(Carregando Índice)... (Carregando Índice)... |
Autores:
Luiz Henrique Martins Castro
Assistente Doutor da Divisão de Clínica Neurológica do HCFMUSP
Pós-Doutorado nas Universidades de Harvard (Neurologia cognitiva e do
comportamento) e Columbia (Neurofisiologia Clínica e Epilepsia)
Médico Chefe do Setor de Vídeo-eletroencefalografia da Divisão de
Clínica Neurológica do HC-FMUSP
Lécio Figueira Pinto
Especialista em Neurologia pelo Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo (HC-FMUSP)
Pós-graduando (doutorado) pela Faculdade de Medicina da USP
Última revisão: 22/08/2009
Comentários de assinantes: 0
Crises epilépticas ocorrem por atividade elétrica anormal, excessiva e síncrona de grupos de neurônios levando a manifestações neurológicas variadas, a depender do local em que ocorrem. As crises podem se manifestar por vários sinais e sintomas, como alterações sensitivas (parestesias, visuais, auditivas, gustativas etc.), autonômicas, motoras (abalos, mioclonias etc.), cognitivas (experienciais, dismnésicas) e do nível de consciência. O termo crise convulsiva reserva-se ao subgrupo de crises epilépticas que se apresentam com manifestações motoras, e atualmente seu uso é desaconselhado.
A probabilidade de um indivíduo apresentar uma crise epiléptica em qualquer momento da vida é estimado entre 5% a 10%. Em cerca de 25% das crises, um fator causal pode ser identificado. Essas crises são denominadas crises agudas sintomáticas (ou crises provocadas). Os principais desencadeantes identificáveis de crises agudas sintomáticas são distúrbios no equilíbrio hidroeletrolítico ou ácido-básico (geralmente associadas a doenças clínicas), hipoglicemia ou hiperglicemia, hipóxia, medicamentos (por exemplo, quinolonas), intoxicação exógena por drogas que rebaixem o limiar epiléptico, abstinência de drogas sedativas ou insulto neurológico agudo (vascular, infeccioso etc.). Crises agudas sintomáticas tendem a não mais recorrer, uma vez eliminado o fator desencadeante.
Outras vezes, não se identifica fator causal para a crise epiléptica, e muitos desses pacientes não voltarão a ter crises. Crise única (ou isolada) refere-se a crise epiléptica não provocada (crises que ocorram em intervalo inferior a 24 horas).
Epilepsia é uma doença crônica caracterizada por crises epilépticas recorrentes não provocadas. O paciente deve apresentar pelo menos duas crises espontâneas, sem evidência de desencadeantes agudos de crises epilépticas. Segundo o último consenso da ILAE (International League Against Epilepsy), a epilepsia é definida como desordem cerebral caracterizada por uma predisposição persistente, que leva ao aparecimento de crises epilépticas e a suas conseqüências neurobiológicas, cognitivas e psicossociais. A definição requer a ocorrência de duas ou mais crises epilépticas espontâneas.
Segundo a Organização Mundial da Saúde, a epilepsia é a doença cerebral mais comum. Estudos epidemiológicos em Rochester (Minnesota, Estados Unidos) indicam uma incidência ajustada de 3,1% até a idade de 80 anos. Estudos de base populacional em diversos locais estimam uma prevalência entre 0,9 e 57 casos/1.000 habitantes e uma incidência entre 26 e 90 casos/100.000 habitantes.
Estudos brasileiros indicam prevalência de 16,5/1.000 em Porto Alegre e 11,9/1.000 em São Paulo. A prevalência de epilepsia ativa (pacientes que apresentem crises ou necessitem tratamento crônico para controle de crises) é menor, estimada em 0,5% nos Estados Unidos e em 1,5% a 2% na América Latina, 0,54% em São Paulo e 0,51% no Rio de Janeiro, o que demonstra que parte dos pacientes pode entrar em remissão, ao menos por algum período (epilepsia inativa). Crises isoladas são ainda mais comuns. A incidência cumulativa de crises para toda a vida é de cerca de 9%-11%. Aproximadamente uma em cada dez pessoas apresentará, em algum momento da vida, uma crise epiléptica. A maioria dos pacientes que apresentam uma crise epiléptica não tem epilepsia.
A epilepsia pode manifestar-se em qualquer fase da vida. Nos países industrializados observam-se dois picos de incidência: no primeiro ano de vida (decorrente de processos pré, peri e pós-natais) e após a sétima década de vida (decorrente, entre outros, de doenças neurológicas degenerativas e de lesões vasculares). A incidência e prevalência de epilepsia na faixa etária acima dos 70 anos é duas a três vezes maior que na infância. Em São Paulo, a prevalência de epilepsia é maior em idosos (0,85%).
