(Carregando Índice)... (Carregando Índice)... |
Você está em:
Inicial  acp-medicine Endocrinologia
acp-medicine Endocrinologia
Última revisão: 13/01/2014
Comentários de assinantes: 0
John D. Brunzell, MD, FACP
Professor Emeritus, Department of Medicine, Division of Metabolism, Endocrinology and Nutrition, University of Washington, Seattle, WA
R. Alan Failor, MD
Clinical Professor, Department of Medicine, Division of Metabolism, Endocrinology and Nutrition, University of Washington, Seattle, WA
Artigo original: Brunzell JD, Failor RA. Diagnosis and treatment of dyslipidemia. ACP Medicine. 2010;1-23.
[The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2011 Decker Intellectual Properties Inc. All Rights Reserved.]
Agradecimentos: Figuras 1 e 4 – Andy Christie. Figuras 2, 3, 5 e 6 – Seward Hung.
Tradução: Soraya Imon de Oliveira.
Revisão técnica: Dr. Rodrigo Antônio Brandão Neto.
Os distúrbios do metabolismo de lipídios aliados à prevalência das dietas ricas em gordura, obesidade e inatividade física resultaram em uma epidemia de doença aterosclerótica nos Estados Unidos e em outros países industrializados. A interação de distúrbios genéticos com estes fatores ambientais adversos leva ao desenvolvimento precoce de aterosclerose. Nos Estados Unidos, a mortalidade associada à doença arterial coronariana (DAC), particularmente entre os indivíduos com menos de 60 anos de idade, está em declínio desde 1970. Entretanto, a doença cardiovascular aterosclerótica ainda é a causa mais comum de morte entre homens e mulheres.1,2
Antigamente, a hiperlipidemia era definida pela elevação dos níveis de lipoproteínas na população. O reconhecimento de que um nível baixo de HDL e a presença de pequenas partículas densas de LDL são clinicamente importantes na patofisiologia dos distúrbios lipídicos levou ao uso do termo “dislipidemia” para descrever uma gama de distúrbios na qual estão incluídos os níveis anormais (altos e baixos) de lipoproteínas, bem como as anormalidades na composição destas partículas. As dislipidemias são clinicamente importantes, sobretudo porque contribuem para a aterogênese. A pancreatite e a doença do fígado esteatótico são manifestações menos comuns, porém clinicamente significativas, de distúrbios lipídicos.
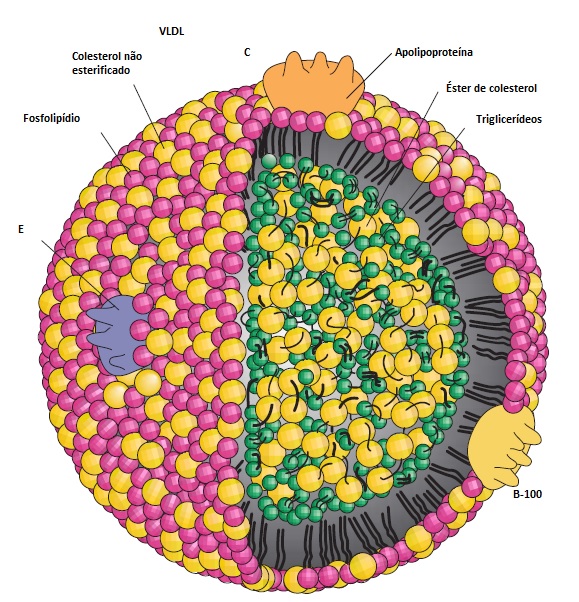
As lipoproteínas são complexos macromoleculares esféricos contendo lipídio e proteína [Figura 1]. Os lipídios de importância clínica presentes no sangue são o colesterol (não esterificado e esterificado) e os triglicerídeos (moléculas que consistem em 3 ácidos graxos fixos a uma estrutura de glicerol). O colesterol exerce 3 funções primárias: atua na estrutura das membranas celulares, na síntese de hormônios esteroides e na formação dos ácidos biliares. As principais funções dos triglicerídeos são o armazenamento de energia (no tecido adiposo) e o uso da energia (pelos músculos). Como a gordura não pode ser prontamente dissolvida no plasma, o colesterol e o triglicerídeo são transformados em moléculas miscíveis pela incorporação às lipoproteínas (p. ex., lipoproteína de densidade muito baixa [VLDL], LDL e HDL). As apolipoproteínas são o componente proteico das lipoproteínas, com função no transporte e no processo de distribuição dos lipídios, atuando de 3 modos: servindo de elementos estruturais, atuando como ligantes de receptores e exercendo o papel de cofatores regulatórios [Tabela 1].

Figura 1. As lipoproteínas transportam triglicerídeos e colesterol hidroinsolúveis pela circulação sanguínea. Todas as lipoproteínas contendo apolipoproteína (apo) B possuem uma estrutura similar àquela demonstrada para as lipoproteínas de densidade muito baixa (VLDL). O núcleo (core) é constituído de triglicerídeos e colesteril éster, enquanto a monocamada superficial é constituída de fosfolipídios, colesterol não esterificado e proteína sob a forma de apolipoproteínas. A VLDL contém apolipoproteínas B-100, C-I, C-II e E. A lipoproteína de baixa densidade (LDL), que transporta a maior parte do colesterol encontrado no sangue, contém principalmente apo B-100.
Tabela 1. Principais apolipoproteínas e suas funções
|
Apo |
Função |
|
Apo A-I |
Proteína estrutural da HDL; ativa a lecitina-colesterol aciltransferase |
|
Apo A-II |
Proteína estrutural da HDL |
|
Apo B-48 |
Proteína estrutural do quilomícron |
|
Apo B-100 |
Proteína estrutural da VLDL, IDL e LDL; ligante do receptor de LDL |
|
Apo C-II |
Ativador de LPL |
|
Apo C-III |
Potencial inibidor das funções da apo C-II e da apo E |
|
Apo E |
Ligante do receptor de remanescentes de quilomícron e do receptor de LDL |
|
Apo(a) |
Função desconhecida; antagoniza o plasminogênio |
Apo = Apolipoproteína; HDL = lipoproteína de alta densidade; IDL = lipoproteína de densidade intermediária; LDL = lipoproteína de baixa densidade; LPL = lipoproteína lipase; VLDL = lipoproteína de densidade muito baixa.
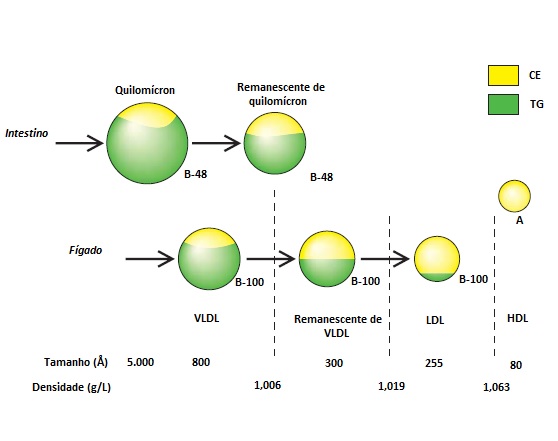
Uma partícula madura de lipoproteína é uma esfera que consiste em um núcleo (core) central lipídico (triglicerídeos e colesteril-éster) cercado por uma monocamada superficial de fosfolipídios, colesterol não esterificado e apolipoproteínas [Figura 1]. Do ponto de vista operacional, as lipoproteínas podem ser descritas com base em seu tamanho e nas características de flutuabilidade [Figura 2].
Figura 2. Características de tamanho e flutuabilidade das lipoproteínas. Os quilomícrons, que são constituídos em grande parte por triglicerídeos (TG), são as maiores e mais flutuantes dentre as moléculas de lipoproteína. As partículas de lipoproteína de alta densidade (HDL) são substancialmente menores e mais densas, além de serem compostas na maior parte por colesteril éster (CE).
LDL = lipoproteína de baixa densidade; VLDL = lipoproteína de densidade muito baixa.
Os quilomícrons são as maiores partículas de lipoproteína. Sua principal proteína estrutural é a apolipoproteína B-48 (apo B-48). O grosso (˜ 80%) do core lipídico consiste em triglicerídeos. Sintetizados e secretados a partir do intestino, os quilomícrons transportam colesterol exógeno, ácidos graxos e vitaminas lipossolúveis absorvidas dos alimentos digeridos [ver Via exógena, adiante].
A partícula rica em triglicerídeos (˜ 80% do core lipídico consiste em triglicerídeos) é sintetizada no fígado, transporta os triglicerídeos até a periferia e é precursora das lipoproteínas de densidade intermediária (IDL) e da LDL. A principal proteína estrutura desta lipoproteína é a apo B-100 [ver Via exógena, adiante].
O restante da VLDL são partículas com densidade de IDL. Esta se forma depois que o triglicerídeo presente na VLDL é hidrolisado pela lipoproteína lipase (LPL). Cerca de metade do conteúdo de partículas de IDL do corpo é depurado do plasma no fígado. A outra metade remanescente sofre processamento adicional e origina LDL [ver Via exógena, adiante]. Na prática clínica, a avaliação dos níveis de LDL inclui a determinação do conteúdo de colesterol nas frações de IDL e de LDL.
Esta lipoproteína resulta do processamento hepático dos remanescentes da VLDL. O core é rico em colesteril-éster e contribui para a maioria do colesterol circulante no sangue. A LDL exerce um papel importante no desenvolvimento da aterosclerose [ver Catabolismo da lipoproteína de baixa densidade (LDL), adiante].
A HDL é formada a partir do colesterol não esterificado e dos fosfolipídeos removidos dos tecidos periféricos e da superfície das proteínas ricas em triglicerídeos [ver Função e regulação da lipoproteína de alta densidade (HDL), adiante]. A principal proteína estrutural é a apo A-I, e o core consiste predominantemente em colesteril éster. A HDL media o retorno da lipoproteína e do colesterol tecidual ao fígado para excreção, no processo referido como transporte reverso de colesterol. Outra função da HDL é transportar a apo E e a apo C-II de/para os quilomícrons e a VLDL.
Após uma refeição, as células intestinais absorvem os ácidos graxos e o colesterol, realizam a esterificação destas moléculas em triglicerídeo e colesteril-éster e incorporam estes ao core dos quilimícrons.1 O triglicerídeo predomina em relação ao colesteril-éster no core do quilomícron. Os quilomícrons são secretados no plasma, sendo que a apo C-II localizada na superfície dos quilomícrons ativa a LPL ligada ao endotélio. A LPL, por sua vez, hidrolisa os triglicerídeos do core do quilomícron e libera ácidos graxos livres que são captados pelo tecido adiposo (armazenamento) e pelos músculos (energia). Durante a lipólise, o quilomícron diminui de tamanho, e alguns de seus componentes de superfície são transferidos para a HDL. O restante da partícula constitui a partícula remanescente de quilomícron. Esta, então, adquire apo E oriunda da HDL e é subsequentemente captada pelo fígado, após a ligação aos sítios de reconhecimento da apo E. A partícula é degradada e, com isso, libera o colesterol da dieta no fígado.
O fígado secreta VLDL rica em triglicerídeos no plasma, onde a VLDL adquire apo C-II da HDL. Assim como os quilomícrons, a VLDL interage com a LPL no endotélio capilar, e o triglicerídeo do core é hidrolisado para fornecer os ácidos graxos que seguirão para o tecido adiposo e os músculos.3 Cerca de metade dos remanescentes de VLDL catabolizados (densidade de IDL) é captada por receptores hepáticos que se ligam à apo E para promoverem degradação. A outra metade – partículas de apo B-100, depletadas de triglicerídeos em relação ao conteúdo de colesteril-éster – é convertida pelo fígado em LDL rica em colesteril-éster. Conforme a IDL é convertida em LDL, a apo E é liberada, e permanece apenas a apolipoproteína B-100 na partícula. Cada partícula desta cascata, desde a VLDL até a LDL, contém uma molécula de apo B-100.
No metabolismo tanto dos quilomícrons como da VLDL, a apo C-II permite a hidrólise dos triglicerídeos pela LPL, enquanto a apo E acelera a captação hepática dos remanescentes. Uma das principais diferenças referentes ao metabolismo destas partículas está no fato de os quilomícrons conterem uma forma truncada de apo B (isto é, apo B-48), enquanto a VLDL contém a forma completa (isto é, apo B-100). Outra diferença é que os remanescentes do quilomícron são degradados depois de serem absorvidos pelo fígado, enquanto muitos dos remanescentes da VLDL tendem mais a serem processados nos sinusoides hepáticos e se transformarem em LDL.
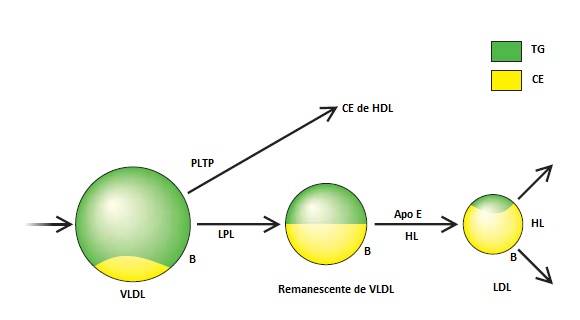
Existem 4 etapas fisiológicas principais, clinicamente importantes, na cascata de lipoproteínas, que vai da VLDL até a LDL – a saber: (1) montagem da VLDL, (2) hidrólise pela LPL, (3) catabolismo de remanescentes, e (4) catabolismo da LDL [Figura 3].3 Os defeitos que ocorrem em qualquer etapa desta cascata podem acarretar hiperlipidemia. Tais defeitos podem ser genéticos ou adquiridos (isto é, secundários a uma doença ou efeitos de fármacos), ou podem resultar de uma interação entre fatores genéticos e adquiridos.
Figura 3. Cascata da apolipoproteína B-100 (apo B-100). A lipoproteína de densidade muito baixa (VLDL) é secretada pelo fígado e apresenta uma apo B na superfície, além de triglicerídeo (TG) e colesteril éster (CE) no core. O core do TG é hidrolisado pela lipoproteína lipase (LPL) e transforma-se em remanescente de lipoproteína que é reconhecido pelo fígado – em parte, pela apo E. O remanescente de lipoproteína é ainda mais processado e origina a lipoproteína de baixa densidade (LDL), que possui um core rico em colesterol e uma apo B na superfície. A partícula de LDL pode ser removida por receptores de LDL hepáticos ou por receptores periféricos. À medida que o core da VLDL é hidrolisado, o colesterol não esterificado e os fosfolipídios são transferidos para a lipoproteína de alta densidade (HDL) por ação da proteína transferidora de fosfolipídio (PLTP), para se transformarem no CE da HDL.
HL = lipase hepática.
A apo B-100 é constitutivamente sintetizada no retículo endoplasmático do hepatócito, e grande parte destas moléculas é degradada também no retículo endoplasmático. O triglicerídeo é adicionado às moléculas sobreviventes de apo B-100 que, então, serão secretadas como VLDL. Seu transporte é feito via complexo de Golgi, onde as moléculas adquirem o conteúdo lipídico adicional do core, formando a partícula de VLDL nascente. Esta, por sua vez, é secretada no plasma, onde adquire as apoliproteínas (p. ex., apo C-II e apo E) a partir da HDL.3
As anormalidades na secreção de VLDL podem ocorrer em 2 formas genéticas de hiperlipidemia: hipertrigliceridemia familiar (HTGF) e hiperlipidemia combinada familiar (HLCF). A HTGF é caracterizada pela produção excessiva de triglicerídeos contidos em um número normal de partículas de VLDL. Como resultado desta produção excessiva, cada partícula contém uma quantidade excessiva de triglicerídeo. Na HLCF, uma quantidade excessiva de apo B-100 é secretada nas particuladas de VLDL ou LDL, e estas partículas tendem a ser menores do que o normal.4
A síndrome metabólica, que é uma condição comum na população em geral, é um componente da maioria dos casos de HLCF e também contribui para a dislipidemia residual observada em pacientes com diabetes melito de tipo 2 tratados com insulina ou secretagogos de insulina. A base molecular da hipersecreção hepática de triglicerídeos ou apo B observada nestes distúrbios é obscura.
Uma deficiência de lipoproteínas contendo apo B é referida como hipo-beta-lipoproteinemia. Uma ausência de apo B é denominada a-beta-lipoproteinemia. A a-beta-lipoproteinemia pode resultar de um defeito envolvendo ambos os genes da apo B, que impede a produção de apo B. E esta condição também pode ocorrer em indivíduos homozigotos para mutações na proteína transportadora de triglicerídeos microssomal, que é decisiva para o transporte da apo B no retículo endoplasmático. A hipo e a-beta-lipoproteinemia homozigotas levam ao desenvolvimento de deficiências de vitaminas lipossolúveis, pois cada uma destas condições resulta na falta de lipoproteínas contendo apo-B, as quais são necessárias ao transporte das vitaminas lipossolúveis. A hipo-beta-lipoproteinemia, que é caracterizada por níveis de apo B equivalentes a 50% do normal, pode ser causada por um defeito em um único gene da apo B.5
Lipoproteína(a). A lipoproteína(a) [Lp(a)] é uma classe específica de partículas de lipoproteína que são sintetizadas no fígado e possuem uma composição lipídica similar a da LDL. A Lp(a) difere da LDL quanto à presença da apolipoproteína(a) [apo(a)], que é uma proteína com estrutura homóloga à do plasminogênio.6 A proteína apo(a) liga-se via ligações dissulfeto à apo B-100, para formar a Lp(a). Níveis elevados de Lp(a) têm ação pró-trombótica e aterogênica.6 Os níveis de Lp(a) no plasma são quase totalmente determinados pela variação no gene de Lp(a).
Remoção de triglicerídeo mediada pela lipoproteína lipase. A LPL é sintetizada no tecido adiposo e nos músculos e, em seguida, transportada para a superfície luminal do revestimento endotelial dos capilares adjacentes, onde atua sobre as lipoproteínas ricas em triglicerídeos. Os ácidos graxos liberados durante o processamento das partículas ricas em triglicerídeos (isto é, quilomícrons e VLDL) podem ser usados para a obtenção de energia pelos músculos, ou podem ser reesterificados em triglicerídeos e armazenados nos adipócitos para uso futuro.7 A apo C-II, ativadora da LPL, é transportada nas lipoproteínas ricas em triglicerídeos – quilomícrons e VLDL.
