobalamina (vitamina B12) sem anemia concomitante.124 Pacientes com deficiência severa de cobalamina inicialmente se queixam de parestesia. A sensação do toque e a sensibilidade à temperatura podem apresentar diminuição mínima. Comprometimento da memória e depressão podem ser proeminentes.124 A doença pode progredir, envolvendo a coluna dorsal, causando ataxia e fraqueza. O exame físico revela uma marcha de base ampla, sinal de Romberg, lentificação dos reflexos e perda do sentido de posição, além de uma sensação de vibração (em especial no teste com diapasão de 256 Hz). Se o distúrbio não for detectado e tratado, as colunas laterais são afetadas e o paciente desenvolve fraqueza, incapacidade de andar, clônus sustentado, hiperreflexia e sinal de Babinski. Visto que os nervos periféricos, bem como as colunas dorsal e lateral, são envolvidos, tais manifestações neurológicas às vezes são denominadas degeneração combinada subaguda ou doença sistêmica combinada subaguda.
A deficiência de cobalamina parece ser a causa de vários distúrbios neuropsiquiátricos, com sintomas como parestesia, ataxia, fraqueza dos membros, perturbações da marcha, defeitos de memória, alucinações e alterações de personalidade e humor.124 Estes sintomas, contudo, não podem ser facilmente responsabilizados pelo tipo de lesões na medula espinal que ocorrem em pacientes com deficiência de cobalamina. Pesquisadores tentaram determinar se um defeito na síntese de metionina ou uma anormalidade envolvendo o metabolismo do ácido propiônico seriam responsáveis pela neuropatia associada à deficiência de cobalamina [Figura 5b], porém o mecanismo exato continua obscuro. As evidências advindas destes estudos sustentam o comprometimento da metionina sintase como causa da neuropatia.114,124 Um estudo no qual vários metabólitos da cobalamina foram quantificados descobriu que apenas níveis elevados de cisteína plasmática eram preditivos de disfunção neurológica.125
Avaliação diagnóstica. A avaliação da suspeita de deficiência de cobalamina geralmente é realizada em 2 estágios: registro da existência de deficiência de vitamina e determinação de sua causa (p. ex., anemia perniciosa, má absorção, falta na dieta). Muitas vezes, é possível estabelecer o diagnóstico (1) medindo a concentração sérica de cobalamina, (2) avaliando metabólitos específicos, e (3) utilizando o teste de Schilling para estabelecer a ocorrência de má absorção de cobalamina. Infelizmente, este teste não está mais disponível na maior parte dos Estados Unidos, embora haja interesse em reativar seu uso como um teste importante para distinguir as etiologias associadas daquelas não associadas à má absorção, na deficiência da cobalamina.126
A macrocitose (volume corpuscular médio [VCM] maior que 100 fL) constitui a principal característica da deficiência de cobalamina, mas pode ser mascarada por distúrbios concomitantes, como a deficiência de ferro. Quando a macrocitose não é evidente ao exame do esfregaço periférico, pode ser facilmente detectada durante a realização das contagens de hemácias com contador de partículas eletrônico. O esfregaço periférico mostra a presença de macro-ovalócitos, hemácias com “cauda de peixe”, neutrófilos hipersegmentados e, ocasionalmente, hemácias nucleadas [Figura 2]. O achado de um PMN isolado apresentando 6 lobos ou de 5% dos PMNs apresentando 5 lobos constitui uma forte evidência de anemia megaloblástica. Em casos severos, o paciente apresenta granulocitopenia e trombocitopenia. Quando há uma deficiência de ferro severa concomitante à macrocitose, a expressão morfológica total da megaloblastose é bloqueada, embora os metamielócitos gigantes na medula e os PMNs hipersegmentados no sangue periférico ainda estejam presentes.
Os níveis plasmáticos de cobalamina e os níveis eritrocitários de ácido fólico devem ser medidos quando o VCM é maior que 100 fL. Quando obtidos, um aspirado e uma biópsia de medula tipicamente revelam a presença de uma enorme hiperplasia eritroide megaloblástica com metamielócitos gigantes.114 A hipercelularidade detectada ao exame de medula óssea pode ser tão expressiva e os megaloblastos tão imaturos que os médicos clínicos ocasionalmente ainda cometem o erro diagnóstico de identificar uma leucemia.114,127
A abordagem padrão para determinar a causa de uma deficiência de cobalamina comprovada baseia-se tradicionalmente no teste de Schilling. Este teste mede a absorção de cobalamina marcada com cobalto 57 (57Co). Após a administração de 1 mcg de cobalamina radiomarcada por via oral, 1 mmcg de cobalamina não marcada é administrado por via parenteral. A dose parenteral satura as transcobalaminas I e II, de modo que uma porção significativa do material absorvido é liberada e excretada na urina. Se a quantidade de 57Co-cobalamina medida em uma amostra de urina de 24 horas coletada corretamente for inferior a 10% da dose administrada por via oral, significa que a absorção de cobalamina é precária.
A má absorção de cobalamina pode ser demonstrada por meio do teste de Schilling alimentar, que encontra-se clinicamente indisponível. Este teste é realizado com ovos de galinhas inoculadas com cobalamina radioativa128 e indica se a quantidade de ácido-pepsina é insuficiente para quebrar o complexo cobalamina-enzima e disponibilizar cobalamina livre para se unir ao FI. Havendo uma forte suspeita de anemia perniciosa em paciente cujo teste de Schilling resultou aparentemente normal e cujos níveis de cobalamina plasmática não estejam diagnosticamente baixos, outras etapas devem ser seguidas para confirmar o diagnóstico, incluindo o exame da morfologia das hemácias, quantificação de anticorpos anti-FI ou realização de um estudo terapêutico utilizando cobalamina administrada por via parenteral. A quantificação dos níveis séricos de homocisteína e ácido metilmalônico tem sido cada vez mais utilizada, pois os níveis destas duas substâncias aumentam em consequência da deficiência de cobalamina [Figura 5].129
Se o teste de Schilling inicial apontar uma excreção diminuída de cobalamina, uma segunda fase do teste pode ser conduzida, com o objetivo de corrigir a absorção de cobalamina produzida pela anemia perniciosa. Nesta fase do teste, administra-se FI suplementar por via oral para normalizar a absorção da cobalamina, a menos que o FI não seja totalmente ativo e o paciente secrete anticorpos contra o FI ou esteja tomando fármacos que interfiram na absorção de cobalamina. Em nenhum dos casos, todavia, o FI suplementar será efetivo em pacientes com má absorção intestinal. É importante reconhecer que a deficiência de cobalamina prolongada compromete as células epiteliais intestinais e, consequentemente, a absorção. Portanto, o segundo estágio do teste deve ser realizado somente após várias semanas de terapia de reposição de cobalamina. Se o resultado obtido no segundo estágio do teste de Schilling for anormalmente baixo, isto sugere que o paciente apresenta má absorção intestinal, como pode ocorrer na psilose, na insuficiência pancreática ou nas síndromes de alça cega.
Uma alternativa ao uso do teste de Schilling consiste em fazer o teste de detecção de anticorpos anti-FI, cuja sensibilidade varia de 50 a 84% e a especificidade é alta.130 Os anticorpos anticélula parietal gástrica são menos sensíveis e específicos.