Para definir a etiologia da epilepsia, é necessária análise do conjunto de dados clínicos (tipo ou tipos de crise apresentados pelo paciente, idade de início das crises, história familiar de epilepsia, presença de doença neurológica prévia), e dos dados de eletroencefalograma (EEG) e neuroimagem.
As crises epilépticas podem ser classificadas em focais (ou parciais) e generalizadas. Nas focais, as descargas anormais ocorrem em áreas circunscritas do córtex cerebral, levando a manifestações devido à disfunção da área acometida. Crises focais podem propagar-se levando ao acometimento de outras áreas do córtex cerebral. Essa propagação pode levar ao acometimento de grande parte ou da totalidade do córtex (crise secundariamente generalizada).
As crises primariamente generalizadas caracterizam-se por alteração eletroencefalográfica acometendo desde o início amplas áreas corticais, e, possivelmente, subcorticais.
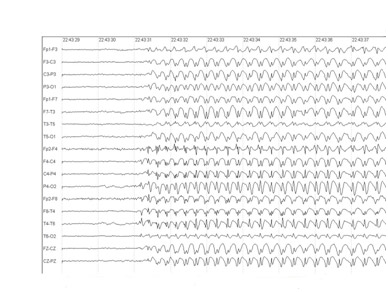
Nas crises de ausência típicas, ocorrem descargas em espícula-onda a 3 Hz (Figura 1), e o paciente apresenta parada súbita e transitória da atividade com duração de poucos segundos, podendo ocorrer automatismos orais e manuais, piscamento, alterações do tônus e sinais autonômicos, principalmente em episódios mais prolongados. A hiperventilação por
Figura 1: EEG mostrando paroxismos de complexos espícula-onda a aproximadamente 3 Hz de projeção generalizada, característico das crises de ausência típica.

Nas crises mioclônicas, ocorrem contrações musculares súbitas e breves, descritas como choques, com correlação no EEG (poliespícula-onda ou multiespícula de projeção generalizada, por vezes predominando nas regiões anteriores), geralmente ao despertar, adormecer e após privação de sono, que podem ser desencadeadas por fotoestimulação intermitente. As crises tônicas são caracterizadas por contração muscular mantida de todo o corpo ou segmento corporal; geralmente afetam a musculatura axial e duram segundos a minutos, enquanto nas atônicas ocorre a perda do tônus postural com queda lenta se o indivíduo estiver de pé.
Nas crises tônico-clônico generalizadas (TCG) ocorre perda súbita de consciência, seguida de uma fase inicial tônica, em que o paciente pode apresentar “grito epiléptico” devido à saída forçada de ar pela contração do diafragma. Evolui com movimentos clônicos dos quatro membros, apnéia, sialorréia e pode apresentar mordedura da língua e liberação esfincteriana. Ao final da crise, ocorre relaxamento muscular, rebaixamento do nível de consciência ou confusão mental, respiração ruidosa e ocasionalmente liberação de esfíncteres. O EEG mostra na fase tônica uma atividade epileptiforme generalizada e rítmica a 10 Hz, bilateral, síncrona e simétrica, evoluindo com padrão de poliespícula-onda lenta na fase clônica.
Síndromes epilépticas caracterizam grupos de doentes com associação de um ou mais tipos de crises, padrão eletroencefalográfico crítico e intercrítico, clínica (englobando vários aspectos como idade de início e alterações neurológicas) e aspectos genéticos semelhantes. Elas podem ser classificadas quanto à etiologia em idiopáticas, sintomáticas ou criptogências (também chamadas de “possivelmente sintomáticas”).
As síndromes epilépticas “idiopáticas” caracterizam-se por uma hiperexcitabilidade cortical possivelmente secundária a alterações na função de canais iônicos (canalopatias), muitas vezes de caráter familiar. São síndromes idade-específicas, acometem pacientes sem outras alterações neurológicas (indivíduos normais), apresentam crises e EEG típicos e curso geralmente benigno, entrando em remissão espontânea na maioria dos casos.
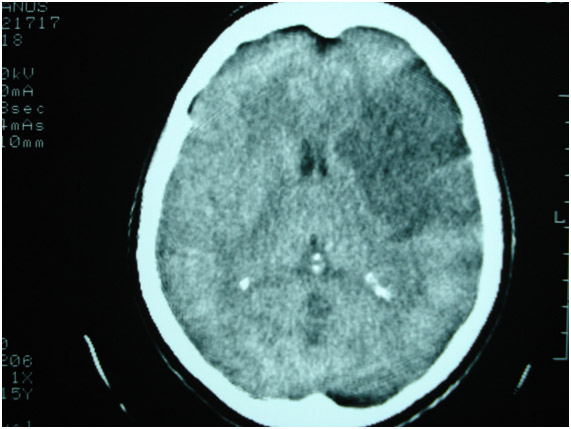
As epilepsias sintomáticas são causadas por lesões corticais adquiridas em qualquer momento da vida, como a esclerose de hipocampo, afecções congênitas ou malformativas, doenças infecciosas e parasitárias do sistema nervoso central (SNC), lesões vasculares (Figura 2), traumáticas, degenerativas ou neoplásicas. A crise epiléptica pode ser, algumas vezes, a primeira manifestação clínica de uma lesão cerebral antiga, muitas vezes assintomática até o momento.
