(Carregando Índice)... (Carregando Índice)... |
Última revisão: 11/11/2015
Comentários de assinantes: 0
Reproduzido de:
Formulário Terapêutico Nacional 2010: Rename 2010 [Link Livre para o Documento Original]
Série B. Textos Básicos de Saúde
MINISTÉRIO DA SAÚDE
Secretaria de Ciência, Tecnologia e Insumos Estratégicos
Departamento de Assistência Farmacêutica e Insumos Estratégicos
Brasília / DF – 2010
Letícia Figueira Freitas
Rachel Magarinos-Torres
Na Rename 2010: item 11
t Pó para solução oral
t Reposição hidreletrolítica no tratamento da desidratação.
t Tratamento de diarreia aguda.
t Usar com cuidado nos casos de:
– insuficiência renal (ver Apêndice D).
– hipernatremia (a reidratação deve ser lenta, em cerca de 12 horas).
t A reidratação oral não é apropriada nos casos de obstrução gastrintestinal ou quando a terapia de reidratação parenteral é indicada, como em desidratação grave ou vômito intratável.
t Dose de 75 mL/kg, por via oral, em pequenos volumes no período de 4 horas. Pode-se aumentar a quantidade caso a criança continue com episódios frequentes de diarreia.
t Dose de 200 mL, por via oral após cada episódio de diarreia.
t Pode ser administrado à velocidade de 20 mL/kg/hora, por sonda nasogástrica, no período de 6 horas.
t Dose de 200 a 400 mL (de acordo com a perda de fluido), por via oral após cada episódio de diarreia.
Nota: A reidratação oral deve continuar até que cesse a diarreia; volumes maiores podem ser dados quando os episódios de diarreia na criança são muito frequentes; em caso de vômito, suspender a reidratação por 10 minutos, recomeçando em velocidade e quantidade menores e com maior frequência; suplementação de zinco deve ser feita após 4 horas de reidratação, tão logo a criança volte a comer; em casos suspeitos de cólera deve-se aumentar a concentração de sódio.
t Podem ocorrer vômitos após administração rápida.
t A administração de soluções muito concentradas, ou nos portadores de insuficiência renal, pode resultar em hipernatremia e hiperpotassemia.
t A solução deve ser preparada somente com água filtrada ou, preferentemente, fervida e fria.
t Respeitar o volume total indicado de 1 L.
t Não ferver a solução depois de preparada.
t Não misturar a solução com outros ingredientes, como açúcar.
t Após a preparação, pode ser armazenada em geladeira, por um período máximo de 24 horas e depois deve ser desprezada.
t Manter à temperatura ambiente, entre 15 e 30 ºC e em lugar seco e fresco.
t Composição por litro após preparo: cloreto de sódio 2,6 g (75 mmol de sódio), glicose anidra 13,5 g (75 mmol de glicose), cloreto de potássio 1,5 g (20 mmol de potássio e 65 mmol de cloreto), citrato de sódio di-idratado 2,9 g (10 mmol de citrato).
t Na falta de glicose anidra e citrato de sódio, pode-se substituir por sacarose (27 g/litro de água) e bicarbonato de sódio (2,5 g/litro de água).
Karen Luise Lang
Na Rename 2010: item 5.5.2.4
t Cápsula 200 mg.
t Tratamento de infecção por HIV em combinação com outros fármacos antirretrovirais.
t Hipersensibilidade ao saquinavir.
t Insuficiência hepática grave (ver Apêndice C).
t Uso concomitante com antiarrítmicos (amiodarona, propafenona, quinidina), ergotamina e análogos, rifampicina, pimozida, midazolam e erva-de-são-joão (Hypericum perforatum).
t Uso como único inibidor de protease na terapia.
t Usar com cuidado nos casos de:
– insuficiência renal grave.
– insuficiência hepática.
– hepatites B e C crônicas, cirrose, alcoolismo, pancreatite recente, hipertrigliceridemia, hemofilia A e B ou diabetes melito.
– consumo de alho, in natura ou em cápsulas (pode reduzir as concentrações plasmáticas de saquinavir).
t Categoria de risco na gravidez (FDA): B (ver Apêndice A).
t 1.000 mg, por via oral, combinado com ritonavir 100 mg, a cada 12 horas.
t 1.000 mg, por via oral, combinado com lopinavir 400 mg, a cada 12 horas.
t 1.200 mg, por via oral, a cada 8 horas, em combinação com outro antirretroviral.
t Pico de concentração plasmática: 3 horas.
t Início de resposta: 2 a 6 semanas.
t Meia-vida: 13 horas.
t Metabolismo: hepático (90%).
t Excreção: fecal (81 a 88%) e renal (1 a 3%).
t Náusea (4 a 20%), diarreia (4 a 20%), dispepsia (4 a 20%), desconforto abdominal (4 a 20%) e pancreatite.
t Astenia (9%), parestesia (4%), neuropatia periférica (5%), ansiedade, depressão, confusão, agitação, insônia, irritabilidade, alucinação, letargia, sonolência e euforia (2%).
t Hepatotoxicidade (menos de 1%).
t Nictúria, cólica renal, infecção e sangramento do trato urinário (2%).
t Acne, dermatite, eczema, foliculite, fotossensibilidade, mudanças na pigmentação da pele, exantema maculopapular, ulceração, urticária, xerodermia, prurido (2%).
t Hiperglicemia, cetoacidose, hipotireoidismo, ginecomastia, lipodistrofia, aumento nos níveis de prolactina, dislipidemia.
t Anemia (2%), neutropenia (2%), trombocitopenia (2%), pancitopenia (2%), hemorragia, tromboflebite e vasoconstrição periférica.
t Pneumonia, bronquite, tosse, dispneia, rinite, sinusite, hemoptise (2%).
t Ácido fusídico pode causar hepatotoxicidade e aumento da concentração plasmática de ambos os fármacos. Recomenda-se evitar a associação.
t Alfentanila, bloqueadores dos canais de cálcio (anlodipino, diltiazem, nifedipino, verapamil, etc.), ciclosporina, colchicina, dapsona, digoxina, everolimo, fentanila, flunarizina, maraviroque, metadona, sildenafila, sirolimo, tacrolimo: têm sua toxicidade aumentada pelo saquinavir. Quando possível, deve-se avaliar a redução da dose dos fármacos associados ao saquinavir.
t Amprenavir e darunavir: podem ter suas concentrações plasmáticas reduzidas. Ajustes na dose de amprenavir podem ser necessários para manter o efeito antiviral. A associação de saquinavir com darunavir deve ser evitada.
t Atazanavir, cetoconazol, cimetidina, claritromicina, delavirdina, indinavir, nelfinavir, omeprazol: o uso concomitante pode levar a aumento de efeito/ toxicidade de saquinavir: Quando possível, deve-se avaliar a redução da dose dos fármacos associados ao saquinavir.
t Carbamazepina, dexametasona, fenitoína, fenobarbital, fosfenitoína, nevirapina, rifampicina, tipranavir: pode haver redução de efeito de saquinavir. Deve-se avaliar a utilização de fármacos alternativos em pacientes em terapia com saquinavir. Caso a associação seja necessária, ajustes na dose de saquinavir podem ser necessários para manter o efeito antiviral.
t Efavirenz: pode resultar na redução da concentração plasmática de ambos fármacos. Não se recomenda a utilização de saquinavir como único inibidor de protease em associação com efavirenz.
t Itraconazol e voriconazol: podem aumentar a concentração plasmática e a incidência de Efeitos adversos de ambos os fármacos. Monitorar o paciente para sinais de toxicidade.
t Loperamida e rifabutina: podem reduzir a concentração plasmática de saquinavir e ter suas concentrações plasmáticas aumentadas. A associação não é recomendada, especialmente em terapias prolongadas.
t Sinvastatina e demais estatinas: o uso concomitante com saquinavir aumenta o risco de desenvolvimento de miopatias e/ou rabdomiólise. A associação não é recomendada, mas caso seja necessária deve-se monitorar sinais de toxicidade.
t Varfarina: o uso concomitante com saquinavir aumenta o risco de sangramento. Caso a associação seja necessária, deve-se monitorar o tempo de protombina e avaliar a redução da dose de varfarina.
t Orientar para ingerir o medicamento 2 horas após as refeições.
t Reforçar orientações sobre prevenção da transmissão do vírus HIV.
t Orientar para o uso durante todo o tempo prescrito, mesmo que haja melhora dos sintomas com as primeiras doses.
t Alertar para o grande número de Efeitos adversos , recomendando avisar o médico antes de usar qualquer outro medicamento.
t Manter à temperatura ambiente, entre 15 a 25 ºC, ao abrigo de ar, luz e em recipiente bem fechado.
Atenção: este fármaco apresenta muitas Efeitos adversos , devendo-se realizar pesquisa específica quanto a este aspecto ao considerar a introdução ou descontinuação do saquinavir e/ou outros medicamentos no esquema terapêutico do paciente.
A segurança e eficácia não estão estabelecidas para crianças e adolescentes menores de 16 anos.
Rosa Martins
Na Rename 2010: itens 14.3 e 14.7
t Comprimidos de 10 mg, 20 mg e 40 mg.
t Prevenção primária e secundária de cardiopatia isquêmica.
t Dislipidemias, associado a dieta.
t Hipersensibilidade à sinvastatina.
t Doença hepática aguda.
t Gravidez. Categoria de risco na gravidez (FDA): X (ver Apêndice A).
t Lactação (ver Apêndice B).
t Usar com cuidado nos casos de:
– uso prolongado de álcool.
– durante grandes procedimentos cirúrgicos, insuficiência renal ou miopatia (há risco aumentado de miopatia/rabdomiólise; suspender o uso se ocorrer tal reação).
– uso concomitante com amiodarona ou verapamil (a dose de sinvastatina não deve exceder a 20 mg/dia).
– uso concomitante com diltiazem (a dose de sinvastatina não deve exceder a 40 mg/dia).
– crianças com menos de 10 anos de idade (a segurança da sinvastatina não foi estabelecida).
t Causas secundárias de hiperlipidemias devem ser afastadas antes de iniciar o tratamento.
t Monitorar a função hepática periodicamente (ver Apêndice C).
t Monitorar a função renal periodicamente (ver Apêndice D).
t Monitorar níveis de creatina cinase; suspender o uso de sinvastatina se ocorrer aumento significativo dos níveis desta enzima.
t Monitorar lipídios séricos após 4 semanas do início do tratamento, e periodicamente com o uso prolongado.
t Dose inicial 10 mg/dia, por via oral, em dose única à noite. Dose de manutenção 10 a 40 mg/dia. Dose máxima 40 mg/dia (10 a 17 anos).
t Dose 20 a 40 mg, por via oral, em dose única à noite. Ajustar dose com intervalo mínimo de 4 semanas. Dose máxima: 80 mg/dia.
Nota: Na insuficiência renal grave (DCE inferior a 10 mg/mL) ou uso concomitante de fibratos: dose inicial de 5 mg/dia, até o máximo de 10 mg/dia.
t Biodisponibilidade: 5%.
t Início de ação: 2 semanas.
t Pico de efeito: 4 a 6 semanas.
t Metabolismo hepático, via CYP3A4; extenso efeito de primeira passagem. Metabolitos ativos.
t Excreção: fecal (60%) e renal (13%).
t Miopatia (15%), rabdomiólise (15%).
t Hepatotoxicidade (7%), elevação de creatina cinase (5%).
t Dor abdominal, náuseas, vômitos e diarreia (20%).
t Distúrbios psiquiátricos (10%), síndrome das pernas inquietas.
t Distúrbios visuais (4%).
t Infecção respiratória alta (2%).
t Hipotensão.
t Alopecia, exantema.
t Disfunção sexual.
t Acenocumarol, ácido fusídico, amprenavir, cetoconazol, ciprofloxacino, claritromicina, colchicina, dasatinibe, eritromicina e outros macrolídeos, fluconazol, indinavir, imatinibe, nefazodona, nelfinavir, nicotinamida (ou niacina, acima de 1 g/dia), risperidona, ritonavir, saquinavir, varfarina e voriconazol: aumentam o efeito/toxicidade da sinvastatina. Usar somente se o potencial benefício superar o risco, monitorar paciente para sinais e sintomas de miopatia e/ou rabdomiólise. Considerar o uso de outra estatina (inibidor da HMGCoA redutase).
t Amiodarona, verapamil: aumentam o efeito/toxicidade da sinvastatina. Reduzir a dose de sinvastatina para no máximo 20 mg/dia. Monitorar paciente para sinais e sintomas de miopatia e/ou rabdomiólise.
t Atazanavir, darunavir, fosamprenavir, itraconazol, lopinavir e tipranavir: aumentam o risco de miopatia e/ou rabdomiólise. Uso concomitante é contraindicado.
t Bosentana, carbamazepina, efavirenz, erva-de-são-joão (Hypericum perforatum), farelo de aveia, fenitoína, pectina, rifampicina e oxcarbazepina: diminuem o efeito da sinvastatina. Monitorar perfil lipídico e considerar ajuste de dose. Monitorar sinais e sintomas específicos.
t Ciclosporina, danazol, genfibrozila: aumentam o efeito/toxicidade da sinvastatina. Reduzir a dose de sinvastatina para no máximo 10 mg/dia. Monitorar paciente para sinais e sintomas de miopatia e/ou rabdomiólise.
t Digoxina: pode ter seu efeito/toxicidade aumentada pela sinvastatina. Monitorar sinais e sintomas específicos.
t Diltiazem: aumenta o efeito/toxicidade da sinvastatina. Reduzir a dose de sinvastatina para no máximo 40 mg/dia. Monitorar paciente para sinais e sintomas de miopatia e/ou rabdomiólise.
t Levotiroxina: pode ter sua efetividade diminuída pela sinvastatina. Monitorar o paciente quanto à efetividade da levotiroxina.
t Orientar para a importância de comunicar ao perceber qualquer sinal de efeito adverso.
t Alertar para não consumir bebidas alcoólicas durante terapia com sinvastatina.
t Informar que a sinvastatina deve ser administrada à noite.
t Em caso de esquecimento de uma dose, usar assim que lembrar. Se estiver perto do horário da próxima dose, omitir a dose anterior, esperar e usar no horário. Nunca usar duas doses juntas.
t Armazenar à temperatura entre 5 e 30 ºC.
Atenção: segurança e eficácia não foram estabelecidas em pacientes com menos de 10 anos de idade. Utilizar com precaução em paciente idoso, devido à maior predisposição a miopatias. Este fármaco apresenta número elevado de Efeitos adversos potencialmente graves, devendo ser realizada pesquisa específica sobre este aspecto antes de introduzir ou descontinuar a sinvastatina ou outro medicamento no esquema terapêutico do paciente.
Rogério Hoefler
Na Rename 2010: item 26
t Solução ácida após diluição: sódio (138 mEq/L); cálcio (3,5 mEq/L); potássio (2 mEq/L); magnésio (1 mEq/L); cloreto (109,5 mEq/L); acetato (3 mEq/L).
t Solução alcalina após diluição: bicarbonato de sódio 8,4% (32 mEq bicarbonato/L).
t Insuficiência renal crônica, para remoção de toxinas urêmicas e correção de alterações ácido-base e hidreletrolíticas, sem restaurar funções renais, endócrinas ou metabólicas (longo prazo).
t Insuficiência renal aguda.
t Remoção de fármacos em intoxicações agudas.
t Usar com cuidado nos casos de:
– idosos
– diabete melito
– insuficiência coronariana
– insuficiência cardíaca
– insuficiência hepática
– desnutrição
t A frequência da diálise varia de acordo com o nível da função renal remanescente. No entanto, a maioria dos indivíduos necessita de duas a três sessões de diálise por semana.
t Cefaleia (5%), febre e calafrios (< 1%).
t Prurido (5%).
t Cãibras (5-20%), lombalgia (2-5%), descalcificação.
t Hipotensão arterial (20-30%), dor torácica (2-5%), hipertensão arterial por hipervolemia, arritmia cardíaca, tamponamento cardíaco.
t Náuseas e vômitos (5-15%), hepatite por contaminação.
t Anemia grave por perda de hemácias, hemólise e embolia gasosa.
t Desnutrição por perda proteica, aumento de peso por excesso de água.
t Reações de hipersensibilidade.
t Lítio: tem sua excreção aumentada pelo bicarbonato de sódio (solução alcalina).
t Inibidores da ECA e diuréticos poupadores de potássio: podem aumentar a concentração sérica de potássio com o uso concomitante da solução ácida contendo potássio.
t Orientar para limitar o consumo de sódio a 2 g por dia.
t Orientar para limitar também o consumo de alimentos ricos em fósforo.
t Recomendar, aos pacientes que não urinam e aos que apresentam concentração sérica de sódio persistentemente baixa ou em progressiva queda, para limitar a ingestão diária de líquidos.
t Conservar à temperatura ambiente, entre 15 e 30 ºC, ao abrigo da luz.
t A solução de hemodiálise consiste em glicose e eletrólitos dissolvidos em água purificada.
t A solução para hemodiálise contém eletrólitos formulados em concentrações similares às do plasma sanguíneo, exceto pelo conteúdo em potássio e substância tampão.
t A solução para hemodiálise é preparada automaticamente na máquina de diálise através da mistura constante de concentrados de hemodiálise e água purificada, numa proporção fixa.
Elaine Silva Miranda
Na Rename 2010: item 24.3
t Solução de iodo 20 mg + iodeto de potássio 40 mg/mL (FN).
t Teste colposcópico para pesquisa de células displásicas ou carcinomatosas de colo uterino.
t Hipersensibilidade a compostos contendo iodo.
t Gravidez (ver Apêndice A).
t Usar com cuidado na lactação.
t Pode interferir nos testes de função da tireoide.
t Após banhar o colo uterino com a solução de Schiller, deve-se observar o resultado: o surgimento de coloração marrom (escura ou clara) significa resultado negativo; a ausência de coloração indica teste positivo. Nos casos de condiloma, o teste de Schiller é sempre negativo.
t Pode causar queimaduras ou irritação de mucosa.
t Manter à temperatura ambiente, até 25 ºC, ao abrigo da luz direta, em lugar seco e em frasco de cor âmbar bem tampado.
t As soluções a 2% de iodo são razoavelmente estáveis. Como o iodo in natura é sublimável, em solução pode ser estabilizado pela adição de iodeto de potássio.
t A formulação com 2% iodo e 4% iodeto torna essa solução particularmente resistente à degradação, mas pode ocorrer com exposição continuada a luz, ar e calor.
Rogério Hoefler
Na Rename 2010: item 26
t Solução injetável com glicose 1,5%. Composição: sódio (140 mEq/L); cálcio (3,5 mEq/L); magnésio (1,5 mEq/L); cloreto (101 mEq/L); lactato (44,6 mEq/L).
t Solução injetável com glicose 4,25%. Composição: sódio (132 mEq/L); cálcio (3,5 mEq/L); magnésio (0,5 mEq/L); cloreto (96 mEq/L); lactato (40 mEq/L).
t Insuficiência renal, para remoção de metabólitos e correção de desequilíbrio eletrolítico e de hipervolemia.
t Peritonite e abscesso abdominal.
t Cirurgia abdominal prévia.
t Doença inflamatória intestinal grave.
t A administração requer utilização de técnicas assépticas para reduzir o risco de infecção.
t A solução deve ser aquecida à temperatura corporal antes de ser utilizada.
t Alguns fármacos podem ser removidos pela diálise.
t Individualizado: de acordo com a condição clínica e dados laboratoriais do paciente.
t Obstrução do cateter; infecção no local do cateter e peritonite.
t Hérnia abdominal.
t Hemoperitôneo.
t Hiperglicemia.
t Desnutrição proteica.
t Conservar à temperatura ambiente, entre 15 e 30 ºC, ao abrigo da luz.
Respeitar o empilhamento máximo indicado pelo produtor.
t A solução com 1,5% de glicose pode ter sua concentração em glicose modificada para 4,25% pela adição quantitativa de solução injetável de glicose a 25% ou 50%.
Atenção: diálise peritoneal é menos eficiente que hemodiálise, mas é preferida em crianças, diabéticos e cardiopatas instáveis. É especialmente útil em pacientes que podem manejar sua condição e moram em locais afastados de centros dialíticos. Esta solução pode ser empregada em diálise peritoneal contínua ambulatorial (CAPD), controlada manualmente pelo paciente, ou em diálise peritoneal automatizada (APD), por máquina de funcionamento contínuo. A exposição à diálise peritoneal por longo prazo pode ocasionar mudanças estruturais progressivas na membrana peritoneal podendo gerar falha de diálise.
Rogério Hoefler
Na Rename 2010: item 9
t Solução injetável (composição): cloreto (109 mEq/L); cálcio (2,7 mEq/L); lactato (27,7 mEq/L); potássio (4 mEq/L); sódio (130 mEq/L).
t Reposição hidreletrolítica.
t Correção do equilíbrio ácido-base.
t Choque hipovolêmico.
t Alcalose metabólica ou respiratória.
t Hipocalcemia.
t Hipocloridria.
t Usar com cuidado nos casos de:
– hipertensão, insuficiência cardíaca, edema periférico ou pulmonar, pré-eclampsia, e outras condições associadas à retenção de sódio.
– cálculo renal ou histórico de cálculo renal (evitar uso por conter cálcio na formulação).
– insuficiência renal ou uso concomitante de doses altas de vitamina D (monitorar cuidadosamente a concentração plasmática de cálcio).
t Para reposição de fluidos e eletrólitos ou para tratar choque hipovolêmico, por infusão intravenosa, a dose é determinada a partir da avaliação clínica e, quando possível, deve-se fazer monitoria de eletrólitos.
t Excesso de sódio e de cálcio é excretado principalmente pelo rim, com pequena parte eliminada nas fezes e suor.
t Edema e alcalose metabólica (se administração excessiva).
t Ceftriaxona sódica (intravenosa): o uso concomitante é contraindicado, especialmente em neonatos, pelo risco de formação de precipitados de ceftriaxona na forma de sal cálcico. A solução de Ringer + lactato não deve ser usada como diluente para ceftriaxona.
t Glicosídeos digitálicos: aumento de efeito, podendo precipitar quadro de intoxicação digitálica.
t Armazenar à temperatura entre 15 e 30 ºC. Evitar congelamento.
t Proteger da luz solar direta.
Rogério Hoefler
Na Rename 2010: item 7.3
t Solução injetável
t Cada mililitro da solução neutraliza, no mínimo, 1,5 DMM (Dose Mínima Mortal) do veneno referência de Tityus serrulatus, 1,5 DMM do veneno referência de Phoneutria nigriventer, e 15 DMN (Dose Mínima Necrosante) de veneno referência de Loxosceles sp.
t Tratamento de envenenamentos por picada de escorpiões do gênero Tityus ou aranhas dos gêneros Phoneutria (ex.: aranha-armadeira) ou Loxosceles (ex.: aranha-marrom).
t Devido ao risco de hipertensão ou hipotensão arterial, arritmia cardíaca, insuficiência cardíaca, choque, taquipneia, dispneia e edema pulmonar agudo, associados aos envenenamentos por Tityus ou Phoneutria, recomenda-se monitoria das funções cardiorrespiratórias.
t A insuficiência renal aguda é complicação grave no acidente por Loxosceles. Cuidados especiais com a hidratação do paciente e cuidado da função renal devem ser instituídos precocemente.
t Em casos graves de acidente por Loxosceles pode ocorrer hemólise intravenosa. Indica-se transfusão sanguínea ou administração de concentrado de hemácias nos casos de anemia intensa.