Os defeitos genéticos que resultam no comprometimento da síntese ou da função da LPL são causas recessivas autossômicas raras de hiperlipidemia. Geralmente, estas mutações surgem em recém-nascidos ou bebês maiores, manifestando-se como hipertrigliceridemia severa. Os pais heterozigotos destas crianças costumam ter hipertrigliceridemia leve. Os defeitos adquiridos de LPL, como aqueles observados em indivíduos não tratados com diabetes ou uremia, são as causas mais comuns de hiperlipidemia. Quando um defeito adquirido de LPL está associado a um distúrbio caracterizado pela entrada excessiva de VLDL, o paciente pode desenvolver uma hipertrigliceridemia marcante. A coexistência de pelo menos 3 distúrbios que aumentam independentemente os níveis de triglicerídeos no plasma (p. ex., HTGF ou HLCF coexistente com diabetes não tratado) pode conduzir a uma acentuada hipertrigliceridemia.7
Catabolismo de remanescentes. Os remanescentes de quilomícron e de VLDL adquirem a apo E oriunda da HDL antes de se ligarem aos receptores hepáticos para serem captadas e degradadas ou adicionalmente processadas em LDL. O gene APOE possui 3 alelos (isto é, APOE*E2, APOE*E3 e APOE*E4) que resultam em 6 combinações possíveis. O produto do alelo APOE*E4 é o que tem maior afinidade pelos receptores hepáticos, seguido do produto do alelo APOE*E3. O produto do alelo APOE*E2 apresenta uma afinidade acentuadamente reduzida pelo receptor hepático.
Os indivíduos homozigotos para o alelo APOE*E2 (E2/E2) apresentam comprometimento severo da captação hepática dos remanescentes das lipoproteínas, com consequente acúmulo dos remanescentes no plasma e níveis muito baixos ou ausência de LDL. De modo interessante, os indivíduos E2/E2 tipicamente apresentam níveis de colesterol normais ou baixos, devido à característica paucidade das partículas de LDL observada neste distúrbio.8 Se, todavia, um indivíduo homozigoto para o alelo APOE*E2 (E2/E2) tiver um defeito – hereditário ou adquirido – que cause entrada excessiva de VLDL, então haverá acúmulo excessivo de remanescentes de VLDL e hiperlipidemia. Isto resulta no desenvolvimento da doença da remoção de remanescentes. Como os remanescentes de quilomícron e VLDL contêm grosseiramente as mesmas concentrações de triglicerídeos e colesterol, a hiperlipidemia associada à doença da remoção de remanescentes é caracterizada por hipercolesterolemia e por hipertrigliceridemia.8
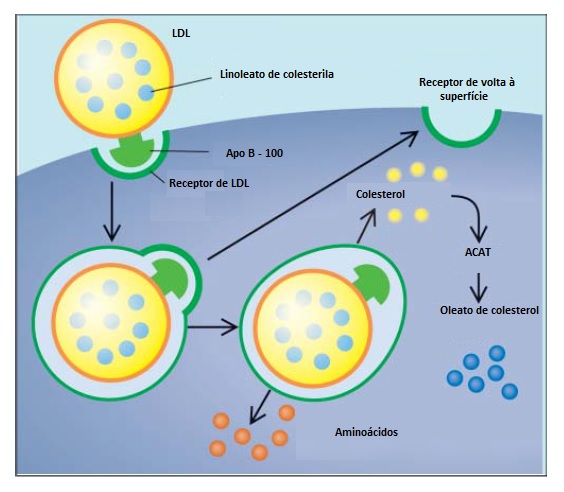
Catabolismo da lipoproteína de baixa densidade (LDL). A etapa final em que pode ocorrer defeito do metabolismo lipoproteico é o catabolismo da LDL. A apo B-100 presente na superfície da LDL liga-se ao receptor existente na superfície celular. A LDL então é absorvida para dentro da célula, onde é catabolizada [Figura 4]. Após a hidrólise dos lipídios do core, o colesterol não esterificado é utilizado pelas células para síntese de membranas, ácidos biliares e hormônios esteroides, bem como para várias ações regulatórias que evitam o acúmulo excessivo de colesterol junto à célula. A vasta maioria das partículas de LDL presentes no plasma é captada pelo fígado via receptor de LDL.
Figura 4. A lipoproteína de baixa densidade (LDL) é absorvida pelas células através do receptor de LDL. Este receptor reconhece a apolipoproteína B-100 (apo B-100), que é a apolipoproteína presente na superfície da LDL. Uma vez internalizada, a lipoproteína é catabolizada, liberando colesterol e aminoácidos. O colesterol livre é convertido em oleato de colesteril, pela enzima acil-coenzima A: colesterol acil transferase (ACAT). O receptor de LDL é reciclado de volta à superfície celular.
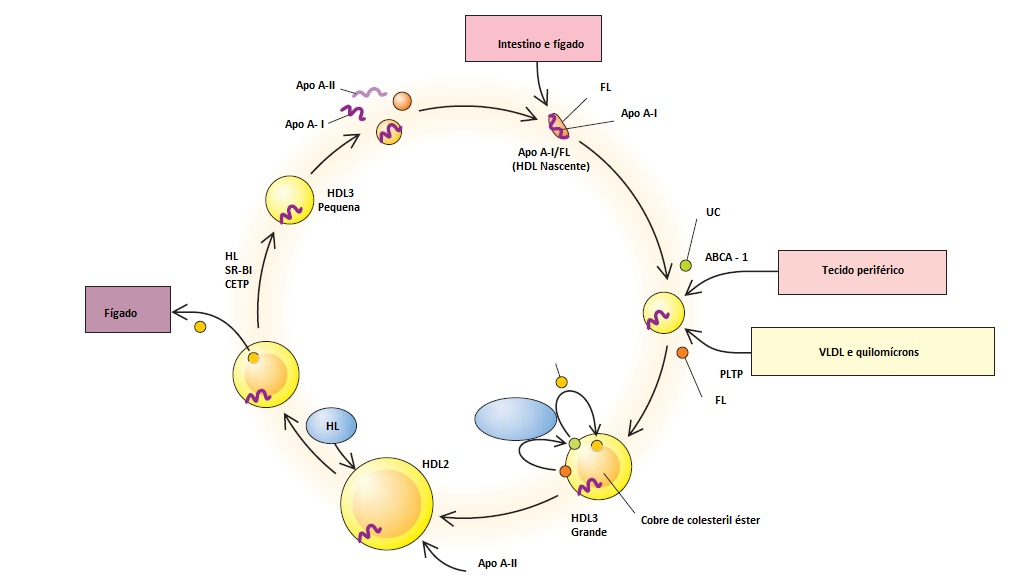
As principais apolipoproteínas da HDL são a apo A-I e a apo A-II, que são formadas no fígado e intestino delgado.10 A apo A-I é secretada com fosfolipídio em uma estrutura estranha, denominada HDL nascente. A maioria das apolipoproteínas e fosfolipídios destinados a se tornarem HDL nascente inicialmente é secretada na superfície dos quilomícrons e VLDL. Depois que a LPL hidrolisa o triglicerídeo em quilomícrons e VLDL, o conteúdo lipídico do core nestas partículas de lipoproteína torna-se menor, e ocorrem redundâncias de colesterol não esterificado e fosfolipídio na camada superficial. Estes componentes de superfície redundantes são transferidos para a HDL pela proteína transferidora de fosfolipídio. As partículas de HDL nascente também captam o excesso de colesterol não esterificado e fosfolipídio existente nos tecidos periféricos via transportador de cassete ligador de trifosfato de adenosina A-1 (ABCA1). Este colesterol de HDL então é esterificado por uma enzima plasmática, a lecitina-colesterol aciltransferase (LCAT). A LCAT é ativada pela apo A-I na superfície da HDL e esterifica o colesterol livre em colesteril-éster, fazendo-o entrar no core. Neste processo, a partícula transforma-se em uma partícula de HDL3 maior e mais flutuante, que progride para uma partícula de HDL2 ainda maior.10,11 Em algum momento, a apo A-II pode ser adicionada à partícula de HDL2 que, então, é destinada ao transporte de colesteril-éster para o fígado via proteína transferidora de colesteril-éster (CETP). A atividade da lipase hepática existente na superfície do fígado hidrolisa o fosfolipídio e o triglicerídeo da partícula de HDL2, promovendo a diminuição do tamanho e da densidade que resulta na partícula de HDL3 e, em seguida, em partículas de HDL ainda menores.11 A reciclagem de uma parte da apo A-I leva à repetição do processo em si [Figura 5].
Figura 5. Via circular da formação e degradação da lipoproteína de alta densidade (HDL).12 A HDL inicialmente é uma apolipoproteína (apo A-I) fosfolipídica (FL) complexa. O colesterol não esterificado (CNE) e os FL são adicionados à HDL nascente por ação do transportador do cassete ligador de trifosfato de adenosina A-1 (ABCA-1) e da proteína transferidora de fosfolipídio (PLTP), para iniciar a formação da partícula de HDL3 de menor tamanho. A lecitina-colesterol aciltransferase (LCAT) transfere um ácido graxo do FL para o CNE e colesteril éster, que se desloca para o core da HDL. Neste processo, a partícula de HDL transforma-se na partícula HDL3 de maior tamanho e mais flutuante, progredindo para uma partícula de HDL2, que é ainda maior. A proteína transferidora de colesteril éster (CETP) contribui para a transferência do colesteril éster da HDL2 para o fígado e diversas lipoproteínas. Com esta perda de colesteril éster, a partícula de HDL sofre colapso e diminui de tamanho. A lipase hepática (HL) hidrolisa o FL o triglicerídeo presentes na partícula de HDL2, promovendo diminuição do tamanho e da densidade, com consequente transformação em HDL3 e, em seguida, em partículas ainda menores de HDL, inclusive apo A-1. A reciclagem de algumas apo A-I resulta na autorrepetição do processo em si. O papel da apo A-II neste processo, em seres humanos, é obscuro.
LPL = lipoproteína lipase; SR-BI = receptor scavenger B-I; VLDL = lipoproteína de densidade muito baixa.
Níveis anormalmente altos ou baixos de HDL colesterol podem ser causados, ainda que em raros casos, por defeitos genéticos. As elevações dos níveis de colesterol de HDL podem resultar de hiper-alfa-lipoproteinemia ou deficiência de CETP. Níveis acentuadamente reduzidos de colesterol de HDL podem ser resultado de uma mutação estrutural em apo A-I; homozigose para a mutação em ABCA1,12 com consequente desenvolvimento da doença de Tangier; ou homozigose para mutações na enzima LCAT, que resulta em deficiência de LCAT e na doença do olho de peixe. Os fatores associados a um aumento dos níveis de HDL incluem o sexo feminino, exercício aeróbico, diminuição do peso, dietas ricas em gordura e certos fármacos (p. ex., álcool, estrogênios, fibratos e ácido nicotínico) [Tabela 2]. Os fatores associados à diminuição dos níveis de HDL incluem sexo masculino, obesidade central, tabagismo, dietas pobres em gordura, hipertrigliceridemia, uremia, heterozigose para doença de Tangier e certos fármacos (p. ex., androgênios, progestinas e alguns agentes anti-hipertensivos) [Tabela 2]. O baixo número de partículas de HDL comumente está associado a níveis aumentados de triglicerídeos, como se observa na síndrome metabólica.
Tabela 2. Classificação do NCEP ATP-III dos níveis de colesterol total, colesterol de LDL, colesterol de HDL e triglicerídeos17
|
Colesterol total (mg/dL)* | |
|
< 200 |
Desejável |
|
200 a 239 |
Limítrofe alto |
|
= 240 |
Alto |
|
Colesterol de LDL (mg/dL) | |
|
< 100 |
Ideal |
|
100 a 129 |
Próximo ou acima do ideal |
|
130 a 159 |
Limítrofe alto |
|
160 a 189 |
Alto |
|
= 190 |
Muito alto |
|
Colesterol de HDL (mg/dL) | |
|
< 40 |
Baixo |
|
= 60 |
Alto |
|
Triglicerídeos (mg/dL) | |
|
< 150 |
Normal |
|
150 a 199 |
Limítrofe alto |
|
200 a 499 |
Alto |
|
= 500 |
Muito alto |
*A elevação do colesterol total pode ser um reflexo de LDL aumentada, HDL aumentada, ou ambas. A LDL aumentada é pró-aterosclerótica, enquanto a HDL elevada é antiesclerótica.
HDL = lipoproteína de alta densidade; LDL = lipoproteína de baixa densidade; NCEP ATP-III = National Cholesterol Education Program Adult Treatment Panel III.
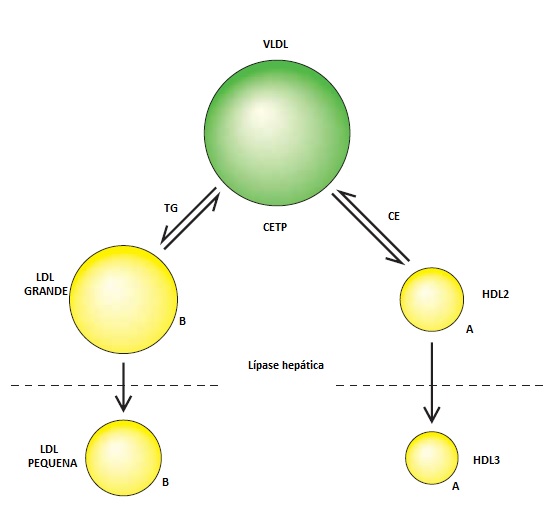
A lipase hepática é sintetizada no hepatócito, liga-se às superfícies endoteliais dos sinusoides hepáticos e atua nas lipoproteínas.13 Depois que a partículas de VLDL ricas em triglicerídeo trocam triglicerídeo por colesteril-éster na LDL e na HDL, a lipase hepática pode hidrolisar o fosfolipídeo e triglicerídeo existente na LDL e na HDL [Figura 6]. Este processo leva à formação de pequenas partículas de LDL densas e converte a HDL2 em HDL3, podendo ser dirigido pela presença de níveis excessivos de VLDL rica em triglicerídeo diante da atividade normal da lipase hepática ou por aumentos dos níveis de lipase hepática. Fatores como o sexo masculino e o acúmulo de tecido adiposo intra-abdominal predispõem ao aumento dos níveis de lipase hepática e estão associados a um aumento dos níveis de partículas pequenas e densas de LDL, bem como à diminuição dos níveis de HDL2. A atividade de lipase hepática aumentada é um fator importante na dislipidemia da síndrome metabólica.13,14 A lipase hepática também pode facilitar o reconhecimento e a captação hepática dos quilomícrons e lipoproteínas remanescentes de VLDL.
Figura 6. Dislipidemia na síndrome metabólica. A lipoproteína de densidade muito baixa (VLDL) rica em triglicerídeos (TG) troca TG por colesteril éster (CE) nas partículas de lipoproteína de baixa densidade (LDL) e de lipoproteína de alta densidade (HDL). Esta mudança de composição de lipoproteína é iniciada pela proteína transferidora de colesteril éster (CETP). A lipase hepática hidrolisa o TG e os fosfolipídios nas partículas grandes de LDL e HDL, diminuindo o tamanho de cada partícula.
As principais consequências clínicas da hiperlipidemia são o desenvolvimento de aterosclerose precoce; pancreatite, que geralmente está associada à síndrome de quilomicronemia; e a doença do fígado esteatótico não alcoólico. A aterosclerose está mais nitidamente associada aos níveis elevados de colesterol de LDL e níveis reduzidos de colesterol de HDL. Tanto na pancreatite como na doença do fígado esteatótico, o distúrbio lipídico subjacente é a hipertrigliceridemia.
É consenso que níveis plasmáticos elevados de LDL e reduzidos de HDL estão associados ao risco aumentado de aterosclerose. O papel da hipertrigliceridemia como fator de risco cardiovascular é mais complexo. A hipertrigliceridemia pode atuar como marcador de outras anormalidades lipoproteicas (p. ex., níveis aumentados de partículas pequenas e densas de LDL; níveis baixos de HDL; ou acúmulo de remanescentes) que fazem parte do padrão dislipidêmico associado à HLCF, diabetes melito de tipo 2 e síndrome metabólica. Nestes contextos, a hipertrigliceridemia constitui um fator preditivo de risco cardiovascular prematuro aumentado. Entretanto, outras formas de hipertrigliceridemia podem não estar associadas à doença cardiovascular prematura [ver Hipertrigliceridemia familiar, adiante]. Os mecanismos precisos pelos quais os níveis elevados de LDL resultam no aumento do risco aterosclerótico são obscuros. Níveis muito altos de partículas de LDL grandes e flutuantes, como ocorre na HF e no defeito familiar da apo B-100, bem como a presença de números mais moderados de partículas pequenas e densas de LDL, estão associados ao risco aumentado de doença cardiovascular. Evidências acumuladas sugerem que a LDL precisa ser modificada para se tornar aterogênica.15 A oxidação da LDL pode aumentar sua aterogenicidade. A LDL oxidada possui muitas propriedades biológicas que podem fazê-la se tornar aterogênica. A aterogenicidade das partículas pequenas e densas de LDL pode resultar da habilidade da LDL de entrar na íntima arterial, onde é retida por moléculas da matriz e sofre oxidação mais prontamente do que as partículas maiores e mais flutuantes de LDL. As propriedades antiaterogênicas da HDL provavelmente estão relacionadas a seu papel no transporte reverso do colesterol, enquanto a HDL pode exercer efeitos anti-inflamatórios e antioxidantes.
A pancreatite está associada à quilomicronemia, geralmente com níveis elevados de VLDL. O mecanismo pelo qual a quilomicronemia causa pancreatite é obscuro. Acredita-se que a pancreatite resulta da liberação de mais ácidos graxos e lisolecitina dos quilomícrons do que a quantidade de moléculas que a lipase pancreática consegue ligar, nos capilares pancreáticos.
A síndrome da quilomicronemia ocasionalmente ocorre quando a LPL é defeituosa, como resultado da variação genética da enzima ou de seu cofator, a apo C-II. Na situação mais comum, a quilomicronemia é causada pela coexistência de uma forma genética de hipertrigliceridemia combinada a um distúrbio adquirido do metabolismo dos triglicerídeos plasmáticos, dentre os quais o mais comum é o diabetes não tratado. Outras condições podem estar implicadas (p. ex., hipotireoidismo e síndrome nefrótica), assim como o uso de fármacos que aumentam os níveis de triglicerídeos.
A síndrome da hiperquilomicronemia está associada a dor abdominal, xantomas eruptivos e perda transiente da memória. Os xantomas eruptivos são mais frequentes nas nádegas e nas superfícies extensoras do membro superior. Uma perda reversível da memória, em particular com relação aos eventos recentes, e a neuropatia periférica, que às vezes mimetiza a síndrome do túnel do carpo, também podem ocorrer. Os vasos da retina ocasionalmente apresentam lipemia retiniana. Quando a síndrome da quilomicronemia não é corrigida, pode levar ao desenvolvimento de pancreatite aguda. A pancreatite aguda pode ser fatal e costuma ser recorrente até que os níveis de triglicerídeos sejam mantidos baixos. O risco de desenvolvimento de pancreatite causada por hipertrigliceridemia severa aumenta de maneira acentuada diante de níveis de triglicerídeos acima de 2.000 mg/dL.