Figura 2: Tomografia de crânio mostrando infarto cerebral em região frontal esquerda.
As síndromes epilépticas criptogênicas, também chamadas de possivelmente sintomáticas, são aquelas em que se acredita existir lesão estrutural, porém esta não pode ser demonstrada pelos métodos atuais de neuroimagem.
Encefalopatias epilépticas ocorrem geralmente em crianças, são caracterizadas por crises refratárias ao tratamento medicamentoso associadas à involução do desenvolvimento neuropsicomotor. Iniciam-se habitualmente até os 5 anos de idade, levando a comprometimento cognitivo. Os principais exemplos são a síndrome de West e a síndrome de Lennox-Gastaut.
As síndromes epilépticas e etiologias mais comuns de epilepsia são apresentadas na Tabela 1.
Tabela 1: Síndromes epilépticas e etiologias
|
Focais |
Idiopáticas |
Epilepsia focal benigna da infância com descargas centro-temporais (rolândica) |
|
Epilepsia da infância com paroxismos occipitais (inícios precoce – Panayatopoulos – e tardio – Gastaut) | ||
|
Sintomáticas |
Esclerose mesial temporal | |
|
Lesões seqüelares: origem vascular (acidente vascular cerebral isquêmico ou hemorrágico), infecciosa (encefalite pelo herpes vírus – Figura 3; cisticercose – Figura 4; abcessos – Figura 5; entre outras) | ||
|
Neoplasias primárias: associadas a tumores de baixo grau de malignidade (ganglioglioma, gangliocitoma, tumores disembrioplásticos primitivos – DNETs, astrocitomas de baixo grau, oligodendrogliomas), astrocitomas malignos, neoplasias metastáticas para o SNC (Figura 6) | ||
|
Distúrbios do desenvolvimento cortical (displasia cortical focal, hemimegalencefalia, heterotopias nodulares periventriculares, polimicrogiria, esquizencefalia, esclerose tuberosa) | ||
|
Pós-traumatismo cranioencefálico | ||
|
Processos degenerativos (por exemplo, demência de Alzheimer) | ||
|
Malformações vasculares (angiomas cavernosos, malformações artério-venosas) | ||
|
Encefalite de Rasmussen | ||
|
Síndrome de Sturge-Weber (angiomatose encéfalo-trigeminal) | ||
|
Possivelmente sintomáticas (criptogênicas) |
Por localização (temporal, frontal, occipital, parietal) | |
|
Idiopáticas |
Epilepsia mioclônica benigna da infância | |
|
Epilepsia ausência da infância | ||