Rogério Hoefler
Na Rename 2010: item 7.3
t Solução injetável (5 mg + 1,5 mg)/mL.
t Cada mililitro neutraliza, no mínimo, 5,0 mg do veneno referência de Bothrops jararaca e 1,5 mg do veneno referência de Crotalus durissus terrificus.
t Tratamento de envenenamentos por picadas de serpentes dos gêneros Bothrops e Crotalus.
t Ver monografias de Soro antibotrópico e Soro anticrotálico.
Rogério Hoefler
Na Rename 2010: item 7.3
t Solução injetável (5 mg + 3 mg)/mL.
t Cada mililitro neutraliza, no mínimo, 5,0 mg do veneno referência de Bothrops jararaca e 3,0 mg do veneno referência de Lachesis muta.
t Tratamento de envenenamentos por picadas de serpentes dos gêneros Bothrops e Lachesis (ex.: surucucu e surucucu-pico-de-jaca).
t A insuficiência renal aguda é complicação grave no acidente botrópico e laquético. Cuidados especiais com a hidratação do paciente e cuidado da função renal devem ser instituídos precocemente.
t Pelo risco potencial de bradicardia, hipotensão e choque, recomenda-se o monitoria das funções vitais nos envenenamentos laquéticos.
t Os acidentes botrópicos e laquéticos podem alterar o tempo de coagulação, promovendo sangramentos espontâneos.
Rogério Hoefler
Na Rename 2010: item 7.3
t Solução injetável 5 mg/mL.
t Cada mililitro neutraliza, no mínimo, 5,0 mg do veneno referência de Bothrops jararaca.
t Tratamento de envenenamentos por picadas de serpentes dos gêneros Bothrops (ex.: jararaca, jararacuçu, urutu, cotiara, caiçaca e outras), Bothriopsis e Porthidium.
t Insuficiência renal aguda é complicação grave no acidente botrópico. Cuidados especiais com a hidratação do paciente e cuidado da função renal devem ser instituídos precocemente.
t Os acidentes botrópicos podem alterar o tempo de coagulação, promovendo sangramentos espontâneos.
Rogério Hoefler
Na Rename 2010: item 7.3
t Solução injetável (375 UI + 275 UI)/mL.
t Cada mililitro da solução neutraliza, no mínimo, 375 UI de toxina botulínica tipo A e 275 UI de toxina botulínica tipo B.
t Tratamento de intoxicações por toxinas secretadas pelos bacilos botulínicos A ou B (Clostridium botulinum A ou B).
t 4 a 20 mL da solução original, diluídos a 1:10 em soro fisiológico, por via intravenosa, de acordo com uma avaliação da quantidade de alimento ingerido, se possível.
t A necessidade de aplicação de uma segunda dose é avaliada pelo aparecimento de sinais ou sintomas da doença.
t 20 mL da solução original, diluídos a 1:10 em soro fisiológico, por via intravenosa. A solução de antitoxina deve ser aplicada lentamente e sempre sob estrita vigilância médica e de enfermagem. A necessidade de administração de doses adicionais, relativas às recomendadas, deverá ser avaliada conforme evolução do quadro clínico.
t Continuar o tratamento até o desaparecimento dos sintomas. Em casos mais graves (midríase, ptose palpebral, disfagia), transportar o paciente para um centro que disponha de pulmão de aço para prevenir a ocorrência de síncope respiratória.
Rogério Hoefler
Na Rename 2010: item 7.3
t Solução injetável 425 UI/mL.
t Cada mililitro da solução neutraliza, no mínimo, 425 UI de toxina botulínica tipo E.
t Tratamento de intoxicação por toxinas secretadas pelo bacilo botulínico E (Clostridium botulinum E).
t 4 a 20 mL da solução original, diluídos a 1:10 em soro fisiológico, por via intravenosa, de acordo com uma avaliação da quantidade de alimento ingerido, se possível.
t A necessidade de aplicação de uma segunda dose é avaliada pelo aparecimento de sinais ou sintomas da doença.
t 20 mL da solução original, diluídos a 1:10 em soro fisiológico, por via intravenosa. A solução de antitoxina deve ser aplicada lentamente e sempre sob estrita vigilância médica e de enfermagem. A necessidade de administração de doses adicionais, relativas às recomendadas, deverá ser avaliada conforme evolução do quadro clínico.
t Continuar o tratamento até o desaparecimento dos sintomas. Em casos mais graves (midríase, ptose palpebral, disfagia), transportar o paciente para um centro que disponha de pulmão de aço para prevenir a ocorrência de síncope respiratória.
Rogério Hoefler
Na Rename 2010: item 7.3
t Solução injetável 1,5 mg/mL.
t Cada mililitro neutraliza, no mínimo, 1,5 mg do veneno referência de Crotalus durissus terrificus.
t Tratamento de envenenamento por picada de serpente do gênero Crotalus (ex.: cascavel).
t Insuficiência renal aguda é complicação grave em acidente crotálico.
Cuidados especiais com a hidratação do paciente e cuidado da função renal devem ser instituídos precocemente.
t Os acidentes crotálicos podem alterar o tempo de coagulação, promovendo sangramentos espontâneos.
Rogério Hoefler
Na Rename 2010: item 7.3
t Solução injetável 1.000 UI/mL.
t Cada mililitro da solução neutraliza, no mínimo, 1.000 UI de toxina diftérica.
t Tratamento de intoxicação por toxinas secretadas pelo bacilo diftérico (Corynebacterium diphtheriae).
t Casos leves e moderados: 20 a 40 mL, por via intramuscular (injetar em vários músculos) ou por intravenosa lenta.
t Casos graves: 40 a 80 mL, por via intramuscular (injetar em vários músculos) ou intravenosa lenta. A necessidade de administração de doses adicionais, relativas às recomendadas, deverá ser avaliada de acordo com a evolução do quadro clínico.
t O uso profilático do soro antidiftérico, em indivíduos não vacinados, pode ser feita a critério médico. Nesse caso, são sugeridas doses de 1 a 2 mL para crianças e 3 a 4 mL para adultos, por via intramuscular ou intravenosa.
Rogério Hoefler
Na Rename 2010: item 7.3
t Solução injetável 1,5 mg/mL.
t Cada mililitro neutraliza, no mínimo, 1,5 mg do veneno referência de Micrurus frontalis.
t Tratamento de envenenamento por picada de serpente do gênero Micrurus (ex.: coral-verdadeira, boicorá).
t A insuficiência respiratória aguda, de instalação rápida, é a mais grave complicação do acidente elapídico. É indispensável o monitoria da função respiratória, considerando a possibilidade de se instalar respiração mecânica. Fármacos anticolinesterásicos são empregados no manejo das paralisias musculares.
Rogério Hoefler
Na Rename 2010: item 7.3
t Solução injetável 1,5 DMM/mL.
t Cada mililitro neutraliza, no mínimo, 1,5 DMM (Dose Mínima Mortal) do veneno referência de Tityus serrulatus.
t Tratamento de envenenamento por picada de escorpião do gênero Tityus.
t Devido ao risco de hipertensão ou hipotensão arterial, arritmia cardíaca, insuficiência cardíaca, choque, taquipneia e dispneia, associados ao envenenamento escorpiônico, recomenda-se monitoria das funções cardiorrespiratórias.
Rogério Hoefler
Na Rename 2010: item 7.3
t Solução injetável 2.400 UI/mL.
t Cada mililitro da solução neutraliza, no mínimo, 2.400 UI de veneno referência de aranhas do gênero Latrodectus.
t Tratamento de envenenamento por picada de aranha do gênero Latrodectus (ex.: viúva-negra).
t Devido ao risco de hipertensão arterial, taquicardia, anormalidades no ECG, distúrbios de condução cardíaca, dispneia, insuficiência respiratória e edema pulmonar, associados aos envenenamentos por Latrodectus, recomenda-se monitoria das funções cardiorrespiratórias.
t Administrar preferentemente por via intravenosa. O conteúdo de uma ampola deve ser diluído em 50 a 100 mL de soro fisiológico ou glicose 5% e infundido em 15 a 30 minutos.
t Uma dose normalmente é suficiente, mas alguns indivíduos podem requerer dose adicional.
t Início de efeito: imediato (intravenoso) e cerca de 5 minutos (intramuscular), embora os sintomas de envenenamento permaneçam por 1 a 3 horas.
t Pico de resposta: 1 a 3 horas (intramuscular).
Rogério Hoefler
Na Rename 2010: item 7.3
t Solução injetável 0,35 mg/mL.
t Cada mililitro da solução neutraliza no mínimo 0,35 mg de veneno de Lonomia obliqua.
t Tratamento de envenenamento por lagarta do gênero Lonomia sp (ex.: oruga, taturana).
t Os acidentes lonômicos podem alterar o tempo de coagulação, promovendo sangramentos espontâneos.
t A insuficiência renal aguda é complicação grave no acidente por Lonomia. Cuidados especiais com a hidratação do paciente e cuidado da função renal devem ser instituídos precocemente.
Rogério Hoefler
Na Rename 2010: item 7.3
t Solução injetável
t Cada mililitro da solução neutraliza, no mínimo, 15 DMN (Dose Mínima Necrosante) de veneno referência de Loxosceles intermedia.
t Tratamento de envenenamento por picada de aranhas do gênero Loxosceles.
t A insuficiência renal aguda é complicação grave no acidente por Loxosceles. Cuidados especiais com a hidratação do paciente e cuidado da função renal devem ser instituídos precocemente.
t Em casos graves de acidente por Loxosceles pode ocorrer hemólise intravenosa. Indica-se transfusão sanguínea ou administração de concentrado de hemácias nos casos de anemia intensa.
Rogério Hoefler
Na Rename 2010: item 7.3
t Solução injetável 200 UI/mL.
t Cada mililitro da solução neutraliza, no mínimo, 200 UI de vírus da raiva.
t Ferimentos graves provocados por animal suspeito (incluindo mordeduras na face, cabeça, mãos e pés; ferimentos múltiplos, profundos ou extensos; lambedura de mucosas).
t Lavar imediatamente o ferimento provocado pela mordedura com água corrente em abundância e sabão.
t Desinfetar o ferimento com antisséptico (ex.: iodopovidona).
t Não suturar as lesões, salvo indicação de sutura reparadora.
t Não utilizar a mesma seringa para aplicação do soro e da vacina antirrábica.
t Não aplicar soro e vacina antirrábica na mesma região anatômica.
t 40 UI/kg, por via intramuscular (injetar em vários músculos), em dose única, não ultrapassando o total de 3.000 UI.
t Parte da dose recomendada, sempre que possível, deverá ser infiltrada ao redor dos ferimentos provocados pela mordedura. O restante deve ser administrado por via intramuscular (glúteo), em região corporal que não seja a mesma da administração da vacina contra raiva. Na impossibilidade de administrar o soro antirrábico de imediato, a vacinação deverá ser iniciada e o soro ser aplicado o mais rapidamente possível, até o 7º dia do início da vacinação. Após este período, o emprego do soro antirrábico não é mais indicado.
Rogério Hoefler
Na Rename 2010: item 7.3
t Solução injetável
t Cada mililitro neutraliza, no mínimo, 1.000 UI de toxina produzida pelo bacilo tetânico (Clostridium tetani).
t Profilaxia e tratamento de intoxicação por toxinas secretadas pelo bacilo Clostridium tetani.
t Profilaxia do tétano: em indivíduos não vacinados contra o tétano (com o toxoide tetânico), com vacinação incompleta ou vacinados há mais de cinco anos sem dose de reforço, aplicar 5.000 UI (5 mL da solução), por via intramuscular, e iniciar a vacinação ou revacinação segundo as doses recomendadas.
t Tratamento do tétano: administrar de 20.000 a 100.000 UI (20 a 100 mL da solução), podendo-se aplicar metade da dose por via intramuscular (distribuída em vários músculos) ou subcutânea e metade por via intravenosa. Em casos mais graves, aplicar no dia seguinte mais 50.000 UI (50 mL da solução) por via intramuscular (distribuídas em vários músculos).
t Profilática ou curativamente, a necessidade de administração de doses adicionais, relativas às recomendadas, deverão ser avaliadas pelo tipo de ferimento suspeito (profundidade, extensão, tecidos necrosados) ou com a evolução do quadro clínico.
Rogério Hoefler
Soros são antitoxinas usadas em imunização passiva como recurso terapêutico pós-exposição. Geralmente provêm de soro de animais e servem para neutralizar as toxinas produzidas em botulismo, raiva, difteria e venenos de cobras, escorpiões e aranhas.
t Usar com cuidado nos casos de:
– antecedentes alérgicos ou hipersensibilidade a soros de origem equina (a infusão intravenosa do soro deverá ser realizada sob estrito monitoria médico, para identificação e pronto atendimento de eventual reação anafilática).
– uso anterior de soro heterólogo (antitetânico, antirrábico, antiofídico) e presença de problemas alérgicos de naturezas diversas (considerar potencial de reações adversas; administrar anti-histamínico e corticosteroide, 15 minutos antes da aplicação do soro).
– ocorrência de reações alérgicas (interromper a soroterapia temporariamente e iniciar tratamento conforme intensidade das reações).
– alimentação prévia e/ou ingestão de bebidas alcoólicas (não contraindicam o uso de soros, mas impõem cuidado mais rigoroso pelo risco de complicações relacionadas a vômitos – aspiração).
– ocorrência de edema intenso e necrólise no local da picada por animal peçonhento (realizar tratamento cirúrgico).
– idosos.
t Uma vez indicado, o soro deve ser administrado o mais rapidamente possível. Quanto mais precoce for a administração da primeira dose do soro, maior o potencial terapêutico.
t Pelo risco de infecção secundária no local da picada, pode-se indicar uso de antimicrobianos e profilaxia contra tétano.
t O soro deve ser aplicado sob supervisão profissional devido ao risco de desencadear reações alérgicas, algumas delas potencialmente graves.
t Não há diferenciação de dose para adultos e crianças; o que define a dose não é faixa etária, mas a gravidade do quadro clínico.
t Exceto quando há orientação específica, os soros antiveneno, diluídos ou não em solução fisiológica, devem ser administrados por via intravenosa lenta, em 20 a 60 minutos. Na impossibilidade de uso desta via, o soro pode ser administrado por via subcutânea.
t A necessidade de administração de doses adicionais, em relação às recomendadas, deverá ser avaliada de acordo com o quadro clínico e o tempo de coagulação. Se o paciente permanecer incoagulável 24 horas após a soroterapia, recomenda-se dose adicional.
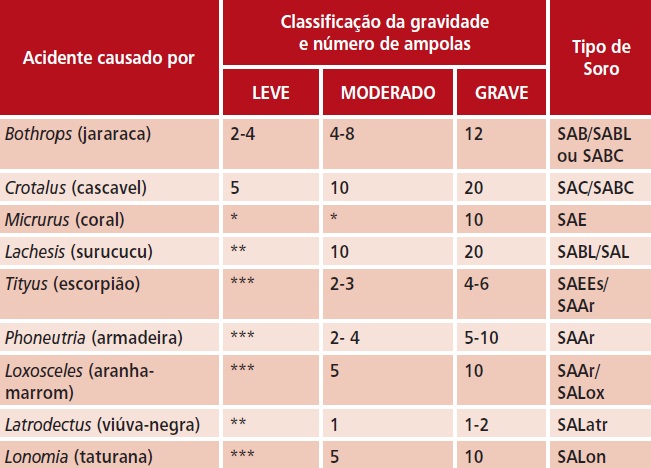
t O Quadro 1, a seguir, apresenta a soroterapia recomendada, conforme classificação quanto à gravidade do envenenamento por animal peçonhento.
Quadro 1. Indicação do número de ampolas de soros antiveneno para tratamento de acidentes por ofídios, aracnídeos e Lonomia

Adaptado da referência 121.
* clinicamente, os acidentes são classificados como graves ou potencialmente graves.
** clinicamente, os acidentes são classificados como moderados ou graves.
*** dispensa soroterapia, indica-se tratamento sintomático e observação hospitalar.
SAC – soro anticrotálico; SABC – soro antibotrópico-crotálico; SABL – soro antibotrópicolaquético; SAL – soro antilaquético; SAB – soro antibotrópico; SAE – soro antielapídico; SAEEs – soro antiescorpiônico; SAAr – soro antiaracnídico; SALox – soro antiloxoscélico;
SALatr – soro antilatrodético; SALonsoro antilonômico.
t Início de ação: imediato (via intravenosa).
t Reações alérgicas de graus variáveis: prurido/rubor cutâneo, urticária, tosse seca, rouquidão, náusea, vômito, crise asmatiforme (frequentes).
t Febre, calafrios, rubor facial e cefaleia.
t Reações graves são pouco frequentes; choque anafilático (0,002%, com uso de soro equino).
t Reações precoces (primeiras 24 horas após administração do soro) têm caráter anafilático ou anafilactoide, podem ser graves e necessitam de cuidados médicos.
t Reações tardias (5 a 24 dias após administração do soro) em geral são benignas. Caracterizam-se por: febre, artralgia, adenomegalia e, raramente, comprometimento neurológico ou renal. São conhecidas como “doença do soro”; o tratamento, de acordo com a intensidade, se dá por meio da administração de corticosteroides, analgésicos e anti-histamínicos.
t Na literatura consultada não há evidências de interação relevante com medicamentos, alimentos ou álcool.
t Informar o paciente sobre o risco de ocorrência da “doença do soro”, que pode ter início tardio (até cerca de 24 dias após a administração do soro). Se ocorrer, deverá retornar ao médico.
t Armazenar os soros sob refrigeração, entre 2 e 8 ºC. Evitar congelamento.
t O conteúdo da ampola deve estar límpido e transparente. Não deve ser usado se houver turvação ou presença de precipitado.
Rosa Martins
Na Rename 2010: itens 14.2 e 14.4.2
t Comprimidos de liberação controlada (succinato) de 25 mg, 50 mg e 100 mg.
t Comprimidos de 100 mg (tartarato)
t Insuficiência cardíaca congestiva de classes II e III.
t Hipertensão arterial sistêmica.
t Hipersensibilidade ao succinato ou tartarato de metoprolol, ou a qualquer componente da formulação.
t Insuficiência cardíaca descompensada.
t Choque cardiogênico.
t Bradicardia sinusal grave.
t Síndrome sinusal (com uso de marcapasso).
t Bloqueio atrioventricular de 2º e 3º graus.
t Usar com cuidado nos casos de:
– uso de anestésicos (risco de depressão miocárdica grave).
– interrupção do tratamento (não suspender abruptamente pelo risco de efeito rebote).
– doença broncoespástica, miastenia grave e feocromocitoma.
– tireotoxicose (metoprolol pode mascarar os sintomas de hipertireoidismo, como taquicardia).
– diabetes melito (metoprolol pode mascarar os sintomas de hipoglicemia).
– doença vascular periférica (metoprolol pode causar ou agravar os sintomas de insuficiência arterial).
– insuficiência hepática (ver Apêndice C).
t Categoria de risco na gravidez (FDA): C (primeiro trimestre) e D (segundo e terceiro trimestres) (ver Apêndice A).
Formulação de liberação controlada
t Dose inicial de 25 mg, por via oral, a cada 24 horas. Em pacientes com insuficiência grave iniciar com 12,5 mg (não partir o comprimido de liberação controlada) ao dia. Esta dose pode, então, ser dobrada a cada 2 semanas, até a mais alta dose tolerada: 200 mg/dia.
Formulação de liberação controlada
t Dose inicial de 25 a 100 mg, por via oral, a cada 24 horas. A dose deve ser ajustada semanalmente, se necessário. Dose máxima: 450 mg/dia
Nota: ao passar da formulação de liberação imediata para a de liberação controlada, começar com a mesma dose diária total e ajustar conforme necessário. A descontinuação do tratamento deve ser feita com redução gradual da dose pelo período de aproximadamente duas semanas, para evitar efeito rebote. Durante a descontinução e por pelo menos duas a três semanas após a descontinuação, o paciente deve minimizar o esforço físico para reduzir o risco de isquemia.
t A presença de alimento aumenta a biodisponibilidade do metoprolol.
t Biodisponibilidade 50% (comprimido de liberação imediata) e 65 a 70% (comprimido de liberação controlada)
t Início da ação: 1 hora.
t Pico de concentração: 1,5 a 2 horas (comprimido de liberação imediata) e 3,3 horas (comprimido de liberação controlada).
t Duração da ação: 3 a 6 horas (comprimido de liberação imediata); cerca de 24 horas (comprimido de liberação controlada).
t Metabolismo hepático: metabólitos inativos. Parece ser 4 vezes mais intenso no último trimestre da gravidez em comparação com o período pós-parto.
t Excreção: renal (95%).
t Meia-vida: 3 a 7 horas.
t Broncoespasmo (1%), dispneia (3%), sibilo (1%).
t Bradiarritmia (3% a 16%), insuficiência cardíaca congestiva (1%), hipotensão (1% a 27%), extremidades frias (1%), palpitação (1%), edema periférico (1%), síncope (1%).
t Prurido (5%), exantema (5%).
t Indigestão (1%), obstipação (1%), diarreia (5%), náusea (<1%), dor abdominal (<1%).
t Cefaleia, depressão (5%), tontura (10%), fadiga (10%).
t Alteração do perfil lipídico, hipoglicemia, hiperglicemia (14%).
t Amiodarona, bupropiona, bloqueadores de canais de cálcio do tipo diidropiridínicos, cimetidina, citalopram, difenidramina, diltiazem, dronedarona, escitalopram, fenelzina, fluoxetina, fentanila, hidralazina, hidroxicloroquina, inibidores de protease (ex.: ritonavir), paroxetina, propafenona, propoxifeno, quinidina, rifapentina, terbinafina, tioridazina, verapamil: podem aumentar o efeito/toxicidade do metoprolol. Monitorar função cardíaca, particularmente em pacientes predispostos a insuficiência cardíaca, pode ser necessário ajuste de dose, monitorar sinais e sintomas específicos.
t Bloqueadores alfa-1 adrenérgicos (primeira dose), clonidina (na retirada) e lidocaína: podem ter o efeito/toxicidade aumentado pelo metoprolol. Monitorar sinais e sintomas específicos.
t Digoxina: aumento do risco de bloqueio atrioventricular e toxicidade digitálica. Monitorar concentrações de digoxina e eletrocardiograma. Ajustar dose de digoxina, se ncessário.
t Erva-de-são-joão (Hypericum perforatum), fenobarbital (barbitúricos), rifamicina e venlafaxina: podem diminuir o efeito do metoprolol. Monitorar sinais e sintomas específicos.
t Hipoglicemiantes: podem ter o efeito mascarado pelo metoprolol e causar hiper ou hipoglicenia. Evitar uso concomitante, preferir betabloqueador cardiosseletivo, monitorar sinais e sintomas específicos.
t Orientar para a importância de comunicar ao perceber qualquer sinal de efeito adverso.
t Orientar para ingerir com alimento ou imediatamente após as refeições.
t Em caso de esquecimento de uma dose, usar assim que lembrar. Se estiver perto do horário da próxima dose, desconsiderar a dose anterior, esperar e usar no horário. Nunca usar duas doses juntas.
t Não interromper o uso deste medicamento abruptamente.
t Pressão alta pode não ter sintoma, não deixar de usar o medicamento sem falar com o médico.
t Manter à temperatura ambiente de 15 a 30 ºC, em recipiente hermético, ao abrigo de umidade e luz direta.
Atenção: proceder a retirada gradual do medicamento em 1 a 2 semanas para evitar hipertensão, taquicardia e isquemia.