A esteatose hepática parece ocorrer em ambas as formas de hipertrigliceridemia, genética e adquirida. Em geral, é causada pela síntese de triglicerídeos hepáticos em concentrações que são excessivas em relação à concentração de apo B sintetizada. Isto leva ao acúmulo de triglicerídeos no fígado, em vez da secreção hepática de triglicerídeos da VLDL. A esteatose hepática também pode ocorrer na hipo-beta-lipoproteinemia, por causa da síntese diminuída de apo B hepática associada a este distúrbio. A doença do esteatose hepática não alcoólica também ocorre diante da síntese aumentada de triglicerídeos hepáticos, diante do comprometimento da síntese de apo B.16 A esteatose hepática não alcoólica foi associada à síndrome metabólica,14 que, por sua vez, está relacionada a obesidade central, resistência à insulina e hipertrigliceridemia.
Qualquer forma severa de hipertrigliceridemia com catabolismo defeituoso da VLDL também pode estar associada ao fígado esteatótico e à hepatoesplenomegalia. De fato, a deficiência familiar de LPL – uma forma de hipertrigliceridemia totalmente causada por um defeito extra-hepático na hidrólise de triglicerídeos – costuma estar associada à doença do fígado esteatótico. Neste contexto, a esteatose hepática não alcoólica regride rapidamente mediante restrição da gordura na dieta. Em alguns pacientes, a esteatose hepática progride para uma esteato-hepatite que está associada à fibrose e necrose. Os motivos que levam a esta progressão não estão esclarecidos. Talvez, seja necessário haver uma 2ª agressão para que estes pacientes desenvolvam uma esteato-hepatite não alcoólica e, então, evoluam para cirrose.
Os níveis de colesterol de LDL são considerados como “limítrofemente elevados” no paciente que apresenta baixo risco de aterosclerose, quando excedem 130 mg/dL. Os níveis de HDL são considerados altos quando estão acima 160 mg/dL [Tabela 2]. Por definição, os níveis de triglicerídeos de um indivíduo são normais,17 enquanto os níveis de colesterol de HDL são variáveis, ainda que frequentemente normais. Os distúrbios lipídicos apresentados por estes pacientes em geral são descobertos durante a avaliação de colesterol de rotina. Embora alguns observadores questionem a relação custo-efetividade da avaliação de homens e mulheres com mais de 20 anos de idade, nos Estados Unidos, a alta prevalência dos níveis de colesterol de LDL elevados justifica o estudo da população, segundo as recomendações do National Cholesterol Education Program (NCEP) e outras autoridades.
Os níveis de colesterol severamente elevados são uma indicação de HF. A habilidade de diagnosticar a HF é valiosa, pois os indivíduos afetados necessitam de terapia farmacológica quando ainda são relativamente jovens [ver Hipercolesterolemia familiar (HF), adiante].
A hipercolesterolemia isolada pode estar presente de maneira intermitente em pacientes com HLCF. Uma história familiar fortemente positiva de doença cardiovascular prematura ou a observação de qualquer um dos outros critérios de HLCF devem fornecer indícios do diagnóstico deste distúrbio [ver Hiperlipidemia combinada familiar (HLCF), adiante]. Nem todos os casos de hipercolesterolemia isolada leve são indicativos de HF ou HLCF. Estes casos podem resultar de interações de fatores adquiridos e ambientais, em particular de fatores dietéticos, com fatores genéticos desconhecidos que conferem suscetibilidade à hipercolesterolemia.
A maioria das diretrizes terapêuticas vigentes baseia-se principalmente nos níveis de colesterol de LDL, pois foi demonstrado que a redução do LDL minimiza a doença cardiovascular em até 50%.18 A diminuição do consumo de colesterol e gorduras saturadas oriundas da dieta em geral promove uma redução modesta dos níveis de colesterol de LDL. Esta redução depende, em parte, da dieta basal. As mudanças de estilo de vida, incluindo a dieta e a perda de peso, serão suficientes para alguns indivíduos terem seus níveis de colesterol reduzidos a uma faixa aceitável. Entretanto, é improvável que esta abordagem seja efetiva para os pacientes com as formas familiares de dislipidemia, como a HF ou a HLCF.
Nos casos de pacientes com as formas familiares da doença ou daqueles para os quais as ações isoladas relacionadas ao estilo de vida falham em trazer os níveis de colesterol de LDL para junto dos valores recomendados pelas diretrizes, devem ser adicionados fármacos redutores de colesterol ao regime terapêutico [ver Terapia farmacológica na dislipidemia, adiante]. A terapia dietética pode também reduzir os níveis de colesterol LDL em 5 a 15%, além das reduções alcançadas com o uso dos fármacos.19 A terapia dietética, portanto, pode levar à redução das dosagens dos fármacos requeridos e deve ser instituída de maneira combinada à terapia farmacológica. A principal classe de fármacos usados para diminuir o colesterol de LDL é a das estatinas. Entretanto, as resinas ligadoras de ácido biliar e os fármacos que bloqueiam a absorção do colesterol são valiosos para os pacientes que não respondem adequadamente apenas às estatinas, podendo ser usados combinados às estatinas ou a outros fármacos.
Uma elevação isolada dos níveis de triglicerídeos pode ser causada por um distúrbio primário do metabolismo lipídico (p. ex., HTGF ou HLCF); pode ser secundária ao uso de fármacos terapêuticos; ou pode, ainda, ser um componente da síndrome metabólica ou do diabetes melito de tipo 2. Diferentemente dos níveis de colesterol, tem sido difícil determinar os níveis de triglicerídeos em que o risco de DAC aumenta ou diminui. É importante determinar a causa da hipertrigliceridemia, pois as abordagens terapêuticas podem ser distintas.
Exemplificando, é importante distinguir entre HTGF (que não confere risco de desenvolvimento precoce de DAC) e HLCF (que está associada a uma alta incidência de aterosclerose prematura).19 Contudo, pode ser difícil distinguir estes distúrbios quando a HLCF está associada à hipertrigliceridemia. Uma história familiar ou pessoal positiva de aterosclerose prematura sugere a HLCF. Além disso, os pacientes com HLCF frequentemente apresentam fatores de risco cardiovascular não lipídicos (isto é, obesidade central, hipertensão, resistência à insulina, comprometimento da tolerância à glicose, níveis aumentados de inibidor do ativador de plasminogênio-1 [PAI-1] e níveis aumentados de marcadores inflamatórios circulantes). A hipertrigliceridemia presente na HLCF indica a existência de números aumentados de partículas de LDL pequenas e densas, além de conferir um risco aumentado de doença cardiovascular prematura.14 De modo similar, a hipertrigliceridemia associada ao diabetes melito de tipo 2 e a síndrome metabólica constituem um importante fator de risco cardiovascular. Outros fatores de risco cardiovascular geralmente estão presentes em pacientes com diabetes melito de tipo 2, síndrome metabólica ou HLCF. Desta forma, a estratégia terapêutica deve considerar outros fatores além do distúrbio lipídico.
Os pacientes com HTGF não parecem apresentar risco significativamente aumentado de desenvolvimento prematuro de DAC. No entanto, estes pacientes apresentam risco aumentado de desenvolvimento da síndrome de hiperquilomicronemia, diante da existência de formas secundárias de hipertrigliceridemia, como a hipertrigliceridemia causada pelo uso de fármacos que elevam os níveis de triglicerídeos [Tabela 3]. A síndrome da hiperquilomicronemia também ocorre na HLCF, combinada a outras causas de hipertrigliceridemia. Em pacientes com pancreatite causada por hipertrigliceridemia, os níveis de triglicerídeos ultrapassam 2.000 mg/dL e podem atingir valores bem mais altos. Recomenda-se que os níveis plasmáticos de triglicerídeos sejam mantidos abaixo de 2.000 mg/dL, para evitar uma pancreatite aguda recorrente. Uma meta segura seria alcançar níveis inferiores a 1.000 mg/dL.
Tabela 3. Efeitos de fármacos selecionados sobre os níveis de lipoproteína
|
Fármaco |
VLDL |
LDL |
HDL |
|
Álcool* |
+ |
0 |
+ |
|
Estrogênios, estradiol* |
+ |
– |
+ |
|
Androgênios, testosterona |
+ |
+ |
– |
|
Progestinas |
– |
+ |
– |
|
Glicocorticoides* |
+ |
0 |
+ |
|
Ciclosporinas |
+ |
+ |
+ |
|
Tacrolimo |
+ |
+ |
+ |
|
Diuréticos tiazida* |
+ |
+ |
– |
|
Betabloqueadores * |
+ |
0 |
– |
|
Bloqueadores de canal de cálcio |
0 |
0 |
0 |
|
Inibidores da enzima conversora de angiotensina |
0 |
0 |
0 |
|
Sertralina* |
Possível + |
+ |
0 |
|
Inibidores de protease* |
+ |
0 |
0 |
|
Valproato e fármacos relacionados |
+ |
0 |
– |
|
Isotretinoína* |
+ |
0 |
– |
*Podem causar hipertrigliceridemia severa e síndrome de quilomicronemia em pacientes com a forma familiar da hipertrigliceridemia ou diabetes melito de tipo 2.
Os pacientes que apresentam elevação dos níveis plasmáticos de colesterol total e triglicerídeos são classificados em 3 categorias. Na 1ª categoria, estão aqueles em que ocorre elevação de VLDL e LDL, como a observada na HLCF. Na 2ª categoria, estão os pacientes com elevação dos níveis de remanescentes de VLDL e de remanescentes de quilomícrons, como ocorre na doença da remoção de remanescente. A 3ª categoria consiste de pacientes com níveis de triglicerídeos bastante altos, nos quais o aumento do colesterol total é resultado do colesterol da VLDL e dos quilomícrons.
Em pacientes com HLCF, observa-se com frequência um aumento dos níveis de triglicerídeos e colesterol de LDL. Estes pacientes apresentam níveis altos de apo B e partículas de LDL pequenas e densas. A terapia destes indivíduos muitas vezes requer o uso de vários fármacos – um para diminuir os níveis de triglicerídeos e outro para reduzir a concentração de partículas pequenas e densas de LDL [ver Terapia farmacológica na dislipidemia, adiante].
Em pacientes com doença de remoção de remanescentes, os níveis de colesterol e triglicerídeos no plasma frequentemente são iguais. Nestas circunstâncias, é importante considerar tal condição. A terapia é voltada para a diminuição da secreção lipoproteica hepática, com o uso de estatinas, fibratos ou niacina.
Em pacientes com hipertrigliceridemia severa, o aumento dos níveis plasmáticos de colesterol total é resultado do colesterol da VLDL e dos quilomícrons. Os fibratos com frequência são o fármaco de escolha. Entretanto, é muito importante determinar a etiologia da hipertrigliceridemia severa e remover qualquer fármaco agressor [Tabela 3] ou tratar qualquer causa secundária de hipertrigliceridemia.
Muitos pacientes (se não a maioria) com hipertrigliceridemia apresentam redução concomitante dos níveis de colesterol de HDL. Sendo assim, o tratamento dos níveis baixos de colesterol de HDL deve ser considerado no contexto do tratamento do distúrbio subjacente (p. ex., HLCF ou diabetes melito de tipo 2) [ver Pacientes com elevação isolada dos níveis de triglicerídeos, anteriormente]. Níveis baixos isolados de colesterol de HDL, da ordem de 20 a 30 mg/dL, raramente são observados na ausência de hipertrigliceridemia concomitante ou de outras alterações nos níveis de lipídios e lipoproteínas. Mesmo assim, estes níveis reduzidos constituem um fator de risco de doença cardiovascular.10 No passado, estas reduções dos níveis de HDL frequentemente não eram detectadas. As estratégias de avaliação utilizadas antigamente eram baseadas na avaliação dos níveis de colesterol total que, por sua vez, muitas vezes não estavam elevados em pacientes com reduções isoladas de HDL. A quantificação específica do colesterol de HDL se faz necessária para identificar estes pacientes. O tratamento dos raros casos de pacientes com níveis baixos isolados de colesterol de HDL ainda é controverso. Foi demonstrado que tanto os fibratos como o ácido nicotínico elevam os níveis de colesterol de HDL.20,21 Embora os fibratos diminuam os níveis de triglicerídeos de VLDL, também elevam os níveis de colesterol de HDL. Muitos estudos sobre a terapia da aterosclerose à base de fibratos foram inconclusivos.22 No entanto, o Veterans Affairs High-Density Lipoprotein Intervention Trial (VA-HIT) demonstrou uma redução dos eventos de cardiopatia coronariana em homens com esta doença que apresentavam baixos níveis de colesterol de HDL.23 Dois estudos recentes sobre o uso de fenofibrato no tratamento de pacientes diabéticos com doença cardiovascular, o Fenofibrate Intervention and Event Lowering in Diabetes (FIELD)24 e o Action to Control Cardiovascular Risk in Diabetes (ACCORD),25 falharam em minimizar a doença cardiovascular, levantando a questão da eficácia dos fibratos no tratamento da cardiopatia coronariana. O ácido nicotínico, que atua em numerosos sítios metabólicos, também aumenta os níveis de colesterol de HDL. O ácido nicotínico diminuiu os casos de morte por causa cardiovascular no Coronary Drug Project, conduzido nos anos 1970.26 Desde então, vários estudos sugeriram que o uso de niacina, seja isolada ou combinada a outros fármacos, eleva os níveis de colesterol de HDL.21 As estatinas podem aumentar ou diminuir os níveis de colesterol de HDL.20
Nota do editor: O estudo VA-HIT demonstrou em um estudo de prevenção secundária, que um aumento de 2 mg/dl dos níveis de HDL colesterol foi suficiente para redução de eventos cardiovasculares de forma significativa, a medicação utilizada foi o gemfibrosil.
Em raras ocasiões, pacientes de meia-idade com aterosclerose estabelecida não apresentam anormalidades lipoproteicas ou lipídicas detectáveis. Em adição ao perfil lipídico padrão, a quantificação da apo B e da Lp(a) muitas vezes revela a existência de anormalidades lipoproteicas mínimas, como números aumentados de partículas pequenas e densas de LDL, nestes pacientes. A avaliação dos fatores de risco não lipoproteicos (p. ex., homocisteína e marcadores inflamatórios, como a proteína C reativa) também pode ser valiosa na avaliação do risco cardiovascular. Embora os níveis de alguns destes fatores de risco possam ser reduzidos com a adoção de várias estratégias (p. ex., hemocisteína com terapia à base de folato), o uso das estatinas no tratamento de indivíduos de todas as categorias de alto risco, em particular daqueles com doença vascular estabelecida, mostrou-se benéfico, mesmo diante de níveis de lipídios aparentemente normais.
Nota do editor: Uma análise demosntrou que em estudo de prevenção primária TEXCAPS/AFSCAPS, a lovastatina demonstrou diminuição de eventos cardiovasculares em subgrupo com níveis baixos de colesterol e níveis aumentados de proteína C reativa.
Os distúrbios primários do metabolismo de lipoproteínas são aqueles originados a partir de defeitos genéticos envolvendo as vias metabólicas das lipoproteínas (isto é, distúrbios familiares causados pela secreção hepática aumentada de lipoproteínas ou por defeitos catabólicos). Os distúrbios que causam aumento da secreção lipoproteica são a síndrome metabólica, HLCF, diabetes melito de tipo 2 e HTGF. As elevações dos níveis de Lp(a) também podem causar aumento da secreção de lipoproteínas. Os distúrbios do catabolismo da LDL são a HF e o defeito familiar da apo B-100. A doença da remoção dos remanescentes é um defeito do catabolismo dos remanescentes.
A síndrome metabólica consiste em uma adiposidade de distribuição central ou obesidade visceral, resistência à insulina, elevações dos níveis plasmáticos de ácidos graxos livres, comprometimento da tolerância à glicose, hipertensão, dislipidemia e estado pró-coagulante anormal. Muitos aspectos desta síndrome comprovadamente predispõem homens e mulheres ao desenvolvimento de DAC prematura.14
Um acúmulo de gordura visceral, em vez de subcutânea, tem sido encontrado em indivíduos com a distribuição central do tecido adiposo corporal, característica da síndrome metabólica. Os homens têm mais gordura visceral do que as mulheres em pré-menopausa, mesmo quando apresentam índices de massa corporal (IMC) compatíveis. Foi sugerido que estas diferenças de gordura visceral e as mudanças associadas nos níveis de lipoproteínas e na pressão arterial poderiam contribuir, em parte, para a diferença de risco de desenvolvimento de DAC prematura observada entre homens e mulheres em pré-menopausa.27 A gordura visceral aumentada está associada à resistência à insulina, hiperinsulinemia, baixos níveis plasmáticos de adiponectina e elevações dos níveis plasmáticos de ácidos graxos livres.28 Foi sugerido que o acúmulo de gordura visceral precede e causa resistência à insulina, bem como a resultante hiperinsulinemia, porque a sensibilidade à insulina aumenta e os níveis de ácidos graxos livres caem quando a gordura visceral é reduzida após a restrição calórica.29,30
Os níveis de insulina, glicose, triglicerídeos, colesterol de HDL, pressão arterial, PAI-1 e outros marcadores inflamatórios encontram-se aumentados e acima da média normal em pacientes com síndrome metabólica. Embora estas variáveis geralmente evoluem para níveis elevados, algumas permanecem dentro da faixa normal-alta em determinados indivíduos afetados. Os níveis de HDL tendem a ser mais baixos do que a média normal. Fatores genéticos e ambientais parecem afetar a distribuição destas variáveis em indivíduos normais e naqueles com síndrome metabólica. Como a síndrome metabólica está associada a múltiplos fatores de risco cardiovascular, indivíduos com esta condição apresentam risco aumentado de desenvolvimento de DAC. Não se sabe se todos os indivíduos que atendem às diretrizes do NCEP para síndrome metabólica31 [ver Diagnóstico, adiante] apresentam risco aumentado de desenvolvimento de DAC prematura. Entretanto, o diabetes melito de tipo 2 e a HLCF são distúrbios específicos, dos quais a síndrome metabólica é um componente.14,32 Estes 2 distúrbios são responsáveis por no mínimo 40 a 50% dos casos de DAC prematura e precisam ser considerados no contexto da síndrome metabólica.
O risco de desenvolvimento do padrão de distribuição de gordura abdominal, dislipidemia, comprometimento do metabolismo da glicose e hipertensão – todos sintomas-sentinela da síndrome metabólica – aumenta com o avanço da idade.33 A obesidade central associada à síndrome metabólica pode ser evidente em adultos jovens após a conclusão do crescimento na adolescência. Entretanto, a obesidade central e a resistência à insulina mais tipicamente se manifestam na meia-idade. Enquanto as elevações dos níveis de colesterol de LDL podem não ser preditivas do aparecimento da aterosclerose em idosos, a obesidade central, hipertensão e resistência à insulina constituem fatores de risco de desenvolvimento de aterosclerose. E a prevalência destes fatores aumenta com o avanço da idade,34-36 possivelmente devido à síndrome metabólica.
