(Carregando Índice)... (Carregando Índice)... |
Última revisão: 30/01/2018
Comentários de assinantes: 0
|
Artigo original: Ekwall, A, MD, PhD. Firestein, G, MD. Rheumatoid Arthritis: Etiology and Pathogenesis [The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2015 Decker Intellectual Properties Inc. All Rights Reserved.] Tradução: Paulo Henrique Machado. Revisão técnica: Dr. Lucas Santos Zambon. |
Anna-Karin H. Ekwall, MD, PhD
Especialista em Reumatologia na University of California San Diego School of Medicine (La Jolla, CA). Pesquisadora/Postdoc em The Sahlgrenska Academy at Gothenburg University (Gothenburg, Sweden).
Gary S. Firestein, MD
Professor de Medicina, Reitor e Vice-Reitor Associado de Medicina Translacional na UC San Diego School of Medicine (La Jolla, CA).
Resumo
A artrite reumatoide (AR) está entre as formas mais comuns de artrite inflamatória crônica. Esse tipo de doença afeta, aproximadamente, 1% de adultos, sendo que é duas as três vezes mais prevalente em mulheres. Não há testes laboratoriais específicos para AR; o diagnóstico depende de uma miríade de sinais e sintomas com suporte de sorologia e de imagens radiográficas. A doença evolui durante muitos anos como consequência de estresse ambiental constante, causando inflamação e ativação imune, seguidas de um colapso na tolerância em indivíduos com históricos genéticos específicos. Essa descrição é uma definição de AR de sua etiologia, incluindo genética, infecções, papel do tabagismo e citrulações de proteínas, e mecanismos epigenéticos; e sua patogênese, incluindo histopatologia sinovial, danos nos ossos e nas cartilagens, imunidade adaptativa e inata, e o papel das citocinas e da sinalização intracelular. Os quadros incluem os critérios da American Rheumatism Association de 1987 para classificação de AR e a classificação de AR de 2010 dos órgãos American College of Rheumatology/European League Against Rheumatism. As figuras mostram proteínas citrulinadas em células das vias respiratórias, secção do sinóvio proliferativo de um paciente com AR clássica, e regiões com erosão na junção entre um sinóvio proliferativo inflamado e o osso.
Anna-Karin H. Ekwall, MD, PhD, e Gary S. Firestein, MD.
Definição da Doença
A artrite reumatoide (AR) está entre as formas mais comuns de artrite inflamatória crônica. Esse tipo de doença afeta, aproximadamente, 1% de adultos, sendo de duas as três vezes mais prevalente em mulheres. Em algumas populações, a AR pode iniciar na infância, embora, de um modo geral, o início ocorra na quinta ou sexta décadas de vida. Não há testes laboratoriais específicos para AR; o diagnóstico depende de uma miríade de sinais e sintomas com suporte de sorologia e de imagens radiográficas.
Tipicamente, o envolvimento das pequenas articulações das mãos e dos pés é importante para o diagnóstico. Os critérios clínicos específicos evoluíram bastante, embora, na prática, o diagnóstico seja feito por meio da observação cuidadosa do padrão de atividade da doença ao longo do tempo. Recentemente, os critérios de classificação foram revisados e o novo sistema dá grande ênfase à sorologia e ao diagnóstico precoce em pacientes com algumas articulações inchadas ou sensíveis.1
Alterações radiográficas clássicas, como as erosões marginais, deixaram de ser consideradas, ao passo que foram incluídas as evidências reveladas pela ultrassonografia na ausência de sinais e sintomas. Ainda não está suficientemente claro se os critérios revisados poderão melhorar substancialmente a especificidade da classificação clínica.
Etiologia
É pouco provável que um único fator etiológico seja responsável por todos os casos de AR em adultos. Ao contrário, a AR provavelmente evolua ao longo de muitos anos, talvez durante décadas, como consequência de estresses ambientais constantes, causando inflamação e ativação imune, seguidas do colapso na tolerância em indivíduos com históricos genéticos específicos.
As associações genéticas mais fortes nos casos de AR envolvem variações no sulco de ligação de antígenos das moléculas do complexo de histocompatibilidade principal de classe II (em inglês, major histocompatibility complex [MHC]) e afeta a imunidade adaptativa, embora outros polimorfismos genéticos ou mecanismos epigenéticos induzidos pelo meio ambiente também possam contribuir para a suscetibilidade à doença.
As influências ambientais estudadas mais intensivamente sobre a suscetibilidade à AR incluem tabagismo e outros agentes irritantes e infecções nas vias respiratórias, como, por exemplo, a periodontite. A exposição constante a esses fatores induz inflamação nos tecidos das mucosas e pode disparar respostas imunes aberrantes.
Além disso, o tabagismo e determinados tipos de infecções foram associados à modificação proteica nos tecidos afetados por meio da conversão da arginina em citrulina. Um grande grupo de pacientes com AR é definido pela presença de anticorpos peptídicos anticitrulinosos (em inglês, anti-citrullinated peptide antibodies [ACPAs]) no sangue. Os ACPAs são altamente específicos para AR e podem ser detectados no soro de pacientes em até 10 anos antes do surgimento da doença clínica.
As influências ambientais vão muito além das exposições tóxicas. Alguns estudos recentes envolvem o microbioma ? somatório de todas as comunidades bacteriológicas ecológicas que povoam locais como a pele humana, a cavidade oral, as vias respiratórias, o trato gastrintestinal e o trato geniturinário ? como fator ambiental importante com forte influência no desenvolvimento de doenças imunomediadas como a AR.
Algumas evidências dão suporte ao conceito de que as inflamações associadas à AR possivelmente tenham origem nos tecidos das mucosas, principalmente nos brônquios, nos pulmões, nas gengivas e nos intestinos, como resultado de respostas aberrantes ou de respostas imunes desreguladas a patógenos e a modificações pós-translacionais a proteínas em tecidos inflamados ou devido a um desequilíbrio entre microbiotas protetores e pró-inflamatórios.2
O processo não ocorre de forma isolada; ao contrário, exige grupos específicos de fatores pré-determinados, notadamente genes herdados que predispõem para a hiper-reatividade imune.
Genética
A estrutura genética desempenha papel importante na suscetibilidade à AR, embora esteja bastante claro que os genes herdados não são os únicos determinantes da suscetibilidade. Por exemplo, gêmeos idênticos são apenas 12 a 15% concordantes para a doença; os parentes de primeiro grau de pacientes com AR têm um aumento de duas a três vezes na incidência da doença.
A ligação genética mais proeminente nos casos de AR envolve um único peptídeo no complexo de histocompatibilidade principal de classe II. Esse epítopo de suscetibilidade está associado à terceira região hipervariável das cadeias DR ß do antígeno leucocitário humano (em inglês, human leukocyte antigen [HLA]), que contém uma sequência específica de aminoácidos de 70 a 74 (glutamina-leucina-arginina-alanina-alanina, também conhecida por QKRAA ou epítopo compartilhado) encontrada em DRB1*0401, DRB1*0404 e em outros alelos relacionados à AR.3
Essa sequência é encontrada na maior parte dos pacientes com AR; ela também está associada ao aumento na gravidade da doença, embora se desenvolva em apenas uma pequena fração de pacientes com a sequência QKRAA. Os indivíduos com esse tipo de epítopo têm um risco aumentado em quatro a seis vezes de desenvolver AR e, o mais interessante, é que a associação entre epítopo compartilhado e AR está fortemente relacionada à expressão de ACPAs.4
Esse tipo de fenômeno é atribuído em parte à descoberta de que os peptídeos contendo citrulina se ligam mais avidamente do que o peptídeo nativo à bolsa ligante de QKRAA contendo moléculas de DRB1 e, consequentemente, podem se apresentar com mais eficiência além de ativar células T.