|
Epilepsia com crises generalizadas de início na adolescência (epilepsia mioclônica juvenil, epilepsia ausência da juventude, epilepsia com crises tônico-clônico generalizadas do despertar) | ||
|
Epilepsia generalizada com crises febris plus (GEFS+) | ||
|
Sintomáticas ou criptogênicas |
Síndrome de West (espasmos epilépticos ou infantis) | |
|
Síndrome de Lennox-Gastaut | ||
|
Epilepsia com crises mioclônico-astáticas (síndrome de Doose) | ||
|
Epilepsia com ausências mioclônicas | ||
|
Distúrbios do desenvolvimento cortical (complexo paquigiria-lissencefalia-heterotopia subcortical em banda) | ||
|
Epilepsia com crises neonatais | ||
|
Síndrome de Dravet (epilepsia mioclônica severa da infância) | ||
|
Epilepsia com estado de mal eletrográfico durante o sono de ondas lentas | ||
|
Afasia epiléptica adquirida (síndrome de Landau-Kleffner) | ||
|
Trauma (recente ou remoto) | ||
|
Hemorragia intracraniana (subdural, epidural, subaracnóidea, intraparenquimatosa) | ||
|
Anormalidades estruturais do SNC (aneurisma, má-formação arteriovenosa, tumores primários ou metastáticos, doenças degenerativas ou doenças congênitas) | ||
|
Infecções (meningite, encefalite, abscesso cerebral, cisticercose) | ||
|
Hiperglicemia ou hipoglicemia | ||
|
Hiponatremia ou hipernatremia | ||
|
Uremia | ||
|
Insuficiência hepática | ||
|
Hipocalcemia | ||
|
Hipomagnesemia | ||
|
Abstinência alcoólica, a sedativos, a barbitúricos | ||
|
Intoxicação exógena (anfetaminas, cocaína, teofilina, antidepressivos tricíclicos, lidocaína, lítio, isoniazida, anticonvulsivantes etc.) | ||
|
Encefalopatia hipertensiva | ||
|
Isquemia grave do SNC (hipoxemia grave, parada cardiorrespiratória) | ||
|
Generalizadas | ||
|
Síndromes epilépticas indeterminadas se focais ou generalizadas | ||
|
Crises sintomáticas agudas | ||
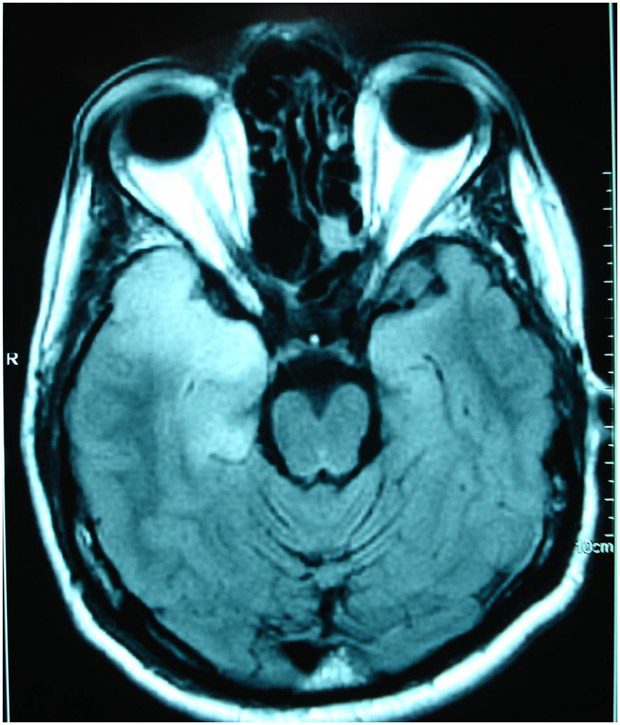
Figura 3: Ressonância magnética de encéfalo mostrando hipersinal em FLAIR em região temporal direita em paciente com meningoencefalite herpética.
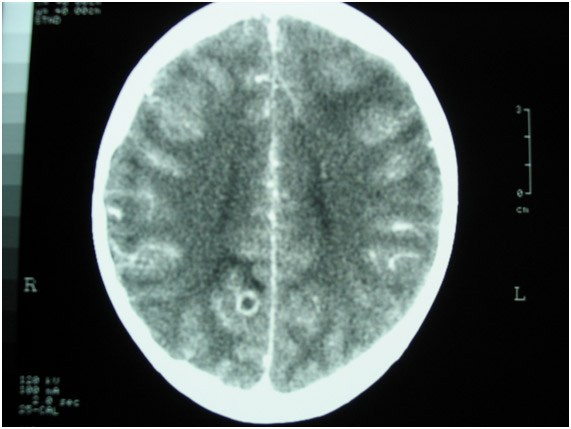
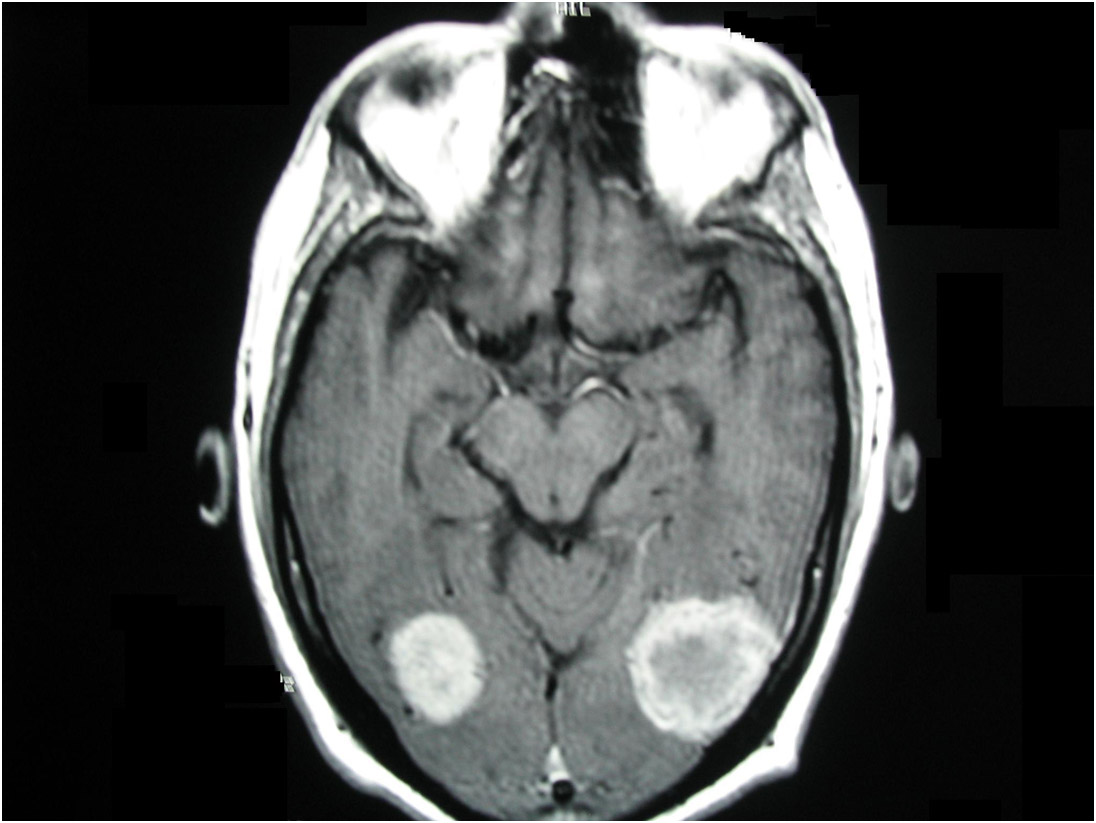
Figura 4: Tomografia de crânio mostrando lesão em região occipital direita com captação de contraste em paciente com neurocisticercose.