Fatores que afetam os resultados do teste. A existência de uma alfa-talassemia concomitante pode minimizar a macrocitose da anemia perniciosa.131 Esta possibilidade deve ser considerada particularmente em casos de pacientes afrodescendentes, entre os quais há uma elevada prevalência de alfa-talassemia (cerca de 30%). A anemia por inflamação crônica ou resultante de perda sanguínea e deficiência de ferro também pode diminuir o grau de macrocitose, porém sem afetar a hipersegmentação neutrofílica. Em um estudo, a deficiência de ferro foi descoberta em 20% dos 121 pacientes com anemia perniciosa;132 em outro, 19% dos pacientes com a mesma anemia não eram anêmicos e 33% não apresentavam macrocitose.133
Níveis séricos de cobalamina falsamente baixos são detectados durante a gravidez e quando há deficiência de ácido fólico.129 Em épocas passadas, o declínio desses níveis geralmente não era considerado importante, exceto quando os valores medidos eram baixos demais (p. ex., < 150 pg/mL). Todavia, foi esclarecido que os pacientes com níveis de até 250 pg/mL (e, talvez, mais altos) podem apresentar deficiência de cobalamina.128,133,134 Felizmente, o achado de macro-ovalócitos ou PMNs hipersegmentados no esfregaço de sangue periférico continua sendo indicador sensível dessa deficiência.
É importante notar que, embora 10 a 20% dos pacientes idosos apresentem níveis baixos de cobalamina, menos de 10% apresentarão evidências clínicas de deficiência dessa vitamina.122
Determinação da causa subjacente. Após identificar a existência de macrocitose e de níveis reduzidos de cobalamina, a causa destas condições deve ser determinada. É importante lembrar que a macrocitose pode resultar de outras circunstâncias além da anemia perniciosa, tais como deficiência de ácido fólico, doença hepática, consumo abusivo de álcool, reticulocitose e ingesta de alguns fármacos (p. ex., antimetabólitos, agentes alquilantes e zidovudina).96,131 A deficiência de cobalamina pode ser causada pela absorção inadequada provocada por anormalidades gástricas (p. ex., anemia perniciosa, gastrite) e doença envolvendo o intestino delgado (p. ex., psilose tropical, doença de Crohn), bem como insuficiência pancreática [Tabela 4].
Tabela 4. Causas da deficiência de cobalamina
|
Dieta inadequada |
|
Dieta estritamente vegetariana |
|
Anemia perniciosa (perda de FI) |
|
Má absorção da cobalamina presente nos alimentos (incapacidade de liberar a cobalamina contida nos alimentos e disponibilizá-la para ligação ao FI) |
|
Acloridria gástrica. |
|
Ressecção/reconstrução gástrica |
|
Gastrite |
|
Infecção por Helicobacter pylori |
|
Alcoolismo crônico |
|
Outras causas de má absorção |
|
Doença envolvendo o intestino delgado |
|
Ressecção ou desvio ileal |
|
Síndrome da alça cega com flora intestinal anormal |
|
Psilose celíaca |
|
Doença de Crohn |
|
Fármacos que interferem na absorção de cobalamina (em geral, após anos de uso) |
|
Neomicina |
|
Biguanidas |
|
Colchicina |
|
Etanol |
|
Ácido aminossalicílico |
|
Omeprazol |
|
Tênias de peixe que competem pela cobalamina |
|
Degradação de coenzimas da cobalamina |
|
Anestesia com N2O |
|
Distúrbios congênitos raros |
|
Deficiência de transcobalamina II |
|
Produção defeituosa de FI |
|
Síndrome de Imerslund-Grasbeck (má absorção seletiva de cobalamina com proteinúria) |
FI = Fator intrínseco.
A remoção de FI, pepsina e componentes secretores de ácido via cirurgia gástrica muitas vezes resulta em deficiência de cobalamina (detectada em 31% dos pacientes analisados em estudo135). Pacientes submetidos a esse procedimento devem passar por exames regulares para quantificação da cobalamina plasmática ou dos níveis de homocisteína, bem como receber suplementação terapêutica de cobalamina por toda a vida, caso apresentem níveis baixos.135
O pâncreas danificado não produz quantidades suficientes de tripsina e quimiotripsina para digestão dos complexos proteína R/cobalamina e liberação da vitamina para formação de complexo com o FI, o que pode provocar a má absorção da cobalamina [ver Doenças do Pâncreas].
Existem outras causas de deficiência. Em vegetarianos, especialmente os radicais, pode haver desenvolvimento de uma profunda anemia megaloblástica nutricional, como resultado da ingesta significativamente baixa de cobalamina, além de deficiências de ácido fólico e ferro.136 Um histórico detalhado do paciente deve indicar a possibilidade de ingesta dietética inadequada dessa vitamina. Bebês nascidos de mães vegetarianas radicais podem se tornar gravemente deficientes em cobalamina, em particular quando são amamentados.119 Entretanto, esta deficiência é surpreendentemente comum nos países menos desenvolvidos e nem sempre tem relação com o vegetarianismo estrito.119 A incidência é particularmente alta entre mulheres grávidas e crianças em idade pré-escolar.119
A reposição específica deve ser iniciada imediatamente após a conclusão do diagnóstico e a obtenção de amostras para determinar os níveis de cobalamina. Pacientes com níveis séricos baixos e anemia macrocítica devem ser submetidos a terapia à base de cobalamina parenteral. O diagnóstico de deficiência é confirmado quando a terapia com cobalamina produz uma reticulocitose em 3 a 4 dias, associada à elevação dos níveis de hemoglobina e à queda do VCM.
Se o paciente apresenta sintomas de anemia severa, é possível realizar a transfusão de hemácias concentradas, que deve ser administrada bem lentamente, a fim de evitar precipitação ou agravamento de insuficiência cardíaca congestiva. Esta é uma das poucas circunstâncias em que a transfusão de uma única unidade pode ser justificada pela possibilidade de produzir um aumento de 25% da capacidade de transporte de oxigênio. Deve ser administrada uma dose alta de cobalamina, visto que a sua retenção administrada por via parenteral é baixa, porém variável. Além disso, a vitamina tem baixo custo e não produz efeitos colaterais danosos. A reação dos reticulócitos tem início em 4 a 6 dias, sendo que a contagem de granulócitos, quando baixa, começa a aumentar no momento em que a reposição é iniciada. A hipersegmentação dos PMNs desaparece em 10 a 14 dias e isto sugere que, nas anemias megaloblásticas, a granulopoiese é afetada pela deficiência de cobalamina em duas etapas distintas: (1) na determinação do número de lobos dos PMNs; e (2) quando os granulócitos amadurecem e deixam a medula. A administração de doses semanais de 1000 mcg de cobalamina por um período de 6 semanas deve ser seguida da administração parenteral de dosagens mensais de 1000 mcmg por toda a vida. A indicação parenteral padrão é a cianocobalamina. Em casos de insuficiência pancreática, pode ser feita administração parenteral de cobalamina ou oral de enzimas pancreáticas. Uma terapia deve ser planejada especificamente para pacientes com má absorção intestinal.
Como uma pequena quantidade de cobalamina é absorvida mesmo na ausência de FI e devido à necessidade de apenas 2,4 mcg/dia, a administração de cobalamina por via oral mostrou-se adequada para fins de reposição em pacientes com anemia perniciosa que, assim, ficam liberados das injeções mensais (2?000 mcg/dia, PO, é a dose recomendada).137
Manifestações clínicas. O paciente com deficiência exibe uma manifestação clínica distinta dos indivíduos que apresentam cobalamina insuficiente;122 ele pode ingerir bebidas alcoólicas de maneira abusiva ou ter uma dieta pobre em ácido fólico. Subsequentemente à instituição da medida que obrigou o enriquecimento dos grãos com ácido fólico, em 1998, nos Estados Unidos, a deficiência diminuiu de forma dramática.138 As manifestações hematológicas são as mesmas produzidas pela falta de cobalamina: anemia macrocítica severa, baixa contagem absoluta de reticulócitos e um esfregaço sanguíneo característico, mostrando a presença de macro-ovalócitos, megaloblastos ocasionais e neutrófilos hipersegmentados. Pacientes com anemia megaloblástica que não apresentam glossite, histórico familiar de anemia perniciosa ou aspectos neurológicos característicos da insuficiência de cobalamina podem apresentar deficiência de ácido fólico.