Embora a associação da obesidade central e da resistência à insulina com a dislipidemia esteja bem estabelecida, a causa subjacente ainda é desconhecida. Um mecanismo que poderia explicar esta associação é o aumento dos níveis de ácidos graxos livres de cadeia longa junto à veia porta. Este aumento inibiria a degradação da apo B hepática no retículo endoplasmático e aumentaria a probabilidade de a apo B ser secretada pelo fígado sob a forma de lipoproteínas contendo triglicerídeos. Isto contribuiria para os níveis aumentados de triglicerídeos e para o número aumentado de partículas de VLDL e LDL encontrado em pacientes com condições insulina-resistentes.37 Outro efeito dos ácidos graxos livres de cadeia longa consiste em aumentar a concentração de lipase hepática na superfície dos hepatócitos. A lipase hepática hidrolisa os triglicerídeos e fosfolipídios presentes na LDL e na HDL, diminuindo o tamanho de cada partícula [Figura 6].14 Entretanto, a CETP também contribui para este processo de remodelamento de lipoproteína. A predominância do efeito da lipase hepática ou da CETP sobre o tamanho e a densidade das partículas de LDL e HDL depende do conteúdo de triglicerídeos e da taxa de secreção de VLDL. As diferenças de tamanho da partícula de LDL e de níveis de HDL2 entre homens e mulheres em pré-menopausa podem ser amplamente responsabilizadas pelas diferenças de gordura visceral entre homens e mulheres.
O National Cholesterol Education Program Adult Treatment Panel III (NCEP ATP-III) sugeriu o uso de 5 variáveis clínicas como critérios diagnósticos para a síndrome metabólica: (1) circunferência da cintura aumentada; (2) níveis aumentados de triglicerídeos; (3) níveis diminuídos de colesterol de HDL; (4) pressão arterial elevada; e (5) níveis altos de glicose plasmática de jejum [Tabela 4].17 Um diagnóstico de síndrome metabólica é estabelecido quando pelo menos 3 destas variáveis clínicas estão presentes. Quando estas 5 variáveis foram avaliadas em um levantamento realizado com 8.814 homens e mulheres adultos, cerca de 24% dos participantes do estudo atenderam aos critérios diagnósticos estabelecidos para a síndrome metabólica.38,39 A Organização Mundial de Saúde (OMS) também estabeleceu critérios para a síndrome metabólica. Atualmente, está sendo conduzida uma tentativa de harmonizar estes 2 conjuntos de critérios.
Tabela 4. Aspectos clínicos da síndrome metabólica17
|
A presença de pelo menos 3 das seguintes variáveis indica um diagnóstico de síndrome metabólica: |
|
Obesidade abdominal: circunferência da cintura > 89 cm (mulheres) ou > 101,6 cm (homens) |
|
Triglicerídeos = 150 mg/dL |
|
Colesterol de HDL < 50 mg/dL (mulheres) ou < 40 mg/dL (homens) |
|
Pressão arterial = 130/85 mmHg |
|
Glicose plasmática de jejum = 11 mg/dL |
HDL = lipoproteína de alta densidade
A obesidade visceral e a resistência à insulina são os principais fatores contribuíntes para o desenvolvimento de dislipidemia associada à síndrome metabólica. As anormalidades lipídicas que estão associadas à síndrome metabólica são: níveis aumentados de triglicerídeos; números aumentados de partículas pequenas e densas de LDL; níveis aumentados de apo B; e níveis diminuídos de colesterol de HDL. Entretanto, em populações normais selecionadas ao acaso, a obesidade visceral e a resistência à insulina estavam associadas apenas a um leve aumento dos níveis de triglicerídeos e a uma pequena diminuição dos níveis de colesterol de HDL.28 Em contraste, a obesidade visceral e a resistência à insulina podem contribuir para o desenvolvimento de uma dislipidemia mais severa quando estão associadas ao diabetes melito de tipo 2 e à HLCF.14
A dislipidemia da síndrome metabólica pode ser diagnosticada pela demonstração de elevações discretas a moderadas dos níveis plasmáticos de triglicerídeos e apo B, níveis reduzidos de colesterol de HDL e níveis normais de colesterol de LDL. Embora os níveis de colesterol de LDL sejam normais em pacientes com este distúrbio, o número de partículas de LDL geralmente está aumentado. Além disso, a forma predominante são as partículas pequenas e densas de LDL, que são pobres em colesterol quando comparadas às partículas grandes e flutuantes de LDL. A presença das partículas pequenas e densas de LDL pode ser determinada via medida direta do tamanho ou da densidade da LDL. A quantificação de rotina dos níveis plasmáticos de apo B na prática clínica é desnecessária para o diagnóstico deste distúrbio. No entanto, a medida da concentração de apo B no plasma indica a presença de números aumentados de partículas pequenas e densas de LDL. De modo semelhante, os níveis de HDL total refletem as alterações dos valores de HDL2, indicando que as subfrações de HDL não precisam ser medidas.40
O exercício aeróbico e uma dieta com baixo teor de gordura saturada são indicados como terapia para a maioria dos pacientes com síndrome metabólica. Quando a síndrome metabólica é severa ou o paciente tem HLCF ou diabetes melito de tipo 2, indica-se a instituição de uma terapia mais agressiva [ver Terapia farmacológica na dislipidemia, adiante].
HLCF é um distúrbio autossômico dominante, responsável por até metade das causas familiares de DAC.41 Foi descrito pela 1ª vez em famílias de sobreviventes de infarto do miocárdio (IM).42-44 A HLCF é caracterizada por elevações dos níveis de triglicerídeos ou colesterol, ou de ambos, em parentes afetados. Além destas elevações, os pacientes com HLCF caracteristicamente apresentam elevações dos níveis de apo B e números aumentados de partículas pequenas e densas de LDL.45
A análise de linkage genético sugere que a herança do fenótipo lipídico na HLCF envolve efeitos genéticos separados46 para a elevação dos níveis de apo B47 e para os números aumentados de partículas pequenas e densas de LDL encontrados nas famílias com HLCF. Evidências adicionais da heterogeneidade genética são fornecidas por estudos que detectaram em 1/3 dos indivíduos com HLCF um nível de atividade de LPL reduzido pela metade no plasma pós-heparinização.48 A obesidade visceral e a resistência à insulina contribuem para a dislipidemia observada na HLCF, mas não podem contribuir para a elevação dos níveis de apo B.49
No Familial Atherosclerosis Treatment Study (FATS), a terapia de redução lipídica intensiva com ácido nicotínico ou lovastatina combinada ao colestipol resultou em diminuição da atividade da lipase hepática, número reduzido de partículas pequenas e densas de LDL, e níveis elevados de colesterol de HDL2, com subsequente regressão da DAC, evidenciada por angiografia.50 A redução lipídica intensiva resultou na regressão subsequente da aterosclerose, em particular em indivíduos com partículas pequenas e densas de LDL que tinham HLCF ou níveis basais elevados de Lp(a).
Uma abordagem agressiva para modificar os fatores de risco cardiovascular reversíveis deve ser conduzida em casos de pacientes afetados pelo distúrbio. A terapia dietética e a modificação terapêutica do estilo de vida com inclusão da atividade física devem ser instituídas, aliadas à terapia farmacológica de redução lipídica [ver Terapia farmacológica na dislipidemia, adiante]51 e a tratamento de outros fatores de risco cardiovascular. A escolha da medicação para dislipidemia depende, até certo ponto, da alteração lipídica primária ser uma hipercolesterolemia, hipertrigliceridemia ou elevações combinadas de colesterol e triglicerídeos. Quando a hipercolesterolemia é a manifestação primária, a abordagem deve ser a mesma adotada para o paciente hipercolesterolêmico [ver Pacientes com elevação isolada dos níveis de colesterol lipoproteína de baixa densidade (LDL), anteriormente]. Se a hipertrigliceridemia for a principal anormalidade, então a abordagem inicial poderia ser aquela usada em casos de pacientes com hipertrigliceridemia isolada. Entretanto, a maioria dos pacientes apresenta elevações dos níveis de triglicerídeos e colesterol de LDL, e necessita de uma terapia combinada. Os regimes podem combinar estatina com niacina, fibrato ou ezetimibe [ver Terapia farmacológica na dislipidemia, adiante].
Os pacientes submetidos ao tratamento para diabetes melito de tipo 2 tipicamente apresentam obesidade central e resistência à insulina. Um defeito na secreção de insulina é encontrado em indivíduos com resistência à insulina que desenvolvem hiperglicemia. Os parentes de 1º grau de indivíduos com diabetes melito de tipo 2 podem apresentar obesidade central e resistência à insulina, ou podem responder à glicose com um secreção diminuída de insulina. Os parentes de 1º grau com obesidade central e que possuem defeito de secreção de insulina invariavelmente desenvolvem diabetes melito de tipo 2. Embora a maioria dos genes que contribuem para a obesidade central, resistência à insulina e secreção defeituosa de insulina seja desconhecida, o diabetes melito de tipo 2 é o exemplo clássico de um distúrbio oligogênico. A determinação de todos os genes envolvidos requer uma detalhada caracterização fenotípica dos subgrupos de indivíduos com diabetes melito de tipo 2.
A dislipidemia do diabetes melito não tratado e da hiperglicemia é discutida adiante, neste capítulo [ver Distúrbios endócrinos causadores de dislipidemia, adiante]. A dislipidemia do diabetes melito de tipo 2 tratado é similar àquela associada à síndrome metabólica e à HLCF, caracterizando-se por aumento discreto dos níveis de triglicerídeos, redução dos níveis de colesterol de HDL2 e números aumentados de partículas pequenas e densas de LDL. O tratamento envolve terapia alimentar, aumento da atividade física e terapia farmacológica de redução lipídica [ver Terapia farmacológica na dislipidemia, adiante].52
A HTGF é um distúrbio hereditário comum, considerado autossômico dominante, que afeta cerca de 1% da população. A HTGF é caracterizada pelo aumento da síntese de triglicerídeos que resulta no enriquecimento das partículas de VLDL com triglicerídeos secretados em quantidades normais. Os indivíduos afetados apresentam nível altos de VLDL, porém níveis baixos de LDL e HDL, além de geralmente serem assintomáticos, exceto quando desenvolvem severa hipertrigliceridemia (isto é, síndrome da quilomicronemia). A HTGF pode estar associada ao risco aumentado de DAC prematura.19 É difícil distinguir a HTGF da hipo-alfa-lipoproteinemia familiar, um distúrbio de classificação pouco definida.47,53
Um diagnóstico é estabelecido pela história familiar e avaliação dos perfis lipoproteicos de jejum do paciente e de seus parentes. Os níveis de triglicerídeos variam de 250 a 1.000 mg/dL em cerca de metade dos parentes de 1º grau. Uma história familiar fortemente significativa de DAC prematura em geral inexiste, e não deve haver elevação dos níveis de LDL.
Os pacientes com HTGF devem perder peso, quando necessário, praticar exercícios regularmente e diminuir a ingesta de ácidos graxos saturados e colesterol. Bebidas alcoólicas, estrogênios exógenos e outros fármacos que aumentam os níveis de VLDL talvez tenham de ser restringidos. O diabetes, quando presente, precisa ser devidamente controlado. A hipertrigliceridemia em pacientes com HTGF muitas vezes responde a estas ações. Quando os níveis de triglicerídeos excedem 500 mg/dL após 6 meses de terapia não farmacológica, a terapia farmacológica com um fibrato deve ser considerada.54 A terapia farmacológica deve ser instituída diante de níveis acima de 1.000 mg/dL.
Os fibratos são os fármacos de escolha para a redução de níveis altos de triglicerídeos em pacientes com HTGF [ver Terapia farmacológica na dislipidemia, adiante]. Na HLCF, a niacina promove vários efeitos benéficos adicionais sobre a concentração de lipídios no sangue – aumenta os níveis de colesterol de HDL; diminui os níveis de partículas pequenas e densas de LDL; e pode reduzir os níveis de Lp(a). Embora exerçam um efeito menos dramático sobre os triglicerídeos do que os fibratos, as estatinas são comprovadamente valiosas para o tratamento de pacientes de alto risco com hipertrigliceridemia moderada e níveis aumentados de partículas pequenas e densas de LDL, como ocorre em pacientes com diabetes melito de tipo 2 e HLCF.
A HF é um distúrbio autossômico dominante causado por uma mutação no gene codificador do receptor proteico da LDL. Na condição homozigota com HF, extremamente rara, existem 2 alelos mutantes no locus do receptor de LDL que tornam o indivíduo afetado quase ou absolutamente incapaz de depurar a LDL da circulação via receptor de LDL.9 Os indivíduos heterozigotos com HF possuem um alelo normal, que lhes confere um receptor com metade da atividade normal. Como o receptor de LDL contribui para a depuração dos remanescentes de VLDL presentes no plasma, uma deficiência deste receptor pode acarretar certo grau de acúmulo de lipoproteínas remanescentes. Altas concentrações de LDL resultam na captação não mediada por receptor da LDL pela matriz extracelular, incluindo a matriz da parede arterial, que leva à formação de xantomas e aterosclerose. A forma heterozigota deste distúrbio exibe uma prevalência aproximada de 1 caso em cada 500 indivíduos, que a torna uma das doenças genéticas mais comuns.55
A hipercolesterolemia pode ser detectada ao nascimento, no sangue do cordão umbilical. Se a HF não for detectada ao nascimento, várias condições associadas podem sugerir o diagnóstico posteriormente, em outras fases da vida. Os xantomas de tendão constituem um sinal altamente específico de HF. Tipicamente, os xantomas surgem por volta dos 20 anos de idade e podem ser encontrados em até 70% dos pacientes com idade mais avançada. Ocasionalmente, os xantomas são observados no tendão patelar. Por serem sutis, sua detecção requer o exame detalhado dos tendões dorsais da mão e do tendão de Aquiles. O xantelasma (xantomas cutâneos na pálpebra) e o arco senil são comuns em pacientes com HF com mais de 30 anos de idade, embora também ocorram em indivíduos normocolesterolêmicos. O arco senil prematuro é observado superior e inferiormente nos olhos, e depois passa a ser totalmente circunferencial.
A DAC desenvolve-se nas fases iniciais, acompanhada de sintomas que costumam se manifestar nos homens durante a 4ª ou 5ª década da vida. Cerca de 5% de todos os casos de IM prematuro ocorrem em pacientes com HF heterozigota.9 Antes do desenvolvimento da terapia à base de estatina, pelo menos 50% dos homens com HF heterozigota sofriam IM em torno dos 60 anos de idade. Nas mulheres, os sintomas tendem a ocorrer cerca de 10 anos mais tarde. Os níveis de colesterol total em pacientes heterozigotos geralmente variam de 350 a 550 mg/dL. Os níveis de triglicerídeos podem estar levemente aumentados, enquanto os níveis de colesterol de HDL sofrem uma redução aproximada de 10% nos indivíduos heterozigotos. A função do receptor de LDL pode ser determinada somente em laboratórios especiais.
A HF heterozigota deve ser suspeita diante da detecção de uma hipercolesterolemia severa com níveis de LDL elevados. Havendo xantomas de tendão, o diagnóstico é quase certo. Na ausência de xantomas de tendão, devem ser investigadas causas secundárias da hipercolesterolemia (p. ex., hipotireoidismo), porém o diagnóstico de HF não é excluído. A obtenção de uma história familiar detalhada deve revelar uma história significativa de DAC prematura e hipercolesterolemia sem hipertrigliceridemia. O distúrbio afeta cerca de metade dos parentes de 1º grau. A presença de hipercolesterolemia e xantomas de tendão em um paciente ou em seu irmão é praticamente diagnóstica, assim como a hipercolesterolemia em uma criança da família. A realização de uma avaliação detalhada dos familiares do paciente é obrigatória, pois 50% dos parentes de 1º grau de um indivíduo afetado também estarão afetados e necessitarão de terapia redutora de lipídios agressiva.56
O tratamento da HF requer intervenção na dieta e terapia farmacológica. A meta do tratamento é diminuir os níveis de colesterol de LDL para menos de 130 mg/dL, ou até menos, caso o paciente tenha DAC. Em casos de pacientes heterozigotos para HF, o tratamento efetivo é possível com o uso de combinações de estatinas, fármacos ativos no intestino e ácido nicotínico. Como os níveis de colesterol de LDL tendem a ser muito altos, a terapia combinada utilizando 2 fármacos muitas vezes se faz necessária, enquanto o uso de 3 fármacos pode vir a ser necessário [ver Terapia farmacológica na dislipidemia, adiante]. Embora a terapia alimentar isolada seja insuficiente para os pacientes com HF heterozigota, a diminuição da ingesta de ácidos graxos saturados e de colesterol pode reduzir os níveis de LDL e também a quantidade de medicação necessária. Isto é particularmente importante no caso das crianças e adolescentes, antes da iniciação da terapia farmacológica. Foi demonstrado que os xantomas de tendão regridem quando os níveis de LDL são mantidos dentro da faixa desejável. A redução agressiva do colesterol de LDL em homens e mulheres heterozigotos para HF pode causar regressão da aterosclerose coronariana.
Uma mutação em apo B-100 que resulta na inibição de sua ligação ao receptor de LDL constitui outra causa genética de elevações dos níveis de LDL. A prevalência deste distúrbio é desconhecida, mas foi estimada em 5 a 10% da prevalência da HF. A estrutura e a função do receptor de LDL permanecem normais. Uma molécula integral de apo B-100 é produzida com a substituição de um único aminoácido, resultando em uma apo B-100 que se liga fracamente aos receptores de LDL, com consequente acúmulo de LDL no plasma.
Os indivíduos afetados são clinicamente indistinguíveis dos pacientes com HF heterozigota: podem apresentar hipercolesterolemia severa, xantomas de tendão e aterosclerose prematura. O tratamento com estatinas parece diminuir os níveis de colesterol de LDL em pacientes com este distúrbio. Existem exames especializados, disponibilizados somente em laboratórios de pesquisa, que são necessários para distinguir entre indivíduos afetados com apo B defeituosa e aqueles com receptores de LDL defeituosos.
A Lp(a) é uma classe específica de partículas lipoproteicas sintetizadas no fígado.6 Um componente importante da Lp(a) é a apo(a), cuja estrutura é homóloga à do plasminogênio – uma proteína essencial para a cascata da coagulação. As concentrações plasmáticas de Lp(a) variam acentuadamente de um indivíduo para outro, indo de níveis indetectáveis até 200 mg/dL. A concentração plasmática de Lp(a) é fortemente controlada por fatores genéticos.
A maioria dos estudos epidemiológicos sugere que a Lp(a) constitui um fator de risco de desenvolvimento de DAC e acidente vascular cerebral (AVC). Se a Lp(a) é aterogênica, isto possivelmente se deve às suas propriedades LDL-símiles: foi demonstrado que a Lp(a) sofre captação endotelial e modificação oxidativa, além de promover a formação de células espumosas. Como a Lp(a) apresenta um alto grau de homologia com o plasminogênio, é possível que exerça algum papel na trombose, via interferência na ligação do plasminogênio à fibrina. Níveis altos de Lp(a) parecem aumentar a aterogenicidade de outros fatores de risco cardiovascular, com o aparecimento antecipado de eventos cardiovasculares.