O Quadro 1 apresenta os critérios da American Rheumatism Association de 1987 para classificação de artrite reumatoide.7
Quadro 1
|
CRITÉRIOS DA AMERICAN RHEUMATISM ASSOCIATION DE 1987 PARA CLASSIFICAÇÃO DE ARTRITE REUMATOIDE |
|
Critérios Rigidez matinal: rigidez matinal nas articulações ou ao redor delas com duração de, pelo menos, 1 hora antes da melhora ideal. Artrite em três ou mais áreas articulares: pelo menos, três áreas articulares que apresentarem simultaneamente inchaço nos tecidos moles ou presença de líquidos (não apenas crescimento ósseo excessivo) observadas pelo médico; as possíveis 14 áreas articulares (nos lados direito e esquerdo) são as articulações IFP, articulações MCF, punhos, cotovelos, joelhos e articulações MTF. Artrite nas articulações das mãos: pelo menos, uma área articular inchada, conforme já mencionado, no punho, na articulação MCF ou na articulação IFP. Artrite simétrica: envolvimento simultâneo das mesmas áreas articulares (assim como na artrite em três ou mais áreas articulares) em ambos os lados do corpo (o envolvimento bilateral das articulações IFP, MCF e MTF é aceitável mesmo sem simetria absoluta). Nódulos reumatoides: nódulos subcutâneos nas proeminências ósseas, nas superfícies extensoras ou nas regiões justa-articulares que são observadas pelos médicos. Fator reumatoide sérico: demonstração de quantidades anormais do fator reumatoide no soro por qualquer método cujo resultado tenha sido positivo em menos de 5% de indivíduos de controle normais. Alterações radiográficas: alterações radiográficas típicas de AR nas radiografias da parte póstero-anterior da mão e do punho, incluindo erosões ou descalcificações ósseas inequívocas localizadas ou mais acentuadamente marcantes em relação às articulações envolvidas (as alterações osteoartríticas isoladamente não são qualificadoras). Exclusões A presença de qualquer um dos fatores a seguir exclui o diagnóstico de AR: Erupção cutânea típica de LES. Alta concentração de células de LES (quatro ou mais em dois esfregaços); entretanto, devido às descobertas frequentes de células de LES em pacientes com AR típica sob o ponto de vista clínico, a sugestão é que esses pacientes sejam colocados em listas separadas. Evidências histológicas de poliarterite nodosa com necrose segmentada de artérias, associada a infiltrações leucocitárias nodulares, que se estende no sentido perivascular, incluindo muitos eosinófilos. Inchaço muscular persistente causado por dermatomiosite ou fraqueza nos músculos do pescoço, tronco e faringe. Escleroderma definitivo (não limitado aos dedos). Quadro clínico típico de febre reumática com envolvimento articular migratório e evidências de endocardite, em especial nos casos em que a condição for acompanhada de nódulos subcutâneos, eritema marginado ou coreia (a titulação elevada de antiestreptolisina não exclui o diagnóstico de AR). Quadro clínico típico de artrite gotosa com ataques agudos de inchaço, vermelhidão e dor em uma ou mais articulações, principalmente se a condição for responsiva à colchicina. Tofos Quadro clínico típico de artrite infecciosa de origem bacteriana ou viral com focos agudos de infecção ou com uma associação próxima com alguma doença de origem infecciosa conhecida; calafrios; febre; envolvimento articular agudo que, em geral, é inicialmente migratório (em especial se os organismos estiverem presentes no líquido articular ou se houver resposta à terapia com antibióticos). Presença de bacilos da tuberculose nas articulações ou evidências histológicas de tuberculose articular. Quadro clínico típico de artrite reativa com uretrite e conjuntivite associadas ao envolvimento articular agudo que, em geral, é inicialmente migratório. Quadro clínico típico da síndrome do ombro-mão com envolvimento unilateral do ombro e da mão e inchaço difuso da mão, seguido de atrofia e contraturas. Quadro clínico típico de osteoartropatia hipertrófica com hipocratismo digital ou periostite hipertrófica, ou ambos, ao longo dos eixos dos ossos longos, principalmente na presença de lesão intrapulmonar. Quadro clínico típico de neuroartropatia com condensação e destruição dos ossos das articulações envolvidas e descobertas neurológicas associadas. Ácido homogentísico na urina detectável por meio de alcalinização. Evidências histológicas de sarcoide ou teste de Kveim positivo. Mielomas múltiplos evidenciados por aumento acentuado nas células plasmáticas na medula óssea ou pela presença da proteína de Bence-Jones na urina. Lesões cutâneas típicas de eritema nodoso. Leucemia ou linfoma com células características no sangue periférico, na medula óssea ou nos tecidos. Quadro clínico típico de espondilite anquilosante, psoríase, colite ulcerativa ou enterite regional. |
IFP: interfalângicas proximais; LES: lúpus eritematoso sistêmico; MCF: metacarpofalângicas; MTF metatarsofalângicas.
Para fins de classificação, considera-se que o paciente tenha AR ou atenda a, pelo menos, quatro dos critérios mencionados. A presença dos primeiros quatro critérios é imprescindível durante, ao menos, 6 semanas. Os pacientes com dois diagnósticos clínicos não são excluídos. Não é necessário fazer a designação de AR clássica, definitiva ou provável.
Portanto, a associação com o complexo de histocompatibilidade principal (MHC) talvez não seja um fator de risco independente para AR. Pelo contrário, aparentemente o MHC está associado à produção de ACPAs, que, por sua vez, são fatores de risco reais para a suscetibilidade e a gravidade da doença.5
O mapeamento detalhado das associações genéticas entre o MHC e AR identificou recentemente sinais ainda mais fortes com as posições 11 e 13 dos aminoácidos, fora do epítopo compartilhado, que se localizam no assoalho do sulco de ligação de antígenos da molécula DRB1 do HLA.6
Essa descoberta dá suporte à hipótese de que a associação entre MHC e suscetibilidade para AR se relaciona, pelo menos em parte, com a ligação de antígenos, em comparação com outras funções das moléculas DRB1. Levando-se em consideração que as moléculas HLA-DR também participam dos contatos da célula apresentadora de antígenos contendo células T, poderia também haver influências adicionais do MHC devido à formatação do repertório de células T ou à apresentação inicial de antígenos para as células T.
Provavelmente, a suscetibilidade para AR seja poligênica e um alvo recente; outros estudos genômicos amplos identificaram vários outros alelos associados à doença. Entre esses alelos, dois deles aumentam duas vezes ou mais o risco de incidência da doença. Determinados haplótipos estendidos da peptidilarginina deiminase tipo 4 (PADI-4), que é um dos quatro genes PADI que convertem arginina em citrulina, estão associados ao desenvolvimento de AR nas populações asiáticas e norte-americana.7
Entretanto, essa associação não foi encontrada em populações na Europa Ocidental. O segundo é um polimorfismo no PTPN22, um gene da fosfatase, que regula a função das células T, e também foi associado a diversas doenças autoimunes, incluindo AR. Paradoxalmente, o alelo associado à doença (R620W) aparenta ser um ganho no polimorfismo funcional.
Esse tipo de alelo é muito raro na Ásia, indicando que diferentes grupos étnicos e raciais possivelmente tenham genes distintos que os predisponham para AR.8,9 O fato de que esses dois genes maiores da suscetibilidade são principalmente importantes em populações étnicas e raciais específicas é um exemplo das dificuldades que se encontra nas tentativas de definir conjuntos fixos de genes como causadores da AR. Pode ser necessária uma combinação de muitos genes e de influências ambientais.10
O Quadro 2 contém os critérios Classificação de Artrite Reumatoide da ACR/EULAR de 2010: algoritmo com base em pontuações para classificação em pacientes elegíveis (>6/10 sugere AR).
Quadro 2
|
CRITÉRIOS DE CLASSIFICAÇÃO DE ARTRITE REUMATOIDE DA ACR/EULAR DE 2010: ALGORITMO COM BASE EM PONTUAÇÕES PARA CLASSIFICAÇÃO EM PACIENTES ELEGÍVEIS (> 6/10 SUGERE AR) | |
|
Critérios |
Pontos |
|
Envolvimento articular* 1 articulação média a grande? 2?10 articulações médias a grandes 1?3 articulações pequenas? (com ou sem envolvimento de grandes articulações) 4?10 articulações pequenas (com ou sem envolvimento de grandes articulações) >10 articulações§ (pelo menos, uma articulação pequena) |
(0?5) 0 1 2 3 5
|
|
Sorologia?¶ FR negativa e ACPA negativo FR baixa positiva e ACPA baixo positivo FR alta positiva ACPA alto positivo
|
(0?3) 0 2 |
|
Reagentes na fase aguda?# CRP normal e VHS normal CRP anormal e VHS anormal Duração dos sintomas** >6 semanas =6 semanas |
(0?1) 0 1 (0?1) 0 1 |
ACPA: anticorpos peptídicos anticitrulinosos; ACR: American College of Rheumatology; AR: artrite reumatoide; CRP: proteína C-reativa; FR: febre reumatoide; Eular: European League Against Rheumatism; VHS: velocidade de hemossedimentação.
Os dados para elaboração desse quadro foram reproduzidos com permissão de Aletaha D, Neogi T, Silman AJ et al. Critérios para classificação de AR: uma iniciativa colaborativa do American College of Rheumatology/European League Against Rheumatism. Arthritis Rheum 2010;62:2569-81.
*Envolvimento articular se refere a qualquer articulação inchada ou sensível no exame ou evidências de sinovite nas imagens por ressonância magnética ou na ultrassonografia. As articulações interfalângicas distais, a primeira articulação carpometacarpal, e a primeira articulação metatarsofalângica são excluídas da avaliação. As categorias de distribuição articular são classificadas de acordo com a localização e o número de articulações envolvidas, sendo que a colocação na categoria mais alta possível se baseia no envolvimento articular.
?Articulações médias a grandes se referem a ombros, cotovelos, quadris, joelhos e tornozelos.
?Articulações pequenas se referem a articulações metacarpofalângicas, articulações interfalângicas proximais, 2?5 articulações metatarsofalângicas, articulações interfalângicas do polegar e punhos.
§Nessa categoria, pelo menos uma entre as articulações envolvidas deve ser uma articulação pequena; as demais articulações incluem qualquer combinação de articulações grandes e articulações pequenas adicionais, assim como outras articulações que eventualmente não constarem de nenhuma outra lista (por exemplo, temporomandibular, acromioclavicular, esternoclavicular).
? Os indivíduos devem ser pontuados com base nesses critérios se, pelo menos, estiver disponível o resultado de um teste sorológico e ao menos de um teste de reagente na fase aguda. Nas situações em que o valor de um teste de reagente na fase aguda não estiver disponível, o resultado deverá ser considerado negativo/normal.