Simone Saad Calil
Na Rename 2010: 3.2, 4, 17.1
t Pó para solução injetável 100 mg e 500 mg
t Processos inflamatórios agudos.
t Processos alérgicos agudos e adjuvante em anafilaxia.
t Asma aguda grave (uso restrito a crianças incapazes de reter a forma oral).
t Hipersensibilidade ao fármaco ou a qualquer componente da formulação.
t Infecções fúngicas sistêmicas.
t Vacinas com vírus vivos ou atenuados, pois a resposta imune pode ser reduzida pela hidrocortisona.
t Usar com cuidado nos casos de:
– úlcera péptica, diabete melito, hipertensão arterial, insuficiência cardíaca, enfarte do miocárdio, psicose, hipotireoidismo, glaucoma e osteoporose.
– insuficiência hepática (ver Apêndice C).
– insuficiência renal (ver Apendice D).
– crianças e adolescentes (risco de retardo de crescimento).
– lactação (ver Apêndice B).
– idosos (maior risco de hipertensão arterial e osteoporose).
t Pode aumentar a susceptibilidade e a gravidade de infecções como varicela e sarampo.
t Pode ativar ou exacerbar tuberculose e estrongiloidíase.
t Fator de risco na gravidez (FDA): C (ver Apêndice A).
t 1 a 5 mg/kg, por via intramuscular ou intravenosa, a cada 24 horas ou divididos a cada 12 horas.
t De 1 mês a 1 ano: 25 mg, por via intramuscular ou intravenosa, a cada 8 horas.
t De 1 a 5 anos: 50 mg, por via intramuscular ou intravenosa, a cada 8 horas.
t De 6 a 12 anos: 100 mg, por via intramuscular ou intravenosa, a cada 8 horas.
t 100 a 500 mg, por via intramuscular ou intravenosa durante 30 segundos a 10 minutos, a cada 8 horas.
Nota: Velocidade de administração intravenosa em crianças: bolo – solução diluída (50 mg/mL) durante 3 a 5 minutos; infusão intermitente – solução diluída (1 mg/mL) durante em 20 a 30 minutos.
t Absorção (intramuscular): imediata.
t Pico de efeito: 1 hora (intramuscular).
t Biotransformação: hepática.
t Eliminação: renal.
t Meia-vida de eliminação: 1-2 horas.
t Catarata, síndrome de Cushing (obesidade do tronco, face de “lua cheia”, comprometimento na cicatrização de feridas, acne, corcova de búfalo), glaucoma, hiperglicemia, insuficiência adrenocortical, tuberculose pulmonar, desequilíbrio de fluidos e eletrólitos.
t Maior susceptibilidade e maior gravidade de infecções.
t Euforia, depressão, hipertensão intracraniana, convulsão, cefaleia.
t Reações alérgicas.
t Úlcera péptica, náusea.
t Necrose asséptica óssea, osteoporose; miopatia proximal.
t Hipertensão arterial.
t Alcaçuz: aumento do risco de Efeitos adversos de corticoides. Cautela emcaso de uso concomitante, pois pode ser necessário reduzir a dose do corticosteroide.
t Alcurônio, atracúrio, galamina e brometo de hexaflurônio: diminuição da efetividade destes; prolongamento da fraqueza muscular e da miopatia.
t Bupropiona: diminuição do limiar convulsivo. O uso concomitante é contraindicado.
t Colestipol, primidona e rifapentina: podem reduzir a efetividade da hidrocortisona.
t Fluoroquinolonas: aumento do risco de ruptura dos tendões. Descontinuar o antibacteriano caso o paciente apresente dor, inflamação ou ruptura de tendão.
t Itraconazol: aumento das concentrações séricas de corticosteroide e do risco de seus efeitos adversos.
t Quetiapina: diminuição das concentrações sanguíneas de quetiapina. Cautela em caso de uso concomitante.
t Vacina contra rotavírus: aumento do risco de infecção pela vacina. O uso concomitante é contraindicado.
t Evitar contato com pessoas doentes.
t Atletas devem consultar autoridades esportivas, pois este medicamento pode ter uso restrito em alguns esportes.
t Não tomar qualquer tipo de vacina ou vacina sem consultar o médico.
t Não suspender abruptamente este medicamento após uso prolongado. Após logo período de terapia, prednisona deverá ser retirada de forma gradual.
t Armazenar à temperatura ambiente, em recipientes bem fechados, e ao abrigo de luz, calor e umidade.
t Observar orientação específica do produtor quanto a reconstituição, diluição, compatibilidade e estabilidade da solução.
t O pó deve ser reconstituído com o exato volume oferecido pelo produtor. Após reconstituição, proteger a solução da luz. A solução reconstituída deve ser utilizada somente se estiver límpida e deve ser descartada após 3 dias.
t Após a reconstituição é possível diluição com glicose 5%, cloreto de sódio 0,9% e solução de Ringer + lactato.
Atenção: o uso deste medicamento não deve ser suspenso sem orientação médica. Pode ser necessária retirada gradual para diminuir risco de insuficiência adrenocortical.
Rogério Aparecido Minini dos Santos
Na Rename 2010: item 3.2
t Pó para solução injetável 500 mg
t Afecções alérgicas agudas.
t Afecções da estrutura hematopoiética.
t Afecções da pele, incluindo dermatite herpetiforme bolhosa, dermatite exfoliativa, micose fungoide, pênfigo, síndrome de Stevens-Johnson, psoríase grave, dermatite seborreica grave.
t Afecções inflamatórias do sistema musculoesquelético, incluindo gota aguda, tenossinovite inespecífica, espondilite anquilosante, bursite subaguda, epicondilite, osteoartrite, artrite psoriásica, artrite reumatoide, artrite reumatoide juvenil.
t Afecções oculares alergo inflamatórias.
t Afecções respiratórias, incluindo sarcoidose sintomática do pulmão e tuberculose pulmonar disseminada e fulminante.
t Distúrbio do sistema endrócrino, incluindo insuficiência da suprarrenal, hiperplasia suprarrenal congênita e hipercalcemia de origem neoplásica.
t Doença de Crohn e colite ulcerativa (exacerbação aguda grave).
t Doença do colágeno.
t Edema cerebral associado a tumor primário ou metastático.
t Profilaxia de edema de laringe pós-extubação.
t Exacerbação aguda de esclerose múltipla.
t Tratamento paliativo de leucemias linfoide aguda e crônica.
t Tratamento paliativo linfoma maligno.
t Tratamento adjunto de pneumonia por Pneumocystis carinii.
t Púrpura trombocitopênica idiopática.
t Púrpura trombocitopênica tromboembólica.
t Profilaxia de reações alérgicas à administração de meios de contrates e muromonabe CD3.
t Profilaxia de síndrome artralgia-mialgia associada à infusão de dose total de ferrodextrana.
t Síndrome nefrótica.
t Triquinose com envolvimento neurológico ou cardíaco.
t Tuberculose meníngea com bloqueio subaracnoídeo.
t Vasculite reumatoide.
t Hipersensibilidade a algum constituinte do produto.
t Neonatos (preparações contendo álcool benzílico).
t Infecções fúngicas sistêmicas.
t Vacinação com vírus vivos ou atenuados.
t Usar com cuidado nos casos de:
– altas doses (pode requerer a administração profilática de antiácidos).
– injeção rápida (risco de colapso cardiovascular).
– cirrose (ver Apêndice C).
– hipotireoidismo.
– hipertensão.
– miastenia gravis, osteoporose.
– herpes simples ocular.
– úlcera péptica, diverticulite e colite ulcerativa.
– tendências psicóticas.
– insuficiência renal (ver Apêndice D).
– tuberculose (ativa ou latente).
– infecções sistêmicas não tratadas com antimicrobiano.
– novas infecções (mascaramento de sinais e sintomas e diminuição da defesa imunológica).
– vacinas.
t Categoria de risco na gravidez (ADEC): A (ver Apêndice A).
t Dose inicial de no mínimo 0,5 mg/kg, por via intravenosa lenta a cada 24 horas, ajustada de acordo com a resposta até alcançar a dose de adultos (10 a 40 mg); para terapia de altas doses, 30 mg/kg, por infusão intravenosa por no mínimo 30 minutos, repetida se necessário a cada 4 a 6 horas por até 72 horas.
t 30 mg/kg, por infusão intravenosa lenta, até de 8 em 8 horas. Dose máxima diária: 1 g.
t Dose inicial 10 a 40 mg, por via intravenosa lenta, podendo ser repetida pelas vias intravenosa ou intramuscular de acordo com a resposta do paciente; para terapia de altas doses, 30 mg/kg, por infusão intravenosa por no mínimo 30 minutos, repetida se necessário a cada 4 a 6 horas por até 72 horas.
t Dose inicial 0,8 mg/kg/dia, por infusão intravenosa lenta, diminuída ou aumentada de acordo com a resposta.
t 30 mg/kg/dia, por infusão intravenosa por 2 a 3 horas, durante 3 dias consecutivos; a mesma dose pode ser repetida semanalmente por 2 a 5 semanas com base na resposta do paciente.
t Dose inicial 10 a 40 mg, por via intravenosa lenta, podendo ser repetida pelas vias intravenosa ou intramuscular de acordo com a resposta do paciente; para terapia de altas doses, 30 mg/kg, por infusão intravenosa por no mínimo 30 minutos, repetida se necessário a cada 4 a 6 horas por até 72 horas.
t Na artrite reumatoide, dose de 1 g, por via intravenosa, em terapia de pulso, pode ser dada mensalmente.
t 1 g, por infusão intravenosa por 30 minutos, em dias alternados na primeira semana, seguido de 500 mg, a cada 7 dias, durante as próximas 3 semanas, e 250 mg, a cada 7 dias, por mais 3 semanas. Depois, dose de manutenção de 125 mg, a cada 7 dias, por pelo menos 6 semanas.
t Dose inicial 0,8 mg/kg/dia, por infusão intravenosa lenta, diminuída ou aumentada de acordo com a resposta.
t 1 g, em pulsos por via intravenosa, durante três dias.
t até 1 g, por infusão intravenosa lenta, a cada 24 horas, durante até 3 dias.
t 40 mg, por infusão intravenosa lenta 6 horas antes da extubação.
t 160 mg, por via intravenosa, a cada 24 horas, durante 7 dias, seguido de 64 mg, por via intravenosa ou intramuscular, em dias alternados, durante 1 mês.
t 40 a 60 mg, por via intravenosa, a cada 6 horas, durante 7 a 10 dias.
t 1 g, por via intravenosa, a cada 24 horas, durante 3 dias.
t 30 mg/kg, por infusão intravenosa por no mínimo 30 minutos, a cada 24 horas, durante 3 dias consecutivos, seguido de 20 mg/kg/dia durante mais 4 dias; dose de manutenção com prednisona 5 mg, por via oral, a cada 24 horas.
t 32 a 500 mg, por via intravenosa, entre 1 e 2 horas antes da administração dos fármacos.
t 125 mg, por via intravenosa, antes e após a infusão.
t 1 g, por via intravenosa, a cada 24 horas, durante 3 dias, seguido por 0,4 mg/ kg, por via oral, durante 27 dias.
t metilprednisolona 500 mg, por infusão intravenosa lenta, seguido de ciclofosfamida 500 mg, por infusão intravenosa lenta, no dia 1; metilprednisolona 1 g, por infusão intravenosa lenta, seguido de ciclofosfamida 15 mg/kg, por infusão intravenosa lenta, nos dias 8, 29 e 50. Manutenção posterior com azatioprina 2 mg/kg/dia ou ciclofosfamida 1,5 mg/kg/dia.
t Metabolismo hepático.
t Excreção renal.
t Meia-vida de eliminação de 2 a 3 horas.
t Hipertensão, insuficiência cardíaca congestiva.
t Atrofia dérmica, impedimento da cicatrização de feridas.
t Retenção de fluidos, retardo no crescimento, hipernatremia, hipopotassemia, síndrome de Cushing, hiperglicemia, insuficiência adrenocortical primária.
t Afecções do trato gastrintestinal, úlcera péptica.
t Anormalidades nos testes de função hepática.
t Risco de infecções.
t Fraqueza muscular, osteoporose.
t Depressão, euforia, elevação da pressão intracraniana, convulsões.
t Catarata, glaucoma.
t Tuberculose pulmonar.
t Ácido acetilsalicílico: pode ter suas concentrações plasmáticas reduzidas a níveis subterapêuticos e exacerbação de efeitos tóxicos gastrintestinais. Monitorar sinais e sintomas de toxicidade gastrintestinal e de redução do efeito do ácido acetilsalicílico.
t Alcaçuz (Glycyrrhiza glabra): pode aumentar o risco de Efeitos adversos da metilprednisolona. Evitar o uso concomitante.
t Alcurônio, atracúrio, galamina: podem ter sua efetividade diminuída e risco de fraqueza muscular prolongada e miopatia. Monitorar a efetividade do bloqueador neuromuscular e, se necessário, ajustar a dose, especialmente quando metilprednisolona é dada em altas doses ou por longos períodos.
t Antibióticos macrolídeos, antifúngicos azólicos, diltiazem e nefazodona: podem aumentar as concentrações plasmáticas de metilprednisolona, exacerbando seus efeitos adversos. Reduzir a dose de metilprednisolona, se necessário.
t Aprepitanto: pode exacerbar os efeitos de metilprednisolona. Ajustar a dose de metilprednisolona, quando doses maiores que 40 mg de aprepitanto forem dadas concomitantemente.
t Carbamazepina, fenitoína, fenobarbital e fosfenitoína: podem diminuir a efetividade de metilprednisolona. Ajustar a dose de metilprednisolona até que o efeito terapêutico seja mantido.
t Ciclosporina: pode ter seus efeitos tóxicos aumentados e elevar as concentrações de metilprednisolona. Monitorar níveis plasmáticos de ciclosporina e sinais de exacerbação do efeito de metilprednisolona. Se necessário, considerar ajuste de dose de ambos os fármacos.
t Fluindiona: pode resultar em aumento do risco de sangramento. Se o uso concomitante for necessário, monitorar tempo de protrombina e considerar ajuste de dose de fluindiona.
t Fluoroquinolonas: podem resultar no aumento do risco de ruptura de tendão. Descontinuar o antibiótico em caso de dor ou inflamação no tendão. O risco permanece após o uso de fluoroquinolonas.
t Primidona: pode diminuir a efetividade de metilprednisolona. Monitorar a eficácia terapêutica da metilprednisolona e ajustar a dose se necessário.
t Quetiapina: pode ter suas concentrações plasmáticas diminuídas. Utilizar com cautela.
t Quinupristina/dalfopristina: pode resultar no aumento do risco de Efeitos adversos da metilprednisolona. Proceder à redução da dose de metilprednisolona.
t Rifampicina: pode resultar na redução da efetividade de metilprednisolona. Se necessário, ajustar a dose de metilprednisolona.
t Vacina rotavírus: aumento do risco de infecção pela vacina. O uso concomitante é contraindicado.
t Varfarina e anticoagulantes cumarínicos: podem ter seus efeitos diminuídos ou pode ocorrer aumento do risco de sangramento. Se o uso concomitante for necessário, monitorar tempo de protrombina, principalmente no início e final do uso. Considerar ajuste de dose do anticoagulante.
t Antes de iniciar o tratamento é importante identificar: história prévia de hipersensibilidade a metilprednisolona, infecções por fungos ou outras infecções não tratadas, gravidez, lactação, hipertensão, diabetes, doenças do estômago, osteoporose, herpes ocular, tuberculose, cardiomiopatias.
t Este medicamento pode inibir a resposta vacinal às vacinas, se as mesmas forem administradas concomitantemente.
t Cuidado especial deve ser tomado quando se administra metilprednisolona em mulheres na pré-menopausa e pós-menopausa (risco elevado de osteoporose).
t A metilprednisolona pode causar diminuição na defesa do organismo contra infecções, portanto evitar o contato com pessoas portadoras de doenças infecciosas.
t Informar ao médico se ocorrer distúrbios mentais ou emocionais, como alteração do humor, estresse, e ansiedade.
t Armazenar os frascos com succinato sódico de metilprednisolona à temperatura entre 20 a 25 ºC e protegidos da luz.
t Observar orientação específica do produtor quanto a reconstituição, diluição, compatibilidade e estabilidade da solução.
t Reconstituir o pó com água para injeção. Após reconstituição, a solução permanece estável por até 24 horas sob temperaturas entre 20 a 25 ºC, e sob refrigeração (cerca de 4 ºC) por uma semana.
t Soluções para infusão são preparadas preferentemente com solução injetável de cloreto de sódio 0,9%, permanecendo estável por 24 horas sob temperaturas entre 20 a 25 ºC em concentração de até 0,125 mg/mL. Acima destas concentrações, seguir a orientação do produtor.
t A estabilidade do succinato sódico de metilprednisolona em soluções injetáveis de glicose 5% e Ringer + lactato é muito variável, neste caso seguir a orientação do produtor.
t As soluções de succinato sódico de metilprednisolona são incompatíveis com vários medicamentos e substâncias em misturas intravenosas. Recomenda-se que seja realizada uma pesquisa específica sobre este aspecto quando se considerar o preparo e administração de misturas intravenosas desse medicamento.
Fernando de Sá Del Fiol e Maria Inês de Toledo
Na Rename 2010: itens 5.1.5 e 5.6.2.3
t Comprimido 500 mg.
t Infecções urinárias agudas não complicadas.
t Toxoplasmose. Contraindicações 1-3 t Porfiria.
t Hipersensibilidade a sulfadiazina ou a outras sulfonamidas.
t Crianças com menos de 2 meses (exceto com toxoplasmose congênita).
t Terceiro trimestre de gravidez. Categoria de risco na gravidez (FDA): C (ver Apêndice A).
t Usar com cuidado nos casos de:
– insuficiência hepática (ver Apêndice C).
– insuficiência renal (ver Apêndice D).
– deficiência de glicose-6-fosfato desidrogenase, distúrbios hematológicos e predisposição a deficiência de folato.
– lactação (ver Apêndice B).
t 75 mg/kg, por via oral, seguido de 150 mg/kg/dia, divididos a cada 4 a 6 horas, durante 3 a 5 dias.
t 25 mg/kg (máximo 1 g), por via oral, a cada 6 horas, combinada a pirimetamina 2 mg/kg (máximo 50 mg) mais folinato de cálcio 15 mg, ambos por via oral, a cada 24 horas, durante 3 dias. Depois, sulfadiazina 25 mg/kg, por via oral, a cada 6 horas, combinada a pirimetamina 1 mg/kg (máximo 25 mg) mais folinato de cálcio 15 mg, ambos por via oral, a cada 24 horas, até completar 4 semanas de tratamento.
t 0,5 a 1 g, por via oral, a cada 6 horas, durante 3 a 5 dias.
t 0,5 a 1 g, por via oral, a cada 6 a 12 horas, combinada a pirimetamina 75 a 100 mg mais folinato de cálcio 15 mg, ambos por via oral, a cada 24 horas, durante 3 dias. Depois, sulfadiazina 0,5 a 1 g, por via oral, a cada 6 a 12 horas, combinada a pirimetamina 25 a 50 mg mais folinato de cálcio 15 mg, ambos por via oral, a cada 24 horas, até completar 4 a 6 semanas de tratamento.
t Pico de concentração sérica: 3 a 6 horas.
t Meia-vida: 10 horas.
t Metabolismo: hepático.
t Eliminação: renal (57%); alcalinização da urina torna mais rápida a excreção.
t Náusea, diarreia, vômito, dor abdominal, estomatite, hepatite e pancreatite.
t Exantema, reações de fotossensibilidade, dermatite exfoliativa, eritema nodoso, síndrome de Stevens-Johnson e necrólise epidérmica tóxica.
t Reações de hipersensibilidade.
t Cefaleia, depressão, convulsão, ataxia, vertigem, insônia e alucinação.
t Granulocitopenia, agranulocitose, anemia aplástica, trombocitopenia, leucopenia, anemia hemolítica, púrpura e hipoprotrombinemia.
t Nefrotoxicidade, cristalúria e nefrite intersticial.
t Icterícia, hepatomegalia, necrólise hepática e alterações de provas funcionais hepáticas (0,1%).
t Hiperbilirrubinemia e kernicterus em recém-nascidos e lactentes, se o fármaco for dado a grávidas no último mês de gravidez, à puérpera que amamenta ou no período perinatal (até 2 meses de vida).
t Acetoexamida, clorpropamida, glipizida, tolazamida, tolbutamida: podem ter seus efeitos hipoglicemiantes aumentados. Evitar o uso concomitante com sulfadiazina, mas caso este seja necessário, monitorar estreitamente os níveis de glicose sanguínea.
t Ciclosporina: pode ter sua efetividade reduzida. Se possível, monitorar as concentrações de ciclosporina.
t Fosfenitoína: o uso concomitante pode resultar em aumento do risco de toxicidade da fosfenitoína. Monitorar o paciente para evidências de toxicidade como nistagmo e ataxia. Caso o paciente apresentar sinais de toxicidade ou receber sulfadiazina por mais de cinco dias considerar a avaliação das concentrações séricas de fenitoína.
t Orientar para ingerir bastante líquido para evitar cristalúria.
t Orientar para o uso durante todo o tempo prescrito, mesmo que haja melhora dos sintomas com as primeiras doses.
t Manter à temperatura ambiente, entre 15 a 30 °C, proteger da luz.
Ana Cláudia de Brito Passos
Na Rename 2010: item 20.2
t Antiinfectantes
t Creme 1%
t Profilaxia e tratamento de infecção em queimaduras.
t Tratamento adjuvante, de curto prazo, para infecção em úlcera de perna e úlcera de decúbito.
t Profilaxia de infecção em áreas de abrasão em enxerto de pele.
t Hipersensibilidade à prata ou a sulfonamidas.
t Bebês até 2 meses de idade (segurança e eficácia não foram estabelecidas).
t Usar com cuidado nos casos de:
– deficiência de glicose 6-fosfato desidrogenase (G6PD).
– insuficiência hepática (ver Apêndice C).
– insuficiência renal (ver Apêndice D).
– lactação (ver Apêndice B).
– ocorrência de leucopenia após 2 a 3 dias de uso (a alteração é auto-limitada; não é necessário suspender o uso da sulfadiazina de prata, mas deve-se fazer contagens sanguíneas).
t Evitar uso em áreas extensas.
t Suspender o tratamento se surgirem alterações hematológicas e erupções cutâneas.
t Categoria de risco na gravidez (FDA): B (dois primeiros trimestres).
t Aplicar uma camada de 1,5 mm, 1 a 2 vezes ao dia, com luva estéril, até a melhora da lesão. A aplicação pode ser mais frequente em casos de lesões em áreas Susceptíveis à remoção por movimentação do paciente. Curativos podem ser colocados sobre o creme, mas usualmente não são necessários.
t Absorção: sulfadiazina – pequena, com aplicação em grandes áreas e/ou períodos prolongados; prata – alguma quantidade pode ser absorvida.
t Metabolização: fígado.
t Eliminação renal: 60%.
t Meia-vida de eliminação: 10 horas. A meia-vida aumenta para 22 horas em pacientes anúricos.
t Alteração da estrutura hematopoiética (raro), leucopenia (raro).
t Prurido, irritação de pele, exantema; erupções cutâneas (infrequente); eritema multiforme.
t Reações alérgicas e argiria (coloração ligeiramente acinzentada ou azulada da pele) (pouco frequentes).
t Reação de imuno-hipersensibilidade.
t Temperatura corpórea acima do normal, distúrbios eletrolíticos.
t Cristalúria, nefrotoxicidade.
t Papaína: aplicação concomitante de papaína e formulações contendo sais de prata pode resultar na inativação da papaína como debridante enzimático. Evitar o contato da pele com formulações de metais pesados, como a sulfadiazina prata, durante o debridamento químico com a papaína.
t Ácido paraminobenzoico ou compostos relacionados: a prata contida na sulfadiazina de prata pode inativar os agentes enzimáticos debridantes.
t Orientar que este medicamento somente pode ser empregado para uso externo e para não aplicar ao redor dos olhos.
t Orientar para lavar as mãos antes e depois de usar o creme.
t Ensinar a remover a pele necrosada e limpar a área antes da aplicação.
t Orientar utilizar luva estéril para aplicação.
t Conservar à temperatura ambiente, em recipientes bem fechados. Manter ao abrigo de luz, calor e umidade. Não congelar.