Dados sugerem que a redução dos níveis de colesterol de LDL em pacientes com altos níveis de Lp(a) pode ser uma estratégia efetiva para retardar a progressão da aterosclerose e prevenir os eventos coronarianos. Os próprios níveis de Lp(a) em si podem ser reduzidos com altas doses de niacina, estrogênio ou tamoxifeno, bem como com aférese de LDL. Há dados insuficientes sobre a eficácia da diminuição dos níveis de Lp(a) por si só em termos de promoção da inibição da aterosclerose ou prevenção de eventos coronarianos.6
A dislipidemia por remanescentes, também denominada hiperlipoproteinemia de tipo III, dis-beta-lipoproteinemia e doença beta-ampla, é definida pela presença de partículas de VLDL que migram na posição beta à eletroforese (as partículas de VLDL normais migram na posição pré-beta). As partículas beta-VLDL são remanescentes de quilomícron e de VLDL.
A doença é causada por uma mutação no gene APOE8 [ver Regulação do metabolismo das lipoproteínas, anteriormente]. Esta mutação leva ao comprometimento da captação hepática das lipoproteínas contendo apo E e suspende a conversão da VLDL e IDL em LDL. Na ausência de fatores genéticos, hormonais ou ambientais adicionais, os remanescentes não se acumulam em quantidade suficiente para causar hiperlipidemia, pois são depurados pelos receptores hepáticos que também se ligam (com menos avidez) à apo B-48 e à apo B-100. A dislipidemia por remanescentes resulta de um defeito na apo E (quase sempre associado ao genótipo E2/E2) concomitante a um 2º defeito, genético ou adquirido, que causa produção excessiva de VLDL (como na HLCF) ou diminuição da atividade do receptor de LDL (como na HF familiar heterozigota ou no hipotireoidismo). O genótipo E2/E2 é encontrado em 1% da população branca e em quase todos os indivíduos com doença da remoção de remanescentes.
Indivíduos com dislipidemia por remanescentes apresentam elevações dos níveis de colesterol e triglicerídeos e são propensos ao desenvolvimento de DAC prematura. Por razões ainda obscuras, estes pacientes apresentam risco particularmente aumentado de desenvolvimento de doença vascular periférica. A hiperlipidemia em geral não se desenvolve antes da fase adulta. Os xantomas palmares (xanthoma striata palmaris) – descolorações amarelo-alaranjadas nas dobras palmares – são patognomônicos da doença de remoção de remanescentes genéticos, mas nem sempre estão presentes. Os xantomas palmares podem ser de difícil visualização e devem ser cuidadosamente procurados, com auxílio de iluminação adequada. Os xantomas tuboeruptivos ocasionalmente são encontrados em sítios de compressão, em particular nos cotovelos, nádegas e joelhos.
A suspeita do diagnóstico da dislipidemia por remanescentes deve ser considerada em casos de pacientes com elevação dos níveis de colesterol total e de triglicerídeos, níveis altos de colesterol de VLDL e IDL, e níveis reduzidos de colesterol de LDL e HDL. Os níveis de colesterol e triglicerídeos variam de 300 a 1.000 mg/dL e são grosseiramente iguais, exceto durante uma exacerbação aguda, quando a hipertrigliceridemia tende a predominar. A VLDL de migração beta é detectada por eletroforese, embora este ensaio atualmente seja realizado apenas em raras ocasiões. A ultracentrifugação mostra uma proporção de colesterol de VLDL por triglicerídeos plasmáticos totais superior a 0,3. O diagnóstico definitivo é estabelecido pela detecção do fenótipo E2/E2 por focalização isoelétrica das lipoproteínas plasmáticas, ou do genótipo por análise de genes.
Em geral, a terapia para a dislipidemia por remanescentes é a mesma adotada para outras formas de hipertrigliceridemia. Uma dieta pobre em gorduras, perda de peso e exercícios podem exercer um efeito significativo sobre os níveis de lipídios. Os fibratos, estatinas e ácido nicotínico foram usados com sucesso no tratamento deste distúrbio. Entretanto, os fármacos que elevam os níveis de triglicerídeos, como as resinas ligadoras de ácidos biliares, devem ser evitados.
A hipertrigliceridemia severa pode surgir na infância, como resultado de deficiência de LPL e, em casos extremamente raros, como deficiência de apo C-II. Estes pacientes apresentam risco de desenvolvimento de pancreatite aguda com hipertrigliceridemia severa e devem ser tratados com restrição severa de gorduras na dieta, até a normalização dos níveis plasmáticos de triglicerídeos, para valores abaixo de 1.000 a 2.000 mg/dL.
A HF homozigota é raríssima e leva ao desenvolvimento de hipercolesterolemia severa, aterosclerose e morte, muitas vezes durante as primeiras 2 décadas de vida. Os pacientes com HF homozigota podem ser beneficiados pela aférese de LDL. Em um extremo, a ausência de lipoproteínas contendo apo B pode resultar de defeitos na síntese de apo B (p. ex., hipo-beta-lipoproteinemia homozigota) ou de defeitos no transporte da apo B para dentro do retículo endoplasmático. Indivíduos com níveis muito baixos de apo B não apresentam risco de desenvolvimento de aterosclerose.
A ausência de HDL pode ocorrer em indivíduos homozigotos para defeitos no ABCA-1 (um transportador de colesterol e fosfolipídios). A condição de heterozigose é uma causa incomum de colesterolemia isolada com níveis baixos de HDL12 (isto é, hipo-alfa-lipoproteinemia).
A hipercolesterolemia poligênica já foi considerada uma condição comum. O termo “hipercolesterolemia poligênica”, era usado em referência à ocorrência de elevações discretas dos níveis de colesterol de LDL na ausência aparente de uma forma familiar de dislipidemia ou de dislipidemia de causa secundária. Esta categoria de dislipidemia continua em colapso, dada a descoberta das variantes de LDL – p. ex., Lp(a) e partículas pequenas e densas de LDL.
A hipertrigliceridemia leve a moderada pode ocorrer na presença de defeitos modestos na LPL. Tipicamente, esta condição manifesta-se como um aumento dos níveis de colesterol de VLDL aliado à diminuição dos níveis de colesterol de HDL. É observada nos parentes obrigatoriamente heterozigotos de crianças com deficiência de LPL. Este defeito pode predispor ao desenvolvimento de DAC prematura.
As dislipoproteinemias secundárias são causadas por defeitos secundários no metabolismo lipoproteico, que resultam em hipercolesterolemia, hipertrigliceridemia ou hiperlipidemia. Os níveis de HDL podem estar ou não baixos. A hipertrigliceridemia secundária aliada a uma forma genética comum de hipertrigliceridemia pode ser severa o bastante para causar quilomicronemia com pancreatite. A dislipoproteinemia também pode ser causada por medicações determinadas.
A hiperglicemia não tratada em pacientes com diabetes melito causa aumento da síntese de VLDL, redução do catabolismo de VLDL acompanhada de redução da atividade da LPL, ou ambas. Estas anormalidades resultam em hipertrigliceridemia e redução dos níveis de HDL. Os níveis de LDL geralmente estão normais. A hiperquilomicronemia de jejum ocorre diante da coexistência da forma primária de hipertrigliceridemia. A VLDL e os quilomícrons competem para interagir com a LPL e pode haver acúmulo de ambas as lipoproteínas. Níveis baixos de HDL resultam de um comprometimento da lipólise das lipoproteínas ricas em triglicerídeos, que fornece os componentes lipídicos para o desenvolvimento de HDL. Estes defeitos ocorrem nos diabetes melito não tratados de tipos 1 e 2. Com o tratamento abrangente do diabetes, os níveis de lipídios devem se aproximar do normal, e, caso isto não ocorra, causas adicionais devem ser investigadas [ver Distúrbios genéticos do metabolismo de lipoproteínas, anteriormente]. Em pacientes diabéticos com hipertrigliceridemia persistente de grau moderado a severo, é conveniente usar um ácido fíbrico, que reduz a secreção de VLDL e intensifica a atividade da LPL. O ácido nicotínico pode ser usado, todavia com cautela, em particular no caso de pacientes com diabetes melito do tipo 2, porque pode exacerbar a hiperglicemia.57 As estatinas são efetivas para reduzir a incidência de eventos coronarianos em pacientes diabéticos.58
O hipotireoidismo pode causar elevação severa dos níveis de LDL, devido à atividade diminuída do receptor de LDL. Além disso, é causa frequente de hipertrigliceridemia e de uma diminuição associada dos níveis de HDL, como resultado da atividade reduzida de LPL. Os remanescentes de quilomícrons e VLDL também podem se acumular e revelar a doença da remoção de remanescentes. A dislipoproteinemia que acompanha o hipotireoidismo é corrigida com reposição do hormônio da tireoide.
Nota do editor: Em pacientes com dislipidemia caracterizada principalmente por LDL elevado, deve-se descartar hipotireoidismo antes de iniciar o tratamento específico da dislipidemia.
Os anticoncepcionais orais que contêm uma combinação de estrogênio e progesterona podem exercer efeitos variáveis sobre as lipoproteínas, dependendo da combinação específica usada. O estrogênio tende a elevar os níveis de VLDL e HDL, e a diminuir os níveis de LDL. As progestinas tendem a diminuir os níveis de VLDL e HDL, e a aumentar os níveis de LDL, embora seus efeitos sejam consideravelmente variáveis. A terapia de reposição de estrogênio pós-menopausa diminui os níveis de LDL e eleva os níveis de HDL. A adição de progesterona para proteção uterina ameniza estes efeitos, contudo sem eliminá-los.59 O estrogênio pode elevar gravemente os níveis de triglicerídeos em mulheres com distúrbio de triglicerídeos primário subjacente, levando ao desenvolvimento de pancreatite. Desta forma, os níveis de triglicerídeo devem ser estreitamente monitorados nestes pacientes.7 A terapia de combinação oral com estrogênio e progesterona foi associada a um aumento discreto da incidência de DAC no Women’s Health Initiative Study.60,61 Neste estudo randomizado envolvendo 16.608 mulheres, o uso da terapia de reposição hormonal oral também foi associado a uma frequência excessiva de câncer de mama. Nas mulheres submetidas a histerectomias, constatou-se que a terapia com estrogênio aumentou o risco de AVC [ver Tratamento da dislipidemia em mulheres, adiante].62 Estes estudos levaram à diminuição do uso da terapia de reposição hormonal pós-menopausa.
A síndrome nefrótica causa aumento da secreção hepática de lipoproteínas contendo apo B-100 (isto é, VLDL) em resposta à perda de albumina e outras proteínas na urina. A síntese hepática de colesterol também está aumentada. Os níveis de LDL tipicamente estão altos, e esta elevação pode ser severa. Os níveis de VLDL podem estar associados a uma redução dos níveis de HDL, conforme a lipólise vai sendo comprometida.63 Os pacientes com síndrome nefrótica apresentam risco aumentado de DAC, sendo que o distúrbio lipídico requer tratamento agressivo. Modificação da dieta, perda de peso e exercícios são ações que podem melhorar os níveis de lipoproteína, contudo a terapia farmacológica se faz necessária para alcançar os níveis de lipoproteína desejáveis. O ácido nicotínico deve ser efetivo no tratamento deste distúrbio, pois inibe a secreção hepática das lipoproteínas contendo apo B-100. No entanto, seu uso com esta finalidade ainda não foi extensivamente estudado. As estatinas são úteis para diminuir os níveis de colesterol de LDL em pacientes com síndrome nefrótica. A terapia farmacológica combinada com estatinas, ácido nicotínico, fibratos ou ezetimibe pode ser necessária para reduzir os níveis de colesterol de LDL e os triglicerídeos [ver Terapia farmacológica na dislipidemia, adiante]. Estudos são necessários para avaliar os efeitos de várias combinações farmacológicas sobre os resultados cardiovasculares.
A insuficiência renal crônica produz hipertrigliceridemia como resultado de uma diminuição de LPL e de triglicerídeo lipase hepática.64 A atividade de LCAT também pode estar diminuída.65 Os níveis de triglicerídeos tipicamente variam de 150 a 750 mg/dL, enquanto os níveis de HDL costumam estar baixos. O risco de DAC está aumentado. A ações sobre a dieta devem ser iniciadas quando o tratamento farmacológico estiver sendo considerado. O gemfibrozil, um fármaco que intensifica a atividade a atividade da LPL, é efetivo para redução dos níveis de triglicerídeos em pacientes com insuficiência renal.66 Neste contexto, em relação aos demais fibratos (p. ex., fenofibrato e clofibrato), o gemfibrozil é preferido por ser parcialmente depurado pelo fígado e, assim, impor um risco menor de miopatia fármaco-induzida, em comparação ao risco imposto pelos fibratos depurados pelos rins. Mesmo assim, como o gemfibrozil é parcialmente excretado por via renal, deve ser administrado em sua menor dose efetiva. O ácido nicotínico e as estatinas foram menos estudados quanto ao uso nesta condição. A terapia combinada com ácido nicotínico, estatinas ou gemfibrozil pode ser necessária para alcançar a meta terapêutica.
A cirrose biliar primária é a causa gastrintestinal mais significativa de dislipidemia. Nos estágios iniciais da cirrose biliar primária, quando ainda resta alguma função hepatocelular, as elevações discretas dos níveis de VLDL e LDL ocorrem devido às elevações dos níveis de lipoproteína remanescente e HDL. A doença hepática terminal com cirrose resulta na elevação severa dos níveis de colesterol, em decorrência da produção aumentada de lipoproteína X – uma partícula de lipoproteína anormal, que contém albumina e outros componentes plasmáticos e é rica em colesterol livre e fosfolipídios. O tratamento deste distúrbio terminal requer transplante de fígado.
Muitos fármacos comumente usados produzem efeitos adversos sobre as lipoproteínas [Tabela 3]. A suspensão do fármaco costuma melhorar os níveis de lipídios. Um aumento dos níveis de colesterol de VLDL, LDL e HDL pode resultar do uso de fármacos para a prevenção da rejeição após o transplante de órgão. A pravastatina é o fármaco de escolha para a redução dos níveis de LDL nestes casos, por suas vias catabólicas exclusivas. Os agentes imunossupressores, como a ciclosporina, competem com a atorvastatina e simvastatina pelo sistema do citocromo P-450 3A4. O uso de agentes antifúngicos também interfere no metabolismo destas estatinas. A dislipidemia predominante observada em pacientes com SIDA é similar à dislipidemia que ocorre em pacientes com síndrome metabólica. É comum haver uma discreta hipertrigliceridemia, e alguns pacientes podem apresentar níveis baixos de colesterol de HDL.67 Contudo, alguns pacientes podem desenvolver hipertrigliceridemia extrema como resultado do uso de fármacos contra o HIV, a qual pode estar associada à pancreatite. A etiologia da dislipidemia na SIDA é complexa: observa-se uma mobilização excessiva de ácidos graxos livres aliada ao desenvolvimento de lipodistrofia e resistência à insulina. Além disso, os pacientes com SIDA tipicamente usam fármacos causadores de dislipidemia. A terapia específica precisa ser individualizada para cada paciente.
O tratamento dos distúrbios lipídicos em indivíduos que não apresentam evidências clínicas de DAC é considerado uma medida de prevenção primária. A prevenção primária baseia-se no princípio de que a modificação dos fatores de risco lipídicos irá alterar a história natural da condição não tratada – a conhecida hipótese do lipídio. A associação existente entre colesterol e DAC é conhecida desde o início da década de 1950. Entretanto, foi somente após a publicação do Lipid Research Clinics Coronary Primary Prevention Trial (LRC-CPPT), em 1984, que foram disponibilizados dados sustentando a hipótese do lipídio.68
O LRC-CPPT envolveu quase 4.000 homens com hipercolesterolemia moderada. Os pacientes foram acompanhados durante 7 anos. O grupo submetido ao tratamento recebeu prescrição de colestiramina, que resultou em níveis de colesterol de LDL 12,6% mais baixos do que os níveis apresentados pelos indivíduos do grupo controle, tratados com placebo. O grupo da colestiramina apresentou uma redução de 19% no número de casos de morte por DAC e de IM não fatais (p < 0,05), embora nenhuma diminuição da mortalidade total tenha sido observada.68 Análises adicionais demonstraram que a extensão do benefício depende da redução dos níveis séricos de colesterol alcançada (refletindo a complacência farmacológica). O uso de um modelo de danos proporcionais indicou que uma diminuição de 25% nos níveis de colesterol total ou uma redução de 35% nos níveis de colesterol de LDL supostamente deveria diminuir o risco de um evento de DAC em 50%.69
O Helsinki Heart Study usou o fibrato gemfibrozil para tratar homens dislipidêmicos que não tinham DAC. Passados 6 anos de seguimento, houve uma redução de 34% na incidência de eventos de DAC no grupo tratado, em comparação ao observado no grupo placebo.70 Mais uma vez, nenhuma diminuição da mortalidade por DAC foi demonstrada.
Tanto no Helsinki Heart Study como no LRC-CPPT, o tamanho da amostra foi calculado com base no poder do estudo de detectar os eventos de DAC e não apenas os resultados fatais. Desta forma, a ausência de um efeito sobre a mortalidade não causou surpresa. Todavia, um aumento (não estatisticamente significativo) dos casos de morte não coronariana nos grupos de tratamento de ambos os estudos foi problemático e confundiu as recomendações para instituição de terapia preventiva primária em casos de pacientes hipercolesterolêmicos.66-68 Até 1995, quando foi publicado o West of Scotland Coronary Prevention Study (WOSCOPS), estas preocupações ainda não haviam sido totalmente abordadas.71
O WOSCOPS avaliou o efeito de 5 anos de tratamento com pravastatina sobre a incidência de IAM não fatal e mortes por DAC em 6.595 homens. Estes eram homens de meia-idade (45 a 64 anos) e moderadamente hipercolesterolêmicos (níveis de colesterol de LDL > 155 mg/dL). O grupo tratado apresentou redução de 20% nos níveis de colesterol total, redução de 26% nos níveis de colesterol de LDL, diminuição de 12% nos níveis de triglicerídeos, e aumento de 5% nos níveis de colesterol de HDL, em comparação ao observado no grupo controle. Com base nos princípios de intensão de cura, estas mudanças estavam associadas a uma redução de 31% no risco de IAM fatal ou morte por DAC (p < 0,001), redução de 32% no risco de morte por todas as causas cardiovasculares (p = 0,033), e redução de 22% no risco de mortalidade total (p = 0,051). Além disso, as intervenções coronarianas (isto é, angiografia, angioplastia e cirurgia de desvio arterial coronariano) foram reduzidas em 31 a 37% (p < 0,01).