¶Negativo se refere a valores da unidade internacional (IU) mais baixos que o valor do limite superior da normalidade (ULN) para testes ou ensaios laboratoriais; positivo baixo se refere aos valores da IU maiores que o ULN, porém três vezes ou mais elevados que o valor do ULN para testes ou ensaios laboratoriais; positivo alto se refere aos valores da IU três vezes maiores que o valor do ULN para testes ou ensaios laboratoriais; nas situações em que a FR estiver disponível apenas como valores positivos ou negativos, o valor positivo deve receber a menor pontuação possível para a condição.
#Normal/anormal é determinado por padrões laboratoriais locais.
**Duração dos sintomas se refere aos autorrelatos dos pacientes sobre o tempo de duração dos sinais ou sintomas de sinovite (p.ex., dor, inchaço, sensibilidade) nas articulações clinicamente envolvidas no momento da avaliação.
Há uma grande variedade de genes adicionais envolvidos nos casos de AR, sendo que mais de 100 foram identificados nos últimos anos.11 A maior parte desses genes aumenta em torno de 1,06 a 1,2 vezes o risco relativo de desenvolver AR. Os genes tendem a se agrupar ao redor da função imune. Por exemplo, os polimorfismos dos genes sinalizadores da citocina, como os genes STAT4 e TRAF1, foram identificados e replicados em diversas coortes.12
Outros genes se relacionam ao recrutamento celular (quimiocinas, tais como a CCL21), coativação de células T (CD28 e CTLA4), receptores de Fc (FCGR2A), e muitos mais. Ainda não está muito claro qual é a combinação de genes necessária ou se polimorfismos específicos se correlacionam com respostas clínicas aos tratamentos à base de terapias-alvo como os bloqueadores do fator de necrose tumoral (em inglês, tumor necrosis fator [TNF]), bloqueadores da coestimulação de células T e depleção de células T.
Infecções
Infecções diretas
Há poucas evidências sobre o envolvimento de uma única entidade infecciosa no sinóvio como causa de AR. Entretanto, acredita-se que alguns organismos patogênicos específicos contribuam para o início da doença. Por exemplo, partículas que se assemelham a vírus foram isoladas em efusões sinoviais em casos de AR, e alguns pacientes portadores desse tipo de doença apresentam evidências de alguma infecção recente produzida pelo parvovírus B19.13
Alguns estudos não mostraram nenhuma correlação entre AR e evidências sorológicas de infecção anterior ou a presença de genes B19 no tecido sinovial, ao passo que outros estudos demonstraram a presença de proteínas do parvovírus B19 no sinóvio de pacientes com AR.14 O parvovírus B19 pode também infectar fibroblastos sinoviais cultivados e aumentar a invasão no interior da matriz cartilaginosa.
Outros vírus que foram isolados do líquido sinovial incluem o vírus da rubéola e o vírus Epstein-Barr (EBV). A supressão de infecções pelo EBV causadas por linfócitos de pacientes com AR chega a ser afetada, possivelmente porque as células T criam respostas insuficientes com níveis baixos de interferon ?.
Embora provavelmente não seja o evento inicial da AR, a infecção pelo EBV contribui potencialmente para estimulações imunológicas persistentes agindo como ativador policlonal de células B, aumentando, por conseguinte, a produção de autoanticorpos.
Os linfócitos de alguns pacientes com AR respondem a uma região da glicoproteína gp110 do EBV que contém a mesma sequência de aminoácidos QKRAA como epítopo de suscetibilidade nas cadeias de HLA-DRß.15 Consequentemente, o mimetismo molecular pode levar a um estado de sub-imunidade em determinados indivíduos infectados pelo EBV.
Outras xenoproteínas, mais notadamente a proteína J do DNA do organismo Escherichia coli, também contêm a sequência QKRAA e poderão contribuir para as respostas contra o complexo de histocompatibilidade principal.
Os retrovírus também podem produzir doenças que se assemelham à AR. A infecção causada pelo vírus linfotrópico sinovial da célula T tipo I (HTLV-I) está associada à artrite crônica, sendo que a transdução in vitro de sinoviócitos com o gene tax do HTLV-I intensifica o processo de crescimento.16 Partículas semelhantes ao retrovírus foram observadas em algumas amostras sinoviais e a expressão das proteínas zinco-dedo, associadas a infecções retrovirais, dão algum suporte a essa hipótese.17
Infecções Indiretas
Infecções nas mucosas como a periondotite poderão levar à imunidade contra proteínas citrulinadas em sítios mucosos. Há uma prevalência e uma gravidade cada vez maiores de periodontite em pacientes com AR, em especial nos casos de doença positiva para ACPAs. A bactéria associada, porphyromonas gingivalis, expressa a peptidilarginina deiminase da P. gingivalis (gPAD), que pode citrulinar o fibrinogênio humano e a a-enolase humana.18
Além disso, os anticorpos gerados contra a enolase bacteriana têm potencial para entrar em reação cruzada com a a-enolase citrulinada endogenamente, representando, por conseguinte, um exemplo de mimetismo molecular.
Microbioma
Nos últimos anos, evidências cada vez maiores indicam que o microbioma intestinal não apenas é responsável pela formatação do sistema imune inato e adaptativo, mas também pode promover inflamações se houver alguma perturbação no equilíbrio entre diferentes espécies de bactérias (disbiose).19
Por exemplo, algumas espécies, como a espécie Bacteroides fragilis, dão suporte a respostas anti-inflamatórias por meio da produção reguladora de células T e de interleucina-10 (IL-10), enquanto que outras espécies são pró-inflamatórias.
Vários modelos animais de artrite são menos graves ou são preservados nas situações em que os camundongos são mantidos em ambientes sem germes, demonstrando que a inflamação possivelmente dependa da presença de um único comensal com residência no intestino.20,21 Alguns estudos recentes indicam que há uma disbiose nos microbiotas intestinais de pacientes com AR logo no início da doença, com grande abundância da espécie Prevotella.22,23
Tabagismo e Citrulinação de Proteínas
Entre todas as influências ambientais sobre a AR, o tabagismo é a mais bem definida talvez porque ative as enzimas peptidilarginina deiminase (PAD) e induza a citrulinação de proteínas na mucosa da via respiratória inflamada.24 Há uma clara interação entre tabagismo e genes para aumentar o risco de AR.4
Embora o tabagismo e os genes de suscetibilidade do alelo HLA-DR aumentem individualmente, algumas vezes, a suscetibilidade à AR, a combinação de tabagismo e a sequência QKRAA do “epítopo compartilhado” aumenta o risco em 40 vezes. É interessante observar que essa interação entre determinados genótipos e o tabagismo está presente apenas em indivíduos positivos para os ACPAs.
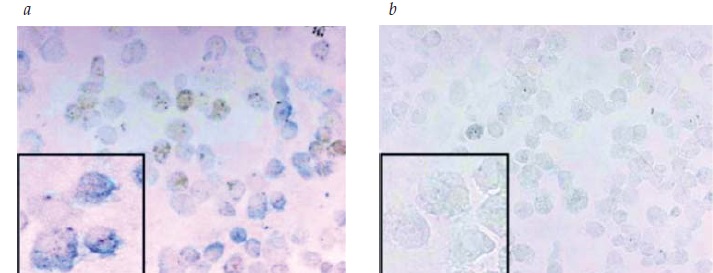
A Figura 1 mostra as proteínas citrulinadas em células das vias respiratórias de um fumante em comparação com um não fumante, detectadas por imuno-histoquímica.

ACPA: anticorpos peptídicos anticitrulinosos.
Figura 1 - Proteínas citrulinadas em células das vias respiratórias de (A) um fumante em comparação com (B) um não fumante, detectadas por imuno-histoquímica (cor azul). Esses peptídeos alterados são apresentados às células T e podem induzir a produção de ACPAs. Essa figura foi reproduzida com permissão de Makrigiannakis D et al.24
Recentemente, foi desenvolvido um novo modelo para o desenvolvimento de AR com base em antígenos alterados e em ACPAs.25 Os peptídeos citrulinados dos pulmões podem ser processados em células apresentadoras de antígenos. Esses peptídeos alterados se ligam mais avidamente aos genes do alelo HLA-DR associados à AR em comparação com os peptídeos nativos, aumentando a eficiência da apresentação às células T.
Esses linfócitos ativados podem estimular a produção de ACPAs por meio das células B, aumentando a prevalência de inflamação pulmonar detectada através de imagens por ressonância nuclear magnética e biópsias do pulmão.26 Observa-se também uma incidência cada vez maior de estruturas germinais centrais com evidência de produção de ACPAs nos brônquios de pacientes com AR.27
Outro estudo mostrou a presença de fatores reumatoides e de ACPAs no esputo de indivíduos sem qualquer histórico ou sinais de artrite,28 indicando que as respostas imunes às proteínas citrulinadas possivelmente sejam eventos pré-clínicos precoces.
Cabe observar que essas respostas não representam necessariamente uma falha nos processos de tolerância imune central, tendo em vista que são direcionadas para os neoepítopos de proteínas modificadas. Em última análise, a disseminação epitópica poderá levar a um colapso de tolerância e produzir sequelas adicionais de autoimunidade direcionada contra a proteína nativa.