Avaliação diagnóstica. É importante obter um meticuloso histórico alimentar, visto que o modismo das dietas, a ingesta alimentar precária e o alcoolismo constituem as causas usuais de deficiência severa desse ácido [Tabela 5]. Na avaliação clínica, devem ser obtidos os valores correspondentes aos níveis séricos de ácido fólico, cobalamina e ácido fólico eritrocitário, sendo que as deficiências dos 2 primeiros, por serem concomitantes, não são distintas com facilidade. O nível de ácido fólico contido nas hemácias reflete as reservas teciduais,139 mas pode estar diminuído em pacientes com deficiência severa de cobalamina. Em casos isolados, esses níveis geralmente estão normais ou elevados em indivíduos deficientes em cobalamina. A deficiência severa e prolongada dessa vitamina acarreta anorexia e perturbações GI, que podem causar insuficiência de ácido fólico alimentar. Como resultado, os níveis séricos de ambos tornam-se baixos e produzem um estado de dupla deficiência.
Uma concentração sérica de ácido fólico inferior a 2 ng/mL é consistente com a deficiência de ácido fólico, do mesmo modo que uma concentração eritrocitária de ácido fólico inferior a 150 ng/mL. Quando os resultados dos exames são inconclusivos ou há necessidade de distinguir uma megaloblastose decorrente de insuficiência de ácido fólico daquela resultante da deficiência de cobalamina, é útil quantificar os níveis séricos de ácido metilmalônico e homocisteína. Quando os testes de detecção de ambos os metabólitos resultam normais (p. ex., níveis de ácido metilmalônico iguais a 70 a 270 nmol/L e concentração total de homocisteína igual a 5 a 14 mcmol/L), exclui-se a possibilidade de deficiência de ambas as vitaminas. Se os níveis de metilmalonato estiverem normais, mas a concentração total de homocisteína estiver aumentada, é provável que o paciente tenha deficiência de ácido fólico e, neste caso, é apropriado iniciar uma investigação para descobrir a causa subjacente.139
Determinação da causa subjacente. A deficiência de ácido fólico é mais frequentemente causada por uma ingesta alimentar insuficiente, mas também pode resultar da absorção inadequada secundária a uma doença ou à administração de um fármaco [Tabela 5]. A ingesta de etanol por indivíduos bem nutridos não provoca o desenvolvimento de megaloblastose; por outro lado, quando há reservas limitadas de ácido fólico, o etanol pode diminuir os níveis séricos desta vitamina e bloquear a resposta de reticulócitos à administração de ácido fólico. O álcool pode bloquear a liberação de ácido fólico a partir dos tecidos para o soro.
Tabela 5. Causas da deficiência de ácido fólico
|
Mecanismo |
Causa |
|
Ingesta absolutamente inadequada |
Alcoolismo |
|
Deficiências nutricionais | |
|
Ingesta relativamente inadequada (resultante de aumento dos requerimentos de ácido fólico) |
Gravidez |
|
Hemólise severa | |
|
Hemodiálise crônica ou diálise peritoneal | |
|
Absorção inadequada |
Psilose tropical |
|
Enteropatia glúten-sensível (não psilose tropical) | |
|
Doença de Crohn | |
|
Linfoma ou amiloidose de intestino delgado | |
|
Enteropatia diabética | |
|
Ressecções ou desvios intestinais | |
|
Interferência fármaco-induzida no metabolismo de ácido fólico |
A ação da di-hidrofolato redutase é bloqueada pelo metotrexato, trimetoprima e pirimetamina |
|
Diminuição da absorção de folatos e depleção de folato nos tecidos causadas pela sulfassalazina | |
|
Interferência de um mecanismo desconhecido produzido pela fenitoína, etanol, fármacos antituberculose e anticoncepcionais orais (?) |
A anemia megaloblástica consequente à administração de fármacos ou gravidez provavelmente é causada pela deficiência de ácido fólico. Muitos agentes antineoplásicos e imunossupressores, como a fluorouracila, hidroxiureia, mercaptopurina, tioguanina, citarabina e azatioprina, provocam megaloblastose. Em mulheres grávidas, a ocorrência dessa anormalidade a princípio pode não ser evidente. Como é comum haver combinação das deficiências de ácido fólico e ferro, a expressão total da doença muitas vezes é bloqueada e a paciente apresenta uma anemia dimórfica, em vez de uma macro-ovulocitose facilmente identificável. A hipersegmentação dos PMNs persiste.114,139
Uma mutação cromossômica pode causar anormalidade no metabolismo do folato. Mulheres homozigotas para este defeito têm risco aumentado de gravidez afetada por defeitos do tubo neural. Uma das enzimas reguladoras dos níveis de homocisteína, a 5,10-metilenotetra-hidrofolato redutase, possui uma variante genética denominada C677T. Indivíduos homozigotos para esta variante apresentam níveis plasmáticos de homocisteína aumentados que podem ser diminuídos por meio da suplementação com folato. Cerca de 5 a 10% da população em geral é constituída por indivíduos homozigotos para esta variante. Mulheres grávidas e não grávidas homozigotas para a mutação C677T apresentam níveis eritrocitários de ácido fólico significativamente mais baixos.140 Estas mulheres podem ser suscetíveis ao desenvolvimento de doença cardiovascular e AVC, e seus bebês podem apresentar defeitos do tubo neural.140,141 Seria recomendável saber antes de uma possível gravidez se uma mulher é homozigota para esta variante e um teste genético seria útil para mulheres com histórico familiar deste tipo de defeito.
Vários distúrbios intestinais causam deficiência de ácido fólico, tais como doença pancreática severa e doença envolvendo o intestino delgado, incluindo má absorção, doença ileal, doença de Crohn, ressecção e desvio [Tabela 5]. Quando a deficiência de folato não possui causa evidente, pode ser prático suspeitar da existência de anormalidade não diagnosticada envolvendo má absorção. Em um estudo prospectivo envolvendo pacientes com deficiência de folato definida por exames laboratoriais, houve uma positividade de 10,9% para anticorpos contra doença celíaca e 4,7% dos pacientes tiveram confirmação histológica dessa doença.142
A terapia padrão para casos de deficiência de ácido fólico consiste na administração de 1 mg/dia, por via oral. A resposta, que se manifesta sob a forma de reticulocitose em 4 a 6 dias, perda da megaloblastose e normalização das contagens sanguíneas, confirma o diagnóstico. A hipersegmentação dos neutrófilos, contudo, desaparece somente após 10 a 14 dias.114 Pacientes com megaloblastose e depressão medular severa secundária à administração de fármacos que bloqueiam a di-hidrofolato redutase (p. ex., pirimetamina e metotrexato) podem ser tratados com ácido folínico. Em casos de toxicidade após a administração de doses altas isoladas de metotrexato, uma única dose equivalente de ácido folínico por via IM (p. ex., miligrama por miligrama) será suficiente. Em casos de toxicidade após a terapia crônica à base de pirimetamina, pode ser feita a administração de ácido folínico – 1 a 5 mg/dia – sem que os efeitos antimaláricos da pirimetamina sejam bloqueados. A megaloblastose causada pela terapia anticonvulsiva pode ser tratada com 1 mg/dia de ácido fólico. Durante a gravidez, recomenda-se adotar uma suplementação que, por sua vez, também pode ser útil para pacientes com hemólise crônica severa.