A diminuição da ocorrência de eventos clínicos começou em 6 meses de randomização e foi independente de outros fatores de risco, como diabetes, tabagismo, pressão arterial, história familiar de DAC e proporção de colesterol total/colesterol de HDL.72 Embora não tenha havido redução de risco sem diminuição dos níveis de colesterol de LDL, uma redução dos níveis de colesterol de LDL de cerca de 24% foi suficiente para promover integralmente os benefícios proporcionados pelo tratamento com estatina. Assim, a redução de LDL isoladamente não responde por todos os benefícios proporcionados pelo tratamento com pravastatina.73 Significativamente, não houve aumento do número de mortes não coronarianas, segundo os relatos dos estudos preventivos primários iniciais que usaram outros fármacos.
O Air Force/Texas Coronary Atherosclerosis Prevention Study (AFCAPS/TexCAPS) foi o 1º estudo amplo sobre intervenção primária a investigar os efeitos da redução dos níveis de colesterol em indivíduos com níveis medianos de colesterol.74 Isto é, a média dos níveis de colesterol total e a média dos níveis de colesterol de LDL estavam mais próximas dos valores médios para a população em geral (221 mg/dL e 150 mg/dL, respectivamente). Além disso, o AFCAPS/TexCAPS foi também o 1º estudo amplo a incluir mulheres (997 mulheres em um total de 6.605 pacientes). A lovastatina foi o agente terapêutico utilizado neste estudo placebo-controlado e randomizado. O colesterol de LDL sofreu uma redução de 25%. O tempo médio de seguimento foi 5,2 anos. O benefício absoluto total foi de 2%, e isto significa que 50 pacientes tiveram de receber tratamento durante 5 anos para que 1 evento fosse evitado. O grupo de tratamento apresentou redução de 28% nos casos de internação cardiovascular, diminuição de 23% nos casos de angioplastia, e redução de 32% nos casos de cirurgia de desvio coronariano. Uma análise da relação custo-efetividade do tratamento com lovastatina demonstrou redução de 27% (US$ 524,00 por paciente) nas despesas de tratamento médico vascular no grupo tratado, em comparação às despesas do grupo placebo.75
Os indivíduos com níveis médios de colesterol também foram avaliados no Anglo-Scandinavian Cardiac Outcomes Trial-Lipid Lowering Arm (ASCOT-LLA).74 Quase 20.000 pacientes hipertensos foram randomizados para receberem 1 dentre 2 regimes anti-hipertensivos. No ramo terapia com agente para dislipidemia deste estudo, 10.305 pacientes com níveis de colesterol total iguais ou abaixo de 251 mg/dL foram randomizados para receberem tratamento com atorvastatina ou placebo. O estudo foi encerrado precocemente porque um benefício significativo foi observado no grupo tratado. O período médio de seguimento foi de 3,3 anos. Este estudo não demonstrou reduções estatisticamente significativas da mortalidade cardiovascular ou da mortalidade por causas diversas. Entretanto, reduções significativas foram encontradas em termos de número total de eventos coronarianos, número total de procedimentos e eventos cardiovasculares, e ocorrência de AVC.76
O efeito da atorvastatina (10 mg/dia) sobre a prevenção primária da doença cardiovascular em pacientes diabéticos foi avaliado no Collaborative Atorvastatin Diabetes Study (CARDS).56 O CARDS randomizou quase 3.000 pacientes diabéticos com níveis de LDL iguais ou inferiores a 160 mg/dL, níveis de triglicerídeos iguais ou inferiores a 600 mg/dL e ao menos 1 da seguintes condições: retinopatia, albuminúria, hábito de fumar cigarros ou hipertensão. O estudo foi interrompido com 2 anos de antecedência em relação à data prevista, porque os critérios predeterminados foram alcançados. O grupo tratado com atorvastatina apresentou redução de 36% na incidência de eventos coronarianos, uma diminuição de 31% nos procedimentos de revascularização coronariana, uma diminuição de 48% na incidência de AVC, e redução de 27% no número de mortes por causas diversas, em comparação ao observado no grupo placebo.
Estes estudos sustentam a instituição da terapia para dislipidemia como ação preventiva primária para pacientes com valores de LDL medianos e altos. Praticamente, não há dados sobre prevenção primária em casos de pacientes com outras anormalidades lipídicas, como níveis baixos de colesterol de HDL isolados ou níveis de triglicerídeos elevados.
A instituição da terapia redutora de lipídios em casos de pacientes com DAC comprovada é considerada uma ação preventiva secundária. Os níveis de lipídio exercem influência significativa sobre as taxas de morte por DAC entre indivíduos com e sem DAC. No entanto, seu impacto é significativamente maior em pacientes com DAC estabelecida.77
Vários estudos investigaram os efeitos de uma intervenção agressiva no estilo de vida sobre pacientes com DAC. O Saint Thomas Atherosclerosis Regression Study (STARS) randomizou homens com DAC e níveis de colesterol total acima de 232 mg/dL para receberem tratamento convencional ou uma dieta com baixos teores de gordura e colesterol. Apesar das modestas alterações nos níveis de lipídios (no grupo submetido à intervenção, a média dos níveis de colesterol de LDL foi de 162 mg/dL), a progressão da DAC diminuiu, e o índice de regressão aumentou no grupo submetido à intervenção. Os sintomas de angina também melhoraram.78
Os efeitos da dieta do Mediterrâneo (conteúdo de ácido alfalinoleico aumentado) foram comparados àqueles produzidos por uma dieta ocidental ponderada, no Lyon Diet Heart Study.79 Todos os participantes do estudo já tinham sofrido um 1º IM. Aqueles que consumiram a dieta do Mediterrâneo apresentaram taxas de pontos terminais primários (morte e IM) e secundários (angina instável, AVC, insuficiência cardíaca) menores do que aqueles que consumiram a dieta ocidental ponderada, em 27 meses. Este efeito persistiu após 4 anos de seguimento. O grupo da dieta do Mediterrâneo apresentou uma taxa de desfechos primários e secundários combinada igual a 2,59 eventos por 100 pacientes ao ano, em comparação aos 9,03 eventos por 100 pacientes ao ano observados no grupo que consumiu a dieta ocidental ponderada.80
Uma variedade de agentes farmacológicos foram usados de modo isolado e também combinados em estudos de prevenção secundária. Alguns estudos adotaram pontos terminais angiográficos ao avaliarem a progressão ou regressão da DAC, enquanto outros usaram desfechos clínicos. O FATS avaliou o efeito de vários regimes redutores de lipídios em homens que apresentavam níveis elevados de apo B. Os 2 regimes mais agressivos (ácido nicotínico-colestipol e lovastatina-colestipol) foram igualmente efetivos. Ambos estavam associados a uma progressão retardada (21% e 25%, respectivamente; vs. 46% no grupo placebo-colestipol) e também a uma maior probabilidade de regressão das estenoses arteriais coronarianas (32% e 39%, respectivamente; vs. 11% no grupo placebo-colestipol). Os desfechos clínicos (morte, IM, piora da angina e revascularização) também foram reduzidos nos grupos submetidos a um tratamento mais agressivo (4,2% e 6,5%, respectivamente; vs. 19% no grupo placebo-colestipol).50 Este foi o 1º dos principais estudos a comprovarem a regressão da DAC com a instituição de uma terapia agressiva para dislipidemia. Uma análise subsequente destes pacientes correlacionou a mudança da severidade da DAC a alterações terapia-induzidas na flutuabilidade da LDL e na atividade da lipase hepática.81
O Scandinavian Simvastatin Survival Study (4S) avaliou 4.444 pacientes com DAC comprovada e hipercolesterolemia moderada a severa no momento basal (concentração de colesterol total variando de 212 a 309 mg/dL).82 Os pacientes foram randomizados para receberem um regime alimentar + sinvastatina ou dieta + placebo. Decorridos 5,4 anos, observou-se uma diminuição significativa da mortalidade total (8% com sinvastatina vs. 12% com placebo), dos eventos coronarianos significativos (19% vs. 28%), dos casos de morte por DAC (diminuição de 42%) e dos eventos cerebrovasculares (2,7% vs. 4,3%). A redução do número de eventos cardiovasculares estava correlacionada com os níveis de colesterol total, com os níveis de colesterol de LDL e com as mudanças em relação ao basal.83
O estudo Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) randomizou cerca de 9.000 homens e mulheres com história recente de IM ou angina instável, para receberem placebo ou pravastatina.84 Este estudo foi interrompido antes do tempo previsto, aos 60 meses, porque os pacientes alcançaram um benefício significativo associado à terapia com pravastatina. Os casos de morte por DAC diminuíram no ramo do estudo submetido ao tratamento (6,4% vs. 8,3%), assim como a mortalidade total (11% vs. 14%), AVC (diminuição relativa de 20%), necessidade de cirurgia de desvio (8,9% vs. 11,3%) e IM (7,4% vs. 10,1%). O benefício alcançado estava principalmente relacionado às alterações dos níveis lipídicos e foi observado em todos os subgrupos predefinidos. A maior redução na incidência de eventos coronarianos foi observada nos pacientes considerados de risco mais alto, conforme a avaliação dos fatores de risco concomitantes.85
O estudo Cholesterol and Recurrent Events (CARE) avaliou 4.159 pacientes com níveis lipídicos relativamente baixos. A média dos níveis de colesterol total foi de 209 mg/dL, enquanto a média dos níveis de colesterol de LDL foi de 139 mg/dL. O tratamento com pravastatina por mais de 5 anos resultou em diminuições significativas das mortes por causa coronariana ou dos casos de IM não fatal (10,2% vs. 13,2% no grupo placebo), da necessidade de revascularização (14,1% vs. 18,8%) e da frequência de AVC (2,6% vs. 3,8%).86 Entretanto, em contraste com os resultados alcançados no 4S e no LIPID, as reduções absolutas ou reduções percentuais dos níveis de LDL apresentaram pouca relação com os eventos coronarianos.85 Os benefícios foram observados apenas nos casos de pacientes cujos níveis basais de LDL estavam acima de 125 mg/dL.87
O Heart Protection Study avaliou mais de 20.000 indivíduos com história de doença cardiovascular (doença coronariana, cerebrovascular ou vascular periférica), diabetes ou hipertensão tratada.88 Desta forma, este estudo foi uma mistura de intervenções primária e secundária. Cerca de 1/3 dos indivíduos tinha níveis basais de colesterol de LDL abaixo de 116 mg/dL, enquanto 25% apresentavam níveis de LDL iniciais na faixa de 116 a 135 mg/dL. Os participantes do estudo foram randomizados para receber simvastatina ou placebo. Após um período de seguimento médio de 5,5 anos, o grupo submetido à redução lipídica apresentou queda de 24% na incidência de eventos cardiovasculares significativos, diminuição de 18% no número de mortes por causa cardiovascular, e redução de 13% na mortalidade por causas diversas, em comparação ao observado no grupo placebo. As reduções percentuais na ocorrência de eventos foram similares em todos os 3 terços de níveis basais de colesterol de LDL e em pacientes com níveis basais de colesterol de LDL abaixo de 100 mg/dL. Estes resultados diferem daqueles relatados no estudo CARE, mas são consistentes com o resultados descritos pelos estudos 4S e LIPID. Os resultados do Heart Protection Study também sugerem que pode não haver um limiar além do qual a terapia redutora de LDL intensificada pare de melhorar o resultado, ao menos em pacientes que apresentam alto risco de sofrerem eventos coronarianos recorrentes.
A terapia redutora de LDL agressiva parece ser mais efetiva do que o tratamento padrão de redução lipídica. O estudo Pravastatin or Atorvastatin Evaluation and Infection Therapy (PROVE-IT) comparou o tratamento padrão de redução de LDL (40 mg/dia de pravastatina) ao tratamento intensivo de redução de LDL (80 mg/dia de pravastatina) em mais de 4.000 pacientes recentemente internados com síndrome coronariana aguda.89 O tempo médio de seguimento foi 24 meses. Os níveis de colesterol de LDL alcançados com o tratamento com atorvastatina foram, em média, 62 mg/dL vs. o valor médio de 96 mg/dL alcançado no grupo tratado com pravastatina. O desfecho composto primário foi a morte por causas diversas, IM, angina instável sem necessidade de internação, revascularização coronariana e AVC. O desfecho primário ocorreu 22,4% dos pacientes no grupo da atorvastatina e 26,3% no grupo da pravastatina. O benefício proporcionado pela terapia agressiva com atorvastatina foi evidente já em 30 dias após a iniciação da terapia e permaneceu consistente ao longo do tempo.
O estudo Reversal of Atherosclerosis with Aggressive Lipid Lowering (REVERSAL) também comparou a terapia redutora de LDL moderada (40 mg/dia de pravastatina) a uma terapia redutora de LDL mais intensiva (80 mg/dia de pravastatina).90 Este estudo usou a ultrassonografia intravascular coronariana, um meio sensível de medir o volume da placa, como medida basal e ponto terminal primário. A mudança percentual média no volume do ateroma foi de 0,4% no grupo da atorvastatina, em comparação aos 2,7% no grupo da pravastatina. Este achado estava correlacionado a níveis médios de colesterol de LDL iguais a 79 mg/dL no grupo da atorvastatina e 110 mg/dL no grupo da pravastatina. Estes resultados deram suporte adicional ao ponto de vista segundo o qual a terapia agressiva para redução de LDL é superior à terapia-padrão de redução de LDL.
Poucos estudos investigaram os benefícios decorrentes da elevação dos níveis de colesterol de HDL na prevenção secundária da DAC. O estudo VA-HIT examinou 2.531 pacientes com DAC comprovada e níveis de colesterol de LDL inferiores a 140 mg/dL, níveis de colesterol de HDL iguais ou superiores a 40 mg/dL e níveis de triglicerídeos iguais ou inferiores a 300 mg/dL.53 Os pacientes foram randomizados para receber gemfibrozil ou placebo. Subsequentemente, os níveis médios de colesterol de HDL no grupo gemfibrozil estavam 6% mais altos do que no grupo placebo, enquanto os níveis médios de triglicerídeos no grupo gemfibrozil estavam 31% mais baixos. Os níveis médios de colesterol de LDL eram de 113 mg/dL em ambos os grupos. O desfecho primário combinado de morte por causa cardíaca e IM não fatal ocorreu em 17% dos pacientes no grupo tratamento e 22% no grupo placebo (redução do risco relativo em 22%). O efeito benéfico do gemfibrozil somente se tornou evidente após 2 anos de randomização.
Foi demonstrado que a terapia combinada, usando uma estatina para diminuir os níveis de colesterol de LDL e niacina para elevar os níveis de colesterol de HDL, confere maior cardioproteção. Em um estudo, os pacientes foram randomizados entre 4 grupos de tratamento: simvastatina + niacina; vitaminas; simvastatina-niacina + antioxidantes; e placebos. No início, os níveis de colesterol de HDL estavam abaixo de 35 mg/dL, e os níveis de LDL colesterol eram inferiores a 145 mg/dL. Os níveis médios de colesterol de LDL e de HDL não sofreram alteração nos grupos antioxidante e placebo, mas mudaram significativamente nos grupos tratados com simvastatina + niacina (redução dos níveis de colesterol de LDL em 42%; elevação dos níveis de colesterol de HDL em 26%). Em 3 anos, a redução da incidência de eventos clínicos nos grupos simvastatina e niacina foi maior do que aquela geralmente relatada pelos estudos que usaram apenas estatinas (risco relativo de 0,1 a 0,4 vs. grupo placebo). Isto sugere que pode haver um benefício associado à elevação dos níveis de colesterol de HDL. Os antioxidantes falharam em promover benefícios adicionais, podendo até atenuar os benefícios proporcionados pela terapia combinada.91
Os fatores de risco de DAC raramente ocorrem de forma isolada, sendo que o risco associado a cada um deles varia bastante em combinação com outros fatores de risco. Esta variabilidade do risco impulsionou o NCEP ATP-III a tentar padronizar as diretrizes de avaliação de risco da DAC. Com o passar do tempo, as diretrizes foram revisadas e passaram a recomendar a adoção de alvos de redução lipídica mais agressivos, como forma de diminuir o risco de DAC [Tabelas 5 e 6]. Esta evolução das diretrizes é o resultado da emergência de dados consistentes que ampliaram nosso conhecimento acerca da dislipidemia, fatores de risco associados e a relação destes com a DAC, bem como a utilidade de novas opções terapêuticas.
Tabela 5. Metas e pontos de corte estabelecidos pelo NCEP ATP-III para as modificações de estilo de vida terapêuticas e terapia farmacológica em diferentes categorias de risco95
Categoria de risco |
Meta de LDL-C |
Iniciar as modificações terapêuticas de estilo de vida |
Considerar a terapia farmacológica |
|
Alto risco: DAC ou equivalente de risco de DAC (doença aterosclerótica não coronariana, diabetes e 2+ fatores de risco de DAC com risco de DAC de 10 anos > 20%)* |
< 100 mg/dL (meta opcional: < 70 mg/dL) |
= 100 mg/dL |
= 100 mg/dL (< 100 mg/dL: considerar opções de fármaco) |
|
Risco moderadamente alto: 2+ fatores de risco de DAC [Tabela 6] com risco de DAC de 10 anos = 10 a 20%* |
< 130 mg/dL (meta opcional: < 100 mg/dL) |
= 130 mg/dL |
= 130 mg/dL (100 a 129 mg/dL: considerar opções de fármaco) |
|
Risco moderado: 2+ fatores de risco de DAC com risco de DAC de 10 anos < 10%* |
< 130 mg/dL |
= 130 mg/dL |
= 160 mg/dL |
|
Baixo risco: 0 a 1 fator de risco de DAC com risco de DAC de 10 anos < 10% |
< 160 mg/dL |
= 160 mg/dL |
= 190 mg/dL (160 a 189 mg/dL: fármaco redutor de LDL opcional) |
DAC = doença arterial coronariana; LDL = lipoproteína de baixa densidade; LDL-C = colesterol de lipoproteína de baixa densidade; NCEP ATP-III = National Cholesterol Education Program Adult Treatment Panel III.
* Calculadoras do risco de 10 anos disponibilizadas on-line: wwwo.nhlbi.nih.gov/guidelines/cholesterol.
Tabela 6. Principais fatores de risco estabelecidos pelo NCEP ATP-III (exclusivos para colesterol de LDL), que modificam as metas de LDL*17
|
Tabagismo |
|
Hipertensão (pressão arterial = 140/90 mmHg ou sob medicação anti-hipertensiva) |
|
Níveis baixos de colesterol de HDL (< 40 mg/dL)† |
|
História familiar de DAC prematura (DAC em parente de primeiro grau, do sexo masculino e com idade < 55 anos; DAC em parente de 1º grau, do sexo feminino e com idade < 65 anos) |
|
Idade (homens > 45 anos; mulheres = 55 anos) |
*O diabetes é considerado um equivalente de risco de DAC.