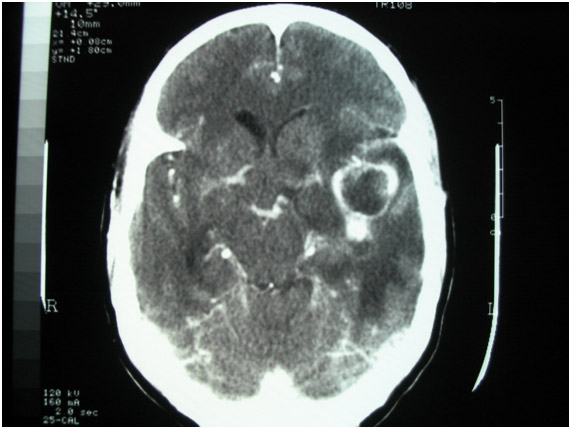
Figura 5: Tomografia de crânio mostrando lesão em região temporal esquerda em paciente com abscesso cerebral.
Figura 6: Ressonância magnética de encéfalo mostrando lesões com hipersinal em T1 após injeção de gadolínio em região têmporo-occipital bilateral em paciente com metástases de melanoma.
Os mecanismos fisiopatológicos das crises epilépticas permanecem pouco conhecidos.
Em alguns modelos, como nas crises de ausência, parece haver disfunção de circuitos tálamo-corticais, com ativação cortical difusa semelhante ao sono não-REM durante a vigília, levando a padrão eletroencefalográfico típico (espícula-onda generalizado a 3 Hz).
Alterações estruturais, com variantes protéicas disfuncionais de subunidades de canais iônicos ou de receptores de neurotransmissores localizados na membrana celular, de origem genética, têm sido reconhecidas como causa de epilepsia idiopática e de outros distúrbios neurológicos paroxísticos (ataxia episódica, enxaqueca hemiplégica familiar, paralisias periódicas) e cardiológicos (síndrome do QT longo). Já foram identificadas canalopatias (em canais de sódio e potássio), além de mutações na codificação de subunidades de receptores GABAérgicos em poucas famílias com fenótipos de epilepsias generalizadas idiopáticas. Esses defeitos determinam anormalidades na excitabilidade elétrica de membranas neuronais, favorecendo despolarização e hiperexcitabilidade neuronal, levando ao aparecimento de crises.
Dentre as crises focais, o modelo mais bem estudado é o da esclerose mesial temporal (EMT), que é a etiologia mais freqüente de crises epilépticas refratárias ao tratamento clínico em adultos. Na EMT observam-se alterações anatômicas em estruturas temporais mesiais, em especial do hipocampo, levando a perda neuronal seletiva, brotamento de axônios de fibras musgosas, reorganização estrutural com formação de circuitos aberrantes (perda de circuitos inibitórios e formação de circuitos excitatórios) e neurogênese. Na epilepsia associada à esclerose de hipocampo (Figuras 7 e 8), freqüentemente se observa a ocorrência de um insulto precipitante inicial, como crises febris complexas na infância (geralmente antes dos 2 anos de idade).
