(Carregando Índice)... (Carregando Índice)... |
Última revisão: 28/02/2014
Comentários de assinantes: 0
Lawrence L.K. Leung, MD
Maureen Lyles D'Ambrogio Professor of Medicine, Department of Medicine, StanfoMaureen Lyles D'Ambrogio Professor of Medicine, Department of Medicine, Stanford University School of Medicine, Stanford, CA; Chief of Staff and Chief, Medical Service, Veterans Affairs Palo Alto Health Care System, Palo Alto, CA
Artigo original: Leung LLK. Thrombotic disorders. ACP Medicine. 2010;1-21.
[The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2011 Decker Intellectual Properties Inc. All Rights Reserved.]
Agradecimentos: Figura 1 – Seward Hung. Figura 2 – Christine Kenney.
Tradução: Soraya Imon de Oliveira.
Revisão técnica: Dr. Euclides Furtado de Albuquerque Cavalcanti.
A trombose é um fenômeno que envolve mais do que coagulação sanguínea excessiva, pois também envolve inflamação vascular. A clássica tríade de Virchow identifica 3 elementos principais na fisiopatologia da trombose: lesão endotelial, diminuição do fluxo sanguíneo e desequilíbrio entre fatores pró-coagulantes e anticoagulantes.
As células endoteliais podem ser ativadas ou lesadas por diversos estímulos, incluindo o traumatismo mecânico, endotoxinas e citocinas, proteases, mediadores inflamatórios, deposição de imunocomplexos, radicais do oxigênio e hipóxia. Cada um destes estímulos afeta múltiplos aspectos da função celular endotelial e, por fim, muda o estado naturalmente antitrombótico da célula para um estado pró-trombótico.
O endotélio vascular, em sua localização exclusiva junto à parede vascular, consegue perceber e responder a diferentes forças mecânicas atuantes na circulação sanguínea. A força de cisalhamento causada pelo atrito produzido pelo fluxo sanguíneo parece ser particularmente importante na modulação das funções endoteliais. Nas áreas de fluxo linear, o sangue movimenta-se seguindo padrões laminares ordenados, de modo pulsátil e regular. Este fluxo sanguíneo laminar estável aparentemente promove um fenótipo endotelial antitrombótico. Nas áreas de fluxo interrompido, como ao nível das bifurcações ou estenoses vasculares, o endotélio pode ficar exposto às alterações significativas dos gradientes de cisalhamento, enquanto as células podem ser ativadas e tornarem-se pró-trombóticas.
O desequilíbrio entre os fatores pró-coagulantes e anticoagulantes pode ser hereditário ou adquirido [Tabela 1]. Alguns fatores de coagulação, como o fator VIII e o fibrinogênio, são reagentes de fase aguda: os níveis plasmáticos destes fatores aumentam de modo significativo com a inflamação aguda, possivelmente conferindo um estado pró-trombótico transiente. Algumas deficiências hereditárias de proteínas anticoagulantes, como o fator V de Leiden e a antitrombina (AT), estão associadas à trombose recorrente. Estas deficiências estão entre as condições hipercoaguláveis clínicas mais bem conhecidas.
Tabela 1. Condições hipercoaguláveis hereditárias e adquiridas
|
Hereditárias |
|
Resistência à PCA/fator V de Leiden |
|
Mutação G20210A no gene da protrombina |
|
Deficiência de proteína C |
|
Deficiência de proteína S |
|
Hiper-homocisteinemia |
|
Níveis altos de fator VIII |
|
Adquiridas |
|
SAF |
|
Condição de hipercoagulação associada a estímulos fisiológicos ou trombogênicos |
|
Idade avançada |
|
Anticoncepcionais orais |
|
Gestação |
|
Cirurgia |
|
Traumatismo |
|
Condição hipercoagulação associada a condições patológicas |
|
Malignidade – síndrome de Trousseau |
|
TIH com trombose |
|
Síndrome nefrótica |
|
Hiperviscosidade (policitemia vera, macroglobulinemia de Waldenström, mieloma múltiplo) |
|
Distúrbios mieloproliferativos (policitemia vera, trombocitemia essencial) |
|
Hemoglobinúria noturna paroxística |
|
Anemia falciforme |
|
Raras ou ainda pouco estabelecidas |
|
Disfibrinogenemia |
|
Hipoplasminogenemia, displasminogenemia |
|
Trombomodulina anormal |
|
Deficiência de fator XII |
|
Níveis elevados de fator VII, fibrinogênio, Lp(a), PAI-1 |
Lp(a) = lipoproteína (a); PAI-1 = inibidor de ativador de plasminogênio-1; PCA = proteína C ativada; SAF = síndrome antifosfolipídica; TIH = trombocitopenia induzida por heparina.
Embora o estado hipercoagulável seja uma condição sistêmica, a trombose ocorre localmente (p. ex., nos membros inferiores). Este aspecto reflete a existência de uma complexa interação entre a predisposição pró-trombótica sistêmica e os mecanismos de controle hemostático locais específicos do leito vascular. A existência de padrões específicos e distintivos de expressão de transcrição gênica nos diferentes leitos vasculares está comprovada.1
As principais questões da avaliação da trombose incluem os seguintes: (1) Qual é a probabilidade de que a trombose seja causada por uma condição de hipercoagulação subjacente? (2) Até que ponto um rastreamento é indicado? (3) Qual é a duração da anticoagulação? As respostas para estas perguntas podem ser encontradas considerando fatores como a idade do paciente no momento da 1ª trombose; presença/ausência de um fator provocador; história familiar e história médica anterior de resposta a situações associadas a um alto risco de trombose; e localização, tipo e gravidade da trombose.
Em um estudo retrospectivo envolvendo 150 famílias com predisposição hereditária à trombose recorrente (trombofilia), a média da idade dos pacientes no momento em que desenvolveram trombose pela 1ª vez foi 35 a 40 anos. Entretanto, o 1º episódio de trombose pode ocorrer já na 2ª década da vida, caso o paciente apresente mais de 1 fator de risco hereditário.2
Os deflagradores comuns da trombose são: cirurgia, traumatismo, gestação, malignidade, imobilização prolongada e infecção. A malignidade pode ser clinicamente evidente ou subclínica. Estas circunstâncias podem provocar trombose até mesmo em indivíduos com sistema de coagulação normal e com frequência revelam uma trombofilia em curso clinicamente silenciosa.2 No entanto, em cerca de 30 a 40% dos casos, nenhum fator provocador é descoberto. Estes casos de trombose idiopática espontânea, sobretudo quando ocorrem em indivíduos jovens, são fortemente sugestivos da existência de uma condição hipercoagulável hereditária subjacente.
Quando a trombose se desenvolve em uma paciente que já passou por outras gestações ou cirurgias (em especial os procedimentos ortopédicos) sem desenvolver nenhuma complicação trombótica, deve ser considerada a hipótese de uma condição de hipercoagulação adquirida. Nestes casos, as condições prováveis são a síndrome antifosfolipídica (SAF) ou a síndrome de Trousseau (ver adiante).
A tromboembolia venosa (TEV) não provocada, comprovada de modo objetivo e que ocorre antes dos 50 anos de idade em um parente de 1º grau é fortemente sugestiva de um distúrbio trombótico hereditário. Entretanto, uma história familiar negativa não exclui a hipótese de uma condição hereditária. A trombose clínica frequentemente representa o auge de múltiplos fatores de risco trombogênicos, dentre os quais apenas um pode ser hereditário e irreversível. Em pacientes com trombose sintomática e condições de hipercoagulação hereditárias bem comprovadas, não é incomum encontrar outros familiares apresentando a mesma deficiência e sem trombose clínica.
Um paciente que sofre de trombose recorrente provavelmente apresenta uma condição de hipercoagulação (hereditária ou adquirida). Entretanto, se um paciente que inicialmente apresenta trombose venosa profunda (TVP) em um membro inferior volta a apresentar sintomas na mesma perna, o problema pode ser uma síndrome pós-flebítica e não trombótica recorrente. A exacerbação aguda da síndrome pós-flebítica, aliada à dor e ao edema aumentados na perna, pode ser difícil de distinguir de uma TVP aguda recorrente. Como medida antecipatória, pode ser útil repetir um exame de ultrassonografia com compressão do membro inferior no momento da conclusão da terapia de anticoagulação oral, após a resolução de um episódio agudo de TVP. A repetição do exame pode fornecer informações basais para comparação futura.
Mais comumente, as tromboses envolvem as veias profundas dos membros inferiores. A trombose em um local atípico, como as veias hepáticas, mesentéricas ou cerebrais (ou a necrose cutânea após a administração de varfarina), aumenta a probabilidade de um estado de hipercoagulação subjacente. Um fator de risco trombofílico é encontrado em cerca de 35% dos pacientes com TVP em membro superior não relacionada ao uso de cateteres venosos.3 A trombose recorrente em sítios arteriais está associada a um diagnóstico diferencial – e, portanto, a um workup – bastante diferente daquele associado à TEV recorrente [Tabela 2]. A maioria das condições hipercoaguláveis hereditárias comuns (p. ex., deficiência de AT ou de fator V de Leiden) está associada à TEV. Estas condições raramente estão associadas às tromboses arteriais, como o ataque isquêmico transiente, acidente vascular cerebral (AVC), isquemia digital e infarto do miocárdio. Alguns estados hipercoaguláveis, como a SAF e a síndrome de Trousseau, estão associados a ambos os tipos de trombose.
Tabela 2. Testes para pacientes com suspeita de condição hipercoagulável
|
Condição subjacente |
Avaliações laboratoriais |
|
Trombose venosa |
Resistência à PCA Fator V de Leiden (teste genético) Ensaio de coagulação (desnecessário, se o teste genético para fator V de Leiden resultar positivo) Mutação G20210 no gene da protrombina AT (ensaio funcional) Proteína C (ensaio funcional) Proteína S Ensaio funcional Ensaio antigênico para proteína S livre |
|
Trombose arterial |
TIH* (ELISA para anticorpos dirigidos contra o complexo heparina/PF4) CID crônica (síndrome de Trousseau)* Distúrbios mieloproliferativos (mutação em JAK2 V617F) Lp(a) |
|
Trombose venosa e/ou trombose arterial |
Homocisteína plasmática (níveis em jejum) Anticorpo antifosfolipídio Ensaios de coagulação para anticoagulante lúpico ELISA para anticorpos IgG e IgM anticardiolipina Disfibrinogenemia (se houver forte suspeita de trombofilia) Ensaio funcional para níveis de fibrinogênio TT, TR |
*No contexto clínico apropriado.
AT = antitrombina; CID = coagulação intravascular disseminada; ELISA = ensaio imunossorvente ligado à enzima; Lp(a) = lipoproteína (a); PCA = proteína C ativada; PF4 = fator plaquetário 4; TIH = trombocitopenia induzida por heparina; TR = tempo de reptilase; TT = tempo de trombina.
Extensão do rastreamento. Com base nas considerações clínicas mencionadas, é possível estimar a probabilidade de que haja trombofilia subjacente em um paciente com trombose [Tabela 3]. Dada a indisponibilidade de estudos custo-efetivos e resultados, é difícil listar as diretrizes práticas estritas referentes à extensão do workup para uma condição hipercoagulável. Em geral, porém, havendo alta probabilidade de existência de uma condição hipercoagulável subjacente, e se os resultados influenciarem o tratamento (p. ex., duração da anticoagulação), então a realização de um rastreamento extenso é justificada. Contudo, o clínico deve estar alerta para a falta de padronização de alguns exames (p. ex., o ensaio da proteína S e os ensaios de anticorpos anticardiolipina) e para as armadilhas de resultados falso-positivos.4
Tabela 3. Aspectos clínicos sugestivos de trombofilia
|
Idade < 50 anos no momento da ocorrência do 1º episódio de trombose |
|
Ausência de fator de risco identificável |
|
História familiar positiva |
|
Trombose recorrente |
|
Sítio de trombose atípico |
A TEV precipitada por um fator provocador evidente (p. ex., o desenvolvimento de TVP em membro inferior após uma cirurgia de substituição de quadril) em geral dispensa a investigação. Em algumas circunstâncias, uma investigação limitada pode ser apropriada para guiar o tratamento futuramente. Exemplificando, no caso de uma jovem que desenvolve TVP na veia femoral superficial durante o uso de um anticoncepcional oral, será informativo avaliar o fator V de Leiden, a mutação G20210A da protrombina, AT, proteína C, proteína S, anticorpos anticardiolipina e anticoagulante lúpico. Contudo, uma TEV não comprovada geralmente é sugestiva de uma condição de hipercoagulação subjacente e justifica a realização da investigação. Neste caso, a extensão da investigação dependerá da manifestação clínica. A hipótese de uma condição de hipercoagulação adquirida deve ser considerada no caso de um paciente idoso com TVP espontânea e sem história prévia de trombose. Em casos como este, as possibilidades diagnósticas incluiriam a SAF ou a síndrome de Trousseau.
Diante de uma história clínica fortemente sugestiva de trombofilia – como no caso do paciente com trombose recorrente ou trombose de localização atípica – é possível argumentar que o rastreamento para uma condição hipercoagulável subjacente é desnecessário, uma vez que o paciente será submetido à anticoagulação prolongada e o resultado não afetará o manejo do caso. Entretanto, a identificação de quaisquer fatores de risco subjacentes irá melhorar a compreensão acerca da doença, tanto para o paciente como para o médico que o está tratando, e orientará o aconselhamento do paciente, sobretudo no que se refere à necessidade de avaliação de seus familiares.
Quando realizar o rastreamento. O clínico precisa saber não só quais exames devem ser solicitados mas também quando solicitá-los. Na trombose aguda, muitos inibidores da cascata de coagulação (p. ex., AT, proteína C) são consumidos, de tal modo que seus níveis plasmáticos podem diminuir, até mesmo em pacientes sem deficiência hereditária. A terapia com heparina pode diminuir os níveis de AT em até 20%, enquanto o tratamento com warfarina diminui os níveis de proteína C e de proteína S. Em geral, é melhor adiar a quantificação destes inibidores até que o episódio trombótico tenha sido completamente resolvido, de preferência decorridas 4 semanas do término da terapia de anticoagulação oral. Os testes para genótipos específicos (p. ex., fator V de Leiden), todavia, podem ser realizados a qualquer momento.
A frequência das diversas condições hipercoaguláveis entre pacientes não selecionados com TEV varia de 1 a 25% [Tabela 4]. É preciso reconhecer que estas trombofilias não conferem um risco trombótico equivalente. O fator V de Leiden e a mutação G20210A da protrombina, que são os 2 fatores de risco mais prevalentes, conferem apenas um modesto aumento do risco relativo de trombose (cerca de 3 a 5 vezes acima do normal). A hiper-homocisteinemia moderada, outro fator de risco comum, também está associada a um aumento modesto do risco. As deficiências heterozigotas de AT, proteína C e proteína S são mais significativas como fatores de risco do que o fator V de Leiden. A deficiência de AT e a SAF são provavelmente os principais fatores de risco. O índice de recorrência em paciente com SAF chega a 50% ao longo de 5 anos, em alguns estudos.
Tabela 4. Frequência e risco relativo de trombose venosa em condições hipercoaguláveis selecionadas*
|
Condição |
Risco relativo† |
Frequência (%)‡ |
|
Deficiência de AT SAF |
Alto Alto |
1 a 2 Desconhecida |
|
Deficiência de proteína C |
Moderado |
3 a 4 |
|
Deficiência de proteína S |
Moderado |
2 a 3 |
|
Fator V de Leiden |
Modesto |
20 a 25 |
|
Mutação G20210 no gene da protrombina |
Modesto |
10 |
|
Hiper-homocisteinemia |
Modesto |
10 |
|
Uso de anticoncepcional oral |
Modesto |
ND |
*A incidência de TEV na população normal está estimada em 0,008% ao ano (0,03% ao ano em pacientes que tomam anticoncepcionais orais).
†O risco modesto é definido por um aumento aproximado de 2,5 a 5 vezes na incidência de tromboembolia, com base nos dados fornecidos pelo Leiden Thrombophilia Study;23 o risco elevado é definido por um aumento aproximado de 3 a 4 vezes em relação ao risco do fator V de Leiden.2
‡Em pacientes não selecionados com TEV.