Os ACPAs presentes em pacientes com AR têm uma grande variedade de especificidades contra proteínas endógenas distintas.29 O papel fundamental das associações genéticas é dar suporte aos estudos de caso-controle demonstrando a alta prevalência (48%) de ACPAs nos soros de parentes de primeiro grau de pacientes com AR, sendo que 41% desses indivíduos eram positivos para a IgA ACPA,30 que é o isótopo da imunoglobulina produzida nos tecidos das mucosas.
Outros Fatores Ambientais
Outros fatores relacionados ao estilo de vida também protegem contra o desenvolvimento de AR. Com embasamento em dados de dois grandes estudos de caso-controle independentes, o uso de álcool foi associado a uma redução significativa no risco de AR.31 O consumo de etanol neutraliza também a interação entre tabagismo e o epítopo compartilhado HLA-DRB1 para o desenvolvimento de AR, embora o mecanismo ainda não tenha sido definido.
O efeito do uso de contraceptivos orais é menos certo; alguns estudos sugerem que não há nenhum risco, ao passo que outros estudos sugerem que ocorre uma redução na prevalência de AR em mulheres que utilizam essa forma de controle da natalidade.
Há também a possibilidade de que eventos estocásticos possam participar na etiologia desse tipo de doença. Por exemplo, uma chance aleatória poderia permitir o escape de clones individuais de células T patogênicas da deleção tímica e permitir o desenvolvimento posterior de autoimunidade com estímulos ambientais apropriados.
Influência Epigenética
Mecanismos epigenéticos como metilação de DNA, modificações nas histonas e produção de micro-RNA podem influenciar profundamente a expressão genética e afetar os processos celulares em resposta aos estímulos ambientais, incluindo fatores dietéticos e estresse.
No que diz respeito à AR, alguns estudos indicam a presença de anomalias na metilação do DNA em células do sangue periférico, embora dados mais recentes tenham demonstrado que há apenas a assinatura da metilação de DNA em sinoviócitos semelhantes a fibroblastos (em inglês, fibroblast-like synoviocytes [FLS]).32
Os FLSs, juntamente com os sinoviócitos semelhantes a macrófagos, formam o revestimento sinovial e as camadas do subrevestimento articular e ajudam a regular a homeostase tecidual por meio da remodelação da matriz extracelular. No que diz respeito à AR, os FLSs são ativados e apresentam comportamento agressivo através da mediação na formação de membranas e da destruição articular.33,34
Alguns estudos envolvendo sinoviócitos semelhantes a fibroblastos em casos de AR registraram a presença de expressão anormal do microRNA (miR146 e miR155) e hipometilação de determinados tipos de genes, como o CXCL12 e o receptor 3 da morte, com impacto potencial sobre a patogênese da AR.
O fato mais intrigante é que, ao se utilizar uma abordagem completa de genomas, demonstrou-se que há uma assinatura de metiloma específica para FLS de AR em comparação com FLS normal e FLS de pacientes com osteoartrite, envolvendo genes e vias que são reconhecidamente importantes nos casos de AR.35,36
A análise das vias de loci metilados diferencialmente mostrou que há uma grande variedade de eventos patogênicos, incluído adesão de células, regulação matricial e sinalização de células. Ainda permanece desconhecida a extensão em que as modificações epigenéticas poderão afetar o comportamento dos FLSs da AR e influenciar o desenvolvimento e a gravidade da doença, embora os dados indiquem a existência de um mecanismo potencial de impressão celular nesse tipo de artrite.
Patogênese
Histopatologia Sinovial e Invasão
Nos casos de AR, o tecido sinovial é acentuadamente hiperplásico, com dobras redundantes, vilosidades frondais e edema. Nos estágios iniciais, a proliferação de vasos sanguíneos e de danos endoteliais é perfeitamente visível. A hiperplasia do revestimento íntimo sinovial (região que mantém contato direto com líquido sinovial) é uma ocorrência provável, mesmo nas situações em que o infiltrado inflamatório no subrevestimento for brando.
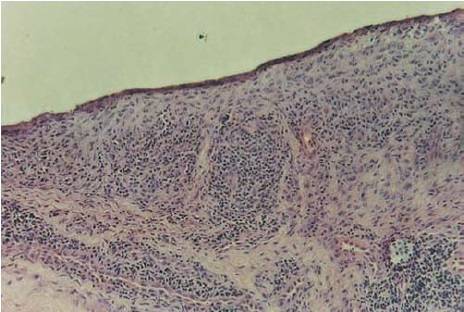
Na medida em que a doença evolui, a hiperplasia no revestimento interno se torna mais proeminente, aumentando em até cinco vezes em relação à espessura normal de uma ou duas camadas celulares, como mostra a Figura 2. A hiperplasia no revestimento sinovial é causada, em parte, pela proliferação local de FLSs tipo B e pela migração de novos sinoviócitos tipo A, semelhantes a macrófagos, a partir da medula óssea e do fluxo sanguíneo para as articulações.
Figura 2 - Esta secção do sinóvio proliferativo de um paciente com AR clássica revela a presença de hiperplasia no revestimento sinovial e uma infiltração e agregação linfocitária no revestimento interno. Coloração da fosfatase alcalina x 250 de amplificação em relação ao original.
As células tipo B expressam a proteína de adesão caderina-11, que é responsável pela agregação de sinoviócitos e pela formação da camada de revestimento da túnica íntima.37 A taxa de morte celular também determina a celularidade tecidual. Muitas células do revestimento da túnica íntima contêm DNA danificado, que normalmente leva à apoptose (morte celular programada). Um número relativamente pequeno de células completa esse processo, possivelmente como resultado de uma apoptose defeituosa, contribuindo para a hiperplasia.38
Nos casos de AR crônica, as células inflamatórias (incluindo células T, células B, macrófagos e células plasmáticas) se acumulam na região do revestimento interno. Os linfócitos se organizam em agregados discretos, embora seja comum a presença de infiltração difusa de células mononucleares ou de tecido fibroso relativamente acelular.
A maior parte das células T é formada por células de memória CD4+ com pequenos núcleos e citoplasmas escassos. Embora as células sejam quiescentes sob o ponto de vista funcional, muitas delas expressam antígenos superficiais, que sugerem ativação prévia. O aumento no número de vasos sanguíneos ainda é uma descoberta importante na fase crônica.
Estudos morfométricos dos vasos capilares sugerem que a rede de capilares é mais desorganizada que o normal, sendo que o volume tecidual ultrapassa a proliferação de vasos sanguíneos. Normalmente, a sinovite reumatoide é acompanhada de um aumento nas efusões sinoviais. A contagem de leucócitos no líquido sinovial nos casos de AR ativa é de, aproximadamente, 10.000/mL (cerca de 70% de neutrófilos).
Ao contrário do sinóvio, há mais células T DC8+ que células T DC4+ nas efusões sinoviais. Os leucócitos polimorfonucleares são arrastados para o interior do líquido articular ao longo de um gradiente formado por substâncias quimiotáticas, tais como o leucotrieno B4, fator de ativação plaquetária, fragmento de complemento C5a e quimiocinas incluindo a IL-8.
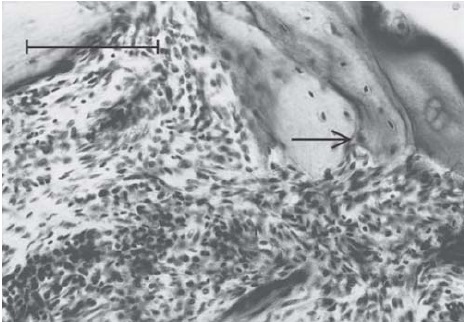
Linfócitos, macrófagos e células de revestimento também são observados no líquido sinovial. Surpreendentemente, o sinóvio nos casos de AR possui uma quantidade muito pequena de neutrófilos, mesmo que eles sejam abundantes nas efusões. A membrana, uma região invasiva do sinóvio que sofre erosão na cartilagem e no osso, como se pode ver na Figura 3, contém macrófagos e células mesenquimais primitivas, porém possui poucos linfócitos.
Figura 3 - Na junção entre um sinóvio reumatoide proliferativo inflamado e o osso, observam-se regiões cobertas por erosão (seta). Essa secção foi colorida com hematoxilina e eosina (escala da barra = 100 mm).
As células invasivas possuem características distintas, como, por exemplo, uma alta expressão da molécula-1 de adesão celular nos vasos e de CD55 (fator de ativação da deterioração).39 Há também descrições de células-tronco mesenquimais no tecido sinovial da AR; essas células expressam proteínas superficiais distintas (por exemplo, receptores de proteínas morfogenéticas e endoglina); além disso, poderão migrar para o interior do sinóvio através dos poros do osso cortical ou do sangue em circulação.40
Danos nos Ossos e nas Cartilagens
Os osteoclastos são responsáveis pelas erosões ósseas, ao passo que os sinoviócitos semelhantes a fibroblastos são as células primárias que degradam a cartilagem e outras estruturas de apoio das articulações.41 O ativador do receptor do fator nuclear kB (RANK) e o ligante do RANK (RANKL) têm papéis importantes na ativação de osteoclastos locais e na destruição óssea; outros papéis contribuintes são desempenhados pelo TNF, pelo fator de estimulação de colônias de macrófagos (M-CSF), pela interleucina-l (IL-6) e por outras citocinas que dão suporte à maturação de osteoclastos.