Fernando de Sá Del Fiol e Maria Inês de Toledo
Na Rename 2010: itens 5.1.5 e 5.4
t Comprimido 400 mg + 80 mg.
t Suspensão oral (40 mg + 8 mg)/mL.
t Solução injetável (80 mg + 16 mg)/mL.
t Infecções por microrganismos sensíveis
t Tratamento pneumocistose.
t Profilaxia de pneumocistose em pessoas com Aids.
t Hipersensibilidade à sulfonamida ou trimetoprima.
t Porfiria.
t Anemia megaloblástica por deficiência de folato.
t Usar com cuidado nos casos de:
– insuficiência renal (ver Apêndice D).
– insuficiência hepática grave (uso não recomendado).
– deficiência de glicose-6-fosfato desidrogenase, predisposição a deficiência de folato, hiperpotassemia, alterações hematológicas, asma e idosos.
– crianças com menos de 6 semanas de vida (uso não recomendado).
t Suspender o uso, imediatamente, se ocorrer exantema e alterações hematológicas.
t Manter adequada hidratação para evitar cristalúria.
t Categoria de risco na gravidez (FDA): C.
t 30 a 50 mg/kg (sulfametoxazol) + 6 a 10 mg/kg (trimetoprima), por via oral, dividido a cada 12 horas, por 5 a 14 dias.
t 40 a 50 mg/kg (sulfametoxazol) + 6 a 10 mg/kg (trimetoprima), por via oral, dividido a cada 6 a 12 horas, por 5 a 14 dias.
t Em infecção do trato urinário inferior aguda não complicada o tratamento deve ser realizado por via oral durante 3 dias.
t 75 a 100 mg/kg (sulfametoxazol) + 15 a 20 mg/kg, por via oral ou intravenosa, dividido a cada 6 a 12 horas, durante 14 a 21 dias.
t 25 mg/kg (sulfametoxazol) + 5 mg/kg (trimetoprima), por via oral, dividido a cada 12 horas, 3 vezes por semana em dias alternados ou consecutivos.
t 800 a 1200 mg (sulfametoxazol) + 160 a 240 mg (trimetoprima), por via oral ou intravenosa, a cada 12 horas, durante 5 a 14 dias.
t 75 a 100 mg/kg (sulfametoxazol) + 15 a 20 mg/kg, por via oral ou intravenosa, dividido a cada 6 a 12 horas, durante 14 a 21 dias.
t 800 mg (sulfametoxazol) + 160 mg (trimetoprima), por via oral, a cada 24 horas ou 3 vezes por semana.
t Apresenta biodisponibilidade oral de 90 a 100%.
t Pico de concentração plasmática: 1 a 4 horas (oral).
t Meia-vida: 8 a 11 horas para o sulfametoxazol e 6 a 17 horas para a trimetoprima.
t Metabolismo: hepático.
t Excreção: renal (10% a 30% para o sulfametoxazol e 50% a 75% para a trimetoprima).
t Exantema, síndrome de Stevens-Johnson, eritema multiforme, dermatite alérgica e necrólise epidérmica tóxica, vasculite.
t Arritmia cardíaca e miocardite.
t Hipouricemia, hiperpotassemia, hipoglicemia, hiponatremia e acidose metabólica.
t Esofagite, pancreatite, enterocolite pseudomembranosa.
t Agranulocitose, anemias aplástica, megaloblástica e hemolítica, neutropenia, leucopenia, pancitopenia, eosinofilia e trombocitopenia.
t Hepatite, icterícia, necrólise hepática e hepatotoxicidade.
t Reações de hipersensibilidade graves, lupus eritematoso sistêmico.
t Ataxia, meningite, cefaleia, parkinsonismo, tremor, convulsão, ansiedade, delírio, depressão e psicose.
t Nefrotoxicidade, urolitíase, cristalúria e nefrite intersticial.
t Náuseas, vômitos e diarreia.
t Acetoexamida, clorpropamida, gliburida, glipizida, tolazamida, tolbutamida: pode aumentar a ação hipoglicêmica. Evitar o uso. Caso necessário, monitorar glicose sanguínea.
t Ácido folínico: pode aumentar o risco de falha terapêutica. Monitorar a eficácia do tratamento.
t Amantadina: pode resultar em toxicidade no sistema nervoso central (insônia, confusão). Monitorar estes sinais.
t Anisindiona: risco aumentado de sangramento. Monitorar o tempo de protrombina, diminuindo a dose do anticoagulante, se necessário.
t Antiarrítmicos da classe 1A (quinidina, disopiramida, procainamida, pirmenol, prajmálio, hidroquinidina): aumento do risco de cardiotoxicidade. Monitorar os níveis do antiarrítmico e ajustar dose, se necessário.
t Antidepressivos tricíclicos: aumento do risco de cardiotoxicidade. Uso concomitante não recomendado.
t Bepridil, cisaprida, dofetilida, mesoridazina, pimozida, terfenadina, tioridazina: aumento do risco de cardiotoxicidade. Associação contraindicada.
t Digoxina: pode aumentar o risco de toxicidade da digoxina. Avaliar sinais de intoxicação digitálica (náuseas, vômitos e arritmias). Diminuir a dose de digoxina, se necessário.
t Enalapril, quinapril: pode resultar em hiperpotassemia. Monitorar potássio plasmático ou substituir o anti-hipertensivo.
t Fenitoína, fosfenitoína: pode aumentar a toxicidade da fenitoína (ataxia, nistagmo, tremores). Monitorar o paciente para sinais de toxicidade e ajustar a dose de fenitoína, caso seja necessário.
t Gemifloxacino: aumento do risco de cardiotoxicidade. Aumento do intervalo QT. Associação não recomendada.
t Lamivudina, zidovudina: pode aumentar as concentrações de lamivudina e zidovudina. Observar Efeitos adversos da lamivudina e zidovudina (distúrbios gastrintestinais, cefaleia, fadiga, mialgia e neutropenia). Alteração nas doses dos fármacos não é recomendada.
t Metotrexato: aumento do risco da toxicidade do metotrexato (mielotoxicidade). Caso haja necessidade da utilização, monitorar parâmetros hematológicos.
t Pirimetamina: aumento no risco de anemia megaloblástica e pancitopenia. Monitorar padrões hematológicos e associar ácido folínico.
t Repaglinida: pode aumentar a concentração plasmática de repaglinida. Monitorar glicose sanguínea e ajustar a dose de repaglinida, se necessário.
t Rifabutina: pode aumentar a toxicidade do sufametoxazol. Usar com precaução e monitorar sinais da intoxicação (exantema, leucopenia, trombocitopenia e alterações em transaminases hepáticas).
t Rosiglitazona: pode aumentar a concentração plasmática de rosiglitazona, com risco de hipoglicemia. Monitorar paciente para o risco de hipoglicemia.
t Varfarina: aumento no risco de sangramento. Monitorar o paciente (tempo de protrombina).
t Orientar para a importância de ingerir bastante líquido, para evitar cristalúria.
t Orientar para não ingerir bebida alcoólica durante o tratamento, pelo risco de reação tipo dissulfiram.
t Orientar para o uso durante todo o tempo prescrito, mesmo que haja melhora dos sintomas com as primeiras doses.
t Proteger a pele da luz solar.
t Armazenar à temperatura ambiente, entre 15 e 30 oC, proteger do calor, umidade e luz direta.
t Observar orientação específica do produtor quanto a diluição, compatibilidade e estabilidade da solução.
t Para cada ampola de 5 mL deve-se adicionar 125 mL de glicose 5%. Após diluição, a solução não deve ser refrigerada. Administrar dentro de 6 horas.
t Em caso de paciente com restrição hídrica, para cada ampola de 5 mL deve-se adicionar 75 mL de glicose 5%. Administrar dentro de 2 horas.
t Descartar a solução se cristalizar.
t A infusão intravenosa deve ser feita durante 60 a 90 minutos. Devem-se evitar infusões rápidas ou injeções em bolo.
Maria Isabel Fischer
Na Rename 2010: itens 3.3 e 16.6
t Comprimido 500 mg
t Distúrbios reumatoides.
t Colite ulcerativa.
t Doença de Crohn.
t Hipersensibilidade à sulfassalazina, salicilatos e sulfonamidas.
t Insuficiência renal grave.
t Crianças com menos de 2 anos de idade.
t Porfiria.
t Obstrução intestinal ou urinária.
t Gravidez (37 a 42 semanas de gravidez).
t Usar com cuidado nos casos de:
– discrasias sanguíneas, alergia ou asma graves.
– história de alergia.
– deficiência de G6PD.
– artrite reumatoide juvenil de curso sistêmico (maior risco de reações tipo doença do soro).
– insuficiência hepática (ver Apêndice C).
– insuficiência renal (ver Apêndice D).
– acetiladores lentos.
– lactação (ver Apêndice B).
t Monitorar função renal regularmente (ver Apêndice D).
t Monitorar função hepática e contagem sanguínea durante os 3 primeiros meses de tratamento.
t A sulfassalazina pode causar deficiência de folato; considerar suplementação de ácido folínico (1 mg/dia).
t Categoria de risco na gravidez (FDA): B (categoria D, se administrado próximo ao parto) (ver Apêndice A).
t Acima de 6 anos: 30 a 50 mg/kg, por via oral, divididos a cada 12 horas. Dose máxima: 2 g/dia.
t Dose inicial 500 mg, por via oral, a cada 12 ou 24 horas, aumentado em 500 mg em intervalos de 1 semana. Dose de manutenção 1 g, a cada 12 horas. Dose máxima diária: 3 g.
t Dose inicial 1 g a 2 g, por via oral, a cada 6 horas, na crise aguda até a remissão dos sintomas. Dose de manutenção 500 mg, por via oral, a cada 6 horas.
t 1 g a 2g, por via oral, a cada 6 horas, na crise aguda até a remissão dos sintomas.
Nota: doses acima de 4 g/dia estão associadas a aumento no risco de toxicidade e de reações adversas. Doses iniciais de 500 mg cada 6 ou 12 horas podem ser recomendadas para diminuir o desconforto gástrico.
t Início de ação: em colite ulcerativa: 3 a 4 semanas; artrite reumatoide: 9 semanas.
t Pico de concentração sérica: 3 horas, sulfapiridina 12 horas.
t Meia-vida de eliminação: 5,7 a 10 horas.
t Absorção: aproximadamente 1/3 da dose ingerida é absorvida.
t Biodisponibilidade: 15%
t Metabolismo: no trato intestinal é clivada via enzimas bacterianas a sulfapiridina e mesalazina.
t Excreção: renal (75% a 91% como sulfassalazina ou metabólitos, dependendo da forma farmacêutica).
t Frequência maior que 10%:
– Cefaleia.
– Fotossensibilidade.
– Exantema.
– Anorexia, náusea, vômito, diarreia, desconforto gástrico.
– Oligospermia reversiva.
t Frequência menor que 3%:
– Dermatite exfoliativa, prurido, necrólise epidérmica, urticária, alopecia.
– Alucinações, vertigem, depressão, insônia.
– Hepatite.
– Reações tipo doença do soro.
– Alterações hematológicas, anemia megaloblástica, anemia hemolítica, anemia aplástica, leucopenia, neutropenia, trombocitopenia.
– Reações de hipersensibilidade: eritema multiforme, síndrome de Stevens Johnson, febre, anafilaxia.
– Nefrite intersticial, urina de cor alaranjada, nefrotoxicidade, proteinúria, cristalúria.
– Ataxia, neuropatia periférica.
– Sintomas tipo lupus eritematoso sistêmico.
t Ciclosporina: pode ter sua eficácia reduzida. O uso concomitante de ciclosporina com sulfonamida oral pode aumentar o risco de nefrotoxicidade. Se a coadministração não puder ser evitada, monitorar frequentemente as concentrações de creatinina sérica e de ciclosporina, principalmente quando a sulfassalazina é adicionada ou descontinuada. Ajustar a dose de ciclosporina conforme a necessidade. Pode ser necessário aumentar as doses de ciclosporina quando administrar com sulfassalazina (para manter as concentrações terapêuticas da ciclosporina) ou diminuir, quando a sulfassalazina é diminuída.
t Digoxina: pode ter sua biodisponibilidade reduzida, possivelmente resultando em efeito terapêutico diminuído. Monitorar as concentrações da digoxina durante a terapia e após a descontinuidade da sulfassalazina. Se ocorrer redução dos níveis ou da resposta clínica da digoxina pode ser necessário descontinuar a sulfassalazina. Pode ser necessário aumentar a dose da digoxina.
t Metotrexato: risco aumentado de hepatotoxicidade. Pacientes devem ser monitorados cuidadosamente para sintomas clínicos de toxicidade hepática; monitorar testes de função hepática. Adicionalmente, sulfonamidas podem aumentar o risco de supressão da medula óssea. Se não puder ser evitado o uso concomitante, monitorar cuidadosamente para toxicidade hematológica.
t Varfarina e outros anticoagulantes: pode haver aumento do efeito anticoagulante, resultando em hemorragia. Monitorar a ação anticoagulante e ajustar a dose se necessário.
t Orientar para usar o medicamento após as refeições ou com alimento para diminuir a irritação gastrintestinal nos primeiros dias de tratamento. Ingerir com um copo cheio de água.
t Manter ingesta hídrica e diurese adequadas para prevenir cristalúria e evitar formação de pedras.
t Não exceder 8 horas de intervalo entre as doses, mesmo à noite.
t Monitorar contagem sanguínea e interromper imediatamente o tratamento se houver suspeita ou evidência de alteração hematológica.
t Pode ocorrer coloração amarelo-alaranjada da urina ou da pele sem significado clínico.
t Contatar o médico imediatamente se ocorrer reações de hipersensibilidade.
t Pacientes devem ser orientados a relatar imediatamente se ocorrer sangramento, infecção, dor de garganta, febre, ou mal-estar, púrpura (hemorragia cutânea), pele ou olhos amarelados fraqueza não usual.
t Pode levar de 4 a 12 semanas até o paciente sentir melhora (artrite reumatoide).
t Evitar exposição a luz solar.
t Armazenar à temperatura ambiente, protegido da luz direta, umidade e calor
Beatriz Garcia Mendes
Na Rename 2010: item 5.5.2.1
t Comprimido 300 mg.
t Solução oral 20 mg/mL.
t Tratamento de infecção por HIV, em combinação com outros antirretrovirais.
t Hipersensibilidade ao abacavir. Em caso de reação de hipersensibilidade, suspender o tratamento e nunca fazer a reexposição devido ao risco de hipotensão grave e morte.
t Insuficiência hepática moderada e grave (ver Apêndice C).
t Usar com cuidado nos casos de:
– acidose lática e insuficiência hepática (suspender o tratamento se achados clínicos sugerirem acidose lática e hepatotoxicidade) (ver Apêndice C).
– mulheres, principalmente obesas e em tratamentos prolongados (são mais propensas a desenvolver hepatopatia); utilizar abacavir em combinação com outros antirretrovirais.
– insuficiência renal e risco de doenças cardiovasculares.
t Categoria de risco na gravidez (FDA): C (ver Apêndice A).
t Dose de 8 mg/kg, por via oral, a cada 12 horas, em combinação com outros antirretrovirais. Dose máxima diária 600 mg.
t Dose de 300 mg, por via oral, a cada 12 horas, ou 600 mg, por via oral, a cada 24 horas, em combinação com outros antirretrovirais.
t Início de efeito: 4 semanas.
t Pico de efeito: 0,7 a 1,7 horas.
t Meia-vida: 1,0 a 1,5 horas.
t Metabolismo: hepático, pela enzima álcool desidrogenase.
t Excreção: renal (aproximadamente 83%) e fecal (16%).
t Ligação a proteínas plasmáticas: 50%.
t Náusea (7 a 19%), vômito (2 a 10%), diarreia (7%), dor abdominal (6%), gastrite (6 a 19%).
t Acidose lática, hepatotoxicidade (6 a 8%), elevação das enzimas hepáticas AST e ALT (5 a 6%), pancreatite.
t Reações de hipersensibilidade (2,3 a 9%) que podem ser fatais, síndrome de Stevens-Johnson (menos de 1%), exantema (5 a 6%), síndrome da reconstituição imune.
t Insônia e outros distúrbios do sono (10%), depressão, ansiedade (5%), convulsão, cefaleia (7 a 13%), fadiga (7 a 12%).
t Febre (6%).
t Necrólise epidérmica tóxica.
t Discrasias sanguíneas.
t Anormalidades metabólicas tais como lipodistrofia, hipertrigliceridemia, hipercolesterolemia, resistância a insulina, hiperglicemia.
t Distúrbios no sistema cardiovascular.
t Etanol: aumento em 41% das concentrações de abacavir, devido a competição pela via metabólica comum da álcool desidrogenase. O consumo de álcool durante o tratamento é contraindicado.
t Metadona: diminuição dos níveis da metadona devido ao aumento de sua depuração. Monitorar sinais e sintomas e proceder ajuste de dose, se necessário.
t Ribavirina: aumenta o risco de acidose lática potencialmente fatal. O uso concomitante deve ser realizado com cautela e apenas se o benefício superar o risco.
t Tipranavir: diminuição dos níveis séricos do abacavir. Recomenda-se monitorar os pacientes quanto a eficácia e proceder ajuste da dose.
t Orientar que este medicamento pode ser ingerido com ou sem alimentos.
t Orientar para não usar bebida alcoólica durante o tratamento.
t Orientar para empregar métodos contraceptivos.
t Reforçar orientações sobre prevenção da transmissão do HIV.
t Orientar para o uso durante todo o tempo prescrito, mesmo que haja melhora dos sintomas com as primeiras doses.
t Manter à temperatura ambiente, entre 15 a 30 ºC.
t A solução oral pode ser refrigerada, mas não congelada.
Atenção: o sulfato de abacavir pode provocar reações graves de hipersensibilidade, potencialmente fatais, que apresentam sintomas sistêmicos respiratórios e/ou gastrintestinais, em geral com febre e sem acometimento de mucosas. Sempre que houver qualquer suspeita dessas reações, o uso do medicamento deve ser imediatamente suspenso. Se houver melhora após a retirada, sua reintrodução está contraindicada.
Maria Inês de Toledo e Simone Sena Farina
Na Rename 2010: item 5.1.4
t Solução injetável 50 mg/mL e 250 mg/mL.
t Infecções hospitalares graves, causadas por bacilos gram-negativo aeróbios e Enterococcus sp resistentes a gentamicina.
t Hipersensibilidade à amicacina ou a outros aminoglicosídeos.
t Miastenia grave.
t Usar com cuidado nos casos de:
– neonatos, crianças (meia-vida aumentada) e idosos (diminuição de função renal). Ajustar a dose e monitorar concentração plasmática, função renal e auditiva.
– obesos e pacientes com insuficiência renal (ver Apêndice D). Ajustar a dose e monitorar concentração da amicacina.
t Evitar o uso prolongado.
t Não usar concomitantemente com outros fármacos ototóxicos, nefrotóxicos ou neurotóxicos.
t O uso concomitante de anestésicos e/ou bloqueadores neuromusculares pode resultar em paralisia respiratória.
t Categoria de risco na gravidez (FDA): D.
t Dose inicial: 10 mg/kg/dia por via intravenosa. Dose de manutenção: 7,5 mg/ kg a cada 12 horas. Dose máxima diária: 15 mg/kg. Admite infusão intravenosa, durante 60-120 minutos.
t 15 mg/kg por dia, por via intramuscular ou intravenosa, em dose única diária ou divididos a cada 8 ou 12 horas, durante 7 a 10 dias.
t 15 mg/kg por dia, por via intramuscular ou intravenosa lenta, divididos a cada 12 horas. Para infecções de maior gravidade: 22,5 mg/kg por dia, divididos a cada 8 horas, durante 7 a 10 dias. Dose máxima: 15 mg/kg/dia ou 1,5 g/dia.
Observações
t Infundir por via intravenosa durante 30 a 60 minutos.
t Pico de concentração sérica: 1 hora (intramuscular), 30 minutos (intravenosa).
t Meia-vida plasmática: 2 a 3 horas (adultos com função renal normal)
t Excreção: renal (98%, em forma inalterada).
t É removida por hemodiálise.
t Ototoxicidade vestibular e coclear (3% a 5%), perda auditiva (0,5%, às vezes irreversível) e/ou dificuldade de equilíbrio, zumbidos.
t Náuseas, vômitos, estomatites.
t Nefrotoxicidade (2% a 25%).
t Bloqueio neuromuscular, paralisia muscular aguda e apneia em pacientes submetidos a medicamentos anestésicos e bloqueadores neuromusculares periféricos, fraqueza.
t Erupções cutâneas.
t Colite associada ao uso de antibiótico, hipomagnesemia na terapia prolongada.
t Eosinofilia.
t Bloqueadores neuromusculares não-despolarizantes: podem resultar em aumento ou prolongamento do bloqueio neuromuscular, podendo acarretar depressão respiratória e paralisia. Evitar o uso concomitante, se possível; se o mesmo for necessário, monitorar rigorosamente a função respiratória.
t Ibuprofeno: aumento da exposição à amicacina.
t Vancomicina: aumento do risco de nefrotoxicidade.
t Não usar concomitantemente a outros fármacos ototóxicos, nefrotóxicos ou neurotóxicos.
t 1,3 g de sulfato de amicacina = 1 g de amicacina.
t A solução é incolor e estável à temperatura ambiente por até 2 anos. Pode escurecer para amarelo pálido, devido à oxidação, sem afetar a atividade.
t Observar orientação específica do produtor quanto a diluição, compatibilidade e estabilidade da solução.
t A solução pode ser preparada por adição de 500 mg a 100 ou 200 mL de solução diluente estéril, como solução salina 0,9%, glicose 5%, Ringer + lactato ou qualquer outra solução compatível.
t Incompatível com: penicilinas (ampicilina, benzilpenicilina, carbenicilina, ticarcilina), cefalosporinas (cefazolina, cefepima, cefamandol), ácido clavulânico, solução lipídica 10% e bicarbonato de sódio. Devido ao alto potencial de incompatibilidade da amicacina não se deve misturar em seringa, solução de infusão ou usar a mesma via intravenosa para administrar outros fármacos.
t É estável por 60 dias estocada em geladeira a 4 oC em concentrações que variam de 250 mg/mL a 5 g/L.