Outras causas de crises focais são as malformações corticais em que ocorre alteração da migração neuronal, proliferação e organização cortical, levando a distúrbios do desenvolvimento corticais, como displasias corticais focais, heterotopias e lisencefalia. A epileptogênese nesses distúrbios ainda não é clara, porém é reconhecido que neurônios aberrantes e heterotópicos possuem epileptogenicidade intrínseca.
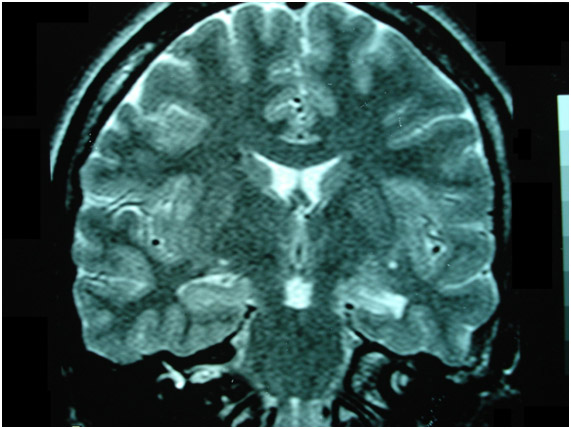
Figura 7: Ressonância magnética de encéfalo mostrando esclerose de hipocampo à esquerda, a causa mais freqüente de epilepsia focal de difícil controle.
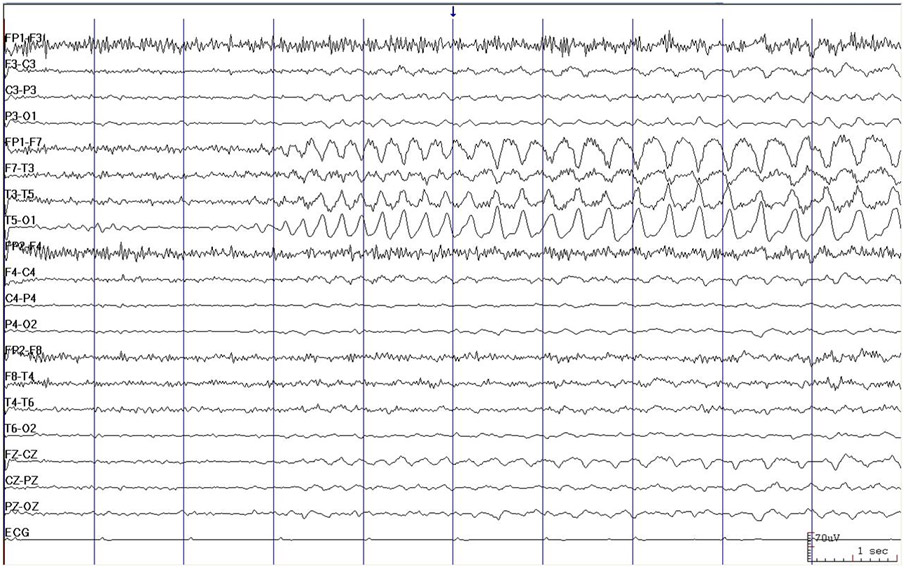
Figura 8: EEG mostrando atividade epileptiforme ritmada na região temporal esquerda, característica de crise de lobo temporal em paciente com esclerose de hipocampo.
Deve-se detalhar o evento, caracterizando os sintomas iniciais e a seqüência em que estes ocorrem, o contexto em que a crise ocorreu (sono, vigília, relação com o despertar, desencadeantes), comprometimento do nível de consciência, fenomenologia clínica, duração, alterações pós-ictais. É importante a presença de acompanhante que tenha presenciado a(s) crise(s) para descrevê-la(s), pois a maioria dos pacientes tem comprometimento parcial ou completo da consciência, impossibilitando descrição pormenorizada dos eventos. A ocorrência de crises exclusivamente durante o sono sugere crises de início focal, mesmo em crises TCG. O desencadeamento de crises pela hiperpnéia e pelo estímulo luminoso é característico de crises de ausência e mioclonias, respectivamente.
Crises TCG ao despertar, freqüentemente associadas a mioclonias e desencadeadas por privação de sono e uso de álcool, por vezes associadas a crises de ausência, são características da epilepsia mioclônica juvenil, que ocorre no contexto das epilepsias generalizadas idiopáticas de início na adolescência.