Na maioria dos pacientes (p. ex., aqueles que não requerem grandes quantidades de ácido fólico, em decorrência de condições como hemólise ou gravidez), a resposta hematológica ocorre após a administração de 200 mcg/dia de ácido fólico. A demanda aumentada durante a gravidez requer a administração de cerca de 200 a 300 mcg/dia.143 Além disso, a suplementação com ácido fólico parece prevenir a ocorrência de defeitos do tubo neural no feto,144 que podem ocorrer no embrião ou bem no início da gestação – até mesmo antes de a gravidez ser confirmada.145 Portanto, recomenda-se que as mulheres em idade fértil ou que estejam planejando engravidar recebam 400 mcg/dia. Mulheres homozigotas para a mutação C677T também devem tomar suplementos. Alimentos básicos, como farinha de trigo e cereais em grão, podem ser enriquecidos com ácido fólico. Entretanto, tem havido uma certa preocupação no sentido de que a suplementação pode mascarar os casos de megaloblastose da anemia perniciosa, levando ao desenvolvimento de neuropatias severas.145
A deficiência de cobre adquirida é rara e pode ser observada com o uso da nutrição parenteral total ou entérica de longa duração,146 bem como em casos de desnutrição, má absorção,147 ou secundariamente ao consumo excessivo de zinco.148 O aumento da concentração de zinco leva à deficiência de cobre em decorrência da regulação positiva da metalotioneína, uma proteína intracelular ligadora de cobre. Como resultado, o cobre é sequestrado dentro dos enterócitos, que subsequentemente sofrem necrose, e depois perdido por meio das fezes.148 O mecanismo de relação entre a deficiência de cobre e a anemia ainda não é totalmente conhecido, no entanto pode ser secundário à diminuição da atividade da oxidase do citocromo mitocondrial, com consequente síntese anormal do heme.149
A anemia causada por deficiência de cobre foi descrita como microcítica, normocítica e macrocítica.150 Neutropenia147 e, menos comumente, trombocitopenia151 também podem estar presentes. A medula óssea pode estar hipo ou hipercelular, e muitas vezes apresenta vacuolização citoplasmática dos precursores mieloides e eritroides.150 Displasia e sideroblastos anelares149 também foram descritos. Os achados neurológicos associados podem incluir uma mielopatia com marcha espástica e ataxia sensorial,152 e sua manifestação pode mimetizar a manifestação de uma deficiência de cobalamina. A repleção de cobre cessa a deterioração neurológica, contudo a melhora neurológica é variável.153 Quando há suspeita de deficiência de cobre, os níveis séricos ou plasmáticos desta molécula devem ser quantificados, ainda que esta medida possa ser relativamente insensível à deficiência de cobre não severa.147
O tratamento inclui a suspensão de qualquer ingesta excessiva de zinco e repleção de cobre. Um regime utilizado com sucesso consistiu na administração endovenosa de cloreto de cobre (14 doses de 2,5 mg/dia) seguida da administração PO de 3 mg de sulfato de cobre, 3 vezes/dia.149 O tratamento deve ser mantido enquanto houver perdas.
As anemias sideroblásticas podem ser hereditárias ou adquiridas e constituem um grupo heterogêneo de distúrbios caracterizados por anemia, presença de sideroblastos anelares na medula e eritropoiese inefetiva.154 As formas adquiridas são subdivididas em variantes clonais e não clonais. Uma variação bastante comum é a síndrome mielodisplásica, denominada anemia refratária com sideroblastos anelares. Além do álcool e dos fármacos (p. ex., isoniazida), as causas secundárias destas doenças permanecem em grande parte desconhecidas.
As anormalidades da síntese do heme provavelmente constituem a causa mais frequente das anemias sideroblásticas hereditárias. Os defeitos moleculares da enzima 5-aminolevulinato sintase foram descritos como causadores desta anormalidade.155,156 Esta enzima inicia a via sintética do heme e seu comprometimento o afeta profundamente ao realizar a síntese. Em outros casos, ocorrem deleções significativas no DNA mitocondrial. Embora o ferro seja introduzido nos precursores eritroides, a síntese do heme está comprometida e, assim, sua incorporação não pode ocorrer. Então, o ferro acumula-se nas cristas mitocondriais.155
O principal aspecto comum a todas as anemias sideroblásticas consiste numa anemia refratária ou progressiva. Entretanto, uma anemia branda que se estenda por toda a vida do paciente pode não ser notada. O diagnóstico de anemia sideroblástica é estabelecido pela detecção de reticulocitopenia, visto que as hemácias presentes no esfregaço com frequência são profundamente hipocrômicas e microcíticas, e podem haver hemácias distorcidas e pontilhado basofílico.157,158 As hemácias também podem ser normo ou macrocíticas, sendo possível observar uma população eritrocitária dimórfica. Ocasionalmente, corpúsculos de Pappenheimer (depósitos de ferro corados com azul da Prússia) estão presentes dentro das hemácias. Sideroblastos anelares são observados no aspirado de medula óssea (normoblastos de medula com densas incrustações de ferro sem ferritina na mitocôndria) [Figura 8]. Devido à eritropoiese inefetiva, a capacidade de ligação de ferro é saturada (usualmente, em torno de 80%) e há aumento dos níveis séricos de lactato desidrogenase. O estudo citogenético da medula óssea pode revelar um dos padrões típicos observados nas síndromes mielodisplásicas. As anemias sideroblásticas podem ser classificadas em 4 grupos: hereditárias e adquiridas (provavelmente não clonais), provavelmente benignas e distúrbios clonais [Tabela 6].
Figura 8. A coloração com azul da Prússia mostra os sideroblastos anelares na medula óssea de um paciente com anemia sideroblástica idiopática.
Tabela 6. Anemias sideroblásticas
|
Tipo |
Distúrbios |
|
Variante hereditária, provavelmente não clonal |
Distúrbios ligados ao sexo; distúrbios autossômicos. |
|
Variante adquirida, provavelmente não clonal |
Deleções de DNA mitocondrial (síndrome de Pearson). |
|
Variante provavelmente não clonal |
Fármaco-induzidos (p. ex., isoniazida ou outros fármacos antituberculose) ou induzidos por intoxicação com chumbo; sideroblastose alcoólica; anemia responsiva à piridoxina. |
|
Distúrbio clonal (síndrome mielodisplásica) |
Anemia refratária com sideroblastos anelares; anemia sideroblástica idiopática adquirida. |
Para fins prognósticos, é importante decidir se o paciente apresenta uma forma clonal ou não clonal de anemia sideroblástica e reconhecer as formas reversíveis de anemia sideroblástica (p. ex., aquelas causadas por alcoolismo, deficiência de ácido fólico e fármacos, como isoniazida e cloranfenicol) e descontinuar o uso de qualquer agente potencialmente agressor.
Os indicadores de mielodisplasia incluem a granulocitopenia, trombocitopenia, granulopoiese medular displásica, megacariócitos bilobados e anormalidades citogenéticas. Em casos raros, os pacientes apresentam uma resposta de reticulócitos e hemoglobina à administração de piridoxina (200 a 600 mg/dia), com ou sem ácido fólico.159 A sobrecarga de ferro ocorre com frequência,160,161 mesmo na ausência de transfusões, e pode causar uma considerável morbidade. Os índices de ferro devem ser monitorados regularmente e, quando houver sobrecarga, a terapia à base de quelação deve ser iniciada.
Stanley L. Schrier, MD, FACP atuou como consultor junto às empresas Tularik, Inc., Amgen, Inc, Fibrogen, Inc, Apotex, Inc. e Receptron, Inc.
Elizabeth A. Price, MD, MPH não possui relações comerciais com os fabricantes de produtos e prestadores de serviços mencionados neste capítulo.
1. Means RT Jr, Krantz SB. Progress in understanding the pathogenesis of the anemia of chronic disease. Blood 1992;80:1639–47.
2. Cash JM, Sears DA. The anemia of chronic disease: spectrum of associated diseases in a series of unselected hospitalized patients. Am J Med 1989;87:638–44.