Beatriz Garcia Mendes
Na Rename 2010: item 5.5.2.4
t Cápsulas 150 mg e 200 mg.
t Tratamento de infecção por HIV em combinação com outros antirretrovirais.
t Hipersensibilidade ao atazanavir.
t Uso concomitante com di-hidroergotamina, ergonovina, indinavir, irinotecano, lovastatina, metilergonovina, midazolam (adiministrado via oral), pimozida, rifampicina, sinvastatina, erva-de-são-joão e triazolam.
t Porfiria aguda.
t Usar com cuidado nos casos de:
– resistência a outros inibidores da protease (risco de resistência cruzada).
– falha virológica prévia (administrar ritonavir simultaneamente).
– distúrbios da condução cardíaca, insuficiência hepática (ver apêndice C), hepatite B e C, cirrose hepática, hemofilia A e B, e diabetes melito.
t Risco de hiperglicemia (monitorar glicemia regularmente).
t Suspender o tratamento se ocorrer exantema intenso ou nefrolitíase.
t Lactação (ver Apêndice B).
t Categoria de risco na gravidez (FDA): B.
t Pacientes virgens de terapia: 400 mg, por via oral, a cada 24 horas, em combinação com outros antirretrovirais.
t Pacientes em falha terapêutica: 300 mg de atazanavir + 100 mg de ritonavir, por via oral, a cada 24 horas, em combinação com outros antirretrovirais.
t Absorção aumentada com ingestão concomitante de alimentos.
t Início de resposta: 2 semanas.
t Pico de concentração plasmática: 2 a 2,5 horas.
t Meia-vida: aproximadamente 7 horas.
t Metabolismo hepático.
t Excreção: fecal (79%) e renal (13%)
t Dislipidemias, lipodistrofia (2 a 9%), hiperglicemia, acidose lática.
t Exantema (20 a 21%), eritema multiforme, síndrome de Stevens-Johnson, alopecia.
t Febre.
t Náusea (20%), vômito, diarreia (15 a 30%), dor abdominal (20%).
t Elevação das enzimas hepáticas e bilirrubina, icterícia esclerótica (2 a 6%), pancreatite.
t Distúrbios na condução cardíaca.
t Cefaleia (20 a 25%), insônia, depressão, neuropatia periférica.
t Urolitíase.
t Mialgia (4%), fadiga.
t Alho: aumento do risco de falha terapêutica e resistência antirretroviral devido à diminuição da concentração plasmática do atazanavir. Evitar o uso concomitante.
t Amiodarona: aumento da concentração plasmática da amiodarona, aumentando o risco de cardiotoxicidade. Monitorar a concentração plasmática da amiodarona bem como a função cardíaca.
t Antagonistas dos receptores H2 da histamina (cimetidina, famotidina): aumento do risco de falha terapêutica devido à diminuição da concentração plasmática do atazanavir. No caso de coadministração, o ajuste de dose é necessário.
t Antiarrítimicos de classe IA (disopiramida, quinidina): aumento das concentrações plasmáticas destes fármacos. Monitorar as concentrações plasmáticas destes fármacos e/ou a função cardíaca dos pacientes.
t Contraceptivos orais com etinilestradiol + norgestimato ou noretisterona: o uso concomitante com atazanavir pode levar a aumento da exposição a progesterona bem como a aumento ou diminuição da exposição ao etinilestradiol. Se a associação de hormônios está sendo usada com atazanavir + ritonavir, o contraceptivo oral deverá conter no mínimo 35 microgramas de etinilestradiol; se está sendo usado apenas com atazanavir, não deverá conter mais que 30 microgramas de etinilestradiol.
t Contraceptivos hormonais na forma farmacêutica de solução injetável, anel vaginal ou adesivo, ou contraceptivos orais contendo outros progestógenos, ou contraceptivos orais contendo menos que 25 microgramas de etinilestradiol: o uso de um método contraceptivo alternativo deve ser considerado, pois não há estudos que descrevam os efeitos da interação com atazanavir, não havendo portanto dados que confirmem a segurança e eficácia contraceptiva em caso de interação.
t Diltiazem: aumenta o risco de cardiotoxicidade. Monitorar possíveis alterações por eletrocardiograma, podendo ser considerada a redução de até 50% da dose de diltiazem.
t Didanosina: diminuição da solubilidade e da exposição ao atazanavir com a formulação de didanosina tamponada; diminuição da exposição à didanosina na forma de comprimido revestido. Administrar o atazanavir com alimentos 2 horas antes ou 1 hora após a formulação tamponada de didanosina ou comprimidos revestidos contendo didanosina.
t Efavirenz: diminuição da concentração plasmática do atazanavir. A coadministração destes medicamentos em pacientes virgens de terapia deve ser realizada da seguinte maneira: atazanavir 400 mg + ritonavir 100 mg, uma vez ao dia com alimentos, e efavirenz 600 mg administrado com estômago vazio, preferentemente ao dormir. Atazanavir sem ritonavir não deve ser coadministrado com efavirenz.
t Everolimo: aumento no risco dos efeitos tóxicos do everolimo. Monitorar a concentração plasmática deste e proceder à redução da dose, se necessário.
t Fentanila: aumento da toxicidade da fentanila (depressão do sistema nervoso central). Ajustar a dose se necessário.
t Fosamprenavir, minociclina: diminuição da concentração plasmática do atazanavir. Monitorar os pacientes quanto à perda de eficácia.
t Inibidores da bomba de prótons (esomeprazol, lansoprazol e omeprazol): aumento de falha terapêutica devido à diminuição da concentração plasmática do atazanavir; se a administração destes inibidores da bomba de prótons é necessária, em pacientes virgens de tratamento a dose do inibidor da bomba de próton não deve exceder a uma dose equivalente a 20 mg de omeprazol e deve ser administrado cerca de 12 horas antes do atazanavir 300 mg/ritonavir 100 mg; não é recomendado administrar concomitantemente inibidores da bomba de prótons em pacientes que já estejam recebendo atazanavir.
t Lidocaína: aumento da concentração plasmática da lidocaína, aumentando o risco de cardiotoxicidade. Monitorar os pacientes através do eletrocardiograma ou através da concentração plasmática da lidocaína.
t Maraviroque: aumento da concentração plasmática do maraviroque. Recomenda-se administrar 150 mg de maraviroque duas vezes ao dia e monitorar o aparecimentos de seus Efeitos adversos (elevação das transaminases hepáticas, tontura e exantema).
t Metadona: aumento no risco de distúrbios de condução cardíaca. Se coadministrados, recomenda-se monitorar a função cardíaca dos pacientes.
t Nevirapina: aumenta o risco de falha terapêutica devido à diminuição da biodisponibilidade do atazanavir, além de aumentar o risco de toxicidade da nevirapina. A coadministração é contraindicada.
t Posaconazol: aumento da concentração plasmática do atazanavir. Monitorar o aparecimento de Efeitos adversos e sinais de toxicidade.
t Rifabutina: aumento na concentração plasmática da rifabutina. Recomendase a redução em até 75% na dose deste medicamento.
t Rosuvastatina: aumento da toxicidade da rosuvastatina, incluindo aumento de miopatia e rabdomiólise. Se o uso de ambos medicamentos foi necessário, recomenda-se diminuir a dose da rosuvastatina além de monitorar os aparecimentos dos sinais e sintomas de toxicidade e/ou os níveis de creatina cinase.
t Saquinavir: aumento da concentração plasmática do saquinavir e risco de toxicidade. Monitorar pacientes quanto ao aparecimento dos Efeitos adversos relacionados à toxicidade do saquinavir.
t Tipranavir: diminuição da concentração do atazanavir e aumento da concentração do tipranavir. A coadministração não é recomendada.
t Trazadona: aumento da concentração plasmática da trazadona e aumento do risco de Efeitos adversos (náuseas, tontura e hipotensão). Monitorar os pacientes quanto ao surgimento dos efeitos sedativos e de hipotensão e considerar uma redução de dose.
t Orientar para ingerir o medicamento com alimento ou leite.
t Orientar para empregar método contraceptivo, mas não utilizar anticoncepcionais hormonais.
t Reforçar orientações sobre prevenção da transmissão do HIV.
t Orientar para o uso durante todo o tempo prescrito, mesmo que haja melhora dos sintomas com as primeiras doses.
t Manter à temperatura ambiente, entre 15 e 30ºC, longe do calor, umidade e luz direta.
Atenção: como sinonímia para atazanavir (nome que correspondente a Denominação Comum Brasileira) também é empregada a abreviatura ATV, entretanto, não se recomenda a prescrição de fármacos por abreviaturas ou siglas.
Thais Furtado de Souza
Na Rename 2010: itens 1.1.3, 8.2 e 21.4
t Solução injetável 0,25 mg/mL.
t Solução oftálmica a 1%.
t Adjuvante em anestesia geral.
t Antídoto em intoxicações exógenas por organofosforados (reversão de efeitos muscarínicos).
t Produção de midríase ou cicloplegia (solução oftálmica).
t Crianças com histórico de reação sistêmica grave com o uso oftálmico de atropina.
t Glaucoma de ângulo fechado.
t Hipersensibilidade ao fármaco.
t Estenose pilórica ou hipertrofia prostática (solução injetável).
t Usar com cuidado nos casos de:
t íleo paralítico, colite ulcerativa, colostomia ou ileostomia, doenças pulmonar, renal, hepática e do trato biliar.
t diarreia, hipertireoidismo ou hipertensão.
t crianças, idosos ou enfraquecidos.
t lactação.
t Evitar na vigência de febre, desidratação, taquicardia e tirotoxicose. t Evitar em pacientes com miastenia grave e neuropatia autonômica. t Uso oftálmico pode ocasionar efeito sistêmico.
t Mesmo doses inferiores a 0,1 mg em crianças, e a 0,5 mg em adultos, ou a administração lenta, podem associar-se a bradicardia paradoxal.
t Categoria de risco na gravidez (FDA): C.
t 0,02 mg/kg, por via intramuscular ou subcutânea, 30 a 60 minutos antes da indução da anestesia. Dose máxima: 0,6 mg.
t 0,02 mg/kg, por via intravenosa, imediatamente antes da indução da anestesia. Dose máxima: 0,6 mg.
t 0,02 mg/kg, por via intravenosa, com dose mínima de 0,1 mg e dose máxima de 0,5 mg em crianças e 1 mg em adolescentes, repetida a cada 5 minutos até uma dose máxima total de 1 mg em crianças e 2 mg em adolescentes.
t 0,02 a 0,05 mg/kg, por via intravenosa ou intramuscular, a cada a 5 a 10 minutos até que se obtenha o efeito atropínico (taquicardia, pele seca e hiperemiada, pupilas dilatadas); depois, repetir as doses a cada 1 a 4 horas, conforme necessário para manter o efeito atropínico, durante 24 horas.
t A via de administração depende da gravidade da intoxicação.
t Crianças maiores de 5 anos: Instilar 1 gota de solução oftálmica a 1% em cada olho, a cada 12 horas, durante 1 a 3 dias antes do procedimento, com uma dose adicional 1 hora antes do procedimento.
t 0,3 a 0,6 mg, por via intramuscular ou subcutânea, 30 a 60 minutos antes da indução da anestesia.
t 0,3 a 0,6 mg, por via intravenosa, imediatamente antes da indução da anestesia.
t Profilaxia e correção de bradicardia no ato cirúrgico
t 0,3 a 0,6 mg, por via intravenosa, repetida a cada 5 minutos até a dose máxima total de 3 mg.
t Intoxicação por organofosforados
t 2 mg, por via intravenosa ou intramuscular, repetida por duas vezes a cada 10 minutos, se necessário. Não administrar mais de 3 doses.
t A via de administração depende da gravidade da intoxicação.
t Instilar 1 gota de solução oftálmica a 1% em cada olho, a cada 12 horas, durante 1 a 2 dias antes do procedimento; ou uma única aplicação de 1 gota de solução a 1% em cada olho 1 hora antes do procedimento.
t Início da ação: 15 a 50 minutos (bradicardia); 30 a 40 minutos (midríase); 60 a 180 minutos (cicloplegia).
t Duração: mais de 5 horas (bradicardia); 7 a 10 dias (midríase); 6 a 12 dias (cicloplegia).
t A absorção por via conjuntival é variável, podendo ocorrer absorção sistêmica.
t Metabolismo: hepático.
t Meia-vida: 4 horas em adultos; 6,5 horas em crianças.
t Excreção: urinária (30 a 40%).
t Irritação no lugar da injeção.
t Diminuição da sudorese.
t Obstipação, xerostomia, náusea, vômito.
t Visão borrada, sensibilidade a luz, aumento da pressão intraocular.
t Arritmias, taquicardia.
t Hipersensibilidade.
t Confusão (comum em idosos).
t Retenção urinária.
t Depressão respiratória.
t Uso prolongado de solução oftálmica pode causar irritação local, conjuntivite, dermatite de contato, toxicidade sistêmica em crianças e idosos.
t Arbutamina: sulfato de atropina altera a frequência cardíaca, o que pode resultar em alteração de exames para arbutamina. Arbutamina não deve ser administrada em pacientes que receberam sulfato de atropina.
t Cloreto de ambenônio: uso concomitante pode resultar na supressão dos sintomas de overdose de cloreto ambenônio. O uso concomitante é contraindicado.
t Cloreto de potássio (comprimido): o efeito anticolinérgico de sulfato de atropina pode causar um atraso na passagem de cloreto de potássio através do trato gastrintestinal, aumentando assim o risco de lesões gastrintestinais. O uso concomitante é contraindicado.
t Informar que pode causar sonolência, tontura, visão borrada e intolerância a luz. Evitar atividades que necessitem estado de alerta.
t Orientar para aumentar consumo de líquido e reduzir a exposição ao calor a fim de evitar desidratação.
t Orientar para a importância de comunicar ao perceber qualquer sinal de reação alérgica.
t Lavar as mãos antes e depois da utilização do colírio. Agitar o frasco antes de usar. Usar técnica adequada de administração. Não encostar o frasco no olho. Não lavar o olho após o uso, manter o olho fechado e ressionar gentilmente por 1 minuto. O olho pode ficar sensível a luz, usar óculos escuros.
t Manter à temperatura ambiente, entre 15 e 30 °C, em recipiente hermeticamente fechado. Evitar congelamento. Proteger da luz.
t Observar orientação específica do produtor quanto a diluição, compatibilidade e estabilidade da solução
t Incompatível com hemitartarato de norepinefrina, hemitartarato de metaraminol e bicarbonato de sódio.
Atenção: muitos medicamentos têm efeito antimuscarínico; uso concomitante de dois ou mais desses medicamentos pode aumentar os efeitos adversos, como xerostomia, retenção urinária, obstipação e também pode levar a confusão, especialmente, nos idosos.
Isabella Campagnuci Knust
Na Rename 2010: item 24.1
t Suspensão oral 1 g/mL.
t Diagnóstico radiológico de anormalidades e defeitos no trato gastrintestinal.
t Obstrução intestinal.
t Estenose pilórica ou lesões que predispõem à obstrução.
t Perfuração intestinal ou condições predisponentes (retocolite ulcerativa, diverticulite, após biópsia colo-retal, retossigmoidoscopia ou radioterapia).
t Hipersensibilidade aos componentes da formulação.
t Gravidez (ver Apêndice A).
t Suspeita de fistula traqueoesofágica.
t Usar com cuidado no caso de lactação (ver Apêndice B).
t Hidratação adequada antes do procedimento para evitar obstipação.
t Oral: a dose deve ser ajustada de acordo com a quantidade que a criança consegue ingerir do contraste.
t Retal: recomenda-se dose proporcional ao tamanho da criança, devendo ser administrada uma quantidade mínima inicialmente, podendo esta dose ser aumentada, caso necessário, até que se possam visualizar bem as estruturas a serem estudadas.
t Oral: 40 a 450 g. Doses maiores podem ser usadas, caso necessário.
t Retal: 150 a 750 g. Doses maiores podem ser utilizadas.
Nota: Via e dose são dependentes do procedimento realizado. A via retal pode produzir alterações eletrocardiográficas.
t Não é absorvido, portanto não sofre biotransformação.
t Eliminação: 100% fecal.
t Obstipação, diarreia, cólica abdominal, flatulência, obstrução e sangramento gastrintestinais, apendicite.
t Perfuração intestinal e peritonite, aderências e granulomas.
t Fibrilação ventricular, bradicardia e assistolia.
t Pneumonite química ou formação de granuloma pulmonar, quando há broncoaspiração acidental.
t Observação: Os insumos empregados nas formulações do sulfato de bário podem causar reações de hipersensibilidade.
t Orientar para aumentar a ingestão hídrica após o procedimento para prevenir obstipação.
t Orientar que, para administração do contraste por via retal, deve ser adotada, no dia que precede o exame, dieta pobre em resíduos, uso de laxantes e jejum de 8 horas.
t Agitar o frasco por 30 segundos antes do uso.
t Pó branco, fino, denso e suave. Praticamente insolúvel em água, solventes orgânicos e hidróxidos ácidos e básicos.
t Manter à temperatura ambiente, entre 15 e 30 ºC, em recipientes bem fechados, ao abrigo de luz e umidade.
t Para exames do intestino grosso, o sulfato de bário em suspensão para uso oral pode ser preparado para ser utilizado como enema ou clister (para administração retal).
Maurício Fábio Gomes
Na Rename 2010: Item 6.1.4
t Pó para solução injetável 15 U (unidades USP).
t Carcinoma de células escamosas de cabeça e pescoço, nasofaringe, colo do útero, pênis e vulva.
t Carcinoma testicular.
t Derrame pleural neoplásico.
t Doença de Hodgkin (adjuvante para cirurgia e radioterapia em tratamento paliativo).
t Sarcoma de Kaposi relacionado a Aids.
t Doença pulmonar grave.
t Hipersensibilidade ou reação idiossincrática a bleomicina.
t Usar com cuidado nos casos de:
– idosos, fumantes e pacientes submetidos a radioterapia prévia (maior risco de fibrose pulmonar).
– pacientes com linfoma (em 1% destes, pode ocorrer reação idiossincrática após primeira ou segunda dose, caracterizada por hipotensão, confusão mental, febre, dores e dispneia; administrar uma dose teste de 1 U ou 2 U, ou menos, 2 a 4 horas antes de iniciar o tratamento).
– insuficiência renal (ver Apêndice D).
– lactação (ver Apêndice B).
t Doses totais acima de 400 U podem causar transtorno respiratório e fibrose pulmonar (10%). Recomendam-se doses cumulativas máximas de 300 U para pacientes entre 60 e 69 anos, 200 U para pacientes entre 70 e 79 anos e 100 U para pacientes com mais de 80 anos.
t A bleomicina apresenta potencial carcinogênico.
t Categoria de risco na gravidez (FDA): D (ver Apêndice A).
t 10 a 20 U/m2 (0,25 a 0,5 U/kg), por via intravenosa ou intramuscular ou subcutânea, de 1 a 2 vezes por semana.
t 30 U/dia, por via intravenosa, nos dias 2, 9 e 16, em combinação com 100 mg/m2 de etoposídeo e 20 mg/m2 de cisplatina, ambos por via intravenosa, nos dias de 1 a 5. Repetir o ciclo a cada 3 semanas.
t 60 U, em dose única, por bolo intrapleural.
t 25 mg/m2 de doxorrubicina, 10 U/m2 de bleomicina, 6 mg/m2 de vimblastina e 350 a 375 mg/m2 de dacarbazina, todas por via intravenosa, nos dias 1 e 15.
t 6 mg/m2 de mecloretamina e 1,4 mg/m2 (até 2 mg) de vincristina, ambos por via intravenosa, no dia 1; procarbazina 100 mg/m2, por via oral nos dias de 1 a 7; prednisona 40 mg/m2, por via oral, dos dias 1 a 14; doxorrubicina 35 mg/ m2, bleomicina 10 U/m2 e vimblastina 6 mg/m2, todas por via intravenosa, no dia 8. Repetir o ciclo a cada 4 semanas.
t O uso combinado dos dois regimes acima pode ser feito de maneira alternada mensalmente.
t Administração intralesional de acordo com o volume do tumor, combinada com eletroquimioterapia: 0,5 U para 100 mm3; 0,75 U para 100-150 mm3; 1 U para 150-500 mm3; 1,5 U para 500-1000 mm3; 2 U para 1000-2000 mm3; 2,5 U para 2000-3000 mm3; 3 U para 3000-4000 mm3; 3,5 U para 4000-5000 mm3 e 4 U para volumes acima de 5000 mm3. Lidocaína a 1% com vasoconstritor deve ser aplicada em torno do local.
Nota: A administração intravenosa de bleomicina deve ser feita durante 10 minutos.
t Pico de concentração sérica: 60 minutos (intramuscular).
t Metabolismo: parede intestinal, fígado, sangue, rins, pele e pulmões.
t Excreção: renal (50% na forma inalterada).
t Meia-vida de eliminação: 1 a 9 horas (intravenosa), 4 horas (subcutânea), 2 a 30 horas (insuficiência renal).
t Alopecia (1% a 10%), perda das unhas, hiperceratose palmar e plantar (50%), eritema (50%), erupções cutânea (8%), estrias, vesiculação, hiperpigmentação (50%), dor a palpação no local da administração intramuscular e subcutânea.
t Tremor, confusão mental, febre (25% a 50%).
t Náusea, vômitos, estomatite, anorexia, mucosite (30%).
t Hipotensão, enfarte do miocárdio, acidente vascular cerebral, síndrome de Raynaud.
t Mielossupressão (raro).
t Hepatotoxicidade.
t Nefrotoxicidade.
t Tosse, dispneia, sibilos, pneumonite (10%), fibrose pulmonar (5% a 10%), hipóxia e morte (1%).
t Reações anafiláticas.
t Fenitoína: pode ter efetividade diminuída pela bleomicina. Se o uso concomitante for necessário, monitorar concentrações plasmáticas de fenitoína e considerar aumento de dose durante a quimioterapia.
t Vacina de rotavírus humano: aumento do risco de infecção pela vacina. O uso concomitante é contraindicado.
t Vacinas com vírus vivos: aumento do risco de infecção pela vacina. Se a vacina for necessária, administrar a vacina depois de três meses da descontinuação da quimioterapia.
t Orientar para evitar uso de tabaco ou álcool, sob risco de desenvolvimento de fibrose pulmonar.
t Alertar para notificar qualquer sinal de problemas respiratórios, como tosse ou falta de ar.
t Utilizar contracepção efetiva durante o tratamento, homens e mulheres.
t Alertar para evitar vacinas (vacinas) durante o uso de bleomicina.
t Em caso de esquecimento de alguma dose contatar o médico.
t 1 U (Unidade USP) corresponde a 1000 UI (Unidades Internacionais).
t Cada mg de bleomicina corresponde de 1,5 a 2 U.
t Estocar o produto sob refrigeração (2 a 8 oC) e ao abrigo da luz.
t Observar orientação específica do produtor quanto a reconstituição, diluição, compatibilidade e estabilidade da solução.
t Solução reconstituída permanece estável por 24 horas.
t Preparo para administração intravenosa: reconstituir o frasco com 15 U em 5 mL de solução injetável de cloreto de sódio 0,9%. Soluções para infusões prolongadas devem ser preparadas e administradas em frascos de vidro.
t Preparo para administração intramuscular e subcutânea: reconstituir o frasco com 15 U com 1 a 5 mL de água para injeção ou solução injetável de cloreto de sódio 0,9%.
t Preparo para administração intrapleural: dissolver o frasco com 15 U em 50 a 100 mL de solução injetável de cloreto de sódio 0,9%.
t Incompatibilidades: peróxido de hidrogênio, soluções de aminoácidos essenciais, riboflavina, dexametasona, furosemida, carbenicilina, cefazolina, cefalotina sódica, nafcilina sódica, benzilpenicilina sódica, metotrexato, mitomicina, succinato sódico de hidrocortisona, aminofilina, ácido ascórbico, terbutalina, soluções injetáveis de glicose a 5%.
t Soluções não utilizadas em 24 horas devem ser descartadas.
t Observar protocolos locais para manipulação de substâncias citotóxicas.