Nas crises focais, os sintomas iniciais indicam a região cortical inicialmente acometida pelas descargas e são fundamentais para a localização. Muitos pacientes não consideram esses sintomas como crises (referindo-os como auras ou “ameaços”) e tendem a não os valorizar pela pouca magnitude quando comparados a crises parciais complexas (CPC) ou TCG. Deve-se questionar ativamente a presença de sensação visceral ascendente, freqüentemente descrita como sensação de opressão iniciada em abdome que ascende até o tórax e por vezes até o pescoço, muito característica de crises originadas na porção mesial do lobo temporal. Taquicardia, sensação de medo e sintomas experienciais ou dismnésicos (déjà vu, jamais vu, despersonalização ou desrealização) são também comuns nesse tipo de crise. Podem ocorrer alterações sensitivas como parestesias, alterações visuais, olfatórias, auditivas e gustatórias. Os pacientes com CPC de lobo temporal descrevem inicialmente suas crises como “ausências” pelo fato de não se lembrarem dela, porém quando questionado o acompanhante que as presenciou é comum a descrição de olhar vago, seguido de automatismos manuais, mastigatórios, postura distônica e manipulação de órgãos sexuais.
Nas crises focais motoras pode ocorrer um fenômeno conhecido como marcha jacksoniana, em que os movimentos clônicos iniciam-se na mão, e, de forma progressiva, acometem o antebraço, braço e face e podem evoluir com perda de consciência e crise tônico-clônico generalizada. Alterações motoras como abalos musculares repetitivos (clonias), posturas distônicas e tônicas assimétricas também devem ser caracterizados.
Em pacientes com perda de consciência deve-se questionar sobre queda ao solo e trauma associado, cor e temperatura da pele, sialorréia, cianose, liberação de esfíncteres vesical e anal. Normalmente, as crises têm curta duração (segundos a minutos), seguidas de período de confusão pós-ictal de duração variável, que pode chegar a vários minutos. A duração das crises é comumente superestimada pelos acompanhantes, que incluem o período total de duração dos eventos.
A anamnese cuidadosa geralmente permite diferenciação entre crises focais e primariamente generalizadas. Nas primeiras, geralmente é possível caracterizar o início da crise pela disfunção cortical em determinada região, localizando a área de início da crise. Outros dados como idade de início, história familiar, tipo de crises, história natural e resposta a medicamentos, antecedentes, aliados a exames complementares, são fundamentais para o diagnóstico sindrômico e etiológico da epilepsia.
O primeiro passo é a avaliação clínica completa, com atenção ao aparelho cardiovascular (ausculta cardíaca, pressão arterial e freqüência cardíaca, palpação de pulsos e ausculta das carótidas), pois pode ser útil no diagnóstico etiológico (arritmia cardíaca predispondo a embolia cerebral e acidente vascular cerebral) e diferencial (síncope devido à hipotensão). Alterações cutâneas também são importantes em alguns casos, como a presença de adenomas sebáceos na face (esclerose tuberosa) e hemangioma facial em território trigeminal (síndrome de Sturge-Weber).
O exame neurológico completo deve ser realizado em todos os casos. A presença de déficits neurológicos focais pode sugerir a lesão responsável pelas crises. O retardo mental é um dos achados mais freqüentes e deve ser valorizado, pois se associa a maior risco de recorrência de crises.
Queixas de memória são comuns em pacientes epilépticos e podem estar relacionadas a descontrole das crises, efeito das medicações, alterações psiquiátricas associadas e alteração estrutural. O exame cognitivo adequado pode evidenciar esses achados.
A investigação de uma primeira crise difere daquela para pacientes com epilepsia. Na primeira crise, normalmente abordada na emergência, devem-se excluir causas de crises sintomáticas agudas, conforme a suspeita clínica e pode incluir exames laboratoriais (hemograma, gasometria arterial, uréia, creatinina, sódio, potássio, cálcio, glicemia etc.), eletrocardiograma (principalmente quando se suspeita de síncope de origem cardíaca, até mesmo porque, nesses casos, podem ocorrer abalos ou até mesmo uma crise epiléptica – síncope convulsiva devido ao baixo fluxo cerebral), avaliação toxicológica, exames de neuroimagem (tomografia de crânio [TC] e/ou ressonância magnética [RM] conforme a disponibilidade, urgência e cooperação do paciente) e coleta de líquor (especialmente na suspeita de meningite/encefalite) (Tabela 2).