AT = antitrombina; ND = não disponível; SAF = síndrome antifosfolipídica; TEV = tromboembolia venosa.
Os pacientes com trombose sintomática frequentemente apresentam mais de 1 fator de risco, que podem exercer um efeito sinérgico no sentido de aumentar o risco de trombose. Exemplificando, as mulheres com fator V de Leiden que usam anticoncepcionais orais têm um risco de TEV 35 vezes maior do que o observado na população em geral, sendo que cada fator de risco está associado a um risco aproximado de 5 vezes.
A frequência da deficiência de AT sintomática na população em geral foi estimada em aproximadamente 1:500 a 1:5.000 indivíduos.5 A deficiência é transmitida segundo um padrão autossômico dominante. Não há relatos de deficiência de AT homozigota, provavelmente devido à incompatibilidade da condição com o desenvolvimento fetal normal.
Existem 2 tipos de deficiência de AT hereditária. O tipo I é uma deficiência quantitativa, determinada por meio de ensaios antigênicos e funcionais. Várias mutações moleculares foram caracterizadas na deficiência de AT de tipo I, incluindo as deleções genéticas parciais e as substituições de nucleotídeo único que causam mutações nonsense ou missense, com consequente sinalização de parada prematura no processo de translação proteica.
A deficiência de tipo II é qualitativa. Os níveis plasmáticos de antígeno AT permanecem normais. O defeito subjacente em geral é uma alteração de um único nucleotídeo que produz mutaçõesmissense , dando origem a uma proteína disfuncional. Muitas destas proteínas apresentam diminuição da afinidade pela trombina (tipo IIa) ou pela heparina (tipo IIb).6
Em casos raros, a deficiência de AT é adquirida. Esta condição pode ocorrer após a administração de heparina por via intravenosa por mais de 3 dias ou, ainda, após a terapia com L-asparaginase. Esta deficiência também pode desenvolver-se em pacientes que sofrem de coagulação intravascular disseminada (CID), doença hepática severa ou síndrome nefrótica.
A AT inativa o fator Xa e a trombina ao formar um complexo estequiométrico estável com cada uma destas moléculas. A AT é encontrada em concentrações plasmáticas suficientes para inativar toda a trombina formada em determinado volume de plasma. Entretanto, esta inativação é feita de modo bastante lento, exceto quando é ativada pelo heparan sulfato existente na superfície da célula endotelial ou pela heparina administrada. Os pacientes com deficiência de AT hereditária apresentam evidência de uma contínua ativação de fator X e geração de trombina (sustentada pelos elevados níveis plasmáticos de fragmento F1.2 de protrombina), mesmo quando são clinicamente assintomáticos.
Os pacientes com deficiência de AT apresentam incidência aumentada de trombose venosa, geralmente deflagrada por um estímulo pró-trombótico, como cirurgia, infecção, imobilização ou traumatismo. Esta associação sugere que a sobreposição de um estímulo pró-trombótico fornecido por uma condição de hipercoagulação subclínica subjacente leva ao desenvolvimento de trombose clínica.
As manifestações clínicas típicas são TVP em membros inferiores, embolia pulmonar e, ocasionalmente, trombose de veia mesentérica. Não há evidências convincentes sugestivas de que a deficiência de AT aumente o risco de trombose arterial.7
Os pacientes afetados costumam ter história familiar de tromboses recorrentes, que em geral surgem na 3ª década da vida e muitas vezes estão associadas a cirurgia ou a traumatismo. A gestação e o uso de anticoncepcionais orais também aumenta o risco de trombose em pacientes com deficiência de AT. A tendência ao desenvolvimento de trombose aumenta com o avanço da idade: ao redor dos 50 anos, apenas 50% dos pacientes com deficiência de AT são assintomáticos.2
O nível de AT deve ser determinado por um ensaio funcional, em vez do ensaio antigênico, para que seja possível avaliar ambos os tipos de deficiência (I e II). Os pacientes com deficiência de AT apresentam uma diminuição notavelmente modesta da concentração da proteína: os valores medidos por bioensaio e imunoensaio variam de 25 a 60% do valor normal, na doença de tipo I.
O estudo de uma ampla família com deficiência de AT indica que o uso da profilaxia anticoagulante prolongada não é justificado para os portadores assintomáticos desta deficiência.8 Os portadores assintomáticos devem receber profilaxia anticoagulante nas situações que comprovadamente aumentam o risco de trombogênese, como em casos de cirurgia abdominal.7 Entretanto, depois de passarem por um evento trombótico, os pacientes provavelmente passam a necessitar de terapia com warfarina pelo resto da vida, pois o risco de TEV recorrente é alto (estimado em 13 a 17% ao ano).9 A warfarina atualmente é a base da terapia de longa duração destinada aos pacientes com deficiência de AT e tromboembolia recorrente, embora diversos anticoagulantes orais novos provavelmente recebam aprovação do Food and Drug Administration (FDA) em um futuro próximo.
Os episódios agudos de trombose devem ser tratados com heparina. Como a deficiência de AT pode tornar a heparina relativamente inefetiva, o médico deve estar alerta para a resistência à heparina. Em pacientes tratados com heparina não fracionada, a resistência manifesta-se como um prolongamento mínimo do tempo de tromboplastina parcial (TTPA), que ocorre mesmo com a administração de doses terapêuticas. Quando a heparina de baixo peso molecular (HBPM) é usada, como geralmente ocorre, os níveis de anti-fator Xa (anti-FXa) devem ser checados para garantir que um efeito anticoagulante terapêutico seja alcançado.
Se houver resistência à heparina mesmo com o uso de doses maiores, a heparina deve ser administrada com concentrados de AT purificada ou plasma fresco congelado. A AT humana purificada tem meia-vida aproximada de 60 horas, enquanto a AT humana recombinante tem meia-vida aproximada de 10 horas. Estas preparações podem ser usadas para sustentar um paciente com deficiência de AT no decorrer de uma cirurgia ou parto e devem trazer os níveis de AT para próximo de 100%, dependendo dos níveis basais de AT do indivíduo. Os níveis de AT devem ser checados, e a infusão tem de se repetida a intervalos de 24 horas, para manter os níveis de AT normais por 5 a 7 dias após o parto ou cirurgia.
Em uma paciente com deficiência de AT, o manejo da gestação torna-se difícil. Como a varfarina pode causar deformações fetais e hemorragia neonatal, as pacientes devem ser tratadas com doses integrais de heparina não fracionada ou HBPM. As pacientes tratadas com HBPM devem ter a medicação trocada por heparina não fracionada com uma antecedência de 1 a 2 semanas em relação ao parto, para que a rápida reversão da anticoagulação seja mais facilmente realizada, caso necessário. Se não for possível alcançar um efeito terapêutico (medido pelo TTPA com heparina não fracionada ou pelos níveis de anti-FXa com HBPM), então a AT deve ser infundida. Isto geralmente é desnecessário. A anticoagulação deve ser prontamente reiniciada após o parto.
Uma deficiência ou defeito envolvendo as proteínas C ou S resulta em perda da capacidade de inativar o excesso de fator VIIIa e fator Va, que são os dois principais cofatores responsáveis pela regulação da amplificação da cascata da coagulação. Os níveis de proteína C estão baixos em pacientes com CID e hepatopatia, provavelmente porque a ativação da hemostasia consome este fator.
A deficiência de proteína C homozigota causa trombose letal em bebês (púrpura neonatal fulminante). A forma heterozigota desta deficiência está associada a uma prevalência de 1 em 200 a 300 indivíduos, na população em geral. A expressão clínica da deficiência de proteína C heterozigota é variável: muitos indivíduos com deficiência heterozigota, assim como aqueles com níveis de proteína C normais a baixos em decorrência de outras causas, não desenvolvem trombose.10 No entanto, outros pacientes com deficiência heterozigota mostram uma tendência definida ao desenvolvimento de trombose venosa, apesar de apresentarem níveis de proteína C equivalentes a 40 a 50% do normal. Esta variabilidade fenotípica sugere a ocorrência de interações genéticas múltiplas e sustenta a hipótese de que a trombose clínica observada nestes pacientes pode resultar de uma combinação de deficiência de proteína C e uma ou mais mutações pró-trombóticas.11 A trombose venosa cerebral provavelmente responde pelos casos de infarto hemorrágico encefálico que ocorrem em adultos jovens com deficiência de proteína C.
A deficiência de proteína S também leva ao desenvolvimento de trombose venosa, inclusive de trombose na veia mesentérica. A gestação e o uso de anticoncepcionais orais diminuem os níveis de proteína S e isto, em alguns casos, pode explicar a tromboembolia observada em tais circunstâncias.12 A deficiência de proteína S adquirida também ocorre em pacientes com síndrome nefrótica, que perdem a proteína S na urina.13 Relatos de caso associaram a deficiência de proteína S à necrose cutânea induzida pela varfarina.14
Os ensaios funcionais e antigênicos para proteína C são atualmente disponibilizados na maioria dos laboratórios de coagulação. Os ensaios funcionais são preferíveis para fins diagnósticos. O diagnóstico da deficiência de proteína S é complicado pelo fato de a proteína S poder ser encontrada no plasma sob 2 formas distintas: uma forma livre, que funciona como cofator para a proteína C ativada (PCA); e uma forma ligada, em que a proteína S está complexada à proteína ligadora de C4 (C4BP), uma proteína da cascata do complemento. No complexo, a proteína S não é funcional como proteína anticoagulante. A concentração plasmática de proteína S geralmente excede a concentração plasmática de C4BP em 30 a 40%, e, por este motivo, cerca de 30 a 40% da proteína S é encontrada na forma livre, enquanto o restante está complexado à C4BP. Quando um paciente tem deficiência leve de proteína S, o resultado é uma deficiência seletiva da proteína S livre. Contudo, a deficiência mais severa resulta na deficiência de ambas formas, livre e ligada, de proteína S.15 Sendo assim, a quantificação do antígeno da proteína S livre, em vez do antígeno da proteína S total, é o ensaio de escolha para diagnóstico da deficiência de proteína S.16 Foram desenvolvidos ensaios funcionais para proteína S que, em geral, ainda não estão devidamente padronizados. Os ensaios de coagulação para proteínas C e S podem fornecer resultados falsamente baixos em pacientes com fator V de Leiden.17
A varfarina é o tratamento de escolha para prevenção da trombose, apesar de diminuir os níveis de proteína C ainda mais. Como a meia-vida da proteína C é de apenas 6 a 7 horas (bem mais curta do que a meia-vida da pró-trombina e do fator X), um período de hipercoagulabilidade intensificada segue-se ao início da terapia com varfarina em pacientes com deficiência de proteína C. A heparina deve ser administrada com a varfarina durante o início da anticoagulação, com uma sobreposição de cerca de 5 dias, até que uma relação normalizada internacional (INR) terapêutica seja alcançada por 2 dias consecutivos. A necrose cutânea induzida por varfarina é uma complicação rara da terapia anticoagulante, especialmente quando a terapia com varfarina é iniciada sem cobertura de heparina.
O fator V de Leiden é uma forma mutante do fator V (identificado pela 1ª vez por pesquisadores de Leiden, na Dinamarca) que, uma vez ativada, é relativamente resistente aos efeitos anticoagulantes da PCA. O defeito é transmitido como traço autossômico dominante. Cerca de 5% da população branca em geral é heterozigota para fator V de Leiden. O defeito está quase ausente em outros grupos étnicos.18 Todos os indivíduos com fator V de Leiden compartilham o mesmo haplótipo genético do fator V, indicando a existência de um efeito fundador. Estima-se que a mutação tenha surgido há cerca de 21.000 anos, após a migração da África e separação das raças humanas, consistente com sua predominância observada na população branca.19 Há evidências sugestivas de que o fator V de Leiden pode conferir um risco menor de sangramento severo após o parto e, desta forma, propiciar uma vantagem em termos de sobrevida.20 O fator V de Leiden é a condição de hipercoagulação hereditária mais comum. Sua prevalência em pacientes com trombofilia chega a 20 a 50%.21 Em um amplo estudo de coortes envolvendo pacientes não selecionados que haviam sofrido o 1º episódio de TVP sintomática, o fator V de Leiden foi encontrado em 16% dos pacientes.22 O risco de trombose em indivíduos heterozigotos para o fator V de Leiden é aproximadamente 4 a 8 vezes maior do que o risco apresentado pelos indivíduos normais. O risco relativo aumenta para mais de 30 vezes quando o fator V de Leiden é combinado ao uso de anticoncepcional oral, como resultado de um efeito sinergico. Em mulheres que desenvolvem trombose ao tomarem anticoncepcionais orais, a frequência do fator V de Leiden está em torno de 23%,23 embora o risco absoluto de trombose permaneça baixo.24 A avaliação de rotina para se detectar a mutação do fator V de Leiden antes do início do uso de anticoncepcionais orais não é custo-efetiva e, portanto, não é recomendada.25 O risco relativo de TEV em um indivíduo homozigoto para fator V de Leiden é cerca de 80 vezes maior do em um indivíduo normal.26 A associação do fator V de Leiden às deficiências de proteína C, proteína S ou AT foi descrita em algumas famílias.27-29 De forma geral, embora o fator V de Leiden seja altamente prevalente, constitui um fator de risco modesto de trombose.
Aproximadamente 10 a 15% dos casos associados com a resistência à PCA, definida por uma razão de sensibilidade à PCA (tempo de coagulação com tampão contendo PCA dividido pelo tempo de coagulação apenas com tampão), são atribuídos a outras mutações e defeitos.30,31 Condições como a elevação dos níveis de fator VIII, gestação e uso de anticoncepcional, bem como o anticoagulante lúpico, podem resultar em resistência à PCA.31 A resistência que não é causada pelo fator V de Leiden pode atuar como fator de risco de AVC32,33 e trombose venosa.34,35 O risco geral de trombose venosa por resistência à PCA é similar ou inferior ao risco associado ao fator V de Leiden.36
O fator V de Leiden (ou fator V R506Q) resulta da substituição de um nucleotídeo único que leva à substituição da arginina por glutamina na posição 506.36 A arginina 506 está localizada em um dos 2 sítios de clivagem principais do fator V ativado. O fator V de Leiden ativado expressa atividade pró-coagulante normal, contudo sua degradação pela PCA é cerca de 10 vezes mais lenta do que a degradação do fator V ativado normal (fator Va). Este retardo aumenta a geração de trombina.37 Além disso, o fator V (e não o fator Va) aliado à proteína S atua como cofator da PCA na inibição do complexo fator VIIIa/fator IXa, enquanto o fator V de Leiden atua precariamente como cofator de PCA, com consequente intensificação da geração de fator Xa38 [Figura 1].

Figura 1. A degradação do fator V de Leiden trombina-ativado por ação da proteína C ativada (PCA) é significativamente mais lenta do que a degradação do fator V ativado normal (fator Va). Em consequência, a produção de trombina é intensificada (esquerda). Evidências recentes sugerem que o fator V normal, aliado à proteína S, atua como cofator da PCA na inibição do fator VIIIa (direita). Esta função de cofator da PCA apresentada pelo fator V requer a clivagem do fator V pela PCA na arginina 506. Por este motivo, a função de cofator apresentada pelo fator V de Leiden é precária.
As manifestações clínicas do fator V de Leiden são similares àquelas associadas às deficiências de AT, proteína C e proteína S – principalmente a trombose venosa. Entretanto, a 1ª manifestação trombótica do fator V de Leiden muitas vezes ocorre mais tardiamente do que nas demais condições trombofílicas hereditárias. Cerca de 25% dos homens aparentemente sadios com mais de 60 anos de idade que sofreram um 1º episódio de trombose venosa apresentam fator V de Leiden.39 Uma revisão sistemática recente indica que o fator V de Leiden heterozigoto confere um risco levemente aumentado de TEV recorrente (risco geral = 1,56).40
O fator V de Leiden pode ser identificado de forma rápida e precisa por meio de testes de DNA simples. Estes testes permitem estabelecer o diagnóstico de pacientes sob terapia de anticoagulação com warfarina, bem como de pacientes que possuem anticorpos antifosfolípides coexistentes. Como o fator V de Leiden não é a única causa de resistência à PCA, pode ser conveniente perseguir o diagnóstico com um teste de resistência à PCA em casos seletos, quando o fator V de Leiden for excluído.