3. Means RT Jr. Pathogenesis of the anemia of chronic disease: a cytokine-mediated anemia. Stem Cells 1995;13: 32–7.
4. Lacombe C. Resistance to erythropoietin. N Engl J Med 1996;334:660–2.
5. Voulgari PV, Kolios G, Papadopoulos GK, et al. Role of cytokines in the pathogenesis of anemia of chronic disease in rheumatoid arthritis. Clin Immunol 1999;92:153–60.
6. Ganz T. Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood 2003;102: 783–8.
7. Nemeth E, Tuttle MS, Powelson J, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004;306:2090–3.
8. Wrighting DM, Andrews NC. Interleukin-6 induces hepci-din expression through STAT3. Blood 2006;108:3204–9.
9. Nemeth E, Valore EV, Territo M, et al. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood 2003;101:2461–3.
10. Nemeth E, Rivera S, Gabayan V, et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest 2004;113:1271–6.
11. Nicolas G, Chauvet C, Viatte L, et al. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest 2002; 110:1037–44.
12. Roy CN, Mak HH, Akpan I, et al. Hepcidin antimicrobial peptide transgenic mice exhibit features of the anemia of inflammation. Blood 2007;109:4038–44.
13. Intragumtornchai T, Huebers HA, Eng M, Finch CA. In vivo transferrin-iron receptor relationships in erythron of rats. Am J Physiol 1988;255(2 Pt 2):R326–31.
14. Fitzsimons EJ, Houston T, Munro R, et al. Erythroblast iron metabolism and serum soluble transferrin receptor values in the anemia of rheumatoid arthritis. Arthritis Rheum 2002;47:166–71.
15. Punnonen K, Irjala K, Rajamaki A. Serum transferrin receptor and its ratio to serum ferritin in the diagnosis of iron deficiency. Blood 1997;89:1052–7.
16. Rimon E, Levy S, Sapir A, et al. Diagnosis of iron defi-ciency anemia in the elderly by transferrin receptor-ferritin index. Arch Intern Med 2002;162:445–9.
17. Claster S. Biology of anemia, differential diagnosis, and treatment options in human immunodeficiency virus infection. J Infect Dis 2002;185(Suppl 2):S105–9.
18. Gasche C, Waldhoer T, Feichtenschlager T, et al. Predic-tion of response to iron sucrose in inflammatory bowel disease-associated anemia. Am J Gastroenterol 2001;96: 2382–7.
19. Schroder O, Mickisch O, Seidler U, et al. Intravenous iron sucrose versus oral iron supplementation for the treatment of iron deficiency anemia in patients with inflammatory bowel disease—a randomized, controlled, open-label, multicenter study. Am J Gastroenterol 2005;100:2503–9.
20. Pincus T, Olsen NJ, Russell IJ, et al. Multicenter study of recombinant human erythropoietin in correction of anemia in rheumatoid arthritis. Am J Med 1990;89:161–8.
21. Henry DH, Beall GN, Benson CA, et al. Recombinant human erythropoietin in the treatment of anemia associ-ated with human immunodeficiency virus (HIV) infection and zidovudine therapy. Overview of four clinical trials. Ann Intern Med 1992;117:739–48.
22. Schreiber S, Howaldt S, Schnoor M, et al. Recombinant erythropoietin for the treatment of anemia in inflamma-tory bowel disease. N Engl J Med 1996;334:619–23.
23. Goodnough LT, Skikne B, Brugnara C. Erythropoietin, iron, and erythropoiesis. Blood 2000;96:823–33.
24. Ludwig H, Fritz E, Leitgeb C, et al. Prediction of response to erythropoietin treatment in chronic anemia of cancer. Blood 1994;84:1056–63.
25. Henke M, Laszig R, Rube C, et al. Erythropoietin to treat head and neck cancer patients with anaemia undergoing radiotherapy: randomised, double-blind, placebo-controlled trial. Lancet 2003;362:1255–60.
26. Leyland-Jones B, Semiglazov V, Pawlicki M, et al. Main-taining normal hemoglobin levels with epoetin alfa in mainly nonanemic patients with metastatic breast cancer receiving first-line chemotherapy: a survival study. J Clin Oncol 2005;23:5960–72.
27. Rizzo JD, Somerfield MR, Hagerty KL, et al. Use of epoetin and darbepoetin in patients with cancer: 2007 American Society of Clinical Oncology/American Society of Hema-tology clinical practice guideline update. J Clin Oncol 2008; 26:132–49.
28. Allen DA, Breen C, Yaqoob MM, Macdougall IC. Inhibi-tion of CFU-E colony formation in uremic patients with inflammatory disease: role of IFN-gamma and TNF-alpha. J Investig Med 1999;47:204–11.
29. Rao DS, Shih MS, Mohini R. Effect of serum parathyroid hormone and bone marrow fibrosis on the response to erythropoietin in uremia. N Engl J Med 1993;328:171–5.
30. Mladenovic J. Aluminum inhibits erythropoiesis in vitro. J Clin Invest 1988;81:1661–5.
31. Eschbach JW, Egrie JC, Downing MR, et al. Correction of the anemia of end-stage renal disease with recombinant human erythropoietin. Results of a combined phase I and II clinical trial. N Engl J Med 1987;316:73–8.
32. Drueke TB, Locatelli F, Clyne N, et al. Normalization of hemoglobin level in patients with chronic kidney disease and anemia. N Engl J Med 2006;355:2071–84.
33. Singh AK, Szczech L, Tang KL, et al. Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med 2006;355:2085–98.
34. National Kidney Foundation. KDOQI clinical practice guideline and clinical practice recommendations for anemia in chronic kidney disease: 2007 update of hemoglobin target. Am J Kidney Dis 2007;50:471.
35. Bailie GR, Johnson CA, Mason NA. Parenteral iron use in the management of anemia in end-stage renal disease patients. Am J Kidney Dis 2000;35:1–12.
36. Bailie GR, Clark JA, Lane CE, Lane PL. Hypersensitivity reactions and deaths associated with intravenous iron preparations. Nephrol Dial Transplant 2005;20:1443–9.
37. Singh H, Reed J, Noble S, et al. Effect of intravenous iron sucrose in peritoneal dialysis patients who receive erythropoiesis-stimulating agents for anemia: a random-ized, controlled trial. Clin J Am Soc Nephrol 2006;1: 475–82.
38. Altmann P, Plowman D, Marsh F, Cunningham J. Aluminium chelation therapy in dialysis patients: evidence for inhibition of haemoglobin synthesis by low levels of aluminium. Lancet 1988;1:1012–5.
39. Colman N, Herbert V. Hematologic complications of alcoholism: overview. Semin Hematol 1980;17:164–76.
40. Lindenbaum J. Folate and vitamin B12 deficiencies in alcoholism. Semin Hematol 1980;17:119–29.
41. Latvala J, Parkkila S, Melkko J, Niemela O. Acetaldehyde adducts in blood and bone marrow of patients with ethanol-induced erythrocyte abnormalities. Mol Med 2001; 7:401–5.
42. Baldwin JG Jr. Hematopoietic function in the elderly. Arch Intern Med 1988;148:2544–6.
43. Guralnik JM, Eisenstaedt RS, Ferrucci L, et al. Prevalence of anemia in persons 65 years and older in the United States: evidence for a high rate of unexplained anemia. Blood 2004;104:2263–8.
44. Patel KV, Harris TB, Faulhaber M, et al. Racial variation in the relationship of anemia with mortality and mobility disability among older adults. Blood 2007;109:4663–70.
45. Beutler E, West C. Hematologic differences between African-Americans and whites: the roles of iron deficiency and alpha-thalassemia on hemoglobin levels and mean corpuscular volume. Blood 2005;106:740–5.