O sistema RANKL/RANK é compensado pela osteoprotegerina (OPG) que é um inibidor natural; as articulações de indivíduos com AR são relativamente deficientes em OPG, sendo que essa deficiência contribui para as erosões nos ossos. Em modelos animais de artrite, a administração de OPG diminui acentuadamente a destruição óssea, mesmo nos casos em que a inflamação não seja afetada.42
Os danos cartilaginosos são atribuídos à produção de várias famílias de enzimas, incluindo a protease de serina e as catepsinas, por sinoviócitos que se localizam no revestimento da túnica íntima. As enzimas mais destrutivas são as metaloproteinases (por exemplo, colagenase, estromelisina e gelatinase) e as catepsinas, que podem degradar as proteínas estruturais principais das articulações. Citocinas como a IL-1 e o TNF são indutores potentes da expressão do gene das metaloproteinases.
Embora os inibidores da protease, como os inibidores teciduais da metaloproteinase (TIMPs), sejam expressos pelo revestimento sinovial reumatoide, o equilíbrio entre proteases e inibidores aparentemente favorece as proteases nos casos de AR. Os condrócitos da cartilagem e os neurófilos do líquido sinovial também são fontes de proteases que contribuem para os danos articulares.
A destruição da matriz extracelular pelo sinóvio reumatoide possivelmente seja uma resposta normal ao meio da citocina inflamatória ou o resultado da função sinoviocítica anormal.43 Os sinoviócitos semelhantes a FLSs da AR apresentam um fenótipo agressivo atípico que se caracteriza pelo crescimento independente da adesão e pela perda de inibição de contato in vitro.
Os FLSs cultivados que forem implantados concomitantemente com explantes de cartilagem em camundongos portadores de doença de imunodeficiência combinada grave invadem a matriz de cartilagem, enquanto que o FLS de osteoartrite e os fibroblastos dérmicos normais não fazem esse tipo de invasão.44 É interessante observar também que essas células anormais migram para a cartilagem em outras localizações, com reminiscências de tumores metatastáticos.45
As mutações somáticas nos genes que codificam as proteínas reguladoras mais importantes, como o gene supressor tumoral TP53, ou as mitocôndrias podem contribuir para o fenótipo agressivo do FLS em casos de AR.46 Provavelmente, essas anormalidades sejam produzidas pela alta concentração local de oxigênio reativo e nitrogênio na articulação reumatoide.
Imunidade Adaptativa
Linfócitos
O subgrupo de células T que povoam o sinóvio reumatoide possivelmente forneça algumas indicações sobre a desregulação imune na AR. A maior parte das células T sinoviais têm o fenótipo da célula T helper tipo 1 (Th1), que é marcado pela expressão de receptores de quimiocina como CCR5 e CXCR3.47
Essas células regulam a hipersensibilidade tardia e produzem citocinas clássicas de células T como a IL-2 e o interferon ?. As células T helper tipo 2 (Th2) e as respectivas citocinas (como IL-4 e IL-5) são responsáveis por alergias e doenças atópicas e são acentuadamente reduzidas ou ausentes nas articulações reumatoides.
Um fenótipo interessante das células T envolvido na AR é a célula T helper tipo 17 (Th17) que produz a citocina pró-inflamatória IL-17 e as proteínas relacionadas.48 Essa linhagem de células influencia a autoimunidade, sobretudo em modelos de camundongos da doença. As células T não diferenciadas poderão ser impulsionadas na direção desse fenótipo por meio da exposição à IL-1, à IL-6 e/ou à IL-23, que estão presentes no sinóvio da AR.
O nível da IL-17 é elevado no soro e no líquido sinovial de pacientes com AR, sendo que os níveis correlacionados com a doença e com a presença de ACPAs, assim como a superexpressão de IL-17 in vivo em modelos murinos de artrite, promovem inflamação nas articulações, destruição de cartilagens e erosão nos ossos.49 Entretanto, nos casos de AR, os benefícios das abordagens com foco na IL-17 são modestos em comparação com os efeitos demonstrados em testes de psoríase.
As células T reguladoras (Tregs) são células que diferenciam sob a influência do fator de crescimento transformador ß (em inglês, transformating growth factor-ß [TGF-ß]) e podem suprimir a ativação de outras células T no ambiente local.
Aparentemente, a função das Tregs é reduzida nos casos de AR, o que poderá ser parcialmente revertido pelos agentes bloqueadores do fator de necrose tumoral. Ainda não foi definido se a função de Tregs com defeito representa um defeito primário na AR ou se é o resultado do meio das citocinas reumatoides.50
As células T sinoviais e o líquido sinovial geralmente são menos responsivos que as células T do sangue periférico.51 Por exemplo, a proliferação de linfócitos no líquido sinovial, em resposta a mitógenos ou à retirada de antígenos (por exemplo, toxoide tetânico), é significativamente mais baixa que a proliferação de células T no sangue periférico.
A produção in vitro de citocinas (por exemplo, interferon ? e IL-2) pelas células T do líquido sinovial também é baixa após a estimulação por mitógenos inespecíficos. O mecanismo das respostas das células T com defeito na AR possivelmente esteja relacionado ao equilíbrio irregular do redox intracelular, que, por sua vez, interfere na transdução de sinal do receptor de células T.52
Os antígenos micobacterianos e o gene 60 kDa da proteína de choque térmico são exceções às respostas geralmente baixas das células T observadas nos casos de AR, tendo em vista que a proliferação em resposta a esses antígenos é maior em células de efusões reumatoides do que nas células T do sangue periférico.
Entretanto, as respostas a esses antígenos não são específicas de AR; na realidade, a resposta é ainda mais proeminente em casos de artrite reativa. O comprimento do telômero nas células T reumatoides é menor em comparação com as células T normais, sugerindo que as células T reumatoides foram sequenciadas através de múltiplas divisões ou que tenham apresentado sinais de “envelhecimento” prematuro.
A desregulação imune foi observada nas células T no sangue periférico de pacientes com AR, sobretudo naqueles que também tinham infecção causada pelo EBV. As respostas autoimunes direcionadas para antígenos específicos das articulações podem contribuir para a incidência de sinovite.
Em alguns casos, esse efeito poderá ser intensificado nas situações em que a arginina for convertida em citrulina por meio de PADIs nas articulações. Além do colágeno tipo II, que se localiza na cartilagem hialina, há também envolvimento de outros antígenos articulares. Por exemplo, a imunidade contra células T direcionada contra a proteína cartilaginosa gp39, a proteína de ligação cartilaginosa e os proteoglicanos também foram implicados em casos de AR.
Muitos desses antígenos podem induzir artrite em camundongos ou em ratos nas situações em que esses animais forem imunizados com o antígeno em combinação com adjuvantes. Em alguns casos, as proteínas se tornam mais imunogênicas quando citrulinadas.
Anticorpos peptídicos anticitrulinosos, fatores reumatoides e outros autoanticorpos
Os anticorpos que ligam CPs são importantes para compreender a etiologia e a patogênese de AR. Muitas proteínas são citrulinadas em tecidos sinoviais (incluindo vimentina, fibrinogênio e fibronectina), sendo que a citrulinação ocorre também em modelos animais de artrite.53 PADIs são as enzimas responsáveis pela modificação pós-translacional da arginina, e determinadas isoformas, como PADI-2 e PADI-4, também são altamente expressas nos tecidos de articulações inflamadas.
Essas descobertas sugerem que a PADI tem contribuição importante no processo de desenvolvimento de AR. Embora os antígenos CP sejam geralmente produzidos em outros sítios de inflamação (como nos pulmões de fumantes), os anticorpos direcionados contra CP são muito mais específicos para AR.54 As células B que produzem IgC e ACPAs de IgM estão presentes no tecido sinovial reumatoide.
Titulações elevadas de ACPAs também se correlacionam com doença mais agressiva e mais destrutiva. Embora sejam relativamente específicos para AR, os ACPAs poderão ocorrer em outras doenças, incluindo infecções como tuberculose.
Nem todos os indivíduos positivos para ACPAs desenvolvem artrite, sendo que está bastante claro que a mera presença desses autoanticorpos não é suficiente para indução de inflamação sinovial clinicamente aparente.55 Pode ser necessário um segundo “ataque”, como algum estresse local ou uma infecção articular, que leve à citrulinação local em combinação com aumento na permeabilidade vascular causado por complexos imunes.9 O meio ambiente poderá aumentar o ingresso de ACPAs, a fixação de complementos e o início de inflamações.
O aumento na formação de armadilhas extracelulares de neutrófilos (em inglês, neutrophil extracellular traps [NETs]) foi observado em doenças inflamatórias, incluindo AR. As NETs são redes fibrosas extracelulares compostas de DNA e de moléculas antimicrobianas, incluindo proteínas citrulinadas, que são liberadas de neutrófilos como mecanismo de defesa inato cuja função é matar patógenos.
A formação de NETs, isto é, NETose, é intensificada pelo soro de pacientes com AR e, em particular, por anticorpos de vimentina anti-citrulinada.56 As NETs não apenas externalizam epítopos citrulinados que possam perpetuar respostas imunes aberrantes nas articulações, mas aumentam também as respostas inflamatórias por sinoviócitos que se assemelham a fibroblastos. Outros dados recentes sugerem que a produção de ACPAs, particularmente contra a vimentina citrulinada, poderá ativar diretamente osteoclastos e mediar a erosão óssea.57
Os fatores reumatoides são imunoglobulinas com especificidade de anticorpos para a região Fc da IgG. Os testes laboratoriais padrões utilizados em diagnósticos clínicos detectam apenas os fatores reumatoides da IgM.