O tratamento do fator V de Leiden é similar ao tratamento das deficiências de AT, proteína C e proteína S. Os pacientes que passaram pelo 1º episódio de trombose venosa devem receber terapia de anticoagulação por 6 meses. Depois disso, estes pacientes devem receber terapia de anticoagulação profilática em situações que comprovadamente provoquem trombose. A anticoagulação prolongada deve ser considerada somente em casos de pacientes com trombose recorrente.41
As mulheres jovens que são comprovadamente portadoras do fator V de Leiden devem evitar o uso de anticoncepcionais orais, que aumentam o risco relativo de trombose (embora o risco permaneça baixo em termos de incidência absoluta). O tratamento ideal das portadoras durante a gestação ainda não foi estabelecido. A taxa de TEV é baixa, sendo de aproximadamente 2% na ausência de profilaxia contra trombose.24 O autor recomenda não instituir a profilaxia antitrombótica de forma rotineira durante a gestação. Contudo, eu considero a profilaxia pós-parto com duração de 6 semanas, especialmente quando a paciente apresenta história familiar significativa de trombose. A avaliação de rotina dos familiares das pacientes para fator V de Leiden não é custo-efetiva.25,42
Uma mutação G-A no nucleotídeo localizado na posição 20210 da região 3’ não traduzida do gene da protrombina está associada a uma incidência aumentada de trombose venosa. A prevalência da mutação em indivíduos saudáveis é de cerca de 2,3%. Assim como o fator V de Leiden, esta mutação é bastante rara em asiáticos e africanos. Diferente do fator V de Leiden, a mutação é mais comum em sul-europeus do que em norte-europeus.43 O risco relativo de trombose em indivíduos com esta mutação é de 2,8, e este valor é similar ao do risco relativo apresentado por indivíduos com fator V de Leiden.44 A mutação pode ser encontrada em até 18% dos pacientes com trombose e história familiar de trombose. A manifestação mais comum é a TVP em membro inferior. Estudos prospectivos falharam em demonstrar um risco aumentado de TVP recorrente em pacientes com esta mutação.40,45 Todavia, os portadores heterozigotos para fator V de Leiden e mutação da protrombina apresentam risco aumentado de trombose recorrente.46 A combinação do uso de anticoncepcionais orais e mutação no gene da protrombina está associada a uma incidência aumentada de trombose venosa cerebral em mulheres jovens.47
A homocisteína é um aminoácido altamente reativo que é encontrado normalmente no sangue em níveis de 5 a 15 mcmol/L. Na situação normal, a homocisteína é derivada da metionina por um processo de transmetilação e remetilada em metionina ou convertida em cisteína. O metabolismo da homocisteína requer betaína, cobalamina (vitamina B12), folato e piridoxina (vitamina B6). Níveis acentuadamente altos de homocisteína podem ser, de diversos modos, tóxicos para o endotélio vascular.48,49 A homocisteína pode inibir a expressão da trombomodulina e ativação da proteína C, bem como suprimir a expressão de heparan sulfato endotelial, e estes efeitos resultam em hipercoagulabilidade.50,51 A homocisteína também intensifica a ligação da lipoproteína (a) [Lp(a)] (uma lipoproteína aterogênica) à fibrina e isto pode fornecer uma conexão entre hiper-homocisteinemia, trombose e aterosclerose prematura (ver Lipoproteína (a) [Lp(a)], adiante].52 O dano vascular causado pelos níveis elevados de homocisteína acarreta trombose arterial e venosa e, talvez, aterosclerose acelerada.
A hiper-homocisteinemia pode ser dividida em 3 classes: severa (concentração plasmática de homocisteína > 100 mcmol/L); moderada (25 a 100 mcmol/L); ou leve (16 a 24 mcmol/L). A hiper-homocisteinemia severa em geral é causada por uma deficiência homozigota da enzima cistationina betassintase. Os indivíduos afetados apresentam retardo mental grave, lente ectópica, anormalidades esqueléticas e doença trombótica venosa e arterial severa de aparecimento precoce.53
A hiper-homocisteinemia leve ou moderada resulta de defeitos hereditários ou adquiridos envolvendo a via metabólica da homocisteína. A deficiência heterozigota de cistationina betassintase é bastante comum na população em geral, com uma frequência de 0,3 a 1,4%.53 Um defeito na via de remetilação geralmente é causado por uma variante mutante termolábil da enzima metilenotetraidrofolato redutase (MTHFR), cuja atividade corresponde a cerca de 50% do normal. A condição homozigota possui uma prevalência de 5% na população em geral.54 Entretanto, a forma homozigota da isoforma da enzima termolábil MTHFR não é clinicamente relevante em pacientes cuja dieta inclua folato em quantidade adequada.
As causas comuns de hiper-homocisteinemia adquirida são as deficiências dietéticas de cobalamina, folato ou piridoxina. Um estudo prospectivo constatou que a hiper-homocisteinemia leve é bastante comum em idosos, mesmo diante de concentrações séricas de vitaminas normais.55 A hiper-homocisteinemia adquirida também é comum em pacientes com doença renal em estágio terminal.
A hiper-homocisteinemia leve a moderada está associada à doença cerebrovascular, doença arterial coronariana e doença vascular periférica em indivíduos com menos de 55 anos de idade. Em idosos, a hiper-homocisteinemia está associada à estenose da artéria carótida.56,57 É encontrada em 10% dos pacientes que passaram pelo 1º episódio de TVP.58 Em um estudo prospectivo, foi encontrada uma relação graduada entre níveis plasmáticos elevados de homocisteína e mortalidade de pacientes com doença arterial coronariana.59 Entretanto, a relevância clínica da hiper-homocisteinemia leve foi questionada diante dos resultados negativos obtidos em múltiplas avaliações intervencionistas (ver adiante).
A suspeita de hiper-homocisteinemia severa deve ser considerada diante de pacientes com fenótipo característico (ver anteriormente). Deve-se suspeitar de hiper-homocisteinemia moderada em casos de doença trombótica venosa e arterial – incluindo a doença cerebrovascular, doença arterial periférica e TVP – especialmente em indivíduos jovens.
O diagnóstico de hiper-homocisteinemia geralmente é estabelecido por meio da quantificação dos níveis plasmáticos de homocisteína após o jejum de um dia para outro e relatado como homocisteína plasmática total (faixa normal: 5 a 15 mcmol/L). Os níveis plasmáticos de folato e vitamina B12 também devem ser medidos para excluir a hipótese de hiper-homocisteinemia causada por deficiência de folato ou de vitamina B12.
O uso diário de piridoxina oral (250 mg) e ácido fólico (5 mg) diminui os níveis elevados de homocisteína, trazendo-os para valores abaixo do normal, na maioria dos casos.60 Os pacientes com deficiência de vitamina B12 devem receber suplementos da vitamina. Entretanto, deve ser destacado que os suplementos combinados de ácido fólico e vitaminas B6 e B12 não diminuem comprovadamente o risco de eventos cardiovasculares significativos nem de TEV sintomática com doença vascular.61-63 Nos estudos clínicos que abordaram estes aspectos, a maioria dos pacientes apresentou níveis plasmáticos de homocisteína junto ao quartil superior ou 10º percentil superior, contudo um número relativamente pequeno de pacientes apresentou hiper-homocisteinemia moderada ou acentuada. Assim, ainda é preciso esclarecer se a correção da hiper-homocisteinemia neste subgrupo de pacientes trará algum benefício.
A Lp(a) é uma lipoproteína plasmática que consiste em uma partícula de lipoproteína de baixa densidade (LDL) ligada a uma glicoproteína do tipo plasminogênio, a apolipoproteína (a). As distribuições da Lp(a) são distorcidas na população em geral, sendo que os níveis de Lp(a) podem apresentar uma variação de até 1.000 vezes entre os indivíduos.64 O 95º percentil da concentração plasmática de Lp(a) foi estimado na faixa de 25 a 30 mg/dL. Os níveis de Lp(a) são parcialmente determinados pelos polimorfismos envolvendo o gene LPA codificador do componente apolipoproteína (a). O número de repetições KIV-2 plasminogênio-símile apresenta correlação inversa com os níveis plasmáticos de Lp(a).
Muitos estudos demonstraram que a Lp(a) constitui um fator de risco modesto independentemente de trombose arterial coronariana.65-68 Níveis altos de Lp(a) também parecem atuar como fator de risco moderado de AVC isquêmico em adultos jovens, embora isto seja sustentado por dados não tão fortes.69-71 Ainda é controverso se os níveis altos de Lp(a) representam um fator de risco de TEV.72,73
O mecanismo pelo qual a Lp(a) aumenta o risco de doença coronariana é pouco conhecido. Este mecanismo pode suprimir a geração de plasmina na superfície da célula endotelial,74 inibir a expressão de fator tecidual75 ou afetar o transporte de fosfolipídios oxidados pró-inflamatórios.76 A quantificação da Lp(a) deve ser considerada em pacientes jovens com trombose arterial. Níveis altos de colesterol LDL parecem deflagrar ou exacerbar os fatores de risco associados à Lp(a) elevada. Desta forma, a dieta, o exercício e as abordagens farmacológicas padrão devem ser empregadas no tratamento de pacientes com níveis altos de colesterol LDL.77 Doses altas de niacina (2 a 4 g/dia, via oral [VO]) promovem uma diminuição de 30 a 40% nos níveis elevados de Lp(a).78,79 As doses altas de niacina frequentemente estão associadas ao rubor facial e a cefaleias. Estes efeitos colaterais desagradáveis podem ser melhorados com a instituição de um curso de niacina em doses baixas (p. ex., 300 mg/dia), que subsequentemente são aumentadas ao longo do tempo, ou com o uso de niacina de liberação estendida. A função hepática deve ser checada periodicamente.
Cerca de 300 fibrinogênios anormais (desfibrinogênios ou desfibrinogenemia) foram descritos, e aproximadamente 85 defeitos estruturais foram identificados na desfibrinogenemia. Tais defeitos são mais comumente caracterizados como defeitos funcionais de liberação de fibrinopeptídeo A e polimerização de fibrina e, menos comumente, como defeitos de ligação e ativação de plasminogênio. Cerca de metade das mutações do fibrinogênio não está associada a nenhum sintoma clínico. O sangramento leve ou a trombose recorrente ocorrem aproximadamente com a mesma frequência nas mutações remanescentes.80 A desfibrinogenemia adquirida pode agravar o carcinoma hepatocelular ou a doença hepática crônica. É comum a avaliação em laboratório geral mostrar a existência de uma discrepância entre os níveis antigênico e funcional do fibrinogênio, uma vez que a maioria dos pacientes com desfibrinogênios tem função de coagulação subótima, com prolongamento do tempo de trombina (TT) e do tempo de reptilase (TR). Os fibrinogênios também podem formar coágulos de fibrina resistentes à lise e, deste modo, ser responsáveis pelo desenvolvimento de trombose, a qual, por sua vez, pode coexistir com uma diátese hemorrágica em alguns pacientes.81 A identificação precisa do defeito estrutural requer esforços significativos conduzidos em laboratório de pesquisa. O tratamento da trombose recorrente causada por desfibrinogenemia é o mesmo tratamento fornecido aos pacientes com trombofilia.
Os plaminogênios anormais (displasminogênios ou displasminogenemia), cuja ativação em plasmina é defeituosa, raramente estão associados à trombose. Os pacientes que apresentam este distúrbio têm baixos níveis plasmáticos de plasminogênio demonstrados por ensaios funcionais.82 Níveis aumentados de inibidor de ativador de plasminogênio-1 (PAI-1) e uma atividade fibrinolítica plasmática diminuída foram descritos em pacientes com pré-eclâmpsia.83 O comprometimento da atividade fibrinolítica adquirido pode estar associado à trombose pós-operatória.84 Entretanto, estudos adicionais são necessários para estabelecer o papel da fibrinólise anormal na trombose clínica recorrente.85 Os ensaios antigênicos para ativador de plasminogênio tecidual (t-PA) e PAI-1 são disponibilizados por alguns laboratórios comerciais, mas há também ensaios funcionais específicos que são disponibilizados somente em laboratórios de pesquisa.
Níveis plasmáticos elevados de fator VIII, acima do 90º percentil do normal, estão associados a um aumento aproximado de 5 vezes do risco de TVP.86,87 Em termos de função, a condição está associada a um fenótipo de resistência à PCA e é reconhecida como fator de risco moderado de TEV. Níveis plasmáticos elevados de fibrinogênio e fator VII foram associados à doença arterial coronariana.88,89 No entanto, ainda é necessário realizar estudos adicionais para estabelecer a utilidade clínica da quantificação dos níveis de fibrinogênio e fator VII em pacientes com trombose.
A SAF é causada por autoanticorpos dirigidos contra proteínas associadas a fosfolipídios de carga negativa. Os termos “antifosfolipídio” e “anticardiolipina” são usados como sinônimos. Os anticorpos antifosfolipídios incluem também o anticoagulante lúpico, que é um inibidor identificado pela 1ª vez em pacientes com lúpus eritematoso sistêmico. Como está presente em muitos pacientes sem lúpus, este inibidor por vezes é denominado anticoagulante do tipo lúpico.
A SAF é secundária ao lúpus eritematoso sistêmico e, menos comumente, à artrite reumatoide, à arterite temporal e a outros distúrbios do tecido conectivo. Também está associada à infecção pelo HIV-1 e à hepatite C, doenças linfoproliferativas e certos fármacos (p. ex., fenotiazina e procainamida). Quando nenhum fator de risco pode ser identificado, a síndrome é considerada primária. Em um amplo estudo de coorte envolvendo 1.000 pacientes com SAF, 53% dos pacientes foram classificados como tendo SAF primária e 47% tinham SAF secundária.90
As 2 proteínas-alvo mais comuns dos anticorpos antifosfolípides parecem ser a beta-2-glicoproteína I (beta-2-GPI) e a protrombina. A beta-2-GPI é uma proteína plasmática que se liga com alta afinidade aos fosfolipídios aniônicos. Apresenta uma fraca função anticoagulante in vitro. A beta-2-GPI pode induzir a cardiolipina, em sua forma bilaminar habitual, a assumir um formato hexagonal que é altamente imunogênico.91 O ensaio imunossorvente ligado à enzima (ELISA) para anticorpos anticardiolipina geralmente detecta os anticorpos dirigidos contra o complexo cardiolipina/beta-2-GPI. Foram purificados anticorpos anticoagulante lúpico que reagem de modo específico com a protrombina, mas não reagem com a trombina. Estes anticorpos purificados podem intensificar a ligação da protrombina a uma superfície de células endoteliais cultivadas.92 Outros alvos proteína-fosfolípide também podem estar envolvidos. Os anticorpos antifosfatidiletanolamina são encontrados em muitos pacientes com SAF, e alguns destes anticorpos inibem a função da PCA.93 Foram encontrados anticorpos dirigidos contra a heparina e heparan sulfato, que inibem a neutralização heparina-dependente da trombina pela AT.94 Em um modelo murino de perda de gestação induzida por anticorpo antifosfolipídio, os anticorpos antifosfolípides humanos ligaram-se à placenta, onde ativaram o complemento e levaram, então, à geração de C5a. O Ca5a atrai e ativa neutrófilos, contribuindo para a lesão do trofoblasto e resultante morte fetal.95,96 Com base nesta heterogeneidade de anticorpos antifosfolípides, parece provável que múltiplos mecanismos estejam envolvidos na patogênese da trombose, nesta síndrome.