46. Penninx BW, Guralnik JM, Onder G, et al. Anemia and decline in physical performance among older persons. Am J Med 2003;115:104–10.
47. Penninx BW, Pahor M, Cesari M, et al. Anemia is associ-ated with disability and decreased physical performance and muscle strength in the elderly. J Am Geriatr Soc 2004; 52:719–24.
48. Denny SD, Kuchibhatla MN, Cohen HJ. Impact of anemia on mortality, cognition, and function in community-dwelling elderly. Am J Med 2006;119:327–34.
49. Penninx BW, Pahor M, Woodman RC, Guralnik JM. Anemia in old age is associated with increased mortality and hospitalization. J Gerontol A Biol Sci Med Sci 2006; 61:474–9.
50. Bacigalupo A, Hows J, Gluckman E, et al. Bone marrow transplantation (BMT) versus immunosuppression for the treatment of severe aplastic anaemia (SAA): a report of the EBMT SAA working party. Br J Haematol 1988;70:177–82.
51. Brown KE, Tisdale J, Barrett AJ, et al. Hepatitis-associated aplastic anemia. N Engl J Med 1997;336:1059–64.
52. Tzakis AG, Arditi M, Whitington PF, et al. Aplastic anemia complicating orthotopic liver transplantation for non-A, non-B hepatitis. N Engl J Med 1988;319:393–6.
53. Young NS. Autoimmunity and its treatment in aplastic anemia. Ann Intern Med 1997;126:166–8.
54. Young NS, Maciejewski J. The pathophysiology of acquired aplastic anemia. N Engl J Med 1997;336:1365–72.
55. Solomou EE, Rezvani K, Mielke S, et al. Deficient CD4+ CD25+ FOXP3+ T regulatory cells in acquired aplastic anemia. Blood 2007;110:1603–6.
56. Maciejewski JP, Selleri C, Sato T, et al. A severe and consistent deficit in marrow and circulating primitive hematopoietic cells (long-term culture-initiating cells) in acquired aplastic anemia. Blood. 1996;88:1983–91.
57. Cong YS, Wright WE, Shay JW. Human telomerase and its regulation. Microbiol Mol Biol Rev 2002;66:407–25.
58. Young NS. Pathophysiologic mechanisms in acquired aplastic anemia. Hematol Am Soc Hematol Educ Progr 2006;72–7.
59. Yamaguchi H, Calado RT, Ly H, et al. Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia. N Engl J Med 2005;352:1413–24.
60. Fogarty PF, Yamaguchi H, Wiestner A, et al. Late presenta-tion of dyskeratosis congenita as apparently acquired aplastic anaemia due to mutations in telomerase RNA. Lancet 2003;362:1628–30.
61. Go RS, Lust JA, Phyliky RL. Aplastic anemia and pure red cell aplasia associated with large granular lymphocyte leukemia. Semin Hematol 2003;40:196–200.
62. Young NS. Aplastic anaemia. Lancet 1995;346:228–32.
63. Rosenfeld SJ, Kimball J, Vining D, Young NS. Intensive immunosuppression with antithymocyte globulin and cyclosporine as treatment for severe acquired aplastic anemia. Blood 1995;85:3058–65.
64. Storb R, Blume KG, O’Donnell MR, et al. Cyclophospha-mide and antithymocyte globulin to condition patients with aplastic anemia for allogeneic marrow transplanta-tions: the experience in four centers. Biol Blood Marrow Transplant 2001;7:39–44.
65. Deeg HJ, Leisenring W, Storb R, et al. Long-term outcome after marrow transplantation for severe aplastic anemia. Blood 1998;91:3637–45.
66. Schrezenmeier H, Passweg JR, Marsh JC, et al. Worse outcome and more chronic GVHD with peripheral blood progenitor cells than bone marrow in HLA-matched sibling donor transplants for young patients with severe acquired aplastic anemia. Blood 2007;110:1397–400.
67. Locasciulli A, Oneto R, Bacigalupo A, et al. Outcome of patients with acquired aplastic anemia given first line bone marrow transplantation or immunosuppressive treatment in the last decade: a report from the European Group for Blood and Marrow Transplantation (EBMT). Haematologica 2007;92:11–8.
68. Doney K, Leisenring W, Storb R, Appelbaum FR. Primary treatment of acquired aplastic anemia: outcomes with bone marrow transplantation and immunosuppressive therapy. Seattle Bone Marrow Transplant Team. Ann Intern Med 1997;126:107–15.
69. Frickhofen N, Heimpel H, Kaltwasser JP, Schrezenmeier H. Antithymocyte globulin with or without cyclosporin A: 11-year follow-up of a randomized trial comparing treatments of aplastic anemia. Blood 2003;101:1236–42.
70. Bacigalupo A, Bruno B, Saracco P, et al. Antilymphocyte globulin, cyclosporine, prednisolone, and granulocyte colony-stimulating factor for severe aplastic anemia: an update of the GITMO/EBMT study on 100 patients. European Group for Blood and Marrow Transplantation (EBMT) Working Party on Severe Aplastic Anemia and the Gruppo Italiano Trapianti di Midolio Osseo (GITMO). Blood 2000;95:1931–4.
71. Frickhofen N, Kaltwasser JP, Schrezenmeier H, et al. Treat-ment of aplastic anemia with antilymphocyte globulin and methylprednisolone with or without cyclosporine. The German Aplastic Anemia Study Group. N Engl J Med 1991;324:1297–304.
72. Marsh J. Making therapeutic decisions in adults with aplastic anemia. Hematol Am Soc Hematol Educ Progr 2006;78–85.
73. Socie G, Henry-Amar M, Bacigalupo A, et al. Malignant tumors occurring after treatment of aplastic anemia. European Bone Marrow Transplantation-Severe Aplastic Anaemia Working Party. N Engl J Med 1993;329:1152–7.
74. Di Bona E, Rodeghiero F, Bruno B, et al. Rabbit antithymo-cyte globulin (r-ATG) plus cyclosporine and granulocyte colony stimulating factor is an effective treatment for aplastic anaemia patients unresponsive to a first course of intensive immunosuppressive therapy. Gruppo Italiano Trapianto di Midollo Osseo (GITMO). Br J Haematol 1999; 107:330–4.
75. Scheinberg P, Nunez O, Young NS. Retreatment with rabbit anti-thymocyte globulin and ciclosporin for patients with relapsed or refractory severe aplastic anaemia. Br J Haematol 2006;133:622–7.
76. Gupta V, Gordon-Smith EC, Cook G, et al. A third course of anti-thymocyte globulin in aplastic anaemia is only beneficial in previous responders. Br J Haematol 2005;129: 110–7.
77. Sloand EM, Yong AS, Ramkissoon S, et al. Granulocyte colony-stimulating factor preferentially stimulates proliferation of monosomy 7 cells bearing the isoform IV receptor. Proc Natl Acad Sci U S A 2006;103:14483–8.
78. Teramura M, Kimura A, Iwase S, et al. Treatment of severe aplastic anemia with antithymocyte globulin and cyclo-sporin A with or without G-CSF in adults: a multicenter randomized study in Japan. Blood 2007;110:1756–61.
79. Kojima S, Ohara A, Tsuchida M, et al. Risk factors for evolution of acquired aplastic anemia into myelodysplastic syndrome and acute myeloid leukemia after immunosup-pressive therapy in children. Blood 2002;100:786–90.
80. Socie G, Mary JY, Schrezenmeier H, et al. Granulocyte-stimulating factor and severe aplastic anemia: a survey by the European Group for Blood and Marrow Transplanta-tion (EBMT). Blood 2007;109:2794–6.
81. Marsh JC, Ganser A, Stadler M. Hematopoietic growth factors in the treatment of acquired bone marrow failure states. Semin Hematol 2007;44:138–47.