Os resultados desses testes são positivos em 70 a 80% de pacientes com AR clássica, dependendo do método utilizado. Embora os pacientes com AR clássica possam apresentar resultados negativos nos testes, o resultado positivo de alta titulação indica piores prognósticos com um grau maior de danos nas articulações.
Os fatores reumatoides não são testes específicos para AR. Titulações significativas poderão ser encontradas em pacientes com outras doenças imunomediadas (por exemplo, lúpus eritematoso sistêmico [LES], esclerose sistêmica progressiva e dermatomiosite) e em pacientes portadores de distúrbios inflamatórios crônicos não reumáticos e infecções.
Pessoas idosas saudáveis, em particular as mulheres, geralmente apresentam testes positivos. Os fatores reumatoides possivelmente sejam típicos de respostas imunes precoces a muitas proteínas, facilitando a eliminação de antígenos por macrófagos.
O fator reumatoide IgM é o fator detectado com mais frequência; às vezes, observa-se também a presença do fator reumatoide IgC e, com menos frequência, o fator reumatoide IgA. A presença do fator reumatoide IgC está associada a uma taxa mais elevada de erosões ósseas e de complicações sistêmicas (por exemplo, nódulos, vasculite necrosante).
Os estudos que avaliam as sequências de aminoácidos e de DNA dão algumas dicas sobre a origem dos fatores reumatoides em casos de AR. A maturação dos fatores reumatoides na AR parece ser imune à estimulação antigênica, de acordo com a localização e os tipos de mutações somáticas nos genes da imunoglobulina.58
Entretanto, não está absolutamente certo que a IgC propriamente dita age como antígenos ou se outro epítopo relacionado seja o responsável. Os fatores reumatoides podem ser sintetizados por células B e por células plasmáticas que infiltram no sinóvio de pacientes com AR, incluindo alguns pacientes soronegativos.
Outros autoanticorpos também podem ter alguma influência na AR, incluindo os anticorpos direcionados a antígenos específicos de articulações como o gp39, o RA33 e o p205. Os anticorpos contra a glicose-6-fosfato isomerase (GPI), um antígeno amplamente distribuído, podem produzir artrite em camundongos, sendo também detectados em alguns pacientes com AR e em outros casos de artropatias inflamatórias.59
A importância das células B na AR foi recentemente confirmada pelo papel dos ACPAs e de outros anticorpos na patogênese da doença. A observação de que a depleção de células B pode resultar em melhora clínica também demonstra a importância das células B na AR. A depleção profunda de células B periféricas é acompanhada por uma redução modesta nos fatores reumatoides e nos ACPAs.
Embora não haja uma correlação próxima entre a produção de autoanticorpos e a atividade da doença, a recorrência de sinovite após o tratamento se correlaciona com o retorno de células B periféricas e com a elevação no nível de autoanticorpos potenciais. Provavelmente, a ausência de correlação seja porque as células de longa vida que produzem autoanticorpos não expressam CD20 e não sofrem depleção.
Estudos recentes de biópsia sinovial envolvendo pacientes com AR que haviam sido tratados com rituximabe mostram que houve depleção de células B no sinóvio, ainda que não tão completamente como no sangue periférico.60 A produção de autoanticorpos sinoviais e a depleção de células B nem sempre têm alguma correlação com respostas clínicas. Esses dados sugerem que as células B desempenham outras funções, como, por exemplo, apresentação de antígenos nos órgãos linfoides centrais.
Imunidade Inata
Ativação de complementos
Os complementos são ativados pela interação dos complexos imunes contendo ACPAs, fatores reumatoides ou outros complexos de anticorpos que formam as articulações; a ativação de complementos dispara uma cadeia de eventos tais como produção de anafilotoxinas e fatores quimiotáticos. A seguir, os leucócitos polimorfonucleares engolfam os complexos de complementos do fator reumatoide IgC e liberam enzimas lisossomais e outros produtos.
Os complexos do fator reumatoide IgC, que contêm IgC e componentes de complementos, são identificados de imediato no sinóvio, no líquido sinovial e nas lesões extra-articulares.61 Embora o sinóvio seja uma fonte importante de produção de complementos, os níveis de líquido sinovial reumatoide são baixos por causa do consumo local.
Deposições de imunoglobulina e de complementos foram identificadas em cartilagens avasculares e em outros tecidos colagenosos das articulações reumatoides e podem ter alguma influência na formação das lesões destrutivas da AR. Essas deposições, que são altamente específicas para AR, podem ser atraentes para as membranas invasivas.
Receptores de Reconhecimento de Padrões
Os estímulos constantes que produzem inflamação na AR possivelmente também estejam relacionados à ativação dos receptores de reconhecimento de padrões, conhecidos como receptores Toll-like (TLRs), que se localizam em macrófagos, células dendríticas, mastócitos e neutrófilos; em seguida, essa ativação pode produzir inflamações articulares inespecíficas.
Muitos produtos bacterianos, incluindo peptideoglicanos e lipopolissacarídeos, se acumulam rapidamente no sinóvio;62 esses produtos poderão contribuir para o início da AR. Os ligantes endógenos que ativam os TLRs (por exemplo, resíduos necróticos em efusões sinoviais, proteínas de choque térmico e fibrinogênio) também poderão contribuir para esse processo.
As células apresentadoras de antígenos podem ser carregadas com antígenos articulares, como, por exemplo, o colágeno tipo II ou a vimentina citrulinada, e migrar para os órgãos linfoides centrais, onde poderão ativar as células T, que, por sua vez, estimulam as células B autorreativas. Por conseguinte, os linfócitos podem entrar novamente na circulação, retornar para a articulação e continuar respondendo aos antígenos articulares naturais e modificados.9
Nesse cenário, não é necessária a ação isolada de nenhum agente etiológico. Conforme já descrito, inflamações inespecíficas em pacientes com histórico genético específico poderão produzir respostas imunes locais direcionadas para muitos antígenos articulares e peptídeos modificados. Os antígenos deflagradores podem ser distintos dos responsáveis pela perpetuação da doença, tendo em vista que a “disseminação de antígenos” aumenta o repertório de epítopos que poderão ser modificados e reconhecidos.
Citocinas
O perfil das citocinas das articulações reumatoides foi estudado extensivamente e contribuiu para o desenvolvimento de novos tratamentos de AR. A tendência da presença de citocinas das células T em concentrações relativamente baixas está entre as primeiras descobertas, a despeito do fato de representarem a população sinovial mais abundante. Apesar da presença de células Th1, os níveis clássicos de citocinas Th1, como a IL-2 e o interferon ?, são relativamente baixos, ao passo que é muito difícil detectar a presença das citocinas Th2.63
Conforme já mencionado, uma exceção importante é a família de IL-17 (isto é, IL-17A a F; normalmente, a IL-17A é conhecida por IL-17), que poderá entrar em sinergia com o TNF e a IL-1, e é produzida por células Th17 nas articulações.64 Outras citocinas das células T, como a IL-21 e a IL-29, também foram descritas no sinóvio reumatoide.
As citocinas derivadas de macrófagos e fibroblastos podem ser detectadas facilmente em articulações reumatoides.65 Os exemplos incluem IL-1, IL-6, IL-12, IL-15, IL-18, IL-32, TNF, M-CSF, fator estimulador de colônias de granulócitos-macrófagos (GM-CSF) e muitos outros. As alarminas, como a IL-33 e o grupo 1 da caixa de alta mobilidade (HGMB1), agem como sinais de alerta e também são expressas e podem contribuir para as inflamações sinoviais.
Uma grande quantidade dessas citocinas está envolvida na patogênese da AR e poderá estabelecer redes de sinalização parácrinas ou autócrinas perpetuando a artrite. A terapia anticitocinas (incluindo IL-1, TNF e inibidores da IL-6) é bastante eficaz nos casos de AR grave e mostra a importância desses produtos no desenvolvimento de sinovite crônica. Além disso, as citocinas regulam a produção de proteases, angiogênese e mediadores de moléculas pequenas de inflamação e recrutam células adicionais para a articulação.
O TNF é especialmente importante porque pode induzir a proliferação de sinoviócitos, a produção de colagenase e a liberação de prostaglandina. A expressão excessiva generalizada do TNF pode induzir artrite em camundongos. A IL-5, produzida por macrófagos, compartilha muitas atividades com a citocina IL-2 derivada de células T. A IL-5 aumenta a capacidade das células T para induzir a produção de TNF por macrófagos por meio de um mecanismo independente de antígenos com envolvimento no contato intercelular.66
A IL-18 também está presente em articulações com AR e poderá influenciar as respostas das células T em relação à Th1, enquanto que a IL-1 e IL-6 podem desviar as células na direção de Th17. As células T contribuem também através do contato intercelular direto com macrófagos ou fibroblastos, podendo, consequentemente, induzir a expressão genética de citocinas e da metaloproteinase.