Os episódios trombóticos ocorrem em cerca de 30% dos pacientes com anticorpos antifosfolípides (incidência geral de 2,5 episódios por 100 pacientes-anos).97 Os aspectos clínicos da trombose vascular são proteicos e manifestam-se como trombose arterial e/ou venosa, podendo envolver quaisquer tecidos ou órgãos [Tabela 5]. No estudo de coorte mencionado, cerca de 37% dos pacientes apresentaram trombose venosa, 27% tinham trombose arterial, 15% apresentavam trombose venosa e arterial, e 12% tiveram apenas perda fetal.90 Manifestações clínicas como livedo reticular (um padrão com manchas violáceas persistentes na pele do tronco, braços ou pernas), trombocitopenia leve a moderada (contagem de plaquetas < 100.000/mcL), regurgitação de valva mitral moderada a severa, e insuficiência renal associada à microangiopatia trombótica (na ausência de púrpura trombocitopênica trombótica, síndrome urêmico-hemolítica, hipertensão maligna, vasculite ou outras causas de doença renal crônica) também são encontradas na SAF. Entretanto, estes aspectos não são suficientemente específicos para serem incluídos entre os critérios de classificação.98
Tabela 5. Critérios clínicos e laboratoriais propostos para a SAF98
|
A SAF está presente se pelo menos 1 dos seguintes critérios clínicos e laboratoriais for atendido: |
|
Morbidade gestacional (qualquer 1 das seguintes): |
|
Mais de uma morte fetal inexplicável (após a 10ª semana de gestação) |
|
Parto antes da 34ª semana, acompanhado de hipertensão severa induzida pela gestação |
|
Pelo menos 3 perdas de gestação (antes da 10ª semana de gestação) |
|
Trombose vascular: pelo menos 1 episódio clínico de trombose arterial, venosa ou em pequenos vasos, em qualquer tecido ou órgão. A trombose deve ser confirmada com base em critérios objetivos e validados (por meio de exames de imagem ou histopatologia apropriados). Para a confirmação histopatológica, a trombose deve estar presente na ausência de inflamação significativa na parede vascular |
|
Aspectos clínicos |
|
Venosos (tromboflebite superficial; TVP; embolia pulmonar; trombose venal cerebral ou retinal; trombose venosa renal, esplâncnica e mesentérica) |
|
Arteriais (infarto cerebral isquêmico; isquemia cerebral transiente; amaurose fugaz; enxaqueca; trombose arterial carotídea e vertebrobasilar; síndrome do arco aórtico; embolia e trombose arterial periférica; trombose arterial mesentérica e renal; livedo reticular) |
|
Presença do anticoagulante lúpico em pelo menos 2 ocasiões, com um intervalo mínimo de 12 semanas |
|
Aspectos laboratoriais |
|
TTPA ativado; TVVR diluído; tempo de coagulação do caolin; teste de inibição da tromboplastina tecidual |
|
Presença de anticorpos anticardiolipina (qualquer um dos seguintes) em pelo menos 2 ocasiões, com um intervalo mínimo de 12 semanas: |
|
Anticorpos IgG anticardiolipina (> 40 GPL) |
|
Anticorpos IgM anticardiolipina (> 40 MPL) |
|
Anticorpos anti-beta-2-GPI (IgG e/ou IgM) em títulos > 99º percentil, presentes em pelo menos 2 ocasiões, com um intervalo mínimo de 12 semanas |
TTPA = tempo de tromboplastina parcial; GPI = glicoproteína; GPL = unidades de IgG antifosfolipídio; MPL = unidades de IgM antifosfolipídio; SAF = síndrome antifosfolipídica; TVP = trombose venosa profunda; TVVR = tempo de coagulação do veneno da víbora de Russell.
O diagnóstico de SAF deve ser considerado diante de qualquer paciente que apresente trombose venosa ou arterial idiopática, ou no caso de mulheres com história de abortos recorrentes. O diagnóstico é confirmado pela detecção de anticorpos anticardiolipina por ELISA (ver anteriormente) ou pela detecção do anticoagulante lúpico em ensaios de coagulação. Os critérios de classificação baseados em uma declaração de consenso internacional são listados na Tabela 5.98 Os critérios gerais para o diagnóstico do anticoagulante lúpico são: (1) prolongamento de pelo menos um ensaio de coagulação fosfolípide-dependente; (2) comprovação, fornecida por uma combinação de exames, de que este prolongamento é causado por um inibidor, e não por uma deficiência de fator de coagulação; e (3) confirmação de que o inibidor é dependente de fosfolipídio. Os ensaios de coagulação comumente usados são o TTPA e o tempo de coagulação do veneno da víbora de Russell (TVVR) diluído. Os reagentes do ensaio de TTPA apresentam sensibilidade variável ao anticoagulante lúpico e são influenciados pelas concentrações de alguns fatores plasmáticos (p. ex., fator VIII). Desta forma, um reagente de TTPA insensível ao anticoagulante lúpico deve ser usado no teste. O teste de TVVR diluído é bem mais sensível do que o TTPA, embora seja um teste manual e não tão bem padronizado. Outros testes, como o tempo de coagulação do caolin e o teste de inibição de tromboplastina tecidual, são úteis quando estão disponíveis. A presença de um inibidor requer um estudo misto para que seja demonstrada a ausência de correção com plasma normal. A correção do prolongamento pela adição de fosfolipídio em forma de lisados de plaqueta ou fosfolipídio de fase hexagonal confirma o diagnóstico. Os ensaios de fatores de coagulação podem ser usados em casos duvidosos. O anticoagulante lúpico causa deficiência funcional de vários fatores de coagulação dependentes de fosfolipídio, e não apenas de um único fator em particular.
Os anticorpos anticardiolipina são relatados como sendo IgG (em unidades de IgG antifosfolípide [GPL]) e IgM (em unidades de IgM antifosfolípide [MPL]). A prevalência de níveis elevados de anticorpos IgG e IgM anticardiolipina em populações normais é de aproximadamente 5%. Com a repetição dos testes, observa-se uma prevalência inferior a 2%.84 Na declaração de consenso de classificação internacional revisada, são considerados somente títulos altos de anticorpos IgG e IgM anticardiolipina (> 40 unidades de GPL ou MPL).98-100 A importância dos títulos baixos de anticorpos IgG, anticorpos IgM isolados e anticorpos IgA ainda não foi definida.101,102 Devem ser solicitados ensaios funcionais e antigênicos para fins de avaliação do paciente, uma vez que estes testes não se sobrepõem completamente. Em um estudo sobre a SAF, 88% dos pacientes apresentavam anticorpos anticardiolipina (IgG, IgM, ou ambos) e 54% dos pacientes tinham anticoagulante lúpico. O anticoagulante lúpico foi tipicamente encontrado associado aos anticorpos anticardiolipina, mas também foi encontrado isolado em cerca de 12% dos pacientes.89 Algumas infecções e exposições farmacológicas podem levar ao aparecimento transiente de anticorpos antifosfolípides que, por sua vez, desaparecem após a resolução da infecção ou descontinuação do fármaco [Tabela 6]. Portanto, os exames laboratoriais devem ser repetidos pelo menos 1 vez, com um intervalo de 12 semanas, para confirmar o diagnóstico. Contudo, cerca de 20% dos pacientes com títulos baixos de anticorpos IgG anticardiolipina apresentam títulos mais altos nas repetições dos testes. Os casos de pacientes que sofrem um episódio de trombose pela 1ª vez ou sofrem episódios recorrentes de trombose também justificam a repetição dos testes.101
Tabela 6. Classificação dos anticorpos antifosfolípides162
|
Causas autoimunes |
|
Primárias (não atendem aos critérios para lúpus eritematoso sistêmico) |
|
Secundárias (atendem aos critérios para lúpus eritematoso sistêmico ou outros distúrbios do tecido conectivo) |
|
Induzidas por fármaco (p. ex., fenotiazinas, quinidina, quinina, penicilinas sintéticas, hidralazina) |
|
Causas aloimunes |
|
Infecções (virais, bacterianas, fúngicas) |
|
Malignidades (p. ex., leucemia de células pilosas, doença linfoproliferativa) |
As atuais recomendações terapêuticas para SAF baseiam-se principalmente em estudos observacionais, que sustentam a existência de uma associação entre anticorpos antifosfolípides e trombose, em particular de trombose recorrente.97,100-102 No tratamento da TVP aguda em pacientes com SAF, o monitoramento do efeito da heparina não fracionada pode ser problemático porque o anticoagulante lúpico prolonga o aPTT. O uso de HBPM contorna este problema, pois a HBPM dispensa a titulação da dose e o monitoramento. O paciente deve ser tratado com HBPM e varfarina do modo habitual, com sobreposição de no mínimo 5 dias antes da descontinuação da HBPM.
Uma análise retrospectiva mostra que os pacientes com SAF e história de trombose apresentam alta taxa de recorrência de trombose (na faixa de 50% em um período de 5 anos), quando não são tratados com varfarina por tempo prolongado.103,104 A localização do 1º episódio trombótico (isto é, arterial ou venosa) tende a prever o local em que a recidiva ocorrerá.104 A terapia intensiva com warfarina, para obtenção de uma INR de 3 a 3,5, foi defendida para estes pacientes. Contudo, a base desta recomendação era principalmente uma análise retrospectiva.87 Mais recentemente, 2 estudos clínicos prospectivos demonstraram que a maioria destes pacientes pode ser adequadamente tratada com os níveis convencionais de anticoagulação (isto é, para obter uma INR de 2 a 3).105 Contudo, é evidente que há pacientes com SAF que sofrem de trombose recorrente com o uso da anticoagulação convencional e, portanto, necessitam de um nível mais intensivo de tratamento. O melhor tratamento para o AVC isquêmico associado à SAF ainda não está totalmente estabelecido, pois nestes estudos prospectivos o número total de pacientes com trombose arterial era pequeno. As atuais diretrizes de consenso recomendam o uso de aspirina (81 mg/dia) como 1ª opção.106 Alguns especialistas recomendam clopidogrel (75 mg/dia) ou um curso intensivo de varfarina (INR = 3 a 4).107,108
A duração ideal da terapia de anticoagulação oral para SAF não está totalmente definida. A trombose venosa recorrente ou o AVC isquêmico geralmente justificam o uso prolongado da warfarina. No caso de um paciente que teve o 1º episódio de TVP e apresenta anticorpos antifosfolípides, a terapia com warfarina é indicada por um período mínimo de 6 meses e, talvez, por toda a vida, devido ao alto risco de recorrência.109 A severidade do episódio trombótico específico, a coexistência de quaisquer fatores de risco trombótico reversíveis e os riscos associados à terapia de anticoagulação oral prolongada também devem ser considerados. Foi relatado que o anticoagulante lúpico ocasionalmente pode aumentar o tempo de protrombina (PT) e, em consequência, a IRN, dificultando o monitoramento da terapia com warfarina,110 ainda que isto pareça ser incomum. Quando o TP e a INR aumentam, é útil usar uma tromboplastina reagente insensível ao anticoagulante lúpico.
A anticoagulação é desnecessária em casos de pacientes assintomáticos com anticorpos anticardiolipina ou anticoagulante lúpico e sem história de trombose.
Vários estudos prospectivos confirmaram a existência de uma associação entre abortos recorrentes e presença de anticorpos antifosfolípides. É provável que os anticorpos causem perda da gestação por promoverem trombose placentária.111 Os anticorpos antifosfolípides devem ser quantificados em pacientes com história de perda inexplicável da gestão no 2º ou 3º trimestre; morte fetal; pré-eclâmpsia severa de aparecimento precoce; e retardo do desenvolvimento intrauterino.112 Em contraste, os anticorpos antifosfolípides não estão associados à perda esporádica da gestação em fase inicial,113 que frequentemente resulta de anormalidades genéticas no feto. A relação existente entre os anticorpos antifosfolípides e a infertilidade atualmente é incerta.
O tratamento de gestantes com SAF é difícil, porque a síndrome está associada à trombose e ao risco aumentado de sangramento com a terapia antitrombótica. Uma revisão sistemática mostrou que a terapia combinada à base de aspirina e heparina não fracionada diminui as perdas de gestação em 54%, enquanto a prednisona com aspirina é ineficaz e pode aumentar a prematuridade.114 Subsequentemente, foram realizados 2 estudos menores que demonstraram eficácia equivalente da heparina não fracionada e HBPM neste contexto.115,116 O tratamento deve ser iniciado assim que a gestação for confirmada. A HBPM é preferível à heparina não fracionada para uso prolongado, pois pode ser administrada 1 a 2 vezes/dia e consegue diminuir o risco de osteopenia e trombocitopenia induzida por heparina (TIH) (ver adiante). O autor recomenda administrar aspirina (81 mg/dia) com uma dose profilática de HBPM (40 mg/dia de enoxaparina), caso a paciente não tenha recebido anticoagulação crônica antes de engravidar. Se a paciente estiver sob tratamento crônico com warfarina, esta medicação é suspendida e substituída por uma dose terapêutica de HBPM e aspirina. Alguns especialistas sugerem que, para pacientes com história de abortos precoces (< 10 semanas) recorrentes, o curso de HBPM pode ser suspendido após 20 a 24 semanas.108 A administração de imunoglobulina intravenosa (IgIV) é ineficaz no tratamento da SAF e na gestação.117,118
A TIH é uma reação farmacológica mediada por anticorpo relativamente comum, que ocorre em cerca de 1% dos pacientes tratados com heparina suína e em 5% dos pacientes tratados com heparina bovina.119 A incidência da TIH é bem menor em pacientes tratados com HBPM. Em um subgrupo de pacientes, ocorre a evolução da TIH para um distúrbio potencialmente fatal, caracterizado por trombose venosa e arterial. Notavelmente, tanto a frequência de formação de anticorpos na TIH como as manifestações clínicas da TIH variam de maneira considerável em diferentes populações de pacientes. A incidência da formação de anticorpos na TIH é bem maior após a cirurgia cardíaca do que após a cirurgia ortopédica (50% vs. 15%). Entretanto, a incidência de TIH pós-operatória clinicamente significativa parece ser menor em pacientes submetidos à cirurgia cardíaca do que em pacientes ortopédicos, entre os quais a incidência é de 5%.120,121
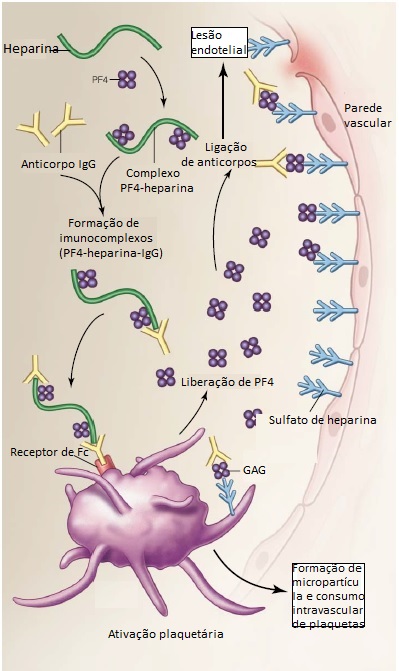
A patogênese da TIH é atribuível à presença de anticorpos IgG que reconhecem um complexo constituído por heparina e fator plaquetário 4 (PF4) [Figura 2].122 O PF4 é uma proteína catiônica que existe normalmente em forma de tetrâmero e é encontrada nos grânulos das plaquetas. Quando liberado dos grânulos, o PF4 liga-se com alta afinidade à molécula negativamente carregada de heparina. O anticorpo IgG liga-se ao complexo PF4-heparina, formando um imunocomplexo. O tamanho relativo, quantidade e estabilidade dos complexos PF4-heparina influenciam a imunogenicidade, sendo que a heparina é a molécula mais eficiente em termos de formação de complexos multimoleculares estáveis ultragrandes com os tetrâmeros de PF4. E HBPM é significativamente menos eficiente, e o fondaparinux tem eficiência ainda menor.123 Isto pode explicar a propensão substancialmente maior ao desenvolvimento de TIH associada à heparina não fracionada. Os imunocomplexos grandes, então, depositam-se nas membranas das plaquetas, onde fazem ligação cruzada com receptores Fc-gama-RIIa e deflagram a ativação e agregação plaquetária. As plaquetas ativadas liberam mais PF4 e intensificam ainda mais a formação de complexos PF4-heparina. Há também evidências de que o PF4 pode se ligar às glicosaminoglicanas (GAG) existentes na superfície das plaquetas e reagir com os anticorpos da TIH.124 A trombocitopenia resulta principalmente da ativação plaquetária intravascular, e não da depuração pelo sistema reticular endotelial.125 As complicações trombóticas que ocorrem na TIH são causadas pela ativação das plaquetas por imunocomplexos, com consequente formação de micropartículas plaquetárias e intensificação da geração de trombina.126 O PF4 também se liga às GAG sulfatadas heparina-símile (p. ex., heparan sulfato), na superfície da célula endotelial. Evidências in vitro indicam que o anticorpo encontrado na TIH consegue se ligar às células endoteliais. Estas células, então, são ativadas e promovem trombose.127 Considerando que apenas um subgrupo de pacientes formadores de anticorpos de TIH desenvolve TIH clínica,121 há fatores adicionais que influenciam o desenvolvimento da trombocitopenia e subsequente trombose. Isto depende em parte dos níveis de PF4 plaquetária total. Os pacientes que apresentam níveis baixos ou normais de PF4 (presente na maioria dos pacientes) exibem níveis relativamente baixos de expressão de PF4 na superfície plaquetária em seguida à ativação das plaquetas (após a lesão vascular). Quando estes pacientes são heparinizados, os complexos PF4-heparina são removidos da superfície das plaquetas, e estas se tornam menos sujeitas a uma nova ativação, caso haja desenvolvimento de anticorpos de TIH. Contudo, em um pequeno subgrupo de pacientes com elevados níveis totais de PF4 plaquetário, observa-se uma alta expressão de PF4 na superfície das plaquetas, e os grandes complexos PF4-heparina ficam retidos na superfície plaquetária após a heparinização. Estes pacientes, portanto, apresentam alto risco de desenvolvimento de TIH clínica.124 Os fatores de risco trombótico concomitantes, que contribuem para a extensão da ativação plaquetária e expressão de PF4 de superfície, também atuam neste processo.