Atenção: fibrose pulmonar é a forma de toxicidade mais grave associada à bleomicina. Sua ocorrência é maior em pacientes idosos que recebem doses totais acima de 400 U.
Simone Sena Farina e Fernando de Sá Del Fiol
Na Rename 2010: item 5.2.2
t Pó para solução injetável 1 g.
t Tratamento de tuberculose quando há intolerância a rifampicina ou a isoniazida
t Hipersensibilidade à estreptomicina e aos aminoglicosídeos.
t Distúrbios auditivos.
t Miastenia grave.
t Gravidez. Categoria de risco na gravidez: D (ver Apêndice A).
t Usar com cuidado nos casos de:
– distúrbio vestibular.
– associação com anestesia ou relaxantes musculares periféricos (aumenta o risco de bloqueio neuromuscular e paralisia respiratória).
– pacientes muito jovens, idosos e com desidratação (são mais predispostos à toxicidade da estreptomicina).
– crianças (evitar o uso pois a injeção provoca dor).
– insuficiência renal (ver Apêndice D).
– hemodiálise (requer suplementação de dose após a hemodiálise).
– lactação (ver Apêndice B).
t Evitar uso concomitante de fármacos neurotóxicos, ototóxicos ou nefrotóxicos.
t 20 a 35 kg: 500 mg, por via intramuscular profunda, a cada 24 horas, durante 2 meses, combinada a isoniazida, pirazinamida e cloridrato de etambutol, seguido de 10 meses de isoniazida e cloridrato de etambutol.
t 36 a 50 kg: 500 mg a 1.000 mg, por via intramuscular profunda, a cada 24 horas, durante 2 meses, combinada a isoniazida, pirazinamida e cloridrato de etambutol, seguido de 10 meses de isoniazida e cloridrato de etambutol.
t Acima de 50 kg: 1.000 mg, por via intramuscular profunda, a cada 24 horas, durante 2 meses, combinada a isoniazida, pirazinamida e cloridrato de etambutol, seguido de mais 10 meses de isoniazida e cloridrato de etambutol.
t 20 a 35 kg: 500 mg, por via intramuscular profunda, a cada 24 horas, durante 2 meses, combinada a rifampicina, pirazinamida e cloridrato de etambutol, seguido de 4 meses de rifampicina e cloridrato de etambutol.
t 36 a 50 kg: 500 mg a 1.000 mg, por via intramuscular profunda, a cada 24 horas, durante 2 meses, combinada a rifampicina, pirazinamida e cloridrato de etambutol, seguido de 4 meses de rifampicina e cloridrato de etambutol.
t Acima de 50 kg: 1.000 mg, por via intramuscular profunda, a cada 24 horas, durante 2 meses, combinada a rifampicina, pirazinamida e cloridrato de etambutol, seguido de 4 meses de rifampicina e cloridrato de etambutol.
Notas:
t O pó deve ser reconstituído em água estéril para injeção, com concentração final de 200 a 400 mg/mL.
t Em casos especiais, com dificuldades de aceitação de medicamento injetável ou para facilitar o uso supervisionado na unidade de saúde, o esquema pode ser alterado para aplicações de 2ª a 6ª feira, durante dois meses, e duas vezes por semana, durante mais 4 meses.
t Pico de concentração sérica: 1 hora.
t Meia-vida de eliminação: 2,5 horas.
t Excreção: urina (29% a 89%) e bile (provavelmente 1%).
t É removido por hemodiálise.
t Aracnoidite, encefalopatia, bloqueio neuromuscular (se uso concomitante com anestésicos e relaxantes musculares), neurite periférica.
t Alteração do nervo óptico, ototoxicidade.
t Nefrotoxicidade.
t Paralisia do trato respiratório (se uso concomitante com anestésicos e relaxantes musculares).
t Eosinofilia, anemia hemolítica, trombocitopenia.
t Parestesia facial, febre.
t Náusea, vômito.
t Dor e abscesso no lugar da injeção.
t Reações de hipersensibilidade.
t Ácido etacrínico, bumetanida: aumento do risco de ototoxicidade. Evitar uso concomitante, especialmente em pacientes com insuficiência renal, comprometimento do ouvido interno e que recebem altas doses. Se uso concomitante for necessário, monitorar função renal e auditiva.
t Bloqueadores neuromusculares não-despolarizantes: aumento do risco de bloqueio neuromuscular, podendo provocar depressão respiratória. Evitar uso concomitante; monitorar condições clínicas do paciente, especialmente respiratória e oxigenação.
t Orientar para o uso durante todo o tempo prescrito, mesmo que haja melhora dos sintomas com as primeiras doses
t Alertar para notificar imediatamente, ao perceber qualquer sinal de efeito adverso, principalmente renal e auditivo.
t Orientar o paciente quanto ao uso concomitante de relaxantes musculares, pois pode ocorrer bloqueio neuromuscular e paralisia respiratória.
t Manter sob refrigeração entre 2 e 8 oC.
t Observar orientação específica do produtor quanto a reconstituição, diluição, compatibilidade e estabilidade da solução.
t Diluir em água estéril para injeção ou solução de cloreto de sódio a 0,9%.
t Após diluição, a solução injetável se mantém estável de 2 a 28 dias, protegida da luz e sob temperatura ambiente, entre 15 e 30 ºC.
Atenção: como sinonímia para estreptomicina (nome correspondente a Denominação Comum Brasileira) também é empregada a abreviatura S, entretanto, não se recomenda a prescrição de fármacos por abreviaturas ou siglas.
Maria Inês de Toledo e Simone Sena Farina
Na Rename 2010: itens 5.1.4 e 21.2
t Solução injetável 10 mg/mL e 40 mg/mL.
t Colírio 5 mg/mL.
t Pomada oftálmica 5 mg/g.
t Infecções hospitalares graves causadas por bacilos gram-negativos aeróbios e Enterococcus sp (preferentemente infecções ginecológicas, abdominais, pielonefrite aguda, pneumonia, e infecções por Pseudomonas aeruginosa).
t Infecções oftálmicas por bactérias sensíveis à gentamicina.
t Hipersensibilidade a gentamicina ou outros aminoglicosídeos.
t Miastenia grave.
t Doença de Parkinson.
t Usar com cuidado nos casos de:
– neonatos, crianças (meia-vida aumentada) e idosos (diminuição de função renal). Ajustar a dose e monitorar concentração plasmática, função renal e auditiva.
– obesos e pacientes com insuficiência renal (ver Apêndice D). Ajustar a dose e monitorar concentração plasmática.
– pacientes com fraqueza muscular, desidratados.
t Evitar o uso prolongado.
t Evitar uso concomitante com outros fármacos ototóxicos ou nefrotóxicos.
t Evitar uso concomitante com anestésicos e bloqueadores da junção neuromuscular.
t Categoria de risco na gravidez (FDA): C (ver Apêndice A).
t Neonatos até 2 semanas: 3 mg/kg, por via intramuscular, por via intravenosa lenta durante pelo menos 3 minutos ou infusão intravenosa, a cada 12 horas.
t De 2 semanas a 12 anos: 2 mg/kg, por via intramuscular, por via intravenosa lenta durante pelo menos 3 minutos ou infusão intravenosa, a cada 8 horas.
t Pomada: aplicar pequena quantidade no(S) olho(S) afetado(S), 2 a 3 vezes ao dia, enquanto necessário.
t Colírio: pingar 1 a 2 gotas no(S) olho(S) afetado(S), a cada hora, enquanto necessário.
t 3 a 5 mg/kg, por via intramuscular, por via intravenosa lenta durante pelo menos 3 minutos ou infusão intravenosa, a cada 24 horas ou dividido a cada 8 horas, durante 7 a 10 dias.
t Pomada: aplicar pequena quantidade no(S) olho(S) afetado(S), 2 a 3 vezes ao dia, enquanto necessário.
t Colírio: pingar 1 a 2 gotas no(S) olho(S) afetado(S), a cada hora, enquanto necessário.
t Concentração terapêutica em infecções bacterianas: 4 a 8 microgramas/mL.
t Pico sérico: 0,5 a 1,5 horas (intramuscular),
t Meia-vida plasmática: 1,5 a 4 horas (adultos com função renal normal).
t Biotransformação: não há.
t Excreção: renal (70% a 100%).
t É removida por hemodiálise (70% a 80% após cada 12 horas).
t Ototoxicidade vestibular e coclear (3% a 5%), perda auditiva (0,5%, às vezes irreversível) e/ou dificuldade de equilíbrio, zumbidos.
t Náuseas, vômitos, estomatites.
t Nefrotoxicidade (2% a 25%)
t Bloqueio neuromuscular, paralisia muscular aguda e apneia em pacientes submetidos a medicamentos anestésicos e bloqueadores neuromusculares periféricos, fraqueza.
t Erupções cutâneas.
t Colite associada ao uso de antibiótico, hipomagnesemia na terapia prolongada.
t Eosinofilia.
t Ácido etacrínico: pode resultar em ototoxicidade. Monitorar periodicamente a função auditiva de pacientes com insuficiência renal ou recebendo altas doses de um dos fármacos envolvidos nesta interferência.
t Bloqueadores neuromusculares despolarizantes: pode resultar em aumento ou prolongamento do bloqueio neuromuscular acarretando depressão respiratória e paralisia. Evitar o uso concomitante, mas se o mesmo for necessário, monitorar rigorosamente a função respiratória.
t Furosemida: pode aumentar a toxicidade da gentamicina (nefro e ototoxicidade). Testar periodicamente a função auditiva de pacientes com insuficiência renal ou recebendo altas doses de um dos fármacos. Se possível, também monitorar concentrações plasmáticas de gentamicina.
t Indometacina: pode aumentar a toxicidade da gentamicina (nefro e ototoxicidade). Se possível, monitorar concentrações plasmáticas de gentamicina.
t Metoxiflurano: pode resultar em insuficiência renal. Monitorar a função renal e ajustar a dose do aminoglicosídeo conforme a resposta clínica.
t Poligelina: aumenta o risco de insuficiência renal. Monitorar função renal durante infusão da solução de poligelina.
t O uso deste medicamento durante a gravidez pode prejudicar o feto. Usar uma forma eficaz de controle de natalidade para não engravidar. Se suspeitar de gravidez durante o uso do medicamento, informe o seu médico imediatamente.
t Manter à temperatura de 2 a 30 ºC.
t Observar orientação específica do produtor quanto a reconstituição, diluição, compatibilidade e estabilidade da solução.
t Compatível com: água para injeção, glicose 5%, manitol 20%, Ringer + lactato e solução salina.
t Incompatível com: penicilinas (ampicilina, benzilpenicilina, carbenicilina, ticarcilina), cefalosporinas (cefazolina, cefepima, cefamandol), ácido clavulânico, solução lipídica 10% e bicarbonato de sódio. Devido ao alto potencial de incompatibilidade da gentamicina não se deve misturar em seringa, solução de infusão ou usar a mesma via intravenosa para administrar outros fármacos.
César Augusto Braum
Na Rename 2010: item 3.3
t Comprimidos de 400 mg.
t Artrite reumatoide.
t Lúpus eritematoso.
t Alterações na retina ou no campo visual.
t Hipersensibilidade ao fármaco.
t Distúrbios neutropênicos.
t Usar com cuidado nos casos de:
– deficiência da enzima glicose-6-fosfato desidrogenase.
– psoríase ou porfiria.
– uso concomitante com fármacos com tendência a causar dermatite ou reações dermatológicas.
– doença gastrintestinal grave.
– doenças neurológicas (especialmente, histórico de epilepsia).
– insuficiência hepática (ver Apêndice C).
– insuficiência renal (ver Apêndice D).
t Exames oftálmicos devem ser realizados antes do início e, periodicamente, durante o tratamento.
t Categoria de risco na gravidez (ADEC): D.
t Dose inicial 400 a 600 mg, por via oral, a cada 24 horas, durante 4 a 12 semanas. Caso não haja melhora após 6 meses, suspender a terapia. Os Efeitos adversos podem requerer uma redução temporária de dose. Após, 5 a 10 dias a dose pode ser aumentada gradualmente até resposta satisfatória.
t Dose de manutenção 200 a 400 mg, por via oral, a cada 24 horas.
t Dose inicial 400 mg, por via oral, a cada 12 ou 24 horas, até remissão.
t Dose de manutenção 200 a 400 mg, por via oral, a cada 24 horas. Aspectos farmacocinéticos clinicamente relevantes 2, 4, 8, 14 t A biodisponibilidade oral é de aproximadamente 74%.
t Pico de concentração plasmática: 3,6 horas.
t Meia-vida de eliminação: 32 a 50 dias.
t Metabolismo: hepático (metabólitos ativos).
t Excreção: renal (16% a 25% em forma inalterada), podendo ser aumentada com acidificação da urina.
t Distúrbios cardíacos (raro).
t Pigmentação de pele e mucosas, alopecia, erupções cutâneas, prurido, pacientes com psoríase podem apresentar piora do quadro, síndrome de Stevens-Johnson, angioedema.
t Náuseas, vômitos, diarreia, anorexia, cólicas abdominais.
t Agranulocitose, neutropenia ou trombocitopenia (raro), anemia aplástica.
t Danos hepáticos.
t Miopatias, fraqueza e atrofia muscular.
t Cefaleia, tontura, irritabilidade, nervosismo, ataxia, convulsões.
t Retinopatia, visão borrada, alteração das cores, desordens de acomodação da visão, queratinopatia.
t Ototoxicidade (raro).
t Broncoespasmo e parada respiratória.
t Metoprolol: uso concomitante com hidroxicloroquina pode aumentar os níveis plasmáticos do metoprolol. Monitorar reações adversas relacionadas à toxicidade do metoprolol, como bradicardia.
t Digoxina: uso concomitante com hidroxicloroquina pode aumentar os níveis séricos de digoxina. Monitorar sinais relacionados à toxicidade da digoxina.
t Orientar para ingerir o medicamento durante as refeições ou com leite para minimizar irritação gastrintestinal.
t Orientar para notificar o surgimento de alterações oftálmicas, hepáticas ou de qualquer natureza.
t Orientar para a exigência de cautela com atividades que exijam atenção, como dirigir e operar máquinas, devido ao risco de surgir tontura e visão borrada.
t Orientar para a necessidade de acompanhamento oftalmológico se for necessário o uso por longo prazo.
t Orientar para tomar o medicamento o quanto antes caso ocorra esquecimento. Não tomar caso esteja próximo do horário da próxima dose. Não tomar dobradas doses.
t Orientar para não usar bebidas alcoólicas devido a irritação gástrica.
t Manter à temperatura ambiente, entre 15 e 30 oC, em local fechado, ao abrigo de umidade e luz.
t 100 mg de sulfato de hidroxicloroquina correspondem a 77,5 mg de hidroxicloroquina base.
t As doses são expressas em sulfato de hidroxicloroquina.
Larissa Niro
Na Rename 2010: itens 9, 10.3, 13.1 e 16.5
t Pó para solução oral 5 a 30 g
t Solução injetável 50% (4,05 mEq Mg++/mL).
t Solução injetável 10% (0,81 mEq Mg++/mL).
t Obstipação intestinal (solução oral).
t Prevenção de convulsões recorrentes em eclampsia.
t Prevenção de desenvolvimento de eclampsia em mulheres com pré-eclampsia grave.
t Tratamento e profilaxia da hipomagnesemia.
t Edema cerebral.
t Intoxicação por bário.
t Bloqueio cardíaco e miocardiopatias (administração parenteral).
t Condições gastrintestinais agudas (administração oral).
t Usar com cuidado nos casos de:
– insuficiência renal (ver Apêndice D).
– coma hepático associado à insuficiência renal (evitar uso do sulfato de magnésio).
– soluções contendo sais de alumínio (uso em neonatos prematuros ou uso prolongado em pacientes com insuficiência renal podem produzir intoxicação por alumínio).
– pacientes com obstrução intestinal, colostomizados ou ileostomizados (não utilizar por via oral).
– miastenia grave.
– controle de hipomagnesemia (administrar inicialmente por dispositivo de infusão controlada e monitorar para sinais de toxicidade pelo magnésio; preferir infusão intravenosa pois injeção intramuscular produz dor local).
– hipersensibilidade a qualquer componente da formulação.
– eclampsia (reservar o uso para convulsões graves que necessitem de controle imediato; usar via intravenosa).
t Categoria de risco na gravidez (FDA): A (ver Apêndice A).
t Monitorar pressão arterial, frequência respiratória, quantidade de urina excretada e sinais de dose(S) excessiva(S)(perda dos reflexos patelares, fraqueza, náuseas, sensação de calor, rubor, afogamento, visão duplicada e fala incompreensível).
t Neonatos: 25 a 50 mg/kg de sulfato de magnésio a 50%, por via intravenosa, a cada 8 a 12 horas.
t Lactentes (manutenção de magnésio): 0,25 a 1,25 g (2 a 10 mEq)/dia, de sulfato de magnésio a 10%, adicionados à terapia de nutrição parenteral.
t Crianças: 25 a 50 mg/kg de sulfato de magnésio a 50%, por via intravenosa, a cada 4 a 6 horas ou 100 a 200 mg/kg, por via oral, a cada 6 horas. Dose máxima diária: 2 g.
Nota: Para injeção intravenosa, diluir uma parte de solução de sulfato de magnésio a 50% em 1,5 partes de água para injeção; concentração final de 20%; infundir não mais que 1 g/minuto.
t 5 a 10 g, em um copo de água, preferivelmente antes do café da manhã. Dose máxima diária: 40 g (320 mEq [160 mmol] de magnésio).
t Dose inicial 4 g de sulfato de magnésio a 50%, por via intravenosa por 5 a 15 minutos, seguido de infusão intravenosa, na velocidade de 1 g/hora, administrada durante 24 horas, ou
t 5 g de sulfato de magnésio a 50%, por via intramuscular profunda, em cada glúteo, seguido de 5 g, por via intramuscular profunda, a cada 4 horas, em nádegas alternadas, durante 24 horas.
t Na ocorrência de convulsões, administrar 2 g de sulfato de magnésio a 50%, por injeção intravenosa.
t Dose inicial 4 g de sulfato de magnésio a 50%, por via intravenosa por 5 a 15 minutos, seguido de infusão intravenosa, na velocidade de 1g/hora, administrada durante 24 horas depois da última convulsão ou,
t 5 g de sulfato de magnésio a 50%, por via intramuscular profunda, em cada glúteo; seguido de 5 g, por via intramuscular profunda, a cada 4 horas, em nádegas alternadas, durante 24 horas depois da última convulsão.
t Convulsões recorrentes podem requerer injeção intravenosa adicional de 2 g de sulfato de magnésio a 50% (4 g se o peso corporal for superior a 70 kg).
t Moderada: 1g de sulfato de magnésio a 10%, por via intramuscular profunda, alternando o local da aplicação, a cada 6 horas em 24 horas.
Grave:
t 5 g de sulfato de magnésio a 10%, por infusão intravenosa, diluídos em 1 L de solução de glicose 5% ou cloreto de sódio 0,9%, durante 3 horas, à velocidade de 3 mL/minuto, ou
t 250 mg/kg de sulfato de magnésio a 50%, por injeção intramuscular profunda, alternando o lugar de aplicação, a cada 4 horas.
t Para manutenção de níveis séricos de magnésio em terapia de nutrição parenteral: 1 a 3 g de sulfato de magnésio a 50% (8 a 24 mEq ou até 12 mmol/ dia) devem ser adicionados às soluções administradas diariamente.
t 1 a 2 g de sulfato de magnésio a 50%, por via intravenosa.
t 2,5 g de sulfato de magnésio a 10%, por via intravenosa.
Nota: Em alguns pacientes pode ser necessária dose de até 6 g/dia, como em paciente com síndrome do intestino curto. Uma solução intravenosa de um sal de cálcio (por exemplo, gliconato de cálcio 10%) deve estar prontamente disponível quando sulfato de magnésio é administrado por via parenteral. Para injeção intramuscular, misturar a solução de sulfato de magnésio a 50% com 1 mL de solução de lidocaína a 2% sem vasoconstritor.
t Biodisponibilidade oral: 33%.
t Duração de atividade anticonvulsivante: 3-4 horas, por via intramuscular; 30 minutos por via intravenosa.
t Distribuição: ossos (50 a 60%), fluido extracelular (1 a 2%).
t Excreção: preponderantemente renal, proporcional às concentrações plasmáticas e à filtração glomerular.
t Dialisável.
t Hipermagnesemia: náuseas, vômitos, irritação gástrica, sede, cólicas intestinais, diarreia se administrado por via oral, hiperemia da pele.
t Anormalidades no ECG, bloqueio cardíaco, parada cardíaca, hipotensão, vasodilatação.
t Distúrbios da coagulação com aumento do tempo de sangramento.
t Hiporreflexia, fraqueza muscular.
t Depressão do SNC, sonolência, confusão, diplopia, fala arrastada, coma.
t Depressão respiratória.
t Reações de hipersensibilidade, como urticária (administração intravenosa).
t Alcaçuz (Glycyrrhiza glabra L.): A administração concomitante por via oral pode resultar no aumento do risco de hipopotassemia. Evitar o uso concomitante.
t Bifosfonatos: a administração concomitante por via oral pode reduzir a absorção dos bifosfonatos. Espaçar os horários de administração dos fármacos.
t Bloqueadores de canais de cálcio (como nifedipino): pode resultar em hipotensão quando o sulfato de magnésio é administrado por via parenteral. Monitorar a pressão arterial quando introduzir ou descontinuar a terapia conjunta com bloqueadores de canais de cálcio.
t Bloqueadores neuromusculares: pode resultar no aumento do bloqueio neuromuscular quando o sulfato de magnésio é administrado por via parenteral. Ajustar a dose dos bloqueadores neuromusculares quando o magnésio for dado em altas doses na toxemia gravídica.
t Labetalol: risco aumentado de bradicardia e redução do débito cardíaco. Monitorar função cardíaca.
t Tetraciclinas: a administração concomitante por via oral pode reduzir a absorção das tetraciclinas.
t Orientar para notificar o surgimento de alergia ao magnésio, tonturas, diarreia intensa.
t Alertar para suspender o uso se ocorrerem cólicas estomacais, náusea, vômito ou escurecimento das fezes.
t Perguntar ao médico ou farmacêutico, antes de usar algum medicamento, vitaminas e produtos herbáceos. Avisar ao médico quando for usar antibióticos ou bebidas alcoólicas.
t Armazenar a solução injetável de sulfato de magnésio heptaidratado a temperatura entre 15 a 30 ºC. Proteger da luz e evitar congelamento.
t Cada grama de sulfato de magnésio heptaidratado corresponde aproximadamente a 98 mg, 4,1 mmol ou 8,12 mEq de magnésio elementar. Cada 10,1 g de sulfato de magnésio anidro são equivalentes a aproximadamente 1 g de magnésio.
t Observar orientação específica do produtor quanto a diluição, compatibilidade e estabilidade da solução.
t A formação de precipitados e outras alterações físico-químicas podem ocorrer quando o sulfato de magnésio é misturado com soluções contendo: álcool (em grandes concentrações), carbonatos e hidróxidos, arsenatos, bário, cálcio, fosfato de clindamicina, metais pesados, succinato sódico de hidrocortisona, fosfatos, anfotericina B, ciclosporina, azatioprina sódica, sulfato de polimixina B, cloridrato de dobutamina, pantoprazol sódico, cefalosporinas, fenitoína sódica, cloridrato de procaína, salicilatos, estrôncio e tartaratos. O potencial de incompatibilidade é influenciado frequentemente por mudanças na concentração dos reagentes e no pH das soluções.
t Pode ocorrer separação de emulsões gordurosas para nutrição parenteral quando as concentrações de magnésio forem superiores a 20 mEq/mL.
t Deve-se ter cuidado extremo na administração parenteral do sulfato de magnésio a fim de evitar concentrações séricas tóxicas. Os pacientes geriátricos requerem frequentemente doses mais baixas devido à função renal ser reduzida.