†Níveis de colesterol de HDL > 60 mg/dL contam como fator de risco negativo; sua presença elimina um fator de risco da contagem total.
DAC = doença arterial coronariana; HDL = lipoproteína de alta densidade; LDL = lipoproteína de baixa densidade; NCEP ATP-III = National Cholesterol Education Program Adult Treatment Panel III.
As diretrizes do ATP enfocam especialmente os níveis de colesterol de LDL como principal fator de risco lipídico. Mais recentemente, os níveis reduzidos de HDL passaram a ser um fator considerado na avaliação do risco. No ATP-III, a síndrome metabólica foi incluída como fator de risco, em uma tentativa de avaliar o risco de DAC em pacientes com obesidade central, apresentando elevações modestas dos níveis de triglicerídeos, níveis baixos de colesterol de HDL, partículas pequenas e densas de LDL, além de diabetes melito de tipo 2 ou HLCF. Em uma tentativa de identificar melhor os indivíduos com maior risco de sofrerem eventos de DAC, o NCEP reconheceu vários equivalentes de DAC. Assim, foram incluídos o diabetes melito, a doença arterial periférica, o aneurisma aórtico abdominal, a doença arterial carotídea sintomática e múltiplos fatores de risco que conferem um risco de DAC de 10 anos superior a 20%.17 A presença destes equivalentes de DAC requer um nível de agressividade terapêutica igual àquele recomendado para os pacientes com DAC estabelecida.
O American College of Physicians (ACP) adotou recomendações um tanto menos agressivas para o tratamento de indivíduos com diabetes melito de tipo 1. O ACP reservou o uso das estatinas para pacientes com diabetes de tipo 2 e outros fatores de risco de DAC.92 As diretrizes ATP-III não diferenciam entre o risco de DAC em pacientes com diabetes de tipo 1 e o risco de DAC em indivíduos com diabetes de tipo 2. É possível argumentar que o risco de desenvolvimento de DAC é maior em pacientes com diabetes de tipo 2 e as diretrizes de tratamento devem diferenciar ambas as entidades.
As diretrizes do ATP-III usam o sistema de escore Framingham para estimar o risco de DAC de 10 anos. Alguns estudos indicam que este sistema de escores superestima o risco em homens norte-americanos de ascendência japonesa, homens hispânicos, mulheres norte-americanas nativas e algumas populações europeias e asiáticas.93-95 Também foi sugerido que o escore de Framingham atribui um peso exagerado à idade como fator de risco. O estudo Pravastatin in Elderly Individuals at Risk of Vascular Disease (PROSPER), que foi o único estudo prospectivo a avaliar a terapia à base de estatina em homens e mulheres com idade acima de 70 anos, demonstrou que a terapia com estatina não promoveu benefício para os pacientes sem aterosclerose preexistente.96 Na presença de DAC, o tratamento de idosos com estatina parece ser benéfico.96 Qualquer distorção de idade presente no sistema de escores de Framingham é eliminada quando este sistema é usado na previsão do risco para pacientes não idosos.
Um estudo internacional e multicentros confirmou a validade da estratificação do risco. Neste estudo, que envolveu mais de 15.000 pacientes com IM oriundos de 52 países, mais de 90% do risco atribuível à população poderia ter sido devido a 9 fatores de risco potencialmente modificáveis.97 A maior parte do risco se deve a uma elevada proporção apo B/apo A1, tabagismo, hipertensão, e diabetes. Estes fatores de risco foram mais significativos em indivíduos mais jovens do que em idosos. Sendo assim, os princípios da prevenção à doença cardiovascular são similares em todo o mundo e têm o potencial de exercerem um impacto significativo.
Várias classes de fármacos podem diminuir os níveis de colesterol de LDL [Tabela 7].18 Antes da introdução das estatinas, na metade da década de 1980, os principais fármacos usados com esta finalidade eram os sequestradores de ácido biliar e a niacina. A introdução das estatinas, com seus poderosos efeitos sobre os níveis de colesterol de LDL, tolerabilidade e relativa ausência de toxicidade, promoveu um avanço significativo no tratamento dos pacientes com hipercolesterolemia. A introdução dos fármacos com atividade intestinal propiciou abordagens adicionais tanto de monoterapia – em especial para indivíduos incapazes de tolerar as estatinas – como, mais particularmente, de terapia combinada. A terapia farmacológica deve ser suficiente para produzir uma redução dos níveis de colesterol de LDL de no mínimo 30 a 40%98 e, possivelmente, de até 50%.99
Tabela 7. Tratamento farmacológico de distúrbios lipídicos
|
Fármaco |
Dosagem* |
Comentários |
|
Resinas ligadoras de ácido biliar |
Começar com 1 pacote (2 g para comprimidos de colestipol) 2 x/dia, e aumentar no decorrer de 1 a 2 semanas até chegar à dose desejada |
Para níveis elevados de LDL, triglicerídeos normais; tomar outro fármaco 1 hora antes ou 4 horas depois; pode ser usado com ácido nicotínico, estatinas ou fibratos |
|
Colestiramina |
Máximo: 24 g/dia, 2 x/dia ou 3 x/dia |
3 x/dia é mais efetivo |
|
Colestipol em pó |
Máximo: 30 g/dia 2 x/dia ou 3 x/dia |
|
|
Comprimidos de colestipol |
Máximo: 16 g/dia |
|
|
Colesevelam |
6 comprimidos de 625 mg/dia, tomados com as refeições, seja como dose única ou divididos em 2 doses; máximo: 7 comprimidos/dia |
Melhor tolerado do que as outras resinas |
|
Ezetimibe |
10 mg/dia |
Pode reduzir os níveis de colesterol de LDL em ˜ 20%, sem aumentar os níveis plasmáticos de triglicerídeos |
|
Fenofibrato |
145 ou 200 mg/dia |
Para níveis altos de triglicerídeos e pacientes com elevação de LDL e triglicerídeos; pode ser usado com resinas ligadoras de ácido biliar ou ácido nicotínico; diminuir a dose em caso de doença renal severa |
|
Gemfibrozil |
600 mg, 2 x/dia |
|
|
Niacina |
Máximo: 6 g/dia; dose raramente usada |
|
|
Cristalina |
Começar com 250 mg, 1 x/dia, após o jantar; aumentar para 0,5 g, 3 a 4x/dia; máximo: 6 g/dia |
Para níveis elevados de LDL, triglicerídeos ou ambos; pode ser usada com resinas ligadoras de ácido biliar, estatinas ou fibratos |
|
Liberação controlada |
1,5 g, na hora de dormir |
|
|
Estatinas |
Várias doses |
Para LDL elevado; possivelmente útil para pacientes com elevação de triglicerídeos e LDL; pode ser usado com resinas ligadoras de ácido biliar ou ácido nicotínico |
*Para obter informações sobre os custos associados ao uso destes fármacos, consulte a tabela de informações do ACP Medicine.
LDL = lipoproteína de baixa densidade.
Estatinas. Atualmente, existem várias estatinas disponibilizadas para uso, e outras novas continuam sendo introduzidas. Até o presente, as estatinas têm se mostrado altamente efetivas nos estudos clínicos, em termos de redução de eventos clínicos, inclusive do AVC. Embora alguns dos benefícios promovidos pelas estatinas tenham sido atribuídos aos conhecidos efeitos pleiotrópicos (p. ex., efeitos anti-inflamatórios) desta classe de fármacos, a extensão da redução dos níveis de colesterol de LDL ainda parece ser o principal determinante da diminuição do risco.
Compostos com atividade intestinal. Os sequestradores de ácido biliar estão entre os fármacos mais antigos a serem disponibilizados para tratamento da hipercolesterolemia e foram a 1ª classe de fármacos a demonstrar que a redução dos níveis de colesterol de LDL estava associada a um menor risco de DAC. No entanto, seu uso foi limitado pela tolerabilidade precária e por seus efeitos modestos na redução do colesterol de LDL. Além disso, os níveis de triglicerídeos tendem a aumentar com o uso em pacientes com níveis plasmáticos basais de triglicerídeos altos. A introdução de um sequestrador de ácido biliar mais tolerável, o colesevelam, resultou em uma melhor complacência com esta classe de fármacos, especialmente quando usado na terapia combinada de pacientes com níveis de colesterol de LDL muito altos (p. ex., pacientes com HF).
Diferente dos sequestradores de ácido biliar, o ezetimibe, um fármaco com atividade intestinal, inibe diretamente a absorção do colesterol. Embora os dados clínicos existentes sejam limitados, parece que o ezetimibe consegue reduzir o colesterol de LDL em cerca de 20%100 e, possivelmente, em até 35%,101 quando usado como monoterapia ou combinado a outros agentes redutores de lipídio. Além disso, o ezetimibe não causa aumento dos níveis plasmáticos de triglicerídeos, como ocorre com os sequestradores de ácido biliar. O ezetimibe foi avaliado em estudos clínicos com pontos terminais cardiovasculares.101,102
Os fármacos preferidos para uso no tratamento da hipertrigliceridemia são os fibratos e a niacina. Esta última é o melhor fármaco atualmente disponível para elevar os níveis de colesterol de HDL. Além disso, a niacina também produz reduções modestas dos níveis de LDL e diminui os níveis de apo B, mas como piora a sensibilidade à insulina, seu uso por pacientes com diabetes melito de tipo 2 é limitado. Os fibratos são os fármacos de escolha para tratamento de pacientes com hipertrigliceridemia marcante, para os quais a meta terapêutica primária é a prevenção da pancreatite e outros aspectos associados à síndrome da quilomicronemia. Os fibratos também são úteis para o tratamento das condições hipertrigliceridêmicas (p. ex., pacientes com formas familiares de hipertrigliceridemia e alguns pacientes com dislipidemia diabética), sobretudo quando os níveis de triglicerídeos estão mais do que levemente aumentados. Estes fármacos promovem, ainda, um efeito benéfico modesto sobre os níveis de colesterol de HDL. Ambos, fibratos e niacina, são úteis na terapia combinada, principalmente com as estatinas.
Os ácidos graxos ômega-3 (p. ex., encontrados nos óleos marinhos) são usados no tratamento da hipertrigliceridemia, especialmente quando outras modalidades de terapia falham em reduzir níveis de triglicerídeos acentuadamente elevados.
Com frequência, torna-se necessário usar combinações de fármacos diante de níveis elevados de colesterol de LDL e de triglicerídeos. A terapia combinada também é útil quando a monoterapia, em especial à base de estatinas, falha em promover os níveis-alvo desejados de lipídios e lipoproteínas, sobretudo de colesterol de LDL. Entre as combinações comumente usadas estão: estatinas + fibratos (embora pouco se saiba sobre os benefícios aditivos destes fármacos em termos de redução dos eventos clínicos) e estatinas + niacina. As estatinas e os sequestradores de ácido biliar também são úteis como combinação, e o uso dos novos inibidores da absorção de colesterol com outras classes de fármacos, em particular as estatinas, provavelmente seja significativo. Em alguns casos, a terapia tripla (p. ex., estatina + niacina + agente de ação intestinal) torna-se necessária.
Numerosos exames de autópsia demonstraram que a aterosclerose coronariana começa na infância e adolescência, e que os níveis de lipoproteína estão consistentemente associados à extensão desta aterosclerose. As crianças de famílias com HF e DAC prematura apresentam níveis de colesterol mais altos e, durante a infância, os níveis de colesterol são fatores preditivos significativos dos níveis de colesterol na fase adulta. Entretanto, muitas das crianças e adolescentes com níveis de colesterol levemente aumentados tornam-se adultos com níveis de colesterol que não são altos o bastante para requerer intervenção.103 A avaliação de todas as crianças para detecção de níveis altos de colesterol conferiria a muitos jovens um rótulo de risco, classificando-os como indivíduos doentes. Todas as crianças com menos de 2 anos de idade seriam beneficiadas por uma dieta com baixo teor de gordura saturada. Esta meta deveria ser parte de qualquer estratégia populacional de controle epidêmico da aterosclerose. Recomenda-se seguir as Dietary Guidelines for Americans e aumentar o nível de atividade física.104 No entanto, a segurança e eficácia a longo prazo da terapia farmacológica ainda não foram estabelecidas para esta faixa etária, sendo que o tratamento deve ser abordado com cautela.51,105
Os médicos devem recomendar aos pacientes com idade inferior a 55 anos e DAC ou distúrbio lipídico comprovado que submetam seus filhos ou netos a exames de colesterol periódicos. Um paciente com distúrbio lipídico geneticamente bem definido deve obter aconselhamento genético apropriado. Os médicos que tratam pacientes com menos de 20 anos de idade apresentando níveis de LDL acentuadamente altos devem insistir à exaustão na adoção de todas as intervenções de estilo de vida, antes de considerarem o uso de medicações. Se estas ações forem inefetivas, então os sequestradores de ácido biliar devem ser usados, e deve-se considerar o encaminhamento do paciente a um especialista clínico.
O tratamento de adultos jovens com níveis elevados de colesterol é controverso. Embora tenha sido proposta a estratégia de compatibilizar a intensidade da intervenção ao nível de risco de aterosclerose, uma avaliação de risco a curto prazo (p. ex., 10 anos) para pacientes adultos jovens pode ser inadequada para estimar o potencial benefício da redução do colesterol. É errado afirmar que todos os tratamentos podem ser adiados com segurança para fases mais tardias da vida ou até a ocorrência de um evento aterosclerótico. A prevenção ao nível populacional e a adoção de intervenções no estilo de vida ainda devem ser favorecidas para os adultos jovens. Contudo, avanços tecnológicos que permitam identificar melhor os pacientes assintomáticos (de qualquer idade) que devem iniciar as etapas da redução do risco se fazem amplamente necessários. Tais avanços talvez permitam identificar ou quantificar com segurança as placas vulneráveis, marcadores de inflamação ou medidas não invasivas de disfunção endotelial.
Antes da menopausa, as mulheres apresentam menor incidência de DAC do que os homens da mesma faixa etária. Ainda que raramente, a DAC ocorre em mulheres em pré-menopausa, em geral associada a múltiplos fatores de risco genéticos e ambientais, como se observa nas pacientes com formas familiares de dislipidemia ou em pacientes diabéticas fumantes.
Após a menopausa, algumas mulheres desenvolvem síndrome metabólica, caracterizada por obesidade visceral, resistência à insulina, hipertensão e dislipidemia.26 Algumas evidências indicam que terapia de reposição de estrogênio consegue reverter estes achados. Entretanto, o Women’s Health Initiative Study demonstrou que terapia combinada oral com estrogênio e progesterona não protegeu as mulheres contra a DAC e, na verdade, acabou produzindo efeitos colaterais.106 O estrogênio, como componente isolado utilizado no Women’s Health Initiative Study, indicou que a terapia à base de estrogênio está associada a um risco modesto de desenvolvimento de DAC, isto é, morte por DAC ou IM). O estudo foi interrompido precocemente em decorrência do risco aumentado de AVC.107
A idade é o fator de risco mais significativo para o desenvolvimento de aterosclerose. A DAC atualmente é uma das principais causas de incapacitação e mortalidade nas populações de idade mais avançada. No entanto, o risco relativo associado a qualquer fator de risco coronariano isolado diminui com o avanço da idade, por causa das comorbidades e da mortalidade não cardiovascular que afetam a população de indivíduos em envelhecimento. Uma das implicações das complexas relações existentes entre os fatores de risco e as comorbidades na patogênese dos eventos relacionados a aspectos coronarianos na idade avançada é representada pelos múltiplos efeitos do tratamento de fatores de risco isolados, como a diminuição dos níveis de colesterol de LDL e dos marcadores de inflamação promovida pelas estatinas. Um número crescente de evidências fornecidas por estudos clínicos indica que a terapia com estatinas é efetiva para idosos. A terapia redutora de lipídios provavelmente é indicada para o tratamento desta população em casos de indivíduos que apresentam alto risco de aterosclerose ou que tenham aterosclerose preexistente.88,96 A intervenção primária com terapia farmacológica em casos de indivíduos que não apresentam alto risco de desenvolvimento de aterosclerose é controversa. No estudo PROSPER, envolvendo indivíduos com idade superior a 70 anos, nenhum benefício foi alcançado com a administração da terapia de estatina aos pacientes sem aterosclerose clínica preexistente. No tratamento de pacientes idosos com dislipidemia, é importante considerar a existência de outras doenças concomitantes e o estado nutricional do paciente, bem como as capacidades dos pacientes de idade avançada.
John D. Brunzell, MD, FACP, atua como consultor junto às empresas GlaxoSmith-Kline, Merck Research Laboratories, Abbott Laboratories e Novartis Pharmaceuticals Corp.
R. Alan Failor, MD, atua como consultor na GlaxoSmithKline.
1. Heron MP, Hoyert DL, Xu J, et al. Preliminary data for 2006. Deaths. Natl Vital Stat Rep 2008;56(16). Available at: http://www.cdc.gov/nchs/data/nvsr/nvsr56/nvsr56_ 16.pdf (accessed January 12, 2010).
2. Cassar A, Holmes DR, Rihal CS, Gersh BJ. Chronic coronary artery disease: diagnosis and management. Mayo Clin Proc 2009;84:1130–46.
3. Havel R, Kane J. Introduction: structure and metabolism of plasma lipoproteins. In: Scriver CR, Beaudet AL, Sly WS, et al, editors. The metabolic and molecular bases of inherited disease. 8th ed. New York: McGraw-Hill; 2001. p. 2705.
4. Brunzell J. Clinical practice. Hypertriglyceridemia. N Engl J Med 2007;357:1009–19. Kane JP, Havel RJ. Disorders of the biogenesis and secretion of lipoproteins containing the B apolipoproteins. In: Scriver CR, Beaudet AL, Sly
5. WS, et al, editors. The metabolic and molecular bases of inherited disease. 8th ed. New York: McGraw-Hill; 2001. p. 2717–52.
6. Utermann G. Lipoprotein (a). In: Scriver CS, Beaudet AL, Sly WS, et al, editors. The metabolic and molecular bases of inherited disease. 8th ed. New York: McGraw-Hill; 2001. p. 2753.
7. Brunzell J, Deeb S. Familial lipoprotein lipase def ciency, apo CII def ciency, and hepatic lipase def ciency. In: Scriver CS, Beaudet AL, Sly WS, et al, editors. The metabolic and molecular bases of inherited disease. 8th ed. New York: McGraw-Hill; 2001. p. 2789.
8. Mahley R, Rall S. Type III hyperlipoproteinemia (dysbetalipoproteinemia): the role of apolipoprotein E in normal and abnormal lipoprotein metabolism. In: Scriver CS, Beaudet AL, Sly WS, et al, editors. The metabolic and molecular bases of inherited disease. 8th ed. New York: McGraw-Hill; 2001. p. 2835.
9. Goldstein JL, Hobbs HH, Brown MS. Familial hypercholesterolemia. In: Scriver CS, Beaudet AL, Sly WS, et al, editors.The metabolic and molecular bases of inherited disease. 8th ed. New York: McGraw-Hill; 2001. p. 2863.