As citocinas derivadas de macrófagos, fibroblastos e células dendríticas também ajudam a organizar a microarquitetura sinovial em agregados linfoides. Por exemplo, a sobrevivência das células B e dos fatores de diferenciação (por exemplo, BLyS e APRIL) ocorre no sinóvio reumatoide.
Além disso, as citocinas (como LIGHT) e as quimiocinas (como CXCL13 e CCL21) orquestram o recrutamento celular em estruturas que possuem centros terminais.67 Portanto, pode ocorrer apresentação de antígenos e intensificação nas respostas imunes sistêmicas aos antígenos articulares.
Sinalização Intracelular
A transdução de sinal é um sistema de informações que transmite informações do ambiente para o interior das células, permitindo a apresentação de respostas apropriadas. Os receptores de citocinas e os sensores de vários patógenos agem em cascatas de moléculas sinalizadoras que integram os estímulos e amplificam as respostas. Um dos mecanismos mais comuns envolve as enzimas denominadas quinases, que transferem o fosfato do trifosfato de adenosina (ATP) para uma proteína ou lipídeo para alterar seu estado funcional.
Uma grande variedade de mecanismos sinalizadores foi envolvida em casos de AR, especialmente em respostas inflamatórias sinoviais. O fator nuclear kappa B (em inglês, nuclear factor kappa B [NF-kB]) é a principal via de sinalização envolvida na sinalização pró-inflamatória para IL-1, TNF e vários ligantes do receptor Toll-like, como os lipopolissacarídeos.
A ativação do NF-kB resulta na produção de muitas citocinas e mediadores, incluindo IL-6 e IL-8.68 Um segundo grupo de quinases sinalizadoras, denominadas quinases de proteínas ativadas por mitógeno (em inglês, mitogen-activated proteins [MAPs]), integra e regula uma série de genes de citocinas e metaloproteinases.
A quinase de MAPs e o NF-kB são ativados em sinóvios de AR e, provavelmente, controlam muitas das citocinas e enzimas degradadoras que contribuem para inflamações e danos nas articulações.69 Em modelos animais, o bloqueio dessas inflamações ou danos diminui a gravidade da doença. A utilidade clínica do bloqueio das vias foi desapontadora devido à falta de eficácia (por exemplo, p38 MAP quinase) ou toxicidade.
O maior sucesso foi obtido tomando-se como alvo a família das quinases de Janus (em inglês, Janus kinases [JAK]). Esse grupo de quinases está envolvido na sinalização do receptor de citocinas, sendo que os inibidores da JAK de moléculas pequenas foram comprovadamente eficazes em casos de AR.70 Várias outras quinases implicadas na AR têm grande potencial como alvos terapêuticos, como as quinases P13, BTK, c-Kit, e muitas outras moléculas sinalizadoras.
Sumário
A AR é uma doença inflamatória sistêmica crônica que se caracteriza pela presença de sinovite poliarticular, geralmente acompanhada pela destruição progressiva de cartilagens e ossos, e pela produção de autoanticorpos como o fator reumatoide e ACPA. Muito provavelmente, a doença evolui durante muitos anos como consequência da exposição a fatores ambientais, como tabagismo e infecções, que causam inflamação nos tecidos das mucosas e aumentam a citrulinação de proteínas.
A ativação subsequente do sistema imune adaptativo e inato resulta em indivíduos com históricos genéticos específicos, em especial o lócus do HLA-DR. Esses genes de suscetibilidade, em associação com a produção de ACPA, aumentam acentuadamente o risco de AR.
O tecido sinovial em AR se caracteriza pela presença de fatores como hiperplasia no revestimento da túnica íntima, acúmulo de células inflamatórias no sub-revestimento e aumento nas efusões sinoviais. Os linfócitos e macrófagos produzem citocinas pró-inflamatórias, como o TNF e a IL-1, e fatores de crescimento.
A ativação dos sinoviócitos semelhantes a fibroblastos apresenta fenótipos agressivos, produzindo enzimas que degradam os tecidos e ativadores de osteoclastos. Ao final, ocorrem a formação e a expansão de tecidos membranosos que invadem e degradam as cartilagens e os ossos. As intervenções terapêuticas com foco nesses eventos patogênicos, como os inibidores da citocina, inibidores de transdução de sinal ou agentes que bloqueiam ou exaurem as células imunes, apresentaram eficácia clínica no tratamento desse tipo de doença.
|
Informações Financeiras: Anna-Karin H. Ekwall, MD, PhD, não tem relações financeiras relevantes a declarar. Gary S. Firestein, MD, é consultor das empresas Pfizer Inc.; Sanofi US; Eli Lilly & Co.; e Janssen Pharmaceuticals Inc. Ele recebeu apoio financeiro para pesquisas da Eli Lilly & Co e da Ono Pharmaceuticals; é acionista da Ignyta Inc. |
Referências
1. Aletaha D, Neogi T, Silman AJ, et al. 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum 2010;62:2569–81.
2. Demoruelle MK, Deane KD, Holers VM. When and where does inflammation begin in rheumatoid arthritis? Curr Opin Rheumatol 2014;26:64–71.
3. Nepom GT, Byers P, Seyfried C, et al. HLA genes associated with rheumatoid arthritis. Identification of susceptibility alleles using specific oligonucleotide probes. Arthritis Rheum 1989;32:15–21.
4. Lundstrom E, Kallberg H, Alfredsson L, et al. Gene-environment interaction between the DRB1 shared epitope and smoking in the risk of anti-citrullinated protein antibody-positive rheumatoid arthritis: all alleles are important. Arthritis Rheum 2009;60:1597–603.
5. van der Helm-van Mil AH, Verpoort KN, Breedveld FC, et al. The HLA-DRB1 shared epitope alleles are primarily a risk factor for anti-cyclic citrullinated peptide antibodies and are not an independent risk factor for development of rheumatoid arthritis. Arthritis Rheum 2006;54:1117–21.
6. Raychaudhuri S, Sandor C, Stahl EA, et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat Genet 2012;44:291–6.
7. Suzuki A, Yamada R, Chang X, et al. Functional haplotypes of PADI4, encoding citrullinating enzyme peptidylarginine deiminase 4, are associated with rheumatoid arthritis. Nat Genet 2003;34:395–402.
8. Vang T, Congia M, Macis MD, et al. Autoimmune-associated lymphoid tyrosine phosphatase is a gain-of-function variant. Nat Genet 2005;37:1317–9.
9. Arend WP, Firestein GS. Pre-rheumatoid arthritis: predisposition and transition to clinical synovitis. Nat Rev Rheumatol 2012;8:573–86.
10. Begovich AB, Carlton VE, Honigberg LA, et al. A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am J Hum Genet 2004;75:330–7.
11. Okada Y, Wu D, Trynka G, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 2014;506:376–81.
12. Plenge RM, Seielstad M, Padyukov L, et al. TRAF1-C5 as a risk locus for rheumatoid arthritis—a genomewide study. N Engl J Med 2007;357:1199–209.
13. Stahl HD, Pfeiffer R, Von Salis-Soglio G, Emmrich F. Parvovirus B19-associated mono- and oligoarticular arthritis may evolve into a chronic inflammatory arthropathy fulfilling criteria for rheumatoid arthritis or spondylarthropathy. Clin Rheumatol 2000;19:510–1.
14. Takahashi Y, Murai C, Shibata S, et al. Human parvovirus B19 as a causative agent for rheumatoid arthritis. Proc Natl Acad Sci U S A 1998;95:8227–32.
15. Albani S, Carson DA. A multistep molecular mimicry hypothesis for the pathogenesis of rheumatoid arthritis. Immunol Today 1996;17:466–70.
16. Nakajima T, Aono H, Hasunuma T, et al. Overgrowth of human synovial cells driven by the human T cell leukemia virus type I tax gene. J Clin Invest 1993;92:186–93.
17. Aicher WK, Heer AH, Trabandt A, et al. Overexpression of zinc-finger transcription factor Z-225/Egr-1 in synoviocytes from rheumatoid arthritis patients. J Immunol 1994;152:5940–8.
18. Lundberg K, Wegner N, Yucel-Lindberg T, Venables PJ. Periodontitis in RA—the citrullinated enolase connection. Nat Rev Rheumatol 2010;6:727–30.
19. Scher JU, Abramson SB. The microbiome and rheumatoid arthritis. Nat Rev Rheumatol 2011;7:569–78.
20. Wu HJ, Ivanov, II, Darce J, et al. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity 2010;32:815–27.
21. Abdollahi-Roodsaz S, Joosten LA, Koenders MI, et al. Stimulation of TLR2 and TLR4 differentially skews the balance of T cells in a mouse model of arthritis. J Clin Invest 2008;118:205–16.
22. Vaahtovuo J, Munukka E, Korkeamaki M, et al. Fecal microbiota in early rheumatoid arthritis. J Rheumatol 2008;35:1500–5.
23. Liu X, Zou Q, Zeng B, et al. Analysis of fecal Lactobacillus community structure in patients with early rheumatoid arthritis. Curr Microbiol 2013;67:170–6.
24. Makrygiannakis D, Hermansson M, Ulfgren AK, et al. Smoking increases peptidylarginine deiminase 2 enzyme expression in human lungs and increases citrullination in BAL cells. Ann Rheum Dis 2008;67:1488–92.
25. Klareskog L, Stolt P, Lundberg K, et al. A new model for an etiology of rheumatoid arthritis: smoking may trigger HLA-DR (shared epitope)-restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheum 2006;54:38–46.