Aline Lins Camargo
Na Rename 2010: itens 1.1.3 e 2.2
t Cápsula de liberação prolongada 60 mg
t Comprimido 30 mg
t Solução injetável 1 mg/mL
t Solução injetável 10 mg/mL
t Solução oral 10 mg/mL
t Dor moderada a grave, aguda e crônica.
t Dor de enfarte do miocárdio e de edema pulmonar agudo.
t Adjuvante de anestesia geral.
t Analgesia pós-operatória.
t Asma grave ou aguda.
t Alcoolismo agudo.
t Hipersensibilidade à morfina.
t Ileo paralítico.
t Pressão intracraniana aumentada.
t Trauma craniano ou tumor cerebral.
t Depressão respiratória aguda.
t Obstrução das vias aéreas superiores.
t Feocromocitoma (injeção).
t Usar com cuidado nos casos de:
– idosos e enfraquecidos (reduzir doses)
– crianças com menos de 3 meses de idade (são mais Susceptíveis à depressão do SNC).
– uso prolongado (leva à dependência física, ocorrendo sintomas graves de abstinência se o uso for interrompido abruptamente).
– asma ou reserva respiratória diminuída
– insuficiência hepática (ver Apêndice C).
– insuficiência renal (ver Apêndice D).
– pós-operatório (monitorar os pacientes para alívio da dor e para efeitos adversos, especialmente depressão respiratória)
t A dose e os intervalos de administração devem ser individualizados de acordo com a gravidade da dor e a resposta do paciente.
t Em crianças, idade e peso também devem ser considerados para seleção de dose.
t Para dor crônica não existe dose máxima ou ótima de morfina. A dose apropriada é aquela que alivia a dor sem causar Efeitos adversos que não sejam possíveis de manejar.
t Categoria de risco na gravidez (FDA): C ou D (se utilizado por períodos prolongados ou em doses elevadas) (ver Apêndice A).
Via oral
t 1 a 12 meses: 80 a 200 microgramas/kg, a cada 4 horas.
t 1 a 2 anos: 200 a 400 microgramas/kg, a cada 4 horas
t 2 a 12 anos: 200 a 500 microgramas/kg (máximo 20 mg), a cada 4 horas.
t 12 a 18 anos: 5 a 20 mg, a cada 4 horas
Vias subcutânea ou intramuscular
t Neonatos: 100 microgramas/kg, a cada 6 horas.
t 1 a 6 meses: 100 a 200 microgramas/kg, a cada 6 horas.
t 6 meses a 2 anos: 100 a 200 microgramas/kg, a cada 4 horas.
t 2 a 12 anos de idade: 200 microgramas/kg, a cada 4 horas.
t 12 a 18 anos: 2,5 a 10 mg, a cada 4 horas.
Via intravenosa lenta
t Neonatos: 25 a 100 microgramas/kg, por via intravenosa, seguida de infusão intravenosa contínua à velocidade de 5 a 40 microgramas/kg/hora
t 1 a 6 meses: 100 a 200 microgramas/kg, por via intravenosa, seguida de infusão intravenosa contínua, à velocidade de 10 a 30 microgramas/kg/hora
t 6 meses a 12 anos: 100 a 200 microgramas/kg, por via intravenosa, seguida de infusão intravenosa contínua à velocidade de 20 a 30 microgramas/kg/hora
t 12 a 18 anos: 2,5 a 10 mg por via intravenosa, seguida de infusão intravenosa contínua à velocidade de 20-30 microgramas/kg/hora
t 150 microgramas/kg, por via intramuscular, 60 a 90 minutos antes da cirurgia.
t 100 microgramas/kg, por via intravenosa, a cada 40 a 60 minutos, durante o procedimento cirúrgico.
t 100 a 200 microgramas/kg (máximo de 10 mg), por via intramuscular, a cada 4 horas, após procedimento cirúrgico.
Nota: A injeção intravenosa deve ser administrada por pelo menos 5 minutos.
t 10 a 30 mg, por via oral, a cada 3 a 4 horas (formulação de liberação imediata), ou
t 10 mg, por via subcutânea ou intramuscular, a cada 2 a 4 horas, ou
t 2,5 a 5 mg, por via intravenosa lenta, a cada 4 horas, ou
t 0,8 a 10 mg/hora, por infusão intravenosa, podendo chegar a 80 mg/hora.
t 150 a 200 microgramas/kg (máximo 10 mg), por via subcutânea ou intramuscular, 60 a 90 minutos antes da cirurgia.
t 100 microgramas/kg, por via intravenosa, a cada 40 a 60 minutos, durante o procedimento cirúrgico.
t 150 a 300 microgramas/kg (máximo de 10 mg), por via intramuscular, a cada 4 horas, após procedimento cirúrgico, ou
t 8 a 10 mg, por infusão intravenosa, em 30 minutos, seguido de 2 a 2,5 mg/ hora.
t 10 mg, por via intravenosa lenta (2 mg/minuto), seguidos de dose adicional de 5 a 10 mg, se necessário.
t 5 a 20 mg, por vias oral, subcutânea ou intramuscular, a cada 4 horas; dose pode ser aumentada de acordo com a necessidade.
t 5 a 10 mg, por via intravenosa lenta (2 mg/minuto).
Nota: Cápsula de liberação prolongada: somente deve ser utilizada após ter sido estabelecida a dose de morfina necessária ao paciente com a apresentação de liberação imediata. A dose deve ser administrada a cada 12 a 24 horas. A dose máxima diária dessa apresentação é de 1.600 mg.
t epidural/intratecal: 15 a 60 minutos
t subcutânea: 10 a 30 minutos
t intravenosa: 5 a 10 minutos
t intramuscular: 10 a 30 minutos
t oral (liberação imediata): 30 minutos
t epidural/intratecal: acima de 24 horas
t subcutânea: 4 a 7 horas.
t intramuscular: 4 a 5 horas.
t oral (liberação imediata): 4 horas
t Metabolismo: hepático.
t Meia-vida de eliminação: 1,5 a 4,5 horas.
t Excreção: renal e fecal.
t Edema periférico (5% a 10%); prurido (acima de 80%); exantema (5% a 10%); sudorese (5% a 10%).
t Dor abdominal (5% a 10%); obstipação (acima de 10%); diarreia (5% a 10%); perda de apetite (5% a 10%); náusea e vômito (7% a 70%); xerostomia (5% a 10%)
t Testes da função hepática anormais (acima de 5%).
t Lombalgia (5% a 10%); astenia (5% a 10%); tontura (6%); sonolência (acima de 10%); cefaleia (acima de 10%); insônia (5% a 10%); parestesias (5% a 10%) ambliopia (acima de 5%); miose; ansiedade (6%); depressão (5% a 10%).
t Retenção urinária (15% a 70%).
t Febre (5% a 10%).
t Soluços (acima de 5%). t Rigidez muscular.
t Parada cardíaca, bradicardia, taquicardia, palpitação, hipotensão ortostática (acima de 5%).
t Confusão; alucinações; choque; síncope (acima de 5%).
t Anafilaxia (rara).
t Aumento da pressão intracraniana.
t mioclônus.
t Dispneia (5% a 10%); depressão respiratória (intratecal: 4% a 7%; epidural: 0,25% a 0,4%).
t Agonistas/antagonistas de opioides (por exemplo, naloxona, buprenorfina, nalbufina): pode resultar em sintomas de retirada dos opioides (cólicas abdominais, náuseas, vômitos, lacrimejamento, rinorreia, ansiedade, inquietação, elevação da temperatura ou piloereção). Antagonistas de opioides devem ser administrados cautelosamente em pessoas com suspeita de dependência física de qualquer agonista de opioide. Caso sinais e sintomas de retirada ocorrerem, pacientes devem ser tratados pela reinstituição da terapia opioide, seguida de redução gradual da dose de opioide combinada com suporte sintomático.
t Barbituratos, benzodiazepínicos, relaxantes musculares de ação central: possível depressão respiratória aditiva. Monitorar paciente para depressão respiratória. Redução da dose de um ou ambos agentes pode ser necessária.
t Ciclosporina: possível aumento do risco de anormalidades e mau funcionamento do sistema neurológico. Pacientes devem ser monitorados para desenvolvimento de complicações neurológicas como ansiedade, insônia, amnésia, confusão grave, etc.
t Cimetidina: pode resultar em toxicidade da morfina (depressão do SNC e respiratório). Monitorar pacientes para sinal de toxicidade de morfina. Ajuste de dose da morfina pode ser necessário.
t Esmolol: uso concomitante pode resultar em toxicidade do esmolol (bradicardia, hipotensão). Os Efeitos adversos podem ser controlados com ajuste da velocidade de administração do esmolol.
t Inibidores da monoamina oxidase (MAO): pode resultar em hipotensão e aumento dos efeitos depressores do SNC e respiratório. Administração concomitante é contraindicada. Instituir intervalo de 14 dias entre os tratamentos.
t Ioimbina: pode resultar em aumento dos efeitos analgésicos e adversos da morfina. Monitorar cuidadosamente os pacientes.
t Naltrexona: pode resultar em sintomas de retirada dos opioides e decréscimo da efetividade do opioide. Administração concomitante é contraindicada. Pacientes devem estar sem usar opioides por no mínimo 7 a 10 dias, antes de iniciar tratamento com naltrexona.
t Rifampicina: possível perda da eficácia da morfina. Monitorar adequado controle da dor. Ajuste da dose de morfina pode ser necessário.
t Somatostatina: pode resultar em redução do efeito analgésico da morfina. Monitorar pacientes para aumento da resposta dolorosa. Terapia alternativa pode ser necessária.
t Orientar para ingerir com alimentos para evitar desconforto gástrico
t Informar que a solução oral de morfina pode ser misturada a sucos de frutas para melhorar o sabor.
t Orientar para ingerir as cápsulas de liberação prolongada de morfina inteiras. Não morder, esmagar ou dissolver.
t Alertar que este medicamento pode causar dependência.
t Orientar para não parar de usar este medicamento abruptamente.
t Orientar para obedecer rigorosamente às determinações de dose e horários. t Alertar para evitar o uso de qualquer outro medicamento que cause sono, como hipno-sedativos, anti-histamínicos, medicamentos para alívio sintomático de gripes e resfriados.
t Não tomar bebidas alcoólicas enquanto estiver utilizando este medicamento.
t Beber bastante líquido e fazer exercícios para evitar obstipação.
t Este medicamento pode causar tontura ou sonolência. Evitar dirigir, usar máquinas ou fazer qualquer atividade que possa ser perigosa se a pessoa não estiver alerta.
t Alertar que é importante notificar imediatamente ao médico se apresentar os seguintes efeitos adversos: sintomas de reações alérgicas (prurido, inchaço na face ou mãos, inchaço ou formigamento na boca ou garganta, aperto no peito, dificuldade de respirar), confusão, diminuição na quantidade e frequência da urina, muita fraqueza, respiração fraca, batimentos cardíacos irregulares, sudorese, pele fria ou úmida, inchaço nas mãos, tornozelos ou pés.
t Comprimidos: armazenar sob temperatura entre 15 a 30 °C, em embalagem bem fechada.
t Solução oral: armazenar sob temperatura entre 15 a 30 °C, em embalagem bem fechada e protegida da luz. Evitar congelamento. Frascos abertos devem ser descartados após 90 dias.
t Cápsula de liberação prolongada: armazenar sob temperatura ambiente controlada (15 a 30 °C), protegida de luz e umidade.
t Solução injetável: armazenar sob temperatura ambiente controlada (15 a 30°C), proteger da luz. Escurecimento da solução indica degradação. Soluções sem preservativos não devem ser autoclavadas. Porções não utilizadas de soluções sem preservativos devem ser desprezadas.
t Observar orientação específica do produtor quanto a reconstituição, diluição, compatibilidade e estabilidade da solução.
t Concentração usual para infusão intravenosa contínua: 0,1 a 1 mg/mL, diluída com solução de glicose a 5%.
t Para administração intravenosa direta, diluir em 4 a 5 mL de água estéril e administrar lentamente (15 mg em 3 a 5 minutos).
t Para administração epidural e intratecal, utilizar soluções sem preservativos.
Helena Lutéscia Luna Coelho
Na Rename 2010: item 17.1
t Aerossol 100 microgramas/dose
t Solução inalante 6 mg/mL (equivalente a 5 mg de salbutamol/mL)
t Solução injetável 0,5 mg/mL
t Tratamento de manutenção e da exacerbação aguda da asma.
t Profilaxia da asma induzida por exercícios.
t Tratamento de doença pulmonar obstrutiva crônica (DPOC).
t Hipersensibilidade ao sulfato de salbutamol ou a qualquer componente da formulação.
t Usar com cuidado nos casos de:
– transtornos convulsivos como epilepsia.
– hipertireoidismo.
– doenças cardiovasculares, insuficiência do miocárdio, arritmias, susceptibilidade a prolongação do intervalo QT e hipertensão.
– asma grave (hipopotassemia pode ser potencializada por hipóxia ou pelo efeito de outros medicamentos antiasmáticos; monitorar o potássio sérico e evitar a indução de hipopotassemia).
– diabete melito (especialmente administração intravenosa; monitorar glicose sanguínea e risco de cetoacidose).
– cetoacidose pré-existente.
– ocorrência de broncoespasmo paradoxal (pode ser fatal; se ocorrer, interromper imediatamente o uso de salbutamol e utilizar tratamento alternativo).
t Categoria de risco na gravidez (FDA): C.
t Solução inalante (gotas):
– 2,5 a 5 mg, por via inalatória em 3 mL de solução salina 0,9%, durante 5 a 15 minutos, até 4 vezes ao dia, se necessário. Dose máxima: 8 mg/dose.
t Aerossol
– 1 a 2 inalações, por via oral, até 4 vezes ao dia.
t Solução inalante (gotas):
– 0,15 mg/kg (dose mínima: 2,5 mg), por via inalatória em 3 mL de solução salina 0,9%, a cada 20 minutos até 3 doses, seguido de 0,15 a 0,3 mg/kg, por via inalatória em 3 mL de solução salina 0,9%, a cada 1 a 4 horas conforme necessário. Dose máxima: 10 mg/dose.
t Aerossol
– 4 a 8 inalações, por via oral, a cada 20 minutos até 3 doses, seguido da mesma dose a cada 1 a 4 horas conforme necessário. Em criança abaixo de 4 anos administrar através de máscara.
t Solução injetável (para asma aguda grave):
– 3 a 5 microgramas/kg, por via subcutânea ou intramuscular, a cada 12 horas
t Aerossol (maiores de 4 anos)
– 2 inalações, por via oral, 15 a 30 minutos antes do exercício.
t Solução inalante (gotas):
– 2,5 a 5 mg, por via inalatória em 3 mL de solução salina 0,9%, durante 5 a 15 minutos, até 4 vezes ao dia, se necessário. Dose máxima: 8 mg/dose.
t Aerossol
– 1 a 2 inalações, por via oral, até 4 vezes ao dia.
t Solução inalante (gotas):
– 2,5 a 5 mg, por via inalatória em 3 mL de solução salina 0,9%, a cada 20 minutos até 3 doses, seguido de 2,5 a 10 mg, por via inalatória em 3 mL de solução salina 0,9%, a cada 1 a 4 horas conforme necessário. Dose máxima: 10 mg/dose.
t Aerossol
– 4 a 8 inalações, por via oral, a cada 20 minutos até 4 horas, seguido da mesma dose a cada 1 a 4 horas conforme necessário.
t Solução injetável (para asma aguda grave):
– 0,25 mg, por via intravenosa lenta, repetida se necessário
– 0,5 mg, por via subcutânea ou intramuscular, a cada 4 horas, se necessário.
t Aerossol
– 2 inalações, por via oral, 15 a 30 minutos antes do exercício.
t Solução inalante (gotas):
– 5 mg, por via inalatória em 3 mL de solução salina 0,9%, durante 5 a 15 minutos, até 6 vezes ao dia.
t Aerossol
– 1 a 2 inalações, por via oral, até 6 vezes ao dia.
t Absorção: forma inalada: 25 minutos; forma nebulizada: 30 minutos.
t Biodisponibilidade: menor que 20%; forma inalada: 10 a 20% da dose atinge as vias aéreas inferiores. O restante é mantido no sistema de entrega (espaçador) ou é engolida e absorvida pelo intestino.
t Início da ação: 5 minutos após a inalação.
t Excreção: predominantemente renal.
t Meia-vida de eliminação: em torno de 5 horas.
t Taquiarritmias; anormalidades no ECG, fibrilação atrial, enfarte do miocárdio, angina.
t Hipopotassemia.
t Hipopotassemia.
t Tremor, nervosismo.
t Eritema multiforme, síndrome de Stevens-Johnson (raro).
t Edema pulmonar.
t Atomoxetina: o uso concomitante de atomoxetina e salbutamol pode resultar no aumento da frequência cardíaca e da pressão arterial.
t Betabloqueadores: o uso concomitante de bloqueadores beta-adrenérgicos e salbutamol pode resultar na redução da efetividade de ambos os fármacos.
t Inibidores da MAO (monoamina oxidase): o uso concomitante com o salbutamol pode resultar no aumento do risco de taquicardia, agitação e hipomania.
t Orientar quanto a utilização correta do aerossol e do espaçador.
t O aerossol (bombinha) deve ser agitado antes do uso e conectado ao espaçador quando da administração em crianças ou em adultos se o médico recomendar. Guardar o inalador à temperatura ambiente, evitando o calor excessivo ou a proximidade com fogo, sob risco de explosão. Não perfurar o inalador.
t Antes de usar o inalador, certifique-se que seu funcionamento está correto, ou seja, verifique se o aerossol está sendo dispersado, cuidado com os olhos durante esta operação. Semanalmente, o bocal do inalador e o espaçador devem ser bem lavados com detergente neutro e deixados para secar naturalmente.
t Quando o inalador for usado ocasionalmente, pode ser mantido sob refrigeração com o bocal bem fechado, e colocado à temperatura ambiente antes de usar.
t O uso do sulfato de salbutamol na forma de nebulização deve estar reservado ao uso hospitalar ou ambulatorial. Somente utilizar em casa, quando a nebulização puder ser realizada por pessoa capacitada, e com treinamento para diluição correta do medicamento, uso correto do nebulizador e com técnicas de higiene e limpeza adequadas da máquina e de seus acessórios.
t Armazenar o sulfato de salbutamol (aerossol sem clorofluorcarbono como propelente) à temperatura ambiente, entre 15 e 25 ºC, longe da umidade. Não expor o inalador a altas temperaturas (aproximadamente 50 ºC).
t Armazenar o sulfato de salbutamol (solução inalante gotas) à temperatura entre 2 e 25 ºC e longe de luz direta.
t Na concentração de 200 microgramas/mL em solução salina 0,9%, o sulfato de salbutamol permanece estável por 7 dias sob temperatura ambiente e sobre refrigeração (situações em que nebulização contínua é necessário).
t Armazenar a solução injetável de sulfato de salbutamol à temperaturas ambiente, entre 15 e 30 ºC, e protegida da luz.
t Observar orientação específica do produtor quanto a diluição, compatibilidade e estabilidade da solução.
t A solução para infusão deve ser preparada a partir da diluição de uma ampola (0,5 mg/mL) de sulfato de salbutamol em 500 mL de solução injetável de cloreto de sódio 0.9% ou glicose 5%, permanecendo estável por 24 horas sob temperatura ambiente.
Larissa Niro
Na Rename 2010: item 6.1.3
t Pó para solução injetável 10 mg
t Carcinoma renal.
t Carcinoma de próstata.
t Carcinoma de bexiga.
t Carcinoma de mama (não responsivo a cirurgia endócrina e terapia hormonal).
t Carcinoma de testículo.
t Carcinoma de não-pequenas células de pulmão.
t Coriocarcinoma.
t Doença de Hodgkin (estádios II e IV).
t Doença de Letterer-Siwe (histiocitose das células de Langerhans).
t Linfoma linfocítico maligno.
t Linfomas disseminados de Hodgkin e não-Hodgkin.
t Micose fungoide.
t Púrpura trombocitopênica idiopática (refratária).
t Tratamento paliativo de sarcoma de Kaposi.
t Tumor de células germinativas de ovário.
t Tumores trofoblásticos.
t Injeção intratecal.
t Hipersensibilidade a vimblastina ou a qualquer componente da formulação.
t Infecções bacterianas não-controladas.
t Supressão grave da medula óssea.
t Lactação (ver Apêndice B).
t Usar com cuidado nos casos de:
– ocorrência de neurotoxicidade (ajustar a dose).
– insuficiência hepática (ver Apêndice C).
– disfunção pulmonar.
– infiltração de células malignas na medula óssea.