Figura 2. Na trombocitopenia induzida por heparina (TIH), os anticorpos IgG reconhecem os complexos de fator plaquetário 4 (PF4)-heparina. Os imunocomplexos PF4-heparina-IgG resultantes ligam-se aos receptores Fc existentes nas plaquetas circulantes. O PF4 também pode ligar-se diretamente às glicosaminoglicanas (GAG) da superfície plaquetária e reagir com o anticorpo patológico da TIH. Qualquer um destes mecanismos resulta na ativação das plaquetas com liberação do PF4 contido nos alfagrânulos das plaquetas, estabelecendo um ciclo de ativação e formação de micropartículas plaquetárias pró-trombóticas. O consumo de plaquetas intravascular e, em menor extensão, a remoção das plaquetas cobertas com imunocomplexos pelo sistema reticuloendotelial acarreta trombocitopenia. O PF4 também se liga à GAG (p. ex., heparan sulfato) existente na superfície das células endoteliais, causando lesão imunomediada, trombose e coagulação intravascular disseminada (CID).
É interessante o fato de que a TIH não exibe as características típicas de uma resposta imune primária (isto é, uma resposta inicial de IgM seguida de uma resposta de IgG mais tardia) nem de uma resposta imune secundária (com persistência de uma resposta forte e duradoura de IgG).128 A condição é consistente com uma resposta de célula B não dependente de célula T e sugere que a resposta imune inicial é dirigida contra o PF4 complexado a um componente heparina-símile endógeno ou exógeno.
A TIH tipicamente se desenvolve decorridos 5 a 10 dias do início da terapia com heparina. Entretanto, em pacientes tratados com heparina nos últimos 100 dias e que estejam sendo novamente tratados, o aparecimento da condição pode ser rápido – dentro de algumas horas após a administração de heparina.129 Contudo, é possível que a TIH somente se manifeste passados no mínimo 19 dias da suspensão da terapia com heparina.130 Este retardo do aparecimento da TIH parece estar associado aos anticorpos IgG, que ativam as plaquetas de modo independente da heparina, mimetizando uma doença autoimune.128
A TIH em geral é definida por uma contagem de plaquetas abaixo de 150 x 109/L ou por uma queda da contagem de plaquetas em mais de 50% em relação à contagem plaquetária basal, no contexto clínico apropriado. A trombocitopenia severa, com contagens de plaquetas abaixo de 20 x 109/L, ocorre em menos de 10% dos pacientes. Em 10 a 15% dos pacientes, apesar da queda de 50% em relação aos níveis de pico, o nadir da contagem de plaquetas está acima de 150 x 109/L.121
No passado, o risco de trombose associada à TIH era considerado mínimo. Entretanto, atualmente se admite que a trombose ocorre em cerca de 1/3 a 1/2 dos pacientes com TIH, sendo que a trombose venosa é mais frequente do que a trombose arterial.125 A trombose pode ocorrer com qualquer contagem plaquetária, inclusive diante de contagens muito baixas.
A TVP, com ou sem embolia pulmonar, é o evento que mais comumente leva ao diagnóstico de TIH. O distúrbio pode ser ainda mais complicado pelo desenvolvimento de gangrena em um membro, sobretudo no contexto do tratamento com varfarina sem cobertura anticoagulante alternativa (ver adiante). A trombose venosa cerebral e a necrose hemorrágica suprarrenal são complicações incomuns da TIH, em que o diagnóstico antecipado e o tratamento urgente podem salvar vidas. A trombose arterial pode manifestar-se como isquemia em um membro, AVC, infarto do miocárdio ou, menos comumente, trombose mesentérica e trombose arterial renal. Alguns pacientes apresentam achados laboratoriais que sustentam um diagnóstico de CID. Pode haver desenvolvimento de lesões cutâneas induzidas por heparina nos sítios de injeção, que variam de pápulas eritematosas dolorosas a uma extensiva necrose dérmica.131
Diferente do que ocorre com os anticorpos induzidos pela quinina, quinidina ou sulfonamidas, que podem persistir por vários anos, os anticorpos induzidos pela heparina parecem ser bastante transientes. Os níveis destes anticorpos diminuem e tornam-se indetectáveis em média em 50 a 85 dias, dependendo do ensaio realizado.129
O diagnóstico da TIH é sustentado pelo achado de agregação plaquetária induzida por heparina em presença do soro do paciente. Contudo, a sensibilidade deste teste pode ser de apenas 50%.132 Em alguns casos, o soro do paciente pode causar agregação espontânea das plaquetas do doador na ausência de heparina, em decorrência da presença de imunocomplexos não relacionados à TIH ou de anticorpos anti-HLA preexistentes, que impossibilita a interpretação correta do resultado do teste. A liberação de serotonina plaquetária induzida por heparina empregando plaquetas lavadas tem alta sensibilidade e especificidade para TIH, mas é disponibilizada somente em alguns laboratórios especializados. Os ELISA para detecção de anticorpos reativos com o complexo PF4-heparina transformaram-se no teste mais comumente usado para TIH. Estes ELISA apresentam uma sensibilidade maior do que a sensibilidade do ensaio de agregação plaquetária e podem ser mais facilmente realizados em um laboratório clínico diagnóstico geral. No entanto, os resultados falso-positivos (isto é, testes que resultam positivos na ausência de TIH ou de trombocitopenia) ocorrem em 10 a 15% dos pacientes médicos e em mais de 20% dos pacientes tratados com heparina para procedimentos cirúrgicos na vasculatura periférica. Os anticorpos anti-PF4-heparina da classe IgG são os mais importantes, enquanto os anticorpos IgM e IgA exercem um papel apenas minoritário.133 A magnitude da resposta de anticorpos também é clinicamente significativa, com a TIH sendo mais provável diante de um resultado de ELISA fortemente positivo (densidade óptica [DO] > 1).134,139 Uma taxa de soroconversão de até 50% foi descrita para pacientes submetidos à cirurgia de desvio cardiopulmonar, limitando sua utilidade nesta situação.136 Como a TIH pode ser agravada por problemas trombóticos sérios, seu diagnóstico deve ser baseado essencialmente em achados clínicos apropriados, e o tratamento deve ser iniciado durante a espera pela confirmação laboratorial.
O tratamento da TIH consiste em suspender a heparina imediatamente e iniciar uma terapia alternativa de anticoagulação. É importante descontinuar o uso de todos os tipos de heparina: existem relatos pouco confiáveis de TIH causada por traços de heparina usados nos jatos de heparina de linhas intravasculares. Mesmo que o paciente tenha apenas uma trombocitopenia leve, sem evidências de trombose, é recomendável suspender o uso de heparina e tratar a condição hipercoagulável subjacente com um anticoagulante alternativo. Esta abordagem agressiva é sustentada por um estudo de coorte retrospectivo, em que houve desenvolvimento de trombose em um período de 30 dias após a suspensão do curso de heparina em metade dos pacientes que inicialmente não apresentavam sintomas clínicos da TIH. 119
A TIH pode desenvolver-se em pacientes que estavam recebendo heparina para tratamento de uma trombose preexistente e agora estejam em processo de troca de medicação para varfarina. Nesta situação, mesmo que o paciente esteja quase alcançando ou já tenha alcançado uma INR terapêutica com a warfarina, é prudente descontinuar o curso deste fármaco e instituir um tratamento agressivo com um anticoagulante alternativo, como um inibidor de trombina direto. Devido à preocupação com a redução inapropriada da dose do inibidor de trombina direto em consequência de um prolongamento do TTPA associado à warfarina, alguns especialistas recomendam a administração de vitamina K para reverter totalmente o efeito da varfarina nesta situação.137
Entre os agentes anticoagulantes alternativos, estão a lepirudina, argatrobana e fondaparinux. Embora seja bem menos imunogênica do que a heparina não fracionada na promoção de TIH,126 a HBPM não pode ser usada com segurança como substituto nos casos em que o paciente desenvolve TIH causada por heparina não fracionada. A HBPM e a heparina não fracionada apresentam extensiva reatividade cruzada (> 90%), em termos de reconhecimento de anticorpos. A HBPM não é uma escolha apropriada para pacientes com TIH.
A hirudina, uma proteína constituída por 65 aminoácidos originalmente extraída da glândula salivar da sanguessuga de uso medicinal (Hirudo medicinalis), é um potente inibidor de trombina direto. A hirudina liga-se diretamente ao sítio ativo da trombina, de modo independente da AT. Além disso, a hirudina não é neutralizada pelo PF4 e exerce uma função anticoagulante que é monitorada por TTPA.
A lepirudina é uma forma recombinante de hirudina que foi aprovada para uso no tratamento da TIH. Em estudos clínicos prospectivos, o uso da lepirudina diminuiu as complicações trombóticas sérias em cerca de 20% (em comparação à taxa aproximada de 40% observada entre os indivíduos-controle, historicamente).138,139 A lepirudina foi administrada como bolo intravenoso (0,4 mg/kg), seguido da infusão contínua a 0,15 mg/kg/h durante 2 a 10 dias, conforme a indicação. A dose foi ajustada para manter um TTPA-alvo equivalente a 1,5 a 5 vezes o normal. Os pacientes apresentaram sangramento mínimo (a partir dos sítios de punção, epistaxe e hematúria), mas não houve sangramento intracraniano. Notavelmente, 40% dos pacientes desenvolveram anticorpos anti-hirudina. Estes anticorpos eram não só neutralizantes, como, na verdade, também intensificavam a potência do fármaco, talvez por retardarem sua depuração – outro motivo para se monitorarem os níveis de TTPA. A julgar pelos estudos de intervenção cardiológica, o risco de sangramento aumentaria substancialmente com o uso concomitante dos agentes trombolíticos. Desta forma, é desaconselhável usar combinações contendo estes agentes. Não existe um antídoto efetivo para a lepirudina. Sua meia-vida aproximada é de 1,3 hora e a depuração é feita pelos rins. Sendo assim, para os pacientes com insuficiência renal, a dose de lepirudina precisa ser ajustada com cuidado, com base nos valores de depuração da creatinina.
A argatrobana, um inibidor de trombina direto sintético, é aprovada para uso na profilaxia ou tratamento da trombose em pacientes com TIH. É administrada por infusão intravenosa contínua de 2 mg/kg/min, para manter um TTPA equivalente a 1,5 a 3 vezes o nível basal (sem exceder 100 segundos ou 10 mg/kg/min). Em um estudo, a argatrobana diminuiu em cerca de 50% a ocorrência das complicações trombóticas graves associadas à TIH, em comparação ao observado nos controles , historicamente, com a maior taxa de sangramento em torno de 7%.140 A meia-vida da argatrobana é de apenas 40 a 50 minutos. Em contraste com a lepirudina, a argatrobana é depurada pelo fígado e, portanto, pode ser administrada com maior facilidade em casos de pacientes com insuficiência renal. Assim como a lepirudina, a argatrobana não possui nenhum antídoto específico, e as complicações hemorrágicas precisam ser assistidas atentamente.
Uma 3ª opção de anticoagulante é o fondaparinux, um pentassacarídeo sintético que ativa a AT e inibe a trombina. O fondaparinux foi aprovado para uso na profilaxia contra a TVP associada a cirurgias ortopédicas. Por seu pequeno tamanho e carga negativa reduzida, o fondaparinux não forma um complexo grande com o PF4 e, assim, não reage com os anticorpos dirigidos contra o complexo PF4-heparina encontrados na TIH. Há relatos de caso descrevendo o uso bem-sucedido do fondaparinux (7,5 mg/dia, por via subcutânea) na TIH.
Os pacientes com TIH sem as complicações trombóticas associadas devem ser tratados com um dos anticoagulantes alternativos, até que as contagens plaquetárias sejam normalizadas (em geral, em 5 a 7 dias). O autor recomenda descontinuar o anticoagulante alternativo neste momento. Entretanto, alguns clínicos experientes optam por manter a anticoagulação empírica por até 1 mês, por considerarem que a TIH representa uma condição de hipercoagulação intensiva. Em pacientes que necessitam de anticoagulação prolongada – seja por apresentarem outras indicações para anticoagulação ou por já terem desenvolvido as complicações trombóticas da TIH – a warfarina é usada no tratamento de longa duração. O curso de varfarina deve ser iniciado somente após o paciente ter recebido uma terapia de anticoagulação adequada com um dos anticoagulantes alternativos. Ambos os fármacos devem ser usados ao mesmo tempo por um período mínimo de 5 dias, antes da descontinuação do curso de anticoagulante alternativo. Tanto a lepirudina como a argatrobana podem aumentar o TP e a INR, interferindo, assim, nos ajustes da dose de warfarina. O TP deve ser reavaliado decorridas 6 horas da descontinuação da lepirudina ou argatrobana, a fim de garantir que uma INR igual a 2 a 3 tenha sido alcançada. Para os pacientes que tenham desenvolvido trombose associada à TIH, a instituição de uma terapia de anticoagulação com duração total de 3 meses deve ser adequada, pois se trata de trombose provocada.
Alguns pacientes com TIH confirmada por sorologia futuramente podem vir a necessitar de cirurgia envolvendo desvio cardiopulmonar. O uso de argatrobana ou hirudina recombinante nestes casos foi descrito em relatos pouco confiáveis.139,141,142 Considerando a natureza transiente dos anticorpos induzidos pela heparina, a reutilização subsequente da heparina é teoricamente razoável. De fato, poucos pacientes voltam a receber heparina depois que os anticorpos induzidos por heparina desaparecem, sem desenvolverem sequelas clínicas significativas.129,143 Mesmo assim, o uso da heparina nesta situação deve ser restrito aos pacientes com indicação que obrigue a isto, como uma cirurgia cardíaca ou vascular, e somente depois de a ausência de anticorpos heparina-dependentes ser confirmada por um ensaio sensível, como o ELISA para PF4-heparina. Do mesmo modo, como a reexposição à heparina pode deflagrar a recorrência dos anticorpos heparina-dependentes, este anticoagulante somente deve ser usado durante o procedimento propriamente dito. Além disso, um curso de anticoagulante alternativo deve ser iniciado no pós-operatório, para fins de profilaxia contra as recidivas da TIH. Os anticorpos anti-heparina-PF4 devem ser checados no pós-operatório.