82. Brodsky RA, Sensenbrenner LL, Jones RJ. Complete remis-sion in severe aplastic anemia after high-dose cyclophos-phamide without bone marrow transplantation. Blood 1996;87:491–4.
83. Brodsky RA, Chen AR, Brodsky I, Jones RJ. High-dose cyclophosphamide as salvage therapy for severe aplastic anemia. Exp Hematol 2004;32:435–40.
84. Tisdale JF, Dunn DE, Geller N, et al. High-dose cyclophos-phamide in severe aplastic anaemia: a randomised trial. Lancet 2000;356:1554–9.
85. Maciejewski JP, Sloand EM, Nunez O, et al. Recombinant humanized anti-IL-2 receptor antibody (daclizumab) pro-duces responses in patients with moderate aplastic anemia. Blood 2003;102:3584–6.
86. Tichelli A, Socie G, Henry-Amar M, et al. Effectiveness of immunosuppressive therapy in older patients with aplastic anemia. European Group for Blood and Marrow Transplantation Severe Aplastic Anaemia Working Party. Ann Intern Med 1999;130:193–201.
87. Kojima S, Inaba J, Kondo M, et al. Unrelated donor marrow transplantation for severe acquired aplastic anemia using cyclophosphamide, antithymocyte globulin, and total body irradiation. Blood 1995;85:291–2.
88. Paquette RL, Tebyani N, Frane M, et al. Long-term outcome of aplastic anemia in adults treated with antithymocyte globulin: comparison with bone marrow transplantation. Blood 1995;85:283–90.
89. Maung ZT, Norden J, Middleton PG, et al. Pure red cell aplasia: further evidence of T cell clonal disorder. Br J Haematol 1994;87:189–92.
90. Casadevall N, Dupuy E, Molho-Sabatier P, et al. Autoanti-bodies against erythropoietin in a patient with pure red-cell aplasia. N Engl J Med 1996;334:630–3.
91. Casadevall N, Nataf J, Viron B, et al. Pure red-cell aplasia and antierythropoietin antibodies in patients treated with recombinant erythropoietin. N Engl J Med 2002;346: 469–75.
92. Charles RJ, Sabo KM, Kidd PG, Abkowitz JL. The patho-physiology of pure red cell aplasia: implications for therapy. Blood 1996;87:4831–8.
93. Park S, Merlat A, Guesnu M, et al. Pure red cell aplasia associated with myelodysplastic syndromes. Leukemia 2000;14:1709–10.
94. Lacy MQ, Kurtin PJ, Tefferi A. Pure red cell aplasia: asso-ciation with large granular lymphocyte leukemia and the prognostic value of cytogenetic abnormalities. Blood 1996;87:3000–6.
95. Fisch P, Handgretinger R, Schaefer HE. Pure red cell aplasia. Br J Haematol 2000;111:1010–22.
96. Walker RE, Parker RI, Kovacs JA, et al. Anemia and eryth-ropoiesis in patients with the acquired immunodeficiency syndrome (AIDS) and Kaposi sarcoma treated with zidovudine. Ann Intern Med 1988;108:372–6.
97. Baker RI, Manoharan A, de Luca E, Begley CG. Pure red cell aplasia of pregnancy: a distinct clinical entity. Br J Haematol 1993;85:619–22.
98. Boven K, Stryker S, Knight J, et al. The increased incidence of pure red cell aplasia with an Eprex formulation in uncoated rubber stopper syringes. Kidney Int 2005;67: 2346–53.
99. Pollock C, Johnson DW, Horl WH, et al. Pure red cell apla-sia induced by erythropoiesis-stimulating agents. Clin J Am Soc Nephrol 2008;3:193–9.
100. Macdougall IC. Epoetin-induced pure red cell aplasia: diagnosis and treatment. Curr Opin Nephrol Hypertens 2007;16:585–8.
101. Zeok JV, Todd EP, Dillon M, et al. The role of thymectomy in red cell aplasia. Ann Thorac Surg 1979;28:257–60.
102. Thompson CA, Steensma DP. Pure red cell aplasia associa-ted with thymoma: clinical insights from a 50-year single-institution experience. Br J Haematol 2006;135:405–7.
103. Palmieri G, Lastoria S, Colao A, et al. Successful treatment of a patient with a thymoma and pure red-cell aplasia with octreotide and prednisone. N Engl J Med 1997;336:263–5.
104. McGuire WA, Yang HH, Bruno E, et al. Treatment of anti-body-mediated pure red-cell aplasia with high-dose intra-venous gamma globulin. N Engl J Med 1987;317:1004–8.
105. Yamada O, Mizoguchi H, Oshimi K. Cyclophosphamide therapy for pure red cell aplasia associated with granular lymphocyte-proliferative disorders. Br J Haematol 1997;97: 392–9.
106. Means RT Jr, Dessypris EN, Krantz SB. Treatment of refractory pure red cell aplasia with cyclosporine A: disappearance of IgG inhibitor associated with clinical response. Br J Haematol 1991;78:114–9.
107. Ramratnam B, Gollerkeri A, Schiffman FJ, et al. Manage-ment of persistent B19 parvovirus infection in AIDS. Br J Haematol 1995;91:90–2.
108. Muller BU, Tichelli A, Passweg JR, et al. Successful treat-ment of refractory acquired pure red cell aplasia (PRCA) by allogeneic bone marrow transplantation. Bone Marrow Transplant 1999;23:1205–7.
109. Verhelst D, Rossert J, Casadevall N, et al. Treatment of erythropoietin-induced pure red cell aplasia: a retrospective study. Lancet 2004;363:1768–71.
110. Bennett CL, Cournoyer D, Carson KR, et al. Long-term outcome of individuals with pure red cell aplasia and antierythropoietin antibodies in patients treated with recombinant epoetin: a follow-up report from the Research on Adverse Drug Events and Reports (RADAR) Project. Blood 2005;106:3343–7.
111. Andrade J, Taylor PA, Love JM, Levin A. Successful reintroduction of a different erythropoiesis-stimulating agent after pure red cell aplasia: relapse after successful therapy with prednisone. Nephrol Dial Transplant 2005;20:2548–51.
112. Fan Q, Leuther KK, Holmes CP, et al. Preclinical evalua-tion of Hematide, a novel erythropoiesis stimulating agent, for the treatment of anemia. Exp Hematol 2006;34: 1303–11.
113. Woodburn KW, Fan Q, Winslow S, et al. Hematide is immunologically distinct from erythropoietin and corrects anemia induced by antierythropoietin antibodies in a rat pure red cell aplasia model. Exp Hematol 2007;35:1201–8.
114. Wickramasinghe SN. Morphology, biology and biochem-istry of cobalamin- and folate-deficient bone marrow cells. Baillieres Clin Haematol 1995;8:441–59.
115. Ingram CF, Davidoff AN, Marais E, et al. Evaluation of DNA analysis for evidence of apoptosis in megaloblastic anaemia. Br J Haematol 1997;96:576–83.
116. Koury MJ, Horne DW. Apoptosis mediates and thymidine prevents erythroblast destruction in folate deficiency anemia. Proc Natl Acad Sci U S A 1994;91:4067–71.
117. Tefferi A, Pruthi RK. The biochemical basis of cobalamin deficiency. Mayo Clin Proc 1994;69:181–6.
118. Pruthi RK, Tefferi A. Pernicious anemia revisited. Mayo Clin Proc 1994;69:144–50.
119. Allen LH. Vitamin B12 metabolism and status during pregnancy, lactation and infancy. Adv Exp Med Biol 1994;352:173–86.
120. Gueant JL, Safi A, Aimone-Gastin I, et al. Autoantibodies in pernicious anemia type I patients recognize sequence 251-256 in human intrinsic factor. Proc Assoc Am Physicians 1997;109:462–9.