10. Tall A, Breslow J, Rubin E. Genetic disorders affecting plasma high-density lipoproteins. In: Scriver CS, Beaudet AL, Sly WS, et al, editors. The metabolic and molecular bases of inherited disease. 8th ed. New York: McGraw-Hill; 2001. p. 2915.
11. Rouhani N, Young E, Chatterjee C, Sparks DL. HDL composition regulates displacement of cell surface-bound hepatic lipase. Lipids 2008;43:793–804.
12. Frikke-Schmidt R, Nordestgaard BG, Jensen GB, et al. Genetic variation in ABC transporter A1 contributes to HDL cholesterol in the general population. J Clin Invest 2004;114:1343.
13. Deeb SS, Zambon A, Carr MC, et al. Hepatic lipase and dyslipidemia: interactions among genetic variants, obesity, gender, and diet. J Lipid Res 2003;44:1279.
14. Carr MC, Brunzell JD. Abdominal obesity and dyslipidemia in the metabolic syndrome: importance of type 2 diabetes and familial combined hyperlipidemia in coronary artery disease risk. J Clin Endocrinol Metab 2004; 89:2601.
15. Griff n BA. Lipoprotein atherogenicity: an overview of current mechanisms. Proc Nutr Soc 1999;58:163.
16. Steinberg D, Pearson TA, Kuller LH. Alcohol and atherosclerosis. Ann Intern Med 1991;114:967.
17. Executive summary of the Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. JAMA 2001;285:2486. Available at: www.nhlbi.nih.gov/guidelines/cholesterol/atp_iii.htm (accessed January 11, 2010).
18. Brown CD, Higgins M, Donato KA, et al. Body mass index and the prevalence of hypertension and dyslipidemia. Obes Res 2000;8:605.
19. Austin MA, McKnight B, Edwards KL, et al. Cardiovascular disease mortality in familial forms of hypertriglyceridemia: a 20-year prospective study. Circulation 2000;101: 2777.
20. Knopp RH. Drug treatment of lipid disorders. N Engl J Med 1999;341:498.
21. Bruckert E, Labreuche J, Amarenco P. Meta-analysis of the effect of nicotinic acid alone or in combination on cardiovascular events and atherosclerosis. Atherosclerosis 2009 Dec 21. [Epub ahead of print]
22. Birjmohun RS, Hutten BA, Kastelein JJ, Stroes ES. Eff cacy and safety of high-density lipoprotein cholesterol-increasing compounds: a meta-analysis of randomized controlled trials. J Am Coll Cardiol 2005;45:185–97.
23. Robins SJ, Collins D, Wittes JT, et al. VA-HIT Study Group. Veterans Affairs High-Density Lipoprotein Intervention Trial. Relation of gemf brozil treatment and lipid levels with major coronary events: VA-HIT: a randomized controlled trial. JAMA 2001;285:1585–91.
24. Scott R, O’Brien R, Fulcher G, et al. Fenof brate Intervention and Event Lowering in Diabetes (FIELD) Study Investigators. Effects of fenof brate treatment on cardiovascular disease risk in 9,795 individuals with type 2 diabetes and various components of the metabolic syndrome: the Fenof brate Intervention and Event Lowering in Diabetes (FIELD) study. Diabetes Care 2009;32:493–8.
25. ACCORD Study Group, Ginsberg HN, Elam MB, Lovato LC, et al. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med 2010;362:1563–74.
26. Clof brate and niacin in coronary heart disease. Coronary Drug Project Research Group. JAMA 1975;231:360.
27. Iribarren C, Go AS, Husson G, et al. Metabolic syndrome and early-onset coronary artery disease: is the whole greater than its parts? J Am Coll Cardiol 2006;48:1800–7.
28. Nieves D, Cnop M, Retzlaff B, et al. The atherogenic lipoprotein prof le associated with obesity and insulin resistance is largely attributable to intra-abdominal fat. Diabetes 2003;52:172.
29. Poirier P, Eckel RH. Obesity and cardiovascular disease. Curr Atheroscler Rep 2002;4:448–53.
30. Ferrannini E, Balkau B, Coppack SW, et al. RISC Investigators. Insulin resistance, insulin response, and obesity as indicators of metabolic risk. J Clin Endocrinol Metab 2007;92:2885–92.
31. Grundy SM. Approach to lipoprotein management in 2001 National Cholesterol Guidelines. Am J Cardiol 2002;90: 11i.
32. Borgman M, McErlean E. What is the metabolic syndrome? Prediabetes and cardiovascular risk. J Cardiovasc Nurs 2006;21:285–90.
33. Grundy SM. Metabolic syndrome pandemic. Arterioscler Thromb Vasc Biol 2008;28:629–36.
34. McLaughlin T, Allison G, Abbasi F, et al. Prevalence of insulin resistance and associated cardiovascular disease risk factors among normal weight, overweight, and obese individuals. Metabolism 2004;53:495–9.
35. Lechleitner M. Obesity and the metabolic syndrome in the elderly—a mini-review. Gerontology 2008;54:253–9.
36. Grundy SM. Metabolic syndrome: connecting and reconciling cardiovascular and diabetes worlds. J Am Coll Cardiol 2006;47:1093–100.
37. Ginsberg HN. Insulin resistance and cardiovascular disease. J Clin Invest 2000;106:453.
38. Ford E, Giles W, Dietz W. Prevalence of the metabolic syndrome among US adults: fi ndings from the Third National Health and Nutrition Examination Survey. JAMA 2002; 287:356.
39. Alexander CM, Landsman PB, Teutsch SM, et al. NCEPdefi ned metabolic syndrome, diabetes, and prevalence of coronary heart disease among NHANES III participants age 50 years and older. Diabetes 2003;52:1210.
40. Lamarche B, Moorjani S, Cantin B, et al. Associations of HDL2 and HDL3 subfractions with ischemic heart disease in men. Thromb Vasc Biol 1997;17:1098.
41. Williams RR, Hopkins PN, Hunt SC, et al. Population-based frequency of dyslipidemia syndromes in coronary-prone families in Utah. Arch Intern Med 1990;150:582.
42. Goldstein JL, Hazzard WR, Schrott HG, et al. Hyperlipidemia in coronary heart disease. I. Lipid levels in 500 survivors of myocardial infarction. J Clin Invest 1973;52: 1533.
43. Goldstein JL, Schrott HG, Hazzard WR, et al. Hyperlipidemia in coronary heart disease. II. Genetic analysis of lipid levels in 176 families and delineation of a new inherited disorder, combined hyperlipidemia. J Clin Invest 1973;52: 1544.
44. Hazzard WR, Goldstein JL, Schrott MG, et al. Hyperlipidemia in coronary heart disease. III. Evaluation of lipoprotein phenotypes of 156 genetically defi ned survivors of myocardial infarction. J Clin Invest 1973;52:1569.
45. Ayyobi AF, McGladdery SH, McNeely MJ, et al. Small, dense LDL and elevated apolipoprotein B are the common characteristics for the three major lipid phenotypes of familial combined hyperlipidemia. Arterioscler Thromb Vasc Biol 2003;23:1289.
46. Coon H, Xin Y, Hopkins PN, et al. Upstream stimulatory factor 1 associated with familial combined hyperlipidemia, LDL cholesterol, and triglycerides. Hum Genet 2005;117: 444–51.
47. Zambon A, Brown BG, Deeb SS, Brunzell JD. Genetics of apolipoprotein B and apolipoprotein AI and premature coronary artery disease. J Intern Med 2006;259:473–80.
48. Babirak SP, Brown BG, Brunzell JD, et al. Familial combined hyperlipidemia and abnormal lipoprotein lipase. Arterioscler Thromb 1992;12:1176.
49. Purnell JQ, Kahn SE, Schwartz RS, et al. Relationship of insulin sensitivity and apoB levels to intra-abdominal fat in subjects with familial combined hyperlipidemia. Arterioscler Thromb Vasc Biol 2001;21:567.
50. Brown G, Albers JJ, Fisher LD, et al. Regression of coronary artery disease as a result of intensive lipid-lowering therapy in men with high levels of apolipoprotein B. N Engl J Med 1990;323:1289.
51. Holmes KW, Kwiterovich PO Jr. Treatment of dyslipidemia in children and adolescents. Curr Cardiol Rep 2005; 7:445–56.
52. Rajpathak SN, Kumbhani DJ, Crandall J, et al. Statin therapy and risk of developing type 2 diabetes: a meta-analysis. Diabetes Care 2009;32:1924–9.
53. Hopkins PN, Heiss G, Ellison RC, et al. Coronary artery disease risk in familial combined hyperlipidemia and familial hypertriglyceridemia: a case-control comparison
i. from the National Heart, Lung, and Blood Institute Family Heart Study. Circulation 2003;108:519–23.
54. Rubins HB, Robins SJ, Collins D, et al. Gemfi brozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial Study Group. N Engl J Med 1999;341: 410.
55. Michaelides AP, Fourlas CA, Pitsavos C, et al. Exercise testing in asymptomatic patients with heterozygous familial hypercholesterolaemia. Coron Artery Dis 2004;15: 461–5.
56. Marks D, Thorogood M, Neil HA, et al. A review on the diagnosis, natural history, and treatment of familial hypercholesterolaemia. Atherosclerosis 2003;168:1.
57. Grundy SM, Vega GL, McGovern ME. Effi cacy, safety, and tolerability of once-daily niacin for the treatment of dyslipidemia associated with type 2 diabetes: results of the assessment of diabetes control and evaluation of the effi cacy of Niaspan trial. Arch Intern Med 2002;162:1568.
58. Colhoun HM, Betteridge DJ, Durrington PN, et al. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): multicentre randomised placebo-controlled trial. Lancet 2004;364:685.
59. Lamon-Fava S, Postfai B, Diffenderfer M, et al. Role of the estrogen and progestin in hormonal replacement therapy on apolipoprotein A-I kinetics in postmenopausal women. Arterioscler Thromb Vasc Biol 2006;26:385–91.
60. Effects of estrogen or estrogen/progestin regimens on heart disease risk factors in postmenopausal women: the Postmenopausal Estrogen/Progestin Interventions (PEPI) trial. The Writing Group for the PEPI trial. JAMA 1995;273:199.
61. Anderson GL, Limacher M, Assaf AR, et al. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women’s Health Initiative randomized controlled trial. JAMA 2004;291:1701.
62. Rossouw JE, Prentice RL, Manson JE, et al. Postmenopausal hormone therapy and risk of cardiovascular disease by age and years since menopause. JAMA 2007;297: 1465–77.
63. Shearer GC, Kaysen GA. Proteinuria and plasma compositional changes contribute to defective lipoprotein catabolism in the nephrotic syndrome by separate mechanisms. Am J Kidney Dis 2001;37(1 Suppl 2):S119–22.
64. Joven J, Villabona C, Vilella E, et al. Abnormalities of lipoprotein metabolism in patients with the nephrotic syndrome. N Engl J Med 1990;323:579.
65. Vaziri ND, Liang K, Parks JS. Down-regulation of hepatic lecithin:cholesterol acyltransferase gene expression in chronic renal failure. Kidney Int 2001;59:2192–6.
66. Dogra G, Irish A, Chan D, Watts G. A randomized trial of the effect of statin and fibrate therapy on arterial function in CKD. Am J Kidney Dis 2007;49:776–85.
67. Khunnawat C, Mukerji S, Havlichek D Jr, et al. Cardiovascular manifestations in human immunodefi ciency virus-infected patients. Am J Cardiol 2008;102:635–42.
68. The Lipid Research Clinics Coronary Primary Prevention Trial results. I. Reduction in incidence of coronary heart disease. JAMA 1984;251:351.
69. The Lipid Research Clinics Coronary Primary Prevention Trial results. II. The relationship of reduction in incidence of coronary heart disease to cholesterol lowering. JAMA 1984;251:365.
70. Frick MH, Elo O, Haapa K, et al. Helsinki Heart Study: primary-prevention trial with gemfi brozil in middle-aged men with dyslipidemia: safety of treatment, changes in risk factors, and incidence of coronary heart disease. N Engl J Med 1987;317:1237.
71. Shepherd J, Cobbe SM, Ford I, et al. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. West of Scotland Coronary Prevention Study Group. N Engl J Med 1995;333:1301.
72. Baseline risk factors and their association with outcome in the West of Scotland Coronary Prevention Study. The West of Scotland Coronary Prevention Study Group. Am J Cardiol 1997;79:756.
73. Infl uence of pravastatin and plasma lipids on clinical events in the West of Scotland Coronary Prevention Study (WOSCOPS). Circulation 1998;97:1440.
74. Downs JR, Clearfi eld M, Weis S, et al. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/ TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA 1998;279:1615.
75. Gotto AM Jr, Boccuzzi SJ, Cook JR, et al. Effect of lovastatin on cardiovascular resource utilization and costs in the Air Force/Texas Coronary Atherosclerosis Prevention Study (AFCAPS/TexCAPS). AFCAPS/TexCAPS Research Group. Am J Cardiol 2000;86:1176.
76. Sever PS, Dahlof B, Poulter NR, et al. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial—Lipid Lowering Arm (ASCOT-LLA): a multicentre randomised controlled trial. Lancet 2003; 361:1149.
77. Karalis DG. Intensive lowering of low-density lipoprotein cholesterol levels for primary prevention of coronary artery disease. Mayo Clin Proc 2009;84:345–52.
78. Watts GF, Lewis B, Brunt JN, et al. Effects on coronary artery disease of lipid-lowering diet, or diet plus cholestyramine, in the St Thomas’ Atherosclerosis Regression Study (STARS). Lancet 1992;339:563.
79. de Lorgeril M, Salen P, Martin JL, et al. Effect of a Mediterranean type of diet on the rate of cardiovascular complications in patients with coronary artery disease: insights into the cardioprotective effect of certain nutriments. J Am Coll Cardiol 1996;28:1103.
80. de Lorgeril M, Salen P, Martin JL, et al. Mediterranean diet, traditional risk factors, and the rate of cardiovascular complications after myocardial infarction: fi nal report of the Lyon Diet Heart Study. Circulation 1999;99:779.
81. Zambon A, Hokanson JE, Brown BG, et al. Evidence for a new pathophysiological mechanism for coronary artery disease regression: hepatic lipase-mediated changes in LDL density. Circulation 1999;99:1959.
82. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet 1994;344:1383.
83. Pedersen TR, Olsson AG, Faergeman O, et al. Lipoprotein changes and reduction in the incidence of major coronary heart disease events in the Scandinavian Simvastatin Survival Study (4S). Circulation 1998;97:1453.
84. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. N Engl J Med 1998;339:1349.
85. Simes RJ, Marschner IC, Hunt D, et al. Relationship between lipid levels and clinical outcomes in the Longterm Intervention with Pravastatin in Ischemic Disease (LIPID) trial: to what extent is the reduction in coronary events with pravastatin explained by on-study lipid levels? Circulation 2002;105:1162.
86. Sacks FM, Pfeffer MA, Moye LA, et al. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events Trial Investigators. N Engl J Med 1996;335:1001.
87. Sacks FM, Moye LA, Davis BR, et al. Relationship between plasma LDL concentrations during treatment with pravastatin and recurrent coronary events in the Cholesterol and Recurrent Events trial. Circulation 1998;97:1446.
88. MCR/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Heart Protection Study Collaborative Group. Lancet 2002;360:7.
89. Cannon CP, Braunwald E, McCabe CH, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004;350:1495.
90. Nissen SE, Tuzcu EM, Schoenhagen P, et al. Effect of intensive compared with moderate lipid-lowering therapy on progression of coronary atherosclerosis: a randomized controlled trial. JAMA 2004;291:1071.
91. Brown BG, Zhao XQ, Chait A, et al. Simvastatin and niacin, antioxidant vitamins, or the combination for the prevention of coronary disease. N Engl J Med 2001;345:1583.
92. Snow V, Aronson MD, Hornbake ER, et al. Lipid control in the management of type 2 diabetes mellitus: a clinical practice guideline from the American College of Physicians. Ann Intern Med 2004;140:644.
93. D’Agostino RB Sr, Grundy S, Sullivan LM, et al. Validation of the Framingham coronary heart disease prediction scores: results of a multiple ethnic groups investigation. JAMA 2001;286:180.
94. Liu J, Hong Y, D’Agostino RB Sr, et al. Predictive value for the Chinese population of the Framingham CHD risk assessment tool compared with the Chinese Multi-Provincial Cohort Study. JAMA 2004;291:2591.
95. Brindle P, Emberson J, Lampe F, et al. Predictive accuracy of the Framingham coronary risk score in British men: prospective cohort study. BMJ 2003;327:1267.
96. Shepherd J, Blauw GJ, Murphy MB, et al. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet 2002;360:1623.
97. Yusuf S, Hawken S, Ounpuu S, et al. Effect of potentially modifi able risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet 2004;364:937.
98. Grundy SM, Cleeman JI, Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004;110:227.
99. Gandhi SK, Järbrink K, Fox KM, Brandrup-Wognsen G. Effectiveness of rosuvastatin in reducing LDL-C and target LDL-C goal attainment in real-world clinical practice. Curr Med Res Opin 2009;25:2817–28.
100.Taylor AJ, Villines TC, Stanek EJ, et al. Extended-release niacin or ezetimibe and carotid intima-media thickness. N Engl J Med 2009;361:2113–22.
101.Sujana Kumar S, Lahey KA, Day A, Lahaye SA. Comparison of the effi cacy of administering a combination of ezetimibe plus fenofi brate versus atorvastatin mono-therapy in the treatment of dyslipidemia. Lipids Health Dis 2009;8:56.
102.Teoh H, Mendelsohn AA, Goodman SG, et al. Guidelines Based Undertaking for Improvement in Dyslipidemia Related Events (GUIDE) Investigators. Usefulness of statin-ezetimibe combination to reduce the care gap in dyslipidemia management in patients with a high risk of atherosclerotic disease. Am J Cardiol 2009;104:798–804.
103.Ford ES, Li C, Zhao G, Mokdad AH. Concentrations of low-density lipoprotein cholesterol and total cholesterol among children and adolescents in the United States. Circulation 2009;119:1108–15.
104.Daniels SR, Greer FR; Committee on Nutrition. Lipid screening and cardiovascular health in childhood. Pediatrics 2008;122:198–208.
105.McCrindle BW. Hyperlipidemia in children. Thromb Res 2006;118:49–58.
106.Rossouw JE, Anderson GL, Prentice RL, et al. Risks and benefi ts of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA 2002; 288:321.
107.Anderson GL, Limacher M, Assaf AR, et al. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women’s Health Initiative randomized controlled trial. JAMA 2004;291:1701.
108.Carlsson CM, Carnes M, McBride PE, et al. Managing dyslipidemia in older adults. J Am Geriatr Soc 1999;47: 1458.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.