– insuficiência renal.
t Administrar somente por via intravenosa.
t Não utilizar em pacientes idosos e com caquexia, ou áreas ulceradas da pele.
t Evitar extravasamento (efeito vesicante).
t O desenvolvimento de nefropatia por ácido úrico em pacientes com linfoma pode ser prevenido pela hidratação adequada e, em alguns casos, pela administração de alopurinol. A alcalinização da urina pode ser necessária se aumentarem as concentrações séricas de ácido úrico.
t Categoria de risco na gravidez (FDA): D (ver Apêndice A).
t 3 mg/m2, por via intravenosa, em combinação com outros agentes antineoplásicos.
t 6 mg/m2, por via intravenosa, em combinação com outros agentes antineoplásicos.
t 6,5 mg/m2, por via intravenosa, como monoterapia.
t Primeira dose 3,7 mg/m2, segunda dose 5,5 mg/m2, terceira dose 7,4 mg/m2, quarta dose 9,25 mg/m2, quinta dose 11,1 mg/m2, por via intravenosa, a intervalos semanais; aumentar a dose semanalmente até a dose máxima de 18,5 mg/m2 ou até que a contagem de células brancas seja reduzida a 3.000 células por mm3; a dose de manutenção semanal deve ser maior do que a dose inicial e menor do que a dose máxima.
t 0,1 mg/kg/dia, por via intravenosa, a cada 3 semanas + alfainterferona 2 a 3 milhões UI, por via subcutânea ou intramuscular, 3 vezes por semana na primeira semana; após, 18 milhões UI por semana nas semanas subsequentes.
t 4 mg/m2/semana, por via intravenosa, durante 6 semanas; intervalo de 2 semanas; repetir o ciclo ou até a normalização do PSA.
t 4 mg/m2/dia, por via intravenosa, uma vez por semana até o dia 29, em combinação com cisplatina 80 a 120 mg/m2. Após, administrar vimblastina a cada 2 semanas até o dia 43 ou até a última dose de cisplatina.
t Regime MVAC: vimblastina 3 mg/m2/dia, por via intravenosa, nos dias 2, 15 e 22; metotrexato 30 mg/m2/dia, por via intravenosa, nos dias 1, 15 e 22; doxorrubicina 30 mg/m2/dia, por via intravenosa, no dia 2; cisplatina 70 mg/ m2/dia, por via intravenosa, no dia 2. Repetir o ciclo a cada 28 dias.
t 0,1 mg/m2 (diluir em 500 a 2.000 mL de solução injetável de cloreto de sódio 0,9%), por infusão intravenosa durante 6 a 8 horas; alternar com vincristina.
t Regime PVB: cisplatina 20 mg/m2/dia, por via intravenosa, durante 5 dias; vimblastina 9 a 12 mg/m2/dia, por injeção intravenosa em bolo no dia 1; bleomicina 20 a 30 UI/m2/dia, por via intravenosa, uma vez por semana. Repetir a cada 3 semanas, por 3 a 4 ciclos.
t Metabolismo: hepático.
t Excreção: biliar e renal (20%).
t Meia-vida de eliminação: 20 horas. Meia-vida: seguindo administração intravenosa rápida: fase inicial: 3.7 minutos; fase intermediária: 1.6 horas; fase terminal: 24.8 horas.
t Não atravessa a barreira hematocerebral em quantidades significantes.
t Irritante aos tecidos, alopecia (>10%).
t Leucopenia (frequente), trombocitopenia e mielossupressão.
t Obstipação, náusea, vômito (moderado).
t Diminuição de sensibilidade e neuropatia periférica.
t Hipertensão (frequente).
t Hiperuricemia.
t Neurotoxicidade (raro).
t Azoospermia.
t Dor óssea, mandíbula, tumor nos tecidos.
t Mal-estar.
t Itraconazol: pode aumentar o risco de neurotoxicidade e íleo paralítico. Se o uso concomitante for necessário, monitorar para desenvolvimento de neurotoxicidade, incluindo parestesias, fraqueza muscular e íleo paralítico. Ajustar a dose de vimblastina.
t Lopinavir/ritonavir: pode resultar em grave toxicidade pela vimblastina. Usar com cautela; monitorar para Efeitos adversos hematológicos e gastrintestinais. Considerar a descontinuação de lopinavir/ritonavir ou a substituição do antirretroviral na presença de sinais de toxicidade pela vimblastina.
t Quinupristina/dalfopristina: pode aumentar o risco de toxicidade pela vimblastina. Se o uso concomitante for necessário, considerar o ajuste da dose de vimblastina ou sua retirada temporária.
t Ritonavir: pode resultar em grave toxicidade pela vimblastina. Se o uso concomitante for necessário, considerar o ajuste da dose de vimblastina e monitorar para sinais de toxicidade a este fármaco.
t Tolterodina: pode ter sua biodisponibilidade aumentada pela vimblastina em indivíduos com déficit de atividade de CYP2D6. Reduzir a dose de tolterodina, se cetoconazol também estiver sendo utilizado concomitantemente a vimblastina e tolterodina.
t Vacina rotavírus humano G1P1[8] (atenuada): aumento do risco de infecção pela vacina. O uso concomitante é contraindicado.
t Vacinas com vírus vivos: aumento do risco de infecção pela vacina. Se a vacina for necessária, administrar a vacina depois de três meses da descontinuação da quimioterapia.
t Voriconazol: pode resultar no aumento das concentrações plasmáticas de vimblastina. Se o uso concomitante for necessário, considerar o ajuste da dose de vimblastina.
t Orientar para aumentar a ingestão líquida durante até 2 dias após a administração de vimblastina.
t Orientar para controlar a obstipação com aumento da ingestão de fibras, vegetais, líquidos e exercícios regulares.
t Orientar para evitar vacinações sem prévia notificação.
t Orientar para evitar contato com pessoas com infecções, especialmente no período de baixa contagem de células sanguíneas.
t Orientar para não interromper o medicamento apesar de náuseas, vômitos. t Avisar o médico imediatamente se ocorrer sangramento ou lesão anormal. t Orientar para ter cautela com objetos cortantes.
t Orientar para não tocar os olhos ou o interior do nariz ao menos que esteja com as mãos lavadas.
t Utilizar contracepção efetiva durante o tratamento, homens e mulheres.
t Armazenar os frascos sob refrigeração (2 e 8 ºC). Proteger da luz.
t Observar orientação específica do produtor quanto a reconstituição, diluição, compatibilidade e estabilidade da solução.
t Reconstituir vimblastina 10 mg com 10 mL de solução injetável de cloreto de sódio 0,9% (com ou sem conservante: álcool benzílico) para concentração final de 1 mg/mL. A solução com conservante permanece estável por 28 dias sob refrigeração.
t Etiquetar o recipiente da solução de sulfato de vimblastina com os seguintes dizeres: “FATAL SE DADA INTRATECALMENTE. APENAS PARA USO INTRAVENOSO” – “NÃO REMOVER ESTA ETIQUETA ATÉ O MOMENTO DA INJEÇÃO”.
t Não diluir a dose em volumes grandes do diluente (100 a 250 mL).
t Não administrar por períodos prolongados (30 a 60 minutos ou mais).
t Incompatível com: anfotericina B, pantoprazol, lansoprazol, cefipima, furosemida.
t A estabilidade é variável quando misturada com cloridrato de doxorrubicina e heparina sódica.
t Não injetar em uma extremidade na qual a circulação é danificada ou potencialmente danificada por condições como neoplasma, flebite ou varicosidade.
t Se ocorrer contaminação acidental, os olhos devem ser imediatamente lavados com água para prevenir irritação e possível ulceração da córnea.
t Observar protocolos locais para manipulação de substâncias citotóxicas.
Atenção: vimblastina deve ser usada apenas por administração intravenosa. Injeção intratecal causa neurotoxicidade fatal. Cuidado especial deve ser tomado durante a administração para evitar extravasamento da solução de vimblastina.
Rogério Aparecido Minini dos Santos
Na Rename 2010: item 6.1.3
t Pó para solução injetável 1 mg.
t Leucemia linfoblástica aguda.
t Linfoma de Hodgkin e não-Hodgkin.
t Rabdomiossarcoma.
t Nefroblastoma
t Neuroblastoma.
t Câncer de pequenas células de pulmão.
t Linfoma das células do manto.
t Micose fungoide
t Hipersensibilidade à vincristina ou a outros alcaloides da vinca.
t Administração intratecal.
t Síndrome de Charcot-Marie-Tooth.
t Usar com cuidado nos casos de:
– doença neuromuscular (ajustar a dose).
– disfunção pulmonar.
– crianças.
– radioterapia concomitante (incluindo os com insuficiência hepática).
– insuficiência hepática (ver Apêndice C); diminuir a dose.
t Pode causar nefropatia aguda por ácido úrico (prevenir com o uso de alopurinol).
t Não manusear os excretas urinários e fecais.
t Evitar extravasamento.
t Categoria de risco na gravidez (FDA): D (ver Apêndice A).
t Até 10 kg: 0,05 mg/kg, por via intravenosa, uma vez por semana; em associação com outros agentes antineoplásicos.
t Acima de 10 kg: 1,5 a 2 mg/m2, por via intravenosa, uma vez por semana; em associação com outros agentes antineoplásicos.
t 1,4 mg/m2, por via intravenosa, uma vez por semana; em associação com outros agentes antineoplásicos.
t 0,03 a 1,4 mg/m2, por via intravenosa, uma vez por semana; em associação com outros agentes antineoplásicos.
Nota: A administração deve feita via intravenosa lenta, por pelo menos 1 minuto; se ocorrer extravasamento, finalizar a dose em outra veia e aplicar hialuronidase (250 U) por via subcutânea para dispersar o medicamento e aplicar calor local por 1 hora, repetindo 4 vezes ao dia, durante 3 a 5 dias.
t Meia-vida: 14,4 a 25,5 horas.
t Metabolismo: hepático.
t Excreção: renal (10 a 20%), fezes (80%).
t Mielosupressão.
t Neuromiopatia.
t Alopecia (20 a 70%).
t Náusea, vômito, obstipação.
t Hipertensão ou hipotensão ortostática.
t Diplopia.
t Depressão do sistema nervoso central, confusão, paralisia do nervo craniano, febre, cefaleia, dificuldades motoras, tontura.
t Filgrastim e sargramostim: aumento do risco de neuropatia periférica (com sargramostim, neuropatia atípica grave). Nos pacientes recebendo filgrastim ou sargramostim, restringir a dose total de vincristina utilizada no primeiro ciclo e monitorar cuidadosamente os pacientes para sintomas de neuropatia periférica.
t Itraconazol: aumento do risco de neurotoxicidade e íleo paralítico. Monitorar o paciente para sinais e sintomas; ajustar a dose da vincristina, se necessário.
t Nifedipino: risco aumentado de toxicidade da vincristina (neuropatia, delírio, tontura). Monitorar a neurotoxicidade, distúrbios no ritmo cardíaco, e efeitos metabólicos como hiperuricemia.
t Posaconazol e voriconazol: o uso concomitante pode resultar em aumento das concentrações de vincristina. Cautela na coadministração; considerar o ajuste de dose da vincristina.
t Quinupristina/dalfopristina: aumento da toxicidade da vincristina (neuropatia, leucopenia, trombocitopenia, tremor). Se for necessário coadministrar quinupristina/dalfopristina, considerar a suspensão ou a redução de dose da vincristina.
t Vacinas com vírus vivos: o uso concomitante de vacinas com vírus vivos pode resultar em aumento do risco de infecção pela vacina com vírus vivos. Em pacientes com leucemia em remissão, a vacinação é permitida após três meses da descontinuação da quimioterapia.
t Vacina oral de rotavírus humano: o uso concomitante de vacina contra rotavírus pode resultar em aumento do risco de infecção pela vacina. A vacinação é contraindicada.
t Valspodar: aumento no risco de toxicidade da vincristina. A redução da dose de vincristina é indicada, mas orientações específicas para tal ainda não foram definidas.
t Varfarina: risco aumentado de sangramento. Monitorar os sinais de sangramento, bem como os parâmetros de coagulação em paciente utilizando varfarina concomitantemente com vincristina. Se necessário, ajustar a dose de varfarina.
t Orientar para aumentar a ingestão líquida durante e até 2 dias após a administração de vimblastina.
t Orientar para controlar a obstipação com aumento da ingestão de fibras, vegetais e líquidos e exercícios regulares.
t Orientar para evitar vacinações sem prévia notificação.
t Orientar para evitar contato com pessoas com infecções, especialmente no período de baixa contagem de células sanguíneas.
t Utilizar contracepção efetiva durante o tratamento, homens e mulheres.
t Armazenar sob refrigeração (2 a 8 ºC) e proteger da luz. Se armazenada a temperatura ambiente (15 a 30 ºC) ou fresca (8 a 15 ºC) permanece estável por 1 mês.
t Observar orientação específica do produtor quanto a reconstituição, diluição, compatibilidade e estabilidade da solução.
t Pode ser diluída em solução glicosada a 5%, solução de cloreto de sódio 0,9% ou Ringer + lactato. Quando diluída em solução glicosada 5% se mantém estável por 24 horas em vidro âmbar ou recipiente de PVC. Manter pH entre 3,5 e 5,5.
t Deve ser diluída a uma concentração máxima de 100 microgramas/mL a um volume de pelo menos 10 mL.
t Etiquetar o recipiente da solução de sulfato de vimblastina com os seguintes dizeres: “FATAL SE DADA INTRATECALMENTE. APENAS PARA USO INTRAVENOSO” – “NÃO REMOVER ESTA ETIQUETA ATÉ O MOMENTO DA INJEÇÃO”.
t Incompatível com solução de bicarbonato de sódio ou qualquer outra solução alcalina.
t Incompatível com cefepime, furosemida, idarubicina, nutrição parenteral total.
t Observar protocolos locais para manipulação de substâncias citotóxicas.
Atenção: a administração por via intratecal é fatal. Vincristina é de uso exclusivo intravenoso.
Maria Isabel Fischer
Na Rename 2010: itens 10.3, 11
t Comprimido mastigável 10 mg
t Solução injetável 1 mg/mL
t Solução injetável 200 microgramas/mL
t Xarope 4 mg/mL
t Nutrição parenteral.
t Suplementação nutricional em estados de deficiência ou de perda de zinco.
t Adjunto para terapia de reidratação oral em doenças diarreicas agudas em crianças.
t Usar com cuidado nos casos de:
– uso prolongado ou em altas doses (pode levar a deficiência de cobre, associada a anemia sideroblástica ou neutropenia).
– falência renal aguda (ver Apêndice D); pode ocorrer acúmulo de zinco.
t Categoria de risco na gravidez (FDA): C (ver Apêndice A).
t 0,5 a 1 mg/kg, por via oral, dividido a cada 8 a 24 horas.
t Prematuros: 300 microgramas/kg/dia, por via intravenosa.
t Crianças até 40 kg: 100 a 250 microgramas/kg/dia, por via intravenosa.
t Crianças acima de 40 kg: 2,5 a 4 mg/kg/dia, por via intravenosa.
t Até 6 meses: 10 mg, por via oral, a cada 24 horas, durante 10 a 14 dias.
t Acima de 6 meses: 20 mg, por via oral, a cada 24 horas, durante 10 a 14 dias.
t 110 a 220 mg, por via oral, a cada 8 horas.
t 2,5 a 4 mg/dia, por via intravenosa.
t Absorção: oral – incompleta e diminuída por fitatos presentes em alimentos como cereais e legumes.
t Biodisponibilidade: oral – 20 a 30%.
t Excreção: basicamente fecal (67%).
t Dor abdominal, dispepsia, náusea, vômito, diarreia, irritação gástrica, gastrite.
t Irritabilidade, cefaleia, letargia.
t Grepafloxacino, moxifloxacino: o uso concomitante pode levar a diminuição da efetividade do antibiótico. A fluoroquinolona deve ser administrada 4 horas antes ou 4 horas (grepafloxacino) e 8 horas (moxifloxacino) depois de produtos contendo zinco.
t Ofloxacino:o uso concomitante pode levar a diminuição da absorção e da eficácia de ofloxacino. Suplementos de zinco ou produtos contendo zinco devem ser administrados 2 horas antes ou 2 horas depois da ofloxacino.
t Sais de ferro: o uso concomitante pode resultar em absorção reduzida de zinco e/ou sais de ferro administrados por via oral. Separar por pelo menos 2 horas a administração de sais de ferro e de zinco.
t Tetraciclinas: reduz absorção e as concentrações séricas do zinco e das tetraciclinas (exposição diminuída a tetraciclina). Evitar o uso concomitante. Administrar a tetraciclina 2 horas antes ou 3 horas após o zinco.
t Comprimidos podem ser dispersos em leite materno, líquidos de reidratação oral ou em água numa colher pequena. Crianças mais velhas podem mastigar o comprimido ou ingerir com água.
t É preferível tomar o medicamento com o estômago vazio, 1 hora antes ou 2 horas depois das refeições, porém, os Efeitos adversos podem ser mais frequentes, neste caso, ingerir com alimento. Evitar ingerir com pão, cereal, farelos, leite, iogurte, queijo ou sorvete.
t Consultar o médico e/ou farmacêutico antes de utilizar outros fármacos, incluindo os de venda livre, vitaminas e produtos à base de plantas. Evitar alimentos e medicamentos que contenham cobre.
t Armazenar em recipiente não metálico, hermeticamente fechado.
t Em soluções intravenosas, zinco pode ser incompatível com sais de morfina. t Zinco, na concentração de 3 mg/mL, se mostrou compatível com solução de nutrição parenteral contendo aminoácidos 3%, glicose 3% e eletrólitos.
Orozimbo Henriques Campos Neto
Na Rename 2010: itens 11 e 15.1
t Comprimido de 40 mg Fe2+
t Solução oral a 25 mg/mL Fe2+
t Tratamento de anemia associada à deficiência de ferro.
t Profilaxia em situações de alto risco para deficiência de ferro (casos de deficiência dietética, síndrome de má-absorção, menorragia, após gastrectomia total ou subtotal)
t Hemossiderose, hemocromatose hemoglobinopatias.
t Qualquer forma de anemia não associada à deficiência de ferro.
t Pacientes que receberam repetidas transfusões sanguíneas.
t Ferroterapia parenteral.
t Hipersensibilidade ao ferro.
t Usar com cuidado nos casos de:
– úlcera péptica, enterite regional, colite ulcerativa, estreitamento intestinal, divertículos (tais condições inflamatórias do trato intestinal podem ser exacerbadas com a administração oral de ferro).
– alcoolismo, insuficiência hepática, insuficiência renal.
– testes laboratoriais (o sulfato ferroso pode causar resultados falso-negativos para testes com glicose oxidase).
– idosos (podem requerer doses orais de ferro maiores que adultos jovens para corrigir anemia).
t Não deve ser administrado por mais de 6 meses.
t Monitorar concentrações plasmáticas de ferritina e ferro para reconhecer e prevenir a hemossiderose.
t A dose excessiva de ferro em crianças (usualmente acidental) é mais comum do que em adultos e pode causar efeitos tóxicos. Neste caso, é necessário atendimento médico imediato e é feita a administração intravenosa de desferroxamina para quelar os íons ferro.
t Prematuros: 2 a 4 mg/kg de ferro elementar (máximo de 15 mg), por via oral, a cada 24 horas ou dividido a cada 12 horas.
t Lactentes e crianças: 3 a 6 mg/kg de ferro elementar, por via oral, a cada 24 horas ou dividido a cada 8 ou 12 horas. Dose máxima diária: 200 mg.
t Com menos de 5 anos: 2 mg/kg de ferro elementar, por via oral, a cada 24 horas. Dose máxima diária: 30 mg de ferro elementar.
t Com mais de 5 anos: 30 mg de ferro elementar, por via oral, a cada 24 horas.
t 50 a 100 mg de ferro elementar, por via oral, a cada 12 horas.
t 60 mg de ferro elementar, por via oral, a cada 24 horas.
t Absorção: irregular e incompleta; a secreção ácida do estômago auxilia a absorção; a porcentagem de absorção é afetada por forma do sal, quantidade administrada, esquema de administração, tamanho do estoque de ferro do organismo e estado de deficiência de ferro (a absorção chega a 25%). Apesar de as preparações de ferro serem mais bem absorvidas no estômago vazio, podem ser administradas após as refeições para reduzir Efeitos adversos gastrintestinais.
t Absorção oral de ferro é pobre em pacientes em diálise peritoneal contínua.
t Tempo para o pico de concentração plasmática (via oral): 2 horas.
t Latência: resposta hematológica aparece em 2 semanas, aumentando a produção de hemoglobina em torno de 2 g/dL nas primeiras 3 semanas de tratamento.
t Meia-vida de eliminação: 6 horas.
t Excreção: quantidade muito pequena de ferro é excretada; a conservação do ferro corporal e a falta de um mecanismo excretor para o excesso de ferro são a causa para a sobrecarga corporal do mineral com a sua ingestão excessiva na terapia e repetidas transfusões.
t Obstipação ou diarreia, fezes escuras, irritação gastrintestinal, pirose.
t Náusea (frequente) e dor epigástrica estes sintomas são dose-dependente.
t Hemossiderose (em terapia prolongada ou administração excessiva).
t Soluções orais podem causar manchas nos dentes.
Nota: Se ocorrerem efeitos adversos, estes podem ser diminuídos por meio de redução da dose, substituição por outro sal de ferro com menor conteúdo de ferro elementar, aumento gradual da dose diária e administração do medicamento com alimento.
t Doxiciclina, minociclina, tetraciclina: redução na efetividade desses antimicrobianos e de sais de ferro. Estabelecer intervalo de pelo menos 3 horas antes ou 2 horas após a administração dos outros medicamentos em relação aos sais de ferro.
t Gatifloxacino: redução na eficácia de gatifloxacino. Estabelecer intervalo de 4 horas entre a administração de sais de ferro e a de gatifloxacino.
t Levodopa: pode aumentar a ocorrência de sintomas da doença de Parkinson. Monitorar paciente e, se houver piora nos sintomas, ajustar dose ou evitar o uso de produtos contendo sais de ferro.
t Levotiroxina: risco de hipotireoidismo. Estabelecer intervalo de pelo menos 4 horas entre a administração de sais de ferro e levotiroxina. Monitorar função tiroideana.
t Lomefloxacino: evitar uso concomitante. Se for necessário, a dose dos sais de ferro deve ser administrada pelo menos 6 horas antes ou 4 horas após a dose de lomefloxacino.
t Metildopa: não é recomendado o uso concomitante a sais de ferro.
t Micofenolato de mofetila, ofloxacino: pode haver redução na eficácia do micofenolato. Estabelecer intervalo de pelo menos 2 horas entre a administração de sais de ferro e o micofenolato de mofetila ou ofloxacino.
t Moxifloxacino: administrar 4 antes ou 8 horas após os sais de ferro.
t Norfloxacino: evitar administração concomitante, fazendo opção por outro antimicrobiano, empregando via intravenosa para norfloxacino ou considerando suspensão temporária dos sais de ferro enquanto usar a quinolona. Se a combinação for necessária, administrar a quinolona 2 horas antes ou 4 a 6 horas após a dose dos sais de ferro. Monitorar intensivamente o paciente para verificar eficácia contínua do antimicrobiano.
t Omeprazol: redução na biodisponibilidade do ferro não-heme. Monitorar o paciente para eficácia do sulfato ferroso se omeprazol for usado concomitantemente. Considerar administração parenteral dos sais de ferro se for inevitável a administração de ambos.
t Penicilamina: dar intervalo de pelo menos 2 horas entre a administração de ferro e da penicilamina.
t Zinco: redução da absorção gastrintestinal de ferro e/ou zinco. Dar intervalo de pelo menos 2 horas entre a administração de ferro e a de zinco.
t Orientar a adoção na dieta de carne vermelha magra, frango, peru, peixe e ácido ascórbico (vitamina C), estimulantes da absorção de ferro não heme.
t Prevenir que ácido fítico (grãos não refinados e soja), polifenóis (chá, café, cacau, vinho tinto), cálcio, fósforo e certas proteínas (de soja, albumina de ovo e caseína) são inibidores da absorção de ferro não heme.
t Reforçar cuidados em situações de hemocromatose, hemossiderose, hemoglobinopatias, outras condições anêmicas, repetidas transfusões sanguíneas, úlcera péptica, colite ulcerativa, entre outros.
t Orientar para ingerir o sulfato ferroso com estômago vazio, 1 hora antes ou 2 horas depois das refeições para aumentar a absorção do ferro. Caso haja desconforto gastrintestinal ingerir após as refeições.
t Tomar com água ou suco de fruta: copo cheio (240 mL) para adultos, meio copo (120 mL) para crianças.
t Ensinar que as preparações líquidas contendo sais de ferro devem ser bem diluídas em água e, se possível, tomadas através de um canudinho para prevenir manchas nos dentes.
t Alertar que o sulfato ferroso não deve ser administrado por mais de 6 meses.
t Alertar para notificar se surgirem efeitos tóxicos ou suspeita de envenenamento.
t Alertar sobre a possível ocorrência de escurecimento das fezes.
t Manter à temperatura ambiente, de 15 a 30 ºC, em recipiente bem fechado.
t Não congelar a solução.
Consta no documento:
“Todos os direitos reservados. É permitida a reprodução parcial ou total desta obra, desde que citada a fonte e que não seja para venda ou qualquer fim comercial.”
O objetivo do site MedicinaNet e seus editores é divulgar este importante documento. Esta reprodução permanecerá aberta para não assinantes indefinidamente.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.