121. Hsing AW, Hansson LE, McLaughlin JK, et al. Pernicious anemia and subsequent cancer. A population-based cohort study. Cancer 1993;71:745–50.
122. Carmel R, Green R, Rosenblatt DS, Watkins D. Update on cobalamin, folate, and homocysteine. Hematology Am Soc Hematol Educ Program 2003:62–81.
123. Carmel R, Spencer CA. Clinical and subclinical thyroid disorders associated with pernicious anemia. Observations on abnormal thyroid-stimulating hormone levels and on a possible association of blood group O with hyperthyroidism. Arch Intern Med 1982;142:1465–9.
124. Weir DG, Scott JM. The biochemical basis of the neuropa-thy in cobalamin deficiency. Baillieres Clin Haematol 1995; 8:479–97.
125. Carmel R, Melnyk S, James SJ. Cobalamin deficiency with and without neurologic abnormalities: differences in homocysteine and methionine metabolism. Blood 2003;101: 3302–8.
126. Carmel R. The disappearance of cobalamin absorption testing: a critical diagnostic loss. J Nutr 2007;137:2481–4.
127. Dokal IS, Cox TM, Galton DA. Vitamin B-12 and folate deficiency presenting as leukaemia. BMJ 1990;300:1263–4.
128. Carmel R, Sinow RM, Siegel ME, Samloff IM. Food cobala-min malabsorption occurs frequently in patients with unexplained low serum cobalamin levels. Arch Intern Med 1988;148:1715–9.
129. Dharmarajan TS, Adiga GU, Norkus EP. Vitamin B12 deficiency. Recognizing subtle symptoms in older adults. Geriatrics 2003;58:30–4, 7–8.
130. Carmel R. Reassessment of the relative prevalences of anti-bodies to gastric parietal cell and to intrinsic factor in patients with pernicious anaemia: influence of patient age and race. Clin Exp Immunol 1992;89:74–7.
131. Chanarin I, Metz J. Diagnosis of cobalamin deficiency: the old and the new. Br J Haematol 1997;97:695–700.
132. Carmel R, Weiner JM, Johnson CS. Iron deficiency occurs frequently in patients with pernicious anemia. JAMA 1987;257:1081–3.
133. Carmel R. Pernicious anemia. The expected findings of very low serum cobalamin levels, anemia, and macrocytosis are often lacking. Arch Intern Med 1988;148: 1712–4.
134. Green R. Screening for vitamin B12 deficiency: caveat emptor. Ann Intern Med 1996;124:509–11.
135. Sumner AE, Chin MM, Abrahm JL, et al. Elevated methyl-malonic acid and total homocysteine levels show high prevalence of vitamin B12 deficiency after gastric surgery. Ann Intern Med 1996;124:469–76.
136. Pippard MJ. Megaloblastic anaemia: geogrAPCVy and diagnosis. Lancet 1994;344:6–7.
137. Kuzminski AM, Del Giacco EJ, Allen RH, et al. Effective treatment of cobalamin deficiency with oral cobalamin. Blood 1998;92:1191–8.
138. Pfeiffer CM, Caudill SP, Gunter EW, et al. Biochemical indicators of B vitamin status in the US population after folic acid fortification: results from the National Health and Nutrition Examination Survey 1999–2000. Am J Clin Nutr 2005;82:442–50.
139. Guidelines on the investigation and diagnosis of cobala-min and folate deficiencies. A publication of the British Committee for Standards in Haematology. BCSH General Haematology Test Force. Clin Lab Haematol 1994;16: 101–15.
140. Molloy AM, Daly S, Mills JL, et al. Thermolabile variant of 5,10-methylenetetrahydrofolate reductase associated with low red-cell folates: implications for folate intake recommendations. Lancet 1997;349:1591–3.
141. Wilcken DE. MTHFR 677C-->T mutation, folate intake, neural-tube defect, and risk of cardiovascular disease. Lancet 1997;350:603–4.
142. Howard MR, Turnbull AJ, Morley P, et al. A prospective study of the prevalence of undiagnosed coeliac disease in laboratory defined iron and folate deficiency. J Clin Pathol 2002;55:754–7.
143. McPartlin J, Halligan A, Scott JM, et al. Accelerated folate breakdown in pregnancy. Lancet 1993;341:148–9.
144. Wald NJ, Bower C. Folic acid, pernicious anaemia, and prevention of neural tube defects. Lancet 1994;343:307.
145. Carmel R. Subtle cobalamin deficiency. Ann Intern Med 1996;124:338–40.
146. Masugi J, Amano M, Fukuda T. Copper deficiency anemia and prolonged enteral feeding. Ann Intern Med 1994;121: 386.
147. Huff JD, Keung YK, Thakuri M, et al. Copper deficiency causes reversible myelodysplasia. Am J Hematol 2007; 82:625–30.
148. Fiske DN, McCoy HE 3rd, Kitchens CS. Zinc-induced sideroblastic anemia: report of a case, review of the litera-ture, and description of the hematologic syndrome. Am J Hematol 1994;46:147–50.
149. Gregg XT, Reddy V, Prchal JT. Copper deficiency mas-querading as myelodysplastic syndrome. Blood 2002;100: 1493–5.
150. Fong T, Vij R, Vijayan A, et al. Copper deficiency: an important consideration in the differential diagnosis of myelodysplastic syndrome. Haematologica 2007;92:1429– 30.
151. Wasa M, Satani M, Tanano H, et al. Copper deficiency with pancytopenia during total parenteral nutrition. JPEN J Parenter Enteral Nutr 1994;18:190–2.
152. Kumar N, Gross JB Jr, Ahlskog JE. Copper deficiency myelopathy produces a clinical picture like subacute combined degeneration. Neurology 2004;63:33–9.
153. Kumar N. Copper deficiency myelopathy (human swayback). Mayo Clin Proc 2006;81:1371–84.
154. Alcindor T, Bridges KR. Sideroblastic anaemias. Br J Haematol 2002;116:733–43.
155. Cotter PD, May A, Li L, et al. Four new mutations in the erythroid-specific 5-aminolevulinate synthase (ALAS2) gene causing X-linked sideroblastic anemia: increased pyridoxine responsiveness after removal of iron overload by phlebotomy and coinheritance of hereditary hemochromatosis. Blood 1999;93:1757–69.
156. Nakajima O, Takahashi S, Harigae H, et al. Heme defi-ciency in erythroid lineage causes differentiation arrest and cytoplasmic iron overload. Embo J 1999;18:6282–9.
157. Bottomley SS, Healy HM, Brandenburg MA, May BK. 5-Aminolevulinate synthase in sideroblastic anemias: mRNA and enzyme activity levels in bone marrow cells. Am J Hematol 1992;41:76–83.
158. Bottomley SS, May BK, Cox TC, et al. Molecular defects of erythroid 5-aminolevulinate synthase in X-linked sideroblastic anemia. J Bioenerg Biomembr 1995;27:161–8.
159. Cotter PD, May A, Fitzsimons EJ, et al. Late-onset X-linked sideroblastic anemia. Missense mutations in the erythroid delta-aminolevulinate synthase (ALAS2) gene in two pyridoxine-responsive patients initially diagnosed with acquired refractory anemia and ringed sideroblasts. J Clin Invest 1995;96:2090–6.
160. Cazzola M, Barosi G, Gobbi PG, et al. Natural history of idiopathic refractory sideroblastic anemia. Blood 1988; 71:305–12.
161. Beris P, Samii K, Darbellay R, et al. Iron overload in patients with sideroblastic anaemia is not related to the presence of the haemochromatosis Cys282Tyr and His63Asp Mutations. Br J Haematol 1999;104:97–9.
"ótima revisão !!! ++++/4"
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.