Alguns pacientes com câncer – sobretudo aqueles com tumores sólidos ocultos de pâncreas, ovário, fígado, cérebro, cólon, pulmão ou mama – podem desenvolver trombose venosa espontânea nos membros superiores e inferiores: a síndrome de Trousseau. Estes pacientes também são mais propensos a desenvolver tromboembolia e trombose arterial recorrente.144 Entre os pacientes com TEV não provocada, observa-se uma incidência aproximada de 10% de câncer descoberto em um período de 12 a 24 meses após o evento trombótico.145-146
A causa subjacente da síndrome de Trousseau é uma forma crônica e compensada de CID. Os pró-coagulantes ativados gerados na CID intensificam a trombose. A coloração imuno-histoquímica de cortes de tumores comumente associados à síndrome de Trousseau muitas vezes revela a presença de fator tecidual na superfície tumoral.147,148 Um pró-coagulante derivado do câncer foi purificado de adenocarcinomas e células leucêmicas. Este pró-coagulante, identificado como uma cisteína protease, pode ativar a cascata de coagulação por meio da ativação direta do fator X.149,150 A interação das células tumorais com monócitos, plaquetas e células endoteliais também pode gerar citocinas inflamatórias e induzir atividade pró-coagulante endotelial e monocítica, exacerbando ainda mais a trombose. As células tumorais podem secretar mucinas solúveis – polissacarídeos complexos capazes de ativar leucócitos, levando à formação de microtrombos de plaqueta-leucócito e geração de trombina.151 A heparina bloqueia a ativação leucocitária promovida pela mucina tumoral, e isto pode explicar parcialmente a eficácia da heparina em relação à warfarina no tratamento desta condição. Pelo menos um destes mecanismos pode mediar a TEV na síndrome de Trousseau.152
Na síndrome de Trousseau, a trombose venosa costuma se manifestar como uma tromboflebite superficial migratória ou TVP nos membros inferiores. A trombose arterial recorrente que ocorre nestes pacientes tem origem em uma endocardite trombótica não bacteriana, em que há deposição de fibrina estéril na valva mitral. O coágulo de fibrina pode embolizar e causar isquemia digital, ataques isquêmicos transientes e AVC.
Na síndrome de Trousseau, os ensaios de coagulação evidenciam uma CID crônica e de baixo grau (isto é, níveis de fibrinogênio e de plaquetas discretamente baixos ou até altos, níveis elevados de D-dímero). O TP e TTPA em geral não sofrem prolongamento. É incomum estes pacientes terem CID evidente.
Uma pergunta é quão agressivamente o diagnóstico de câncer subjacente deve ser procurado em pacientes com TVP idiopática? Estudos anteriores falharam em demonstrar os benefícios e a custo-efetividade de uma abordagem de avaliação extensa.146,153 Segundo uma recente revisão sistêmica, a adoção de uma extensa estratégia de avaliação , incluindo a tomografia computadorizada (TC) de abdome e pelve, aumentou a detecção de cânceres previamente não detectados em cerca de 50 a 70% destes pacientes. No entanto, não foi possível determinar se esta abordagem foi custo-efetiva ou fez alguma diferença em termos de morbidade e mortalidade.154
Atualmente, é prudente obter uma história detalhada, exame físico, radiografia torácica, contagens sanguíneas de rotina e exames de bioquímica. Também pode ser considerada a realização de uma avaliação conforme a idade dos pacientes, para detecção de câncer de cólon (sangue oculto nas fezes ou colonoscopia), testes de antígeno específico da próstata (homens) e realização de mamografia e ultrassonografia pélvica em mulheres.155,156 Testes e exames de seguimento detalhado devem ser conduzidos de acordo com a indicação fornecida pelos exames iniciais.157
O ponto principal do tratamento da síndrome de Trousseau consiste no diagnóstico e tratamento do tumor subjacente. Em um estudo prospectivo, os pacientes com câncer apresentaram um aumento aproximado de 3 vezes na taxa de trombose recorrente, bem como um aumento de 2 vezes na taxa de sangramentos significativos durante o tratamento da TVP com warfarina. Estas complicações ocorreram principalmente durante os primeiros meses de anticoagulação e não refletiram uma sub ou superanticoagulação, mas apresentaram correlação com a extensão e severidade do câncer subjacente.158 A provável explicação para o aumento da recorrência da trombose nestes pacientes está na relativa resistência à warfarina, enquanto o sangramento aumentado pode estar relacionado à ocorrência de sangramento no sítio tumoral primário. Para o tratamento da TEV em pacientes com câncer, todas as diretrizes vigentes recomendam a administração de HBPM, em vez da anticoagulação à base de warfarina. O tratamento deve ter duração mínima de 6 meses, sendo que a terapia indefinida deve ser considerada para indivíduos com câncer ativo e para pacientes que estejam recebendo quimioterapia.159 Em geral, a profilaxia contra TEV é desnecessária para os pacientes ambulatoriais com câncer, com exceção daqueles tratados com regimes quimioterápicos que usam combinações contendo talidomida altamente trombogênica ou lenalidomida. A TEV associada ao cateterismo venoso central ocorre em cerca de 4% dos pacientes com câncer, sendo que a profilaxia anticoagulante não tem efetividade comprovada e, portanto, não é recomendada para uso de rotina.160,161
Os anticoncepcionais orais aumentam o risco de doença tromboembólica em cerca de 4 vezes.162 Estudos epidemiológicos indicam que os anticoncepcionais contendo progestágeno de 3ª geração (p. ex., desogestrel) conferem um risco 2 vezes maior de trombose do que aqueles contendo um fármaco de 2ª geração (p. ex., levonorgestrel).163 Por este motivo, a 1ª opção para as pacientes que vão usar anticoncepcionais orais pela 1ª vez é um composto contendo fármaco de 2ª geração (p. ex., Alesse, Levlite, Levora, Nordette, Triphasil, Trivora). O risco de TEV desaparece quando os fármacos são descontinuados.
Em mulheres em pós-menopausa, a reposição do estrogênio aumenta em cerca de 3 vezes o risco de TEV. Todavia, o risco absoluto é baixo – estima-se que seja de aproximadamente 3,2 a cada 10.000 pacientes-anos. Portanto, a reposição de estrogênio não é contraindicada para pacientes que necessitam de tratamento hormonal para controlar sintomas severos de pós-menopausa. Contudo, as pacientes com história de TVP ou embolia pulmonar apresentam risco aumentado de recorrência e, por isso, devem evitar a terapia de reposição hormonal, quando possível.164
A associação existente entre o uso de estrogênio e a doença tromboembólica ainda é inexplicável.165 O tratamento à base de estrogênio comprovadamente produz alterações nos níveis plasmáticos de muitas proteínas envolvidas na coagulação e na fibrinólise, incluindo diminuições dos níveis de proteína S e AT e aumentos da concentração de plasminogênio. Estas alterações, todavia, em geral são bastante modestas e não são consideradas responsáveis pelo risco de aumento de trombose. Entretanto, em mulheres heterozigotas para o fator V de Leiden, o uso de anticoncepcionais orais aumenta sinergicamente o risco trombótico em cerca de 50 vezes.23 O risco trombótico também sofre um aumento sinérgico moderado (de 15 vezes) diante da combinação da presença do fator V de Leiden com a terapia de reposição hormonal.166
O tratamento agudo do episódio inicial de TEV atribuível a causas provocadas ou a causas não provocadas é o mesmo: heparina seguida de warfarina. A colocação de um filtro na veia cava inferior em geral não é indicada. Em pacientes seletos apresentando TVP proximal aguda e extensa (p. ex., TVP iliofemoral com sintomas por menos de 14 dias), a realização de trombólise endovascular pode ser considerada, desde que haja disponibilidade de especialistas e recursos adequados.167 Estes pacientes apresentam a maior carga de coágulos e o risco mais alto de morbidade pós-trombótica severa (p. ex., edema doloroso crônico e claudicação venosa) quando são tratados apenas com terapia anticoagulante.168
Na TEV, a intensidade e a duração ideal do tratamento com warfarina foram alvo de numerosos estudos clínicos amplos conduzidos ao longo da última década. Com relação à intensidade da anticoagulação oral, uma INR igual a 2 a 3 é comprovadamente ideal, estando associada a taxas menores de recorrência da trombose e uma taxa menor de sangramentos significativos (cerca de 2 a 3% ao ano).169,170 Em comparação, o tratamento de anticoagulação de baixa intensidade com uma meta de INR de 1,5 a 1,9 é menos efetivo em termos de redução das recidivas de trombose (embora seja melhor do que placebo) e não promove diminuição significativa do risco de sangramento.169,170
Com relação à duração da anticoagulação, existem 2 objetivos isolados: (1) concluir o tratamento do episódio agudo de TEV; e (2) prevenir a recorrência de TEV. Para alcançar o 1º objetivo do tratamento agudo da TEV, a limitação da duração da anticoagulação em 4 a 6 semanas é comprovadamente inadequada, além de estar associada ao aumento da recorrência de TEV e, portanto, não é recomendada.171,172 Uma exceção é a TVP distal isolada (isto é, em que o trombo não envolve as veias poplítea e femoral), que é nitidamente provocada por um fator de risco significativo (p. ex., cirurgia de substituição do joelho). É possível tratar esta condição com segurança instituindo uma terapia com duração de 6 semanas.172 As durações de anticoagulação de 3 e 6 meses foram comparadas e foi demonstrado que são equivalentes em termos de risco de TEV recorrente, tendo sido observado um risco menor de sangramento no grupo tratado por 3 meses.173,174 Em conclusão, a terapia de anticoagulação de 3 meses de duração é adequada para o tratamento de episódios agudos de TEV, sendo em geral a duração recomendada para o tratamento da TEV provocada de baixo risco [Tabela 7].
Tabela 7. Diretrizes gerais para o tratamento de pacientes com TEV
|
Risco recorrente |
Tratamento |
|
Alto Trombose idiopática recorrente Um episódio de trombose prejudicial à vida Um episódio de trombose espontânea em localização incomum (p. ex., trombose mesentérica) Um episódio de trombose espontânea associada à SAF Um episódio de trombose com 2 fatores de risco permanentes Um episódio de trombose com síndrome de Trousseau |
Terapia de anticoagulação oral vitalícia: INR = 2 a 3 |
|
Médio Um episódio de trombose com um fator de risco permanente (exceto a síndrome de Trousseau) Trombose idiopática sem fator de risco identificável |
3 a 6 meses de terapia de anticoagulação oral após o 1º episódio de trombose; considerar a anticoagulação prolongada, se o risco de sangramento for baixo e um nível satisfatório de anticoagulação for facilmente alcançável; profilaxia vigorosa em situações de alto risco |
|
Baixo Um episódio de trombose com fator de risco reversível |
3 meses de terapia de anticoagulação oral após o 1º episódio de trombose; profilaxia vigorosa em situações de alto risco |
INR = relação normalizada internacional; SAF = síndrome antifosfolipídica; TEV = tromboembolia venosa.
Para pacientes que sofrem o 1º episódio de TEV não provocada, a duração ideal da terapia de anticoagulação ainda não está totalmente estabelecida. Uma TEV não provocada geralmente é considerada como sendo de risco intermediário, em termos de recorrência de TEV, com uma taxa estimada de cerca de 5 a 15% ao ano após o evento agudo. Desta forma, um painel de especialistas recomenda a instituição de terapia de anticoagulação prolongada para estes pacientes, caso eles de fato apresentem baixo risco de sangramento e uma anticoagulação adequada à base de warfarina seja facilmente alcançável.175 No entanto, a anticoagulação crônica está associada a um risco de sangramento significativo (2 a 3%), com um risco caso-fatalidade aproximado de 9%.176 Há ainda aspectos da qualidade de vida relacionados à anticoagulação contínua, como a frequência de determinações da INR do paciente. Um especialista sugeriu que uma alternativa seria um curso de warfarina de baixa intensidade (INR-alvo de 1,5 a 1,9), que permite diminuir a frequência de monitoramento da INR (a cada 2 meses).177 Assim, apesar do consenso geral de que o tratamento inicial deva ser de 3 a 6 meses de anticoagulação com varfarina, a tomada de decisão terapêutica subsequente (isto é, anticoagulação prolongada com dose integral; anticoagulação de baixa intensidade e longa duração; sem anticoagulação) deve ser individualizada após uma discussão com o paciente, considerando o risco de sangramento, o risco de recorrência e as preferências do paciente [Tabela 7].
A dificuldade para decidir se um paciente com TEV não provocada deve receber anticoagulação estendida advém do fato de que o prolongamento da terapia inicial (p. ex., para 1 a 2 anos) não diminui o subsequente risco recorrente depois que a warfarina é descontinuada,178 bem como da atual incapacidade de identificar com segurança os 10 a 15% dos pacientes mais propensos a desenvolver recidivas. Estudos recentes sugerem que a elevação persistente dos níveis de D-dímero pode discriminar entre pacientes com risco de recorrência maior ou menor. Um resultado negativo no teste de D-dímero, realizado em 3 a 6 semanas após a descontinuação da varfarina, está associado a um baixo risco de recorrência (risco anual aproximado de 3,5%). Contudo, um resultado de D-dímero positivo está associado a um risco anual de recorrência de cerca de 9%.179
Com relação aos pacientes que apresentam alto risco de recorrência, como aqueles com TEV, há um consenso geral de que a anticoagulação prolongada é a terapia apropriada [Tabela 7].
Em estudos clínicos, vários anticoagulantes orais novos, incluindo um inibidor de trombina direto (etexilato de dabigatrana) e vários inibidores de fator Xa (p. ex., rivaroxabana e apixabana) apresentaram a mesma eficácia da HBPM e/ou varfarina na profilaxia ou tratamento da TEV.180 Em geral, estes fármacos novos possuem perfis farmacológicos superiores ao da varfarina, além de apresentarem interações mínimas com alimentos ou outros agentes e dispensarem o monitoramento regular. Fora dos Estados Unidos, alguns destes fármacos foram aprovados para uso na profilaxia contra TEV. É provável que futuramente estes medicamentos exerçam um impacto significativo sobre a forma como a TEV é tratada.
O autor não possui relações comerciais com os fabricantes de produtos ou prestadores de serviços mencionados neste capítulo.
1. Chi JT, Chang HY, Haraldsen G, et al. Endothelial cell diversity revealed by global expression prof ling. Proc Natl Acad Sci U S A 2003;100:10623–8.
2. Martinelli I, Mannucci PM, De Stefano V, et al. Different risks of thrombosis in four coagulation defects associated with inherited thrombophilia: a study of 150 families. Blood 1998;92:2353–8.
3. Linnemann B, Meister F, Schwonberg J, et al. Hereditary and acquired thrombophilia in patients with upper extremity deep-vein thrombosis. Thromb Haemost 2008;100: 440–6.
4. Favaloro EJ, McDonald D, Lippi G. Laboratory investigation of thrombophilia: the good, the bad, and the ugly. Semin Thromb Hemost 2009;35:695–710.
5. Tait RC, Walker ID, Perry DJ, et al. Prevalence of anti-thrombin def ciency in the healthy population. Br J Haematol 1994;87:106–12.
6. Patnaik MM, Moll S. Inherited antithrombin def ciency: a review. Haemophilia 2008;14:1229–39.
7. De Stefano V, Leone G, Mastrangelo S, et al. Clinical manifestations and management of inherited thrombophilia: retrospective analysis and follow-up after diagnosis of 238 patients with congenital def ciency of antithrombin III, protein C, protein S. Thromb Haemost 1994;72:352–8.
8. Demers C, Ginsberg JS, Hirsh J, et al. Thrombosis in antithrombin-def cient persons: report of a large kindred and literature review. Ann Intern Med 1992;116:754–61.
9. van den Belt AGM, Sanson B-J, Simioni P, et al. Recurrence of venous thromboembolism in patients with familial thrombophilia. Arch Intern Med 1997;157:2227–32.
10. Miletich J, Sherman L, Broze G Jr. Absence of thrombosis in subjects with heterozygous protein C def ciency. N Engl J Med 1987;317:991–6.
11. Miletich JP, Prescott SM, White R, et al. Inherited predisposition to thrombosis. Cell 1993;72:477–80.
12. Boerger LM, Morris PC, Thurnau GR, et al. Oral contraceptives and gender affect protein S status. Blood 1987;69: 692–4.
13. Vigano-D’Angelo S, D’Angelo A, Kaufman CE Jr, et al. Protein S def ciency occurs in the nephrotic syndrome. Ann Intern Med 1987;107:42–7.
14. Sallah S, Abdallah JM, Gagnon GA. Recurrent warfarin-induced skin necrosis in kindreds with protein-S def ciency. Haemostasis 1998;28:25–30.
15. Zoller B, Garcia de Frutos P, Dahlback B. Evaluation of the relationship between protein S and C4B-binding protein isoforms in hereditary protein S def ciency demonstrating type I and type III def ciencies to be phenotypic variants of the same genetic disease. Blood 1995;85:3524–31.