26. Demoruelle MK, Weisman MH, Simonian PL, et al. Brief report: airways abnormalities and rheumatoid arthritis-related autoantibodies in subjects without arthritis: early injury or initiating site of autoimmunity? Arthritis Rheum 2012;64:1756–61.
27. Rangel-Moreno J, Hartson L, Navarro C, et al. Inducible bronchus-associated lymphoid tissue (iBALT) in patients with pulmonary complications of rheumatoid arthritis. J Clin Invest 2006;116:3183–94.
28. Willis VC, Demoruelle MK, Derber LA, et al. Sputum autoantibodies in patients with established rheumatoid arthritis and subjects at risk of future clinically apparent disease. Arthritis Rheum 2013;65:2545–54.
29. Klareskog L, Amara K, Malmstrom V. Adaptive immunity in rheumatoid arthritis: anticitrulline and other antibodies in the pathogenesis of rheumatoid arthritis. Curr Opin Rheumatol 2014;26:72–9.
30. Barra L, Scinocca M, Saunders S, et al. Anti-citrullinated protein antibodies in unaffected first-degree relatives of rheumatoid arthritis patients. Arthritis Rheum 2013;65:1439–47.
31. Kallberg H, Jacobsen S, Bengtsson C, et al. Alcohol consumption is associated with decreased risk of rheumatoid arthritis: results from two Scandinavian case-control studie¬s. Ann Rheum Dis 2009;68:222–7.
32. Bottini N, Firestein GS. Epigenetics in rheumatoid arthritis: a primer for rheumatologists. Curr Rheumatol Rep 2013;15:372.
33. Bartok B, Firestein GS. Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunol Rev 2010;233:233–55.
34. Bottini N, Firestein GS. Duality of fibroblast-like synoviocytes in RA: passive responders and imprinted aggressors. Nat Rev Rheumatol 2013;9:24–33.
35. Nakano K, Whitaker JW, Boyle DL, et al. DNA methylome signature in rheumatoid arthritis. Ann Rheum Dis 2013;72:110–7.
36. Whitaker JW, Shoemaker R, Boyle DL, et al. An imprinted rheumatoid arthritis methylome signature reflects pathogenic phenotype. Genome Med 2013;5:40.
37. Kiener HP, Lee DM, Agarwal SK, Brenner MB. Cadherin-11 induces rheumatoid arthritis fibroblast-like synoviocytes to form lining layers in vitro. Am J Pathol 2006;168:1486–99.
38. Firestein GS, Yeo M, Zvaifler NJ. Apoptosis in rheumatoid arthritis synovium. J Clin Invest 1995;96:1631–8.
39. Zvaifler NJ, Firestein GS. Pannus and pannocytes. Alternative models of joint destruction in rheumatoid arthritis. Arthritis Rheum 1994;37:783–9.
40. Corr M, Zvaifler NJ. Mesenchymal precursor cells. Ann Rheum Dis 2002;61:3–5.
41. Lee DM, Kiener HP, Agarwal SK, et al. Cadherin-11 in synovial lining formation and pathology in arthritis. Science 2007;315:1006–10.
42. Gravallese EM, Goldring SR. Cellular mechanisms and the role of cytokines in bone erosions in rheumatoid arthritis. Arthritis Rheum 2000;43:2143–51.
43. Firestein GS. Invasive fibroblast-like synoviocytes in rheumatoid arthritis. Passive responders or transformed aggressors? Arthritis Rheum 1996;39:1781–90.
44. Muller-Ladner U, Kriegsmann J, Franklin BN, et al. Synovial fibroblasts of patients with rheumatoid arthritis attach to and invade normal human cartilage when engrafted into SCID mice. Am J Pathol 1996;149:1607–15.
45. Lefevre S, Knedla A, Tennie C, et al. Synovial fibroblasts spread rheumatoid arthritis to unaffected joints. Nat Med 2009;15:1414–20.
46. Tak PP, Zvaifler NJ, Green DR, Firestein GS. Rheumatoid arthritis and p53: how oxidative stress might alter the course of inflammatory diseases. Immunol Today 2000;21:78–82.
47. Aggarwal A, Agarwal S, Misra R. Chemokine and chemokine receptor analysis reveals elevated interferon-inducible protein-10 (IP)-10/CXCL10 levels and increased number of CCR5+ and CXCR3+ CD4 T cells in synovial fluid of patients with enthesitis-related arthritis (ERA). Clin Exp Immunol 2007;148:515–9.
48. Miossec P, Korn T, Kuchroo VK. Interleukin-17 and type 17 helper T cells. N Engl J Med 2009;361:888–98.
49. van den Berg WB, Miossec P. IL-17 as a future therapeutic target for rheumatoid arthritis. Nat Rev Rheumatol 2009;5:549–53.
50. Ehrenstein MR, Evans JG, Singh A, et al. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFalpha therapy. J Exp Med 2004;200:277–85.
51. Warrington KJ, Takemura S, Goronzy JJ, Weyand CM. CD4+,CD28- T cells in rheumatoid arthritis patients combine features of the innate and adaptive immune systems. Arthritis Rheum 2001;44:13–20.
52. Gringhuis SI, Leow A, Papendrecht-Van Der Voort EA, et al. Displacement of linker for activation of T cells from the plasma membrane due to redox balance alterations results in hyporesponsiveness of synovial fluid T lymphocytes in rheumatoid arthritis. J Immunol 2000;164:2170–9.
53. Vossenaar ER, Nijenhuis S, Helsen MM, et al. Citrullination of synovial proteins in murine models of rheumatoid arthritis. Arthritis Rheum 2003;48:2489–500.
54. Linn-Rasker SP, van der Helm-van Mil AH, van Gaalen FA, et al. Smoking is a risk factor for anti-CCP antibodies only in rheumatoid arthritis patients who carry HLA-DRB1 shared epitope alleles. Ann Rheum Dis 2006;65:366–71.
55. Kuhn KA, Kulik L, Tomooka B, et al. Antibodies against citrullinated proteins enhance tissue injury in experimental autoimmune arthritis. J Clin Invest 2006;116:961–73.
56. Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med 2013;5:178ra40.
57. Harre U, Georgess D, Bang H, et al. Induction of osteoclastogenesis and bone loss by human autoantibodies against citrullinated vimentin. J Clin Invest 2012;122:1791–802.
58. Ermel RW, Kenny TP, Chen PP, Robbins DL. Molecular analysis of rheumatoid factors derived from rheumatoid synovium suggests an antigen-driven response in inflamed joints. Arthritis Rheum 1993;36:380–8.
59. Benoist C, Mathis D. Autoimmunity provoked by infection: how good is the case for T cell epitope mimicry? Nat Immunol 2001;2:797–801.
60. Kavanaugh A, Rosengren S, Lee SJ, et al. Assessment of Rituximab’s Immunomodulatory Synovial Effects (ARISE trial). 1: clinical and synovial biomarker results. Ann Rheum Dis 2008;67:402–8.
61. Monach PA, Hueber W, Kessler B, et al. A broad screen for targets of immune complexes decorating arthritic joints highlights deposition of nucleosomes in rheumatoid arthritis. Proc Natl Acad Sci U S A 2009;106:15867–72.
62. Schrijver IA, Melief MJ, Tak PP, et al. Antigen-presenting cells containing bacterial peptidoglycan in synovial tissues of rheumatoid arthritis patients coexpress costimulatory molecules and cytokines. Arthritis Rheum 2000;43:2160–8.
63. Firestein GS, Zvaifler NJ. How important are T cells in chronic rheumatoid synovitis?: II. T cell-independent mechanisms from beginning to end. Arthritis Rheum 2002;46:298–308.
64. Chabaud M, Lubberts E, Joosten L, et al. IL-17 derived from juxta-articular bone and synovium contributes to joint degradation in rheumatoid arthritis. Arthritis Res 2001;3:168–77.
65. Firestein GS, Alvaro-Gracia JM, Maki R. Quantitative analysis of cytokine gene expression in rheumatoid arthritis. J Immunol 1990;144:3347–53.
66. McInnes IB, Leung BP, Sturrock RD, et al. Interleukin-15 mediates T cell-dependent regulation of tumor necrosis factor-alpha production in rheumatoid arthritis. Nat Med 1997;3:189–95.
67. Kim WJ, Kang YJ, Koh EM, et al. LIGHT is involved in the pathogenesis of rheumatoid arthritis by inducing the expression of pro-inflammatory cytokines and MMP-9 in macrophages. Immunology 2005;114:272–9.
68. Aupperle K, Bennett B, Han Z, et al. NF-kappa B regulation by I kappa B kinase-2 in rheumatoid arthritis synoviocytes. J Immunol 2001;166:2705–11.
69. Inoue T, Hammaker D, Boyle DL, Firestein GS. Regulation of p38 MAPK by MAPK kinases 3 and 6 in fibroblast-like synoviocytes. J Immunol 2005;174:4301–6.
70. Riese RJ, Krishnaswami S, Kremer J. Inhibition of JAK kinases in patients with rheumatoid arthritis: scientific rationale and clinical outcomes. Best Pract Res Clin Rheumatol 2010;24:513–26.
71. Arnett FC, Edworthy SM, Bloch DA, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum 1988;31:315–24.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.