16. Dahlback B. Advances in understanding pathogenic mechanisms of thrombophilic disorders. Blood 2008;112:19–27.
17. Faioni EM, Franchi F, Asti D, et al. Resistance to activated protein C in nine thrombophilic families: interference in a protein S functional assay. Thromb Haemost 1993;70: 1067–71.
18. Rees DC, Cox M, Clegg JB. World distribution of factor V Leiden. Lancet 1995;346:1133–4.
19. Prothrombin 20210G>A is an ancestral prothrombotic mutation that occurred in whites approximately 24 000 years ago. Blood 2006;107:4666–8.
20. Lindqvist PG, Svensson PJ, Marsaal K, et al. Activated protein C resistance (FV:Q506) and pregnancy. Thromb Haemost 1999;81:532–7.
21. Svensson PJ, Dahlback B. Resistance to activated protein C as a basis for venous thrombosis. N Engl J Med 1994;330: 517–22.
22. Simioni P, Prandoni P, Lensing AWA, et al. The risk of recurrent venous thromboembolism in patients with an Arg506 Gln mutation in the gene for factor V (factor V Leiden). N Engl J Med 1997;336:399–403.
23. Vandenbroucke JP, Koster T, Briet E, et al. Increased risk of venous thrombosis in oral-contraceptive users who are carriers of factor V Leiden mutation. Lancet 1994;344: 1453–7.
24. Middeldorp S, Henkens CMA, Koopman MMW, et al. The incidence of venous thromboembolism in family members of patients with factor V Leiden mutation and venous thrombosis. Ann Intern Med 1998;128:15–20.
25. Wu O, Robertson L, Twaddle S, et al. Screening for thrombophilia in high-risk situations: a systematic review and cost-effectiveness analysis. The Thrombosis: Risk and Economic Assessment of Thrombophilia Screening (TREATS) Study. Health Technol Assess 2006;10:1–11.
26. Rosendaal FR, Koster T, Vandenbroucke JP, et al. High-risk of thrombosis in patients homozygous for factor V Leiden (APC-resistance). Blood 1995;85:1504–8.
27. Koeleman BPC, Reitsma PH, Allaart RC, et al. Activated protein C resistance as an additional risk factor for thrombosis in protein C–defi cient families. Blood 1994;84: 1031–5.
28. Zoller B, Berntsdotter A, Garcia de Frutos P, et al. Resistance to activated protein C as an additional genetic risk factor in hereditary defi ciency of protein S. Blood 1995;85: 3518–23.
29. van Boven HH, Reitsma PH, Rosendaal FR, et al. Factor V Leiden (FV R506Q) in families with inherited antithrombin defi ciency. Thromb Haemost 1996;75:417–21.
30. Rodeghiero F, Tosetto A. Risk factors for thromboembolism. Ann Intern Med 1999;130:643–50.
31. Castoldi E, Rosing J. APC resistance: biological basis and acquired infl uences. J Thromb Haemost 2010;8:445–53.
32. van der Bom JG, Bots ML, Haverkate F, et al. Reduced response to protein C is associated with increased risk for cerebral vascular disease. Ann Intern Med 1996;125: 265–9.
33. Fisher M, Fernandez JA, Ameriso SF, et al. Activated protein C resistance in ischemic stroke not due to factor V arginine506?glutamine mutation. Stroke 1996;27:1163–6.
34. Rodeghiero F, Tosetto A. Activated protein C resistance and factor V Leiden mutation are independent risk factors for venous thromboembolism. Ann Intern Med 1999;130: 643–50.
35. de Visser MCH, Rosendaal FR, Bertina RM. A reduced sensitivity to activated protein C in the absence of factor V Leiden increases the risk of venous thrombosis. Blood 1999;93:1271–6.
36. de Visser MCH, Rosendaal FR, Bertina RM. A reduced sensitivity for activated protein C in the absence of factor V Leiden increases the risk of venous thrombosis. Blood 1999;93:1271–6.
37. Kalafatis M, Bertina RM, Rand MD, et al. Characterization of the molecular defect in factor V R506Q. J Biol Chem 1995;270:4053–7.
38. Thorelli E, Kaufman RJ, Dahlbäck B. Cleavage of factor V at Arg 506 by activated protein C and the expression of anticoagulant activity of factor V. Blood 1999;93:2552–8.
39. Ridker PM, Hennekens CH, Lindpaintner K, et al. Mutation in the gene coding for coagulation factor V and the risk for myocardial infarction, stroke, and venous thrombosis in apparently healthy men. N Engl J Med 1995;332: 912–7.
40. Segal JB, Brotman DJ, Necochea AJ, et al. Predictive value of factor V Leiden and prothrombin G20210A in adults with venous thromboembolism and in family members of those with a mutation: a systematic review. JAMA 2009; 301:2472–85.
41. Dahlback B. Resistance to activated protein C caused by the factor V R506Q mutation is a common risk factor for venous thrombosis. Thromb Haemost 1997;78:483–8.
42. Middeldorp S, Meinardi JR, Koopman MM, et al. A prospective study of asymptomatic carriers of the factor V Leiden mutation to determine the incidence of venous thromboembolism. Ann Intern Med 2001;135:322–7.
43. Rosendaal FR, Doggen CJ, Zivelin A, et al. Geographic distribution of the 20210 G to A prothrombin variant. Thromb Haemost 1998;79:706–6.
44. Poort SR, Rosendaal FR, Reitsma PH, et al. A common genetic variation in the 3'-untranslated region of the pro-thrombin gene is associated with elevated plasma pro-thrombin levels and an increase in venous thrombosis. Blood 1996;88:3698–703.
45. The risk of recurrent venous thromboembolism in carriers and non-carriers of the G1691A allele in the coagulation factor V gene and the G20210A allele in the prothrombin gene. DURAC Trial Study Group. Duration of Anticoagulation. Thromb Haemost 1999;81:684–9.
46. De Stefano V, Martinelli I, Mannucci PM, et al. The risk of recurrent deep venous thrombosis among heterozygous carriers of both factor V Leiden and the G20210A prothrombin mutation. N Engl J Med 1999;341:801–6.
47. Martinelli I, Sacchi E, Landi G, et al. High risk of cerebral-vein thrombosis in carriers of a prothrombin-gene mutation and in users of oral contraceptives. N Engl J Med 1998; 338:1793–7.
48. Harker LA, Harlan JM, Ross R. Effect of sulfi npyrazone on homocysteine-induced endothelial injury and arteriosclerosis in baboons. Circ Res 1983;53:731–9.
49. Starkebaum G, Harlan JM. Endothelial cell injury due to copper-catalyzed hydrogen peroxide generation from homocysteine. J Clin Invest 1986;77:1370–6.
50. Lentz SR, Sadler JE. Inhibition of thrombomodulin surface expression and protein C activation by the thrombogenic agent homocysteine. J Clin Invest 1991;88:1906–14.
51. Nishinaga M, Ozawa T, Shimada K. Homocysteine, a thrombogenic agent, suppresses anticoagulant heparan sulfate expression in cultured porcine aortic endothelial cells. J Clin Invest 1993;92:1381–6.
52. Harpel PC, Chang VT, Borth W. Homocysteine and other sulfhydryl compounds enhance thebinding of lipoprotein(a) to fi brin: a potential biochemical link between thrombosis, atherogenesis, and sulfhydryl compound metabolism. Proc Natl Acad Sci U S A 1992;89:10193–7.
53. Mudd SH, Levy HL, Skovby F. Disorders of transulfuration. In: Scriver CR, Beaudet AL, Sly WS, et al, editors. The metabolic basis of inherited disease. New York: McGraw-Hill; 1989. p. 693.
54. Malinow MR. Homocyst(e)ine and arterial occlusive diseases. J Intern Med 1994;236:603–17.
55. Naurath HJ, Joosten E, Riezier R, et al. Effects of vitamin B12, folate, and vitamin B6 supplements in elderly people with normal serum vitamin concentrations. Lancet 1995; 346:85–9.
56. Clarke R, Daly L, Robinson K, et al. Hyperhomocysteinemia: an independent risk factor for vascular disease. N Engl J Med 1991;324:1149–55.
57. Selhub J, Jacques PF, Bostom AG, et al. Association between plasma homocysteine concentrations and extracranial carotid-artery stenosis. N Engl J Med 1995;332:286–91.
58. Den Heijer M, Koster T, Blom HJ, et al. Hyperhomocysteinemia as a risk factor for deep-vein thrombosis. N Engl J Med 1996;334:759–62.
59. Nygard O, Nordrehaug JE, Refsum H, et al. Plasma homocysteine levels and mortality in patients with coronary artery disease. N Engl J Med 1997;337:230–6.
60. Franken DG, Boers GH, Blom HJ, et al. Treatment of mild hyperhomocysteinemia in vascular disease patients. Arterioscler Thromb Vasc Biol 1994;14:465–70.
61. Lonn E, Yusuf S, Arnold MJ, et al. Homocysteine lowering with folic acid and B vitamins in vascular disease. Heart Outcomes Prevention Evaluation (HOPE) 2 Investigators. N Engl J Med 2006;354:1567–77.
62. Ray JG, Kearon C, Yi Q, et al. Homocysteine-lowering therapy and risk for venous thromboembolism: a randomized trial. Heart Outcomes Prevention Evaluation (HOPE) 2 Investigators. Ann Intern Med 2007;146:761–7.
63. den Heijer M, Willems HP, Blom HJ, et al. Homocysteine lowering by B vitamins and the secondary prevention of deep vein thrombosis and pulmonary embolism: a randomized, placebo-controlled, double-blind trial. Blood 2007;109:139–44.
64. Marcovina SM, Albers JJ, Jacobs DR Jr, et al. Lipoprotein(a) concentrations and apolipoprotein(a) phenotypes in Caucasians and African-Americans. The CARDIA study. Arterioscler Thromb 1993;13:1037–45.
65. Bostom A, Cupples A, Jenner J, et al. Elevated plasma lipoprotein(a) and coronary heart disease in men aged 55 years and younger. JAMA 1996;276:544–8.
66. Kamstrup P, Tybjærg-Hansen A, Steffensen R, et al. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA 2009;301:2331–9.
67. Wild SH, Fortmann SP, Marcovina SM. A prospective case-control study of lipoprotein(a) levels and Apo(a) size and risk of coronary heart disease in Stanford Five-City Project participants. Arterioscler Thromb Vasc Biol 1997;17: 239–45.
68. Clarke R, Peden JF, Hopewell JC, et al. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med 2009;361:2518–28.
69. Nagayama M, Shinohara Y, Nagayama T. Lipoprotein(a) and ischemic cerebrovascular disease in young adults. Stroke 1994;25:74–8.
70. Ridker PM, Stampfer MJ, Hennekens CH. Plasma concentration of lipoprotein(a) and the risk of future stroke. JAMA 1995;273:1269–73.
71. The Emerging Risk Factors Collaboration. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA 2009;302: 412–23.
72. von Depka M, Nowak-Gottl U, Eisert R, et al. Increased lipoprotein(a) levels as independent risk factor for venous thromboembolism. Blood 2000;96:3364–8.
73. Vormittag R, Vukovich T, Stain M, et al. Lipoprotein (a) in patients with spontaneous venous thromboembolism. Thromb Res 2007;120:15–20.
74. Hajjar KA, Gavish D, Breslow JL, et al. Lipoprotein (a) modulation of endothelial cell surface fi brinolysis and its potential role in atherosclerosis. Nature 1989;339:303–5.
75. Caplice NM, Panetta C, Peterson TE, et al. Lipoprotein(a) binds and inactivates tissue factor pathway inhibitor: a novel link between lipoproteins and thrombosis. Blood 2001;98:2980–7.
76. Tsimikas S, Brilakis ES, Miller ER, et al. Oxidized phospholipids, Lp(a) lipoprotein, and coronary artery disease. N Engl J Med 2005;353:46–57.
77. Maher VM, Brown BG, Marcovina SM, et al. Effects of lowering elevated LDL cholesterol on the cardiovascular risk of lipoprotein(a). JAMA 1995;274:1771–4.
78. Gurakar A, Hoeg JM, Kostner G, et al. Levels of lipoprotein(a) decline with neomycin and niacin treatment. Atherosclerosis 1985;57:293–301.
79. Scanu AM, Bamba R. Niacin and lipoprotein(a): facts, uncertainties, and clinical considerations. Am J Cardiol 2008;101(8A):44B–7B.
80. Martinez J. Congenital dysfi brinogenemia. Curr Opin Hematol 1997;4:357–65.
81. Miesbach W, Scharrer I, Henschen A, et al. Inherited dysfi brinogenemia: clinical phenotypes associated with five different fi brinogen structure defects. Blood Coagul Fibrinolysis 2010;21:35–40.
82. Aoki N, Moroi M, Sakata Y, et al. Abnormal plasminogen: a hereditary molecular abnormality found in a patient with recurrent thrombosis. J Clin Invest 1978;61:1186–95.
83. Estelles A, Gilabert J, Aznar J, et al. Changes in the plasma levels of type 1 and type 2 plasminogen activators in normal pregnancy and in patients with severe preeclampsia. Blood 1989;74:1332–8.
84. Prins MH, Hirsh J. A critical review of the evidence supporting a relationship between impaired fibrinolytic activity and venous thromboembolism. Arch Intern Med 1991;151:1721–31.
85. Wiman B. Plasminogen activator inhibitor-1 (PAI-1) in plasma: its role in thrombotic disease. Thromb Haemost 1995;74:71–6.
86. Koster T, Blann AD, Briet E, et al. Role of clotting factor VIII in effect of von Willebrand factor on occurrence of deep-vein thrombosis. Lancet 1995;345:152–5.
87. O’Donnell J, Tuddenham EGD, Manning R, et al. High prevalence of elevated factor VIII levels in patients referred for thrombophilia screening: role of increased synthesis and relationship to the acute phase reaction. Thromb Haemost 1997;77:825–8.
88. Meade TW. Fibrinogen in ischemic heart disease. Eur Heart J 1995;16 Suppl:31–4.
89. Meade TW, Mellows S, Brozovic M, et al. Haemostatic function and ischaemic heart disease: principal results of the Northwick Park Heart Study. Lancet 1986;2:533–7.
90. Antiphospholipid syndrome: clinical and immunologic manifestations and patterns of disease expression in a cohort of 1,000 patients. Euro-Phospholipid Project Group. Arthritis Rheum 2002;46:1019–27.
91. Rauch J, Janoff AS. Role of antibodies in understanding the interactions between anti-phospholipid antibodies and phospholipids. In: Harris N, Exner T, Hughes GRV, et al, editors. Phospholipid-binding antibodies. Boca Raton (FL): CRC Press; 1991. p. 108.
92. Rao LVM, Hoang AD, Rapaport SI. Mechanisms and effects of the binding of lupus anticoagulant IgG and pro-thrombin to surface phospholipid. Blood 1996;88:4173–82.
93. Smirnov MD, Triplett DT, Comp PC, et al. On the role of phosphatidylethanolamine in the inhibition of activated protein C activity by antiphospholipid antibodies. J Clin Invest 1995;95:309–16.
94. Shibata S, Harpel PC, Gharavi A, et al. Autoantibodies to heparin from patients with antiphospholipid antibody syndrome inhibit formation of antithrombin III–thrombin complexes. Blood 1994;83:2532–40.
95. Redecha P, Tilley R, Tencati M, et al. Tissue factor: a link between C5a and neutrophil activation in antiphos- pholipid antibody induced fetal injury. Blood 2007;110: 2423–31.
96. Girardi G, Redecha P, Salmon JE. Heparin prevents antiphospholipid antibody-induced fetal loss by inhibiting complement activation. Nat Med 2004;10:1222–6.
97. Finazzi G, Brancaccio V, Ciavarella N, et al. Natural history and risk factors for thrombosis in 360 patients with antiphospholipid antibodies: a four-year prospective study from the Italian registry. Am J Med 1996;100:530–6.
98. Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement of an update of the classifi cation criteria for defi nite antiphospholipid syndrome (APS). J Thromb Haemost 2006;4:295–306.
99. Vila P, Hernandez MC, Lopez-Fernadez MF, et al. Prevalence, follow-up and clinical signifi cance of the anticardiolipin antibodies in normal subjects. Thromb Haemost 1994; 72:209–13.
100.Ginsburg KS, Liang MH, Newcomer L, et al. Anticardiolipin antibodies and the risk for ischemic stroke and venous thrombosis. Ann Intern Med 1992;117:997–1002.
101.Silver RM, Porter TF, van Leeuween I, et al. Anticardiolipin antibodies: clinical consequences of “low titers.” Obstet Gynecol 1996;87:494–500.
102.Selva-O’Callaghan A, Ordi-Ros J, Monegal-Ferran F, et al. IgA anticardiolipin antibodies: relation with other antiphospholipid antibodies and clinical signifi cance. Thromb Haemost 1998;79:282–5.
103.Khamashta MA, Cuardrado MJ, Mujic F, et al. The management of thrombosis in the antiphospholipid-antibody syndrome. N Engl J Med 1995;332:993–7.
104.Rosove MH, Brewer MC. Antiphospholipid thrombosis: clinical course after the fi rst thrombotic event in 70 patients. Ann Intern Med 1992;117:303–8.
105.Crowther MA, Ginsberg JS, Julian J, et al. A comparison of two intensities of warfarin for the prevention of recurrent thrombosis in patients with the antiphospholipid antibody syndrome. N Engl J Med 2003;349:1133–8.
106.Albers GW, Amarenco P, Easton JD, et al. Antithrombotic and thrombolytic therapy for ischemic stroke: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2008;133:630S–69S
107.Giannakopoulos B, Krilis SA. How I treat the antiphospholipid syndrome. Blood 2009;114:2020–30.
108.Tuthill JI, Khamashta MA. Management of antiphospholipid syndrome. J Autoimmun 2009;33:92–8.
109.Kearon C, Gent M, Hirsh J, et al. A comparison of three months of anticoagulation with extended anticoagulation for a first episode of idiopathic venous thromboembolism. N Engl J Med 1999;340:901–7.
110.Moll S, Ortel TL. Monitoring warfarin therapy in patients with lupus anticoagulants. Ann Intern Med 1997;127: 177–85.
111.Rand JH, Wu X-X, Andree HAM, et al. Pregnancy loss in the antiphospholipid-antibody syndrome—a possible thrombogenic mechanism. N Engl J Med 1997;337:154–60.
112.Kutteh WH, Rote NS, Silver R. Antiphospholipid antibodies and reproduction: the antiphospholipid antibody syndrome. Am J Reprod Immunol 1999;41:133–52.
113.Infante-Rivard C, David M, Gauthier R, et al. Lupus anticoagulants, anticardiolipin antibodies, and fetal loss: a case-controlled study. N Engl J Med 1991;325:1063–6.
114.Empson M, Lassere M, Craig JC, et al. Recurrent pregnancy loss with antiphospholipid antibody: a systematic review of therapeutic trials. Obstet Gynecol 2002;99: 135–44.
115.Stephenson MD, Ballem PJ, Tsang P, et al. Treatment of antiphospholipid antibody syndrome (APS) in pregnancy: a randomized pilot trial comparing low molecular weight heparin to unfractionated heparin. J Obstet Gynaecol Can 2004;26:729–34.
116.Noble LS, Kutteh WH, Lashey N, et al. Antiphospholipid antibodies associated with recurrent pregnancy loss: prospective, multicenter, controlled pilot study comparing treatment with low-molecular-weight heparin versus unfractionated heparin. Fertil Steril 2005;83:684–90.
117.Branch DW, Peaceman AM, Druzin M, et al. A multicenter, placebo-controlled pilot study of intravenous immune globulin treatment of antiphospholipid syndrome during pregnancy. The Pregnancy Loss Study Group. Am J Obstet Gynecol 2000;182:122–7.
118.Triolo G, Ferrante A, Ciccia F, et al. Randomized study of subcutaneous low molecular weight heparin plus aspirin versus intravenous immunoglobulin in the treatment of recurrent fetal loss associated with antiphospholipid antibodies. Arthritis Rheum 2003;48:728–31.
119.Warkentin TE. Heparin-induced thrombocytopenia: a ten-year retrospective. Annu Rev Med 1999;50:129–47.
120.Bauer TL, Arepally G, Konkle BA, et al. Prevalence of heparin-associated antibodies without thrombosis in patients undergoing cardiopulmonary bypass surgery. Circulation 1997;95:1242–6.
121.Warkentin TE. Heparin-induced thrombocytopenia: a clinicopathologic syndrome. Thromb Haemost 1999;82: 439–47.
122.Amiral J, Bridey F, Dreyfus M, et al. Platelet factor 4 complexed to heparin is the target for antibodies generated in heparin-induced thrombocytopenia. Thromb Haemost 1992;68:95–6.
123.Rauova L, Poncz M, McKenzie SE, et al. Ultralarge complexes of PF4 and heparin are central to the pathogenesis of heparin-induced thrombocytopenia. Blood 2005;105: 131–8.
124.Rauova L, Zhai L, Kowalska MA, et al. Role of platelet surface PF4 antigenic complexes in heparin-induced thrombocytopenia pathogenesis: diagnostic and therapeutic implications. Blood 2006;107:2346–53.
125.Chong BH Murray B, Berndt MC, et al. Plasma P-selectin is increased in thrombotic consumptive platelet disorders. Blood 1994;83:1535–41.
126.Warkentin TE, Levine MN, Hirsh J, et al. Heparin-induced thrombocytopenia in patients treated with low-molecularweight heparin or unfractionated heparin. N Engl J Med 1995;332:1330–5.
127.Visentin GP, Ford SE, Scott JP, et al. Antibodies from patients with heparin-induced thrombocytopenia/thrombosis are specifi c for platelet factor 4 complexed with heparin or bound to endothelial cells. J Clin Invest 1994; 93:81–8.
128.Greinacher A. Heparin-induced thrombocytopenia. J Thromb Haemost 2009;7 Suppl 1:9–12.
129.Warkentin TE, Kelton JG. Temporal aspects of heparin-induced thrombocytopenia. N Engl J Med 2001;344: 1286–92.
130.Warkentin TE, Kelton JG. Delayed-onset heparin-induced thrombocytopenia and thrombosis. Ann Intern Med 2001; 135:502–6.
131.Sallah S, Thomas DP, Roberts HR. Warfarin and heparin-induced skin necrosis and the purple toe syndrome: infrequent complications of anticoagulant treatment. Thromb Haemost 1997;78:785–90.
132.Greinacher A, Amiral J, Dummel V, et al. Laboratory diagnosis of heparin-associated thrombocytopenia and comparison of platelet aggregation test, and platelet factor 4/heparin enzyme linked immunosorbent assay. Transfusion 1994;34:381–5.
133.Greinacher A, Juhl D, Strobel U, et al. Heparin-induced thrombocytopenia: a prospective study on the incidence, platelet-activating capacity and clinical signifi cance of antiplatelet factor 4/heparin antibodies of the IgG, IgM, and IgA classes. J Thromb Haemost 2007;5:1666–73.
134.Zwicker JI, Uhl L, Huang WY, et al. Thrombosis and ELISA optical density values in hospitalized patients with heparin-induced thrombocytopenia. J Thromb Haemost 2004;2: 2133–7.
135.Warkentin TE, Sheppard JI, Moore JC, et al. The quantitative interpretation of optical density measurements using PF4 dependent enzyme immunoassays. J Thromb Haemost 2008;6:1304–12.
136.Warkentin TE, Greinacher A. Heparin-induced thrombocytopenia and cardiac surgery. Ann Thorac Surg 2003;76: 2121–31.
137.Warkentin T, Greinacher A, Koster A, Lincoff M. Treatment and prevention of heparin-induced thrombocytopenia: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2008;133:340S–80S.
138.Greinacher A, Volpol H, Potzsch B. Recombinant hirudin in the treatment of patients with heparin-induced thrombocytopenia (HIT). Blood 1996;88 Suppl:281a.
139.Schiele F, Vuillemenot A, Kramarz P, et al. Use of recombinant hirudin as antithrombotic treatment in patients with heparin-induced thrombocytopenia. Am J Hematol 1995;50:20–5.
140.Lewis BE, Wallis DE, Berkowitz SD, et al. Argatroban anticoagulant therapy in patients with heparin-induced thrombocytopenia. Circulation 2001;103:1838–43.
141.Matsuo T, Yamada T, Yamanashi T, et al. Anticoagulant therapy with MD805 of a hemodialysis patient with heparin-induced thrombocytopenia. Thromb Res 1990;58: 663–6.
142.Gillis S, Merin G, Zahger D, et al. Danaparoid for cardiopulmonary bypass in patients with previous heparin-induced thrombocytopenia. Br J Haematol 1997;98:657–9.
143.Potzsch B, Klovekorn WP, Madlener K. Use of heparin during cardiopulmonary bypass in patients with a history of heparin-induced thrombocytopenia. N Engl J Med 2000; 343:515.
144.Callander N, Rapaport SI. Trousseau’s syndrome. West J Med 1993;158:364–71.
145.Prandoni P, Lensing AWA, Buller HR, et al. Deep-vein thrombosis and the incidence of subsequent symptomatic cancer. N Engl J Med 1992;327:1128–33.
146.Sorensen HT, Mellemkjaer L, Steffensen FH, et al. The risk of a diagnosis of cancer after primary deep venous thrombosis or pulmonary embolism. N Engl J Med 1998;338: 1169–73.
147.Callander NS, Varki N, Rao LVM. Immunohistochemical identifi cation of tissue factor in solid tumors. Cancer 1992; 70:1194–201.
148.Contrino J, Hair G, Kreutzer DL, et al. In situ detection of tissue factor in vascular endothelial cells: correlation with malignant phenotype of human breast disease. Nat Med 1996;2:209–15.
149.Falanga A, Gordon SG. Isolation and characterization of cancer procoagulant: a cysteine proteinase from malignant tissue. Biochemistry 1985;24:5558–67.
150.Falanga A, Consonni R, Marchetti M, et al. Cancer procoagulant and tissue factor are differentially modulated by all-trans-retinoic acid in acute promyelocytic leukemic cells. Blood 1998;92:143–51.
151.Wahrenbrock M, Borsig L, Le D, et al. Selectin-mucin interactions as a probable molecular explanation for the association of Trousseau syndrome with mucinous adenocarcinomas. J Clin Invest 2003;112:853–62.
152.Varki A. Trousseau’s syndrome: multiple defi nitions and multiple mechanisms. Blood 2007;110:1723–9.
153.Prins MH, Hettiarachchi RJK, Lensing AWA, et al. Newly diagnosed malignancy in patients with venous thromboembolism: search or wait and see? Thromb Haemost 1997; 78:121–5.
154.Carrier M, Le Gal G, Wells PS, et al. Systematic review: the Trousseau syndrome revisited: should we screen extensively for cancer in patients with venous thromboembolism? Ann Intern Med 2008;149:323–33.
155.Silverstein RL, Nachman RL. Cancer and clotting: Trousseau’s warning. N Engl J Med 1992;327:1163–4.
156.Buller H, Ten Cate JW. Primary venous thromboembolism and cancer screening. N Engl J Med 1998;338:1221–2.
157.Cornuz J, Pearson SD, Creager MA, et al. Importance of findings on the initial evaluation for cancer in patients with symptomatic idiopathic deep venous thrombosis. Ann Intern Med 1996;125:785–93.
158.Prandoni P, Lensing AW, Picciolo A, et al. Recurrent venous thromboembolism and bleeding complications during anticoagulant treatment in patients with cancer and venous thrombosis. Blood 2002;100:3484–8.
159.Khorana AA, Streiff MB, Farge D, et al. Venous thromboembolism prophylaxis and treatment in cancer: a consensus statement of major guidelines panels and call to action. J Clin Oncol 2009;27:4919–26.
160.Verso M, Agnelli G, Bertoglio S, et al. Enoxaparin for the prevention of venous thromboembolism associated with central vein catheter: a double-blind, placebo-controlled, randomized study in cancer patients. J Clin Oncol 2005; 23:4057–62.
161.Karthaus M, Kretzschmar A, Kroning H, et al. Dalteparin for prevention of catheter-related complications in cancer patients with central venous catheters: fi nal results of a double-blind, placebo-controlled phase III trial. Ann Oncol 2006;17:289–96.
162.Triplett DA. Protean clinical presentation of antiphospholipid-protein antibodies (APA). Thromb Haemost 1995;74: 329-37.
163.Helmerhorst FM, Bloemenkamp KWM, Rosendaal FR, et al. Oral contraceptives and thrombotic disease: risk of venous thromboembolism. Thromb Haemost 1997;78: 327–33.
164.Hoibraaten E, Qvigstad E, Arnesen H, et al. Increased risk of recurrent venous thromboembolism during hormone replacement therapy—results of the randomized, double-blind, placebo-controlled Estrogen in Venous Thromboembolism Trial (EVTET). Thromb Haemost 2000;84:961–7.
165.Vandenbroucke JP, Rosing J, Bloemenkamp KW, et al. Oral contraceptives and the risk of venous thrombosis. N Engl J Med 2001;344:1527–35.
166.Rosendaal FR, Vessey M, Rumley A, et al. Hormonal replacement therapy, prothrombotic mutations and the risk of venous thrombosis. Br J Haematol 2002;116:851–4.
167.Mewissen MW, Seabrook GR, Meissner MH. Catheter-directed thrombolysis for lower extremity deep venous thrombosis: report of a national multicenter registry. Radiology 1999;211:39–49.
168.Delis KT, Bountouroglou D, Mansfi eld AO. Venous claudication in iliofemoral thrombosis: long-term effects on venous hemodynamics, clinical status, and quality of life. Ann Surg 2004;239:118–26.
169.Ridker PM, Goldhaber SZ, Danielson E, et al. Long-term, low-intensity warfarin therapy for the prevention of recurrent venous thromboembolism. N Engl J Med 2003;348: 1425–34.
170.Kearon C, Ginsberg JS, Kovacs MJ, et al. Comparison of low-intensity warfarin therapy with conventional-intensity warfarin therapy for long-term prevention of recurrent venous thromboembolism. N Engl J Med 2003; 349:631–9.
171.Levine MN, Hirsh J, Gent M, et al. Optimal duration of oral anticoagulant therapy: a randomized trial comparing four weeks with three months of warfarin in patients with proximal deep vein thrombosis. Thromb Haemost 1995;74: 606–11.
172.Schulman S, Rhedin AS, Lindmarker P, et al. A comparison of six weeks with six months of oral anticoagulant therapy after a fi rst episode of venous thromboembolism. N Engl J Med 1995;332:1661–5.
173.Pinede L, Ninet J, Duhaut P, et al. Comparison of 3 and 6 months of oral anticoagulant therapy after a fi rst episode of proximal deep vein thrombosis or pulmonary embolism and comparison of 6 and 12 weeks of therapy after isolated calf deep vein thrombosis. Circulation 2001;103:2453–60.
174.Campbell IA, Bentley DB, Prescott RJ, et al. Anticoagulation for three versus six months in patients with deep vein thrombosis or pulmonary embolism, or both: randomised trial. BMJ 2007;334:674–80.
175.Kearon C, Kahn SR, Agnelli G, et al. Antithrombotic therapy for venous thromboembolic disease: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2008;133:454S–545S.
176.Linkins LA, Choi P, Douketis JD. What is the clinical impact of bleeding in patients with venous thromboembolism who are receiving oral anticoagulant therapy? An assessment of case-fatality and risk of major bleeding. Ann Intern Med 2003;139:893–900.
177.Bauer KA. Annotiation: Low intensity warfarin: is it clinically useful in venous thromboembolism management? Br J Haematol 2004;127:155–8.
178.Agnelli G, Prandoni P, Santamaria MG, et al. Three months versus one year of oral anticoagulant therapy for idiopathic deep venous thrombosis. Warfarin Optimal Duration Italian Trial Investigators. N Engl J Med 2001; 345:165–9.
179.Verhovsek M, Douketis JD, Yi Q, et al. Systematic review: D-dimer to predict recurrent disease after stopping anticoagulant therapy for unprovoked venous thromboembolism. Ann Intern Med 2008;149:481–90.
180.Gross PL, Weitz JI. New antithrombotic drugs. Clin Pharmacol Ther 2009;86:139–46.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.