Tabela 2: Crise epiléptica, situação clínica e exames complementares
|
Situação clínica |
Exames complementares |
|
Assim que chega à emergência ou pronto-atendimento |
Glicemia capilar imediata (dextro) |
|
Epiléptico, em uso de anticonvulsivante e que parou de tomar por conta própria a medicação há poucos dias e não há nada novo |
Em geral, não há necessidade; prescrever a medicação de que o paciente faz uso |
|
Epiléptico, em uso regular da medicação, mas com novas crises |
Incluir dosagem sérica do anticonvulsivante em uso |
|
Uma ou mais crises, primeira crise |
Avaliação de causas clínicas: hemograma, plaquetas, exames de coagulação, função renal, hepática, glicemia, sódio, potássio, cálcio, magnésio e gasometria arterial |
|
TC ou RM |
Crise epiléptica sem causa aparente pelos exames iniciais |
|
Suspeita de doenças do SNC | |
|
RM é muito melhor que TC | |
|
TC é mais disponível na urgência para descartar doenças neurológicas (por exemplo, tumor, hemorragia etc.) | |
|
Análise liquórica |
Na ausência de contra-indicação, após TC, em casos de febre, confusão persistente, suspeita de meningite, meningoencefalite, carcinomatose meníngea etc. |
|
Eletroencefalograma |
Na emergência: em casos de estado de mal epiléptico ou na suspeita de estado de mal (confusão persistente, após intubação e bloqueio neuromuscular etc.) |
|
Na epilepsia: em geral, deve-se realizar em todos os casos |
Nos pacientes com epilepsia, a investigação pode incluir exames séricos, EEG, vídeo-EEG, exames de neuroimagem, exames funcionais e de metabolismo neuronal.
É elemento imprescindível para o diagnóstico. O EEG deve ser realizado em vigília e sono, com métodos de sensibilização como fotoestimulação intermitente e hiperpnéia. As alterações neste encontradas podem ser de natureza epileptiforme (focais ou generalizadas) ou inespecíficas (alentecimento focal – indicando disfunção cortical focal, não necessariamente de natureza epileptiforme – ou difuso da atividade elétrica cerebral, um achado inespecífico que indica disfunção cortical difusa).
A ausência de anormalidades epileptiformes ao EEG não exclui epilepsia, pois as descargas intercríticas podem ou não ocorrer no período em que o exame foi realizado. As anormalidades ao EEG devem ser interpretadas no contexto clínico apropriado (ocorrência ou não de crise), pois mesmo indivíduos normais podem apresentar alterações (epileptiformes ou não) ao EEG. Em pacientes com suspeita de crises e EEG normal, este deve ser repetido, em condições técnicas adequadas, pois a realização de até três exames aumenta a sensibilidade do método em detectar anormalidades. O EEG é imprescindível para classificação do tipo de crise, classificação sindrômica, além de contribuir para decisões terapêuticas.
São necessários na maioria dos pacientes, excetuando aqueles com história sugestiva de epilepsia idiopática, exame neurológico normal e achados de EEG compatível com o diagnóstico.
A TC de crânio é limitada na avaliação, prestando-se apenas a casos de urgência para afastar tumores, hemorragia etc., em locais onde outros métodos não estejam disponíveis ou exista contra-indicação para realização de RM, como próteses metálicas, marca-passo e clipes metálicos. A injeção de contraste é feita em casos selecionados.
A RM de crânio é o exame de escolha na investigação de epilepsia, método mais adequado para o diagnóstico de EMT, distúrbios do desenvolvimento cortical, tumores de crescimento lento, e pequenas malformações vasculares como angiomas cavernos etc. O emprego de técnicas de aquisição de imagem direcionadas à investigação da epilepsia aumenta a sensibilidade em se detectar lesões nesses casos. Nos casos de epilepsia, é obrigatório o estudo de estruturas temporais, com cortes finos, perpendiculares ao eixo maior do hipocampo, incluindo a técnica de FLAIR (inversão da recuperação com atenuação de fluidos). O emprego de técnicas de reconstrução volumétrica, com alto contraste entre substância branca e cinzenta (IR-volume), aumenta a sensibilidade de detecção de distúrbios do desenvolvimento cortical. Técnicas de gradiente aumentam a sensibilidade em detectar lesões calcificadas ou lesões hemorrágicas, com depósito de hemossiderina.
São estudos de imagem funcional, que avaliam o padrão de perfusão tecidual, empregados para auxiliar a localização dos focos epilépticos em casos de epilepsia de difícil controle medicamentoso. Estudos ictais baseiam-se no fato de que crises focais associam-se a hiperfluxo na região de início da crise. A injeção do rádio-isótopo, administrada preferencialmente durante registro por vídeo-EEG, deve ser realizada tão próxima do início da crise quanto possível. Exames inter-ictais apresentam baixa sensibilidade, podendo demonstrar hipoperfusão regional em 30% a 50% dos casos de epilepsia de difícil controle medicamentoso.
O PET cerebral permite o estudo de metabolismo cerebral, com o emprego de glicose marcada com material radioativo. Pode demonstrar áreas de hipometabolismo regional em casos de epilepsia focal. É empregado em casos de epilepsia de difícil controle, no contexto da avaliação pré-cirúrgica.
Consiste na monitorização contínua do paciente por meio de registro por vídeo associado a registro eletroencefalográfico contínuo, com intuito de registrar o evento. É particularmente útil no diagnóstico diferencial de fenômenos paroxísticos, na caracterização clínico-eletrográfica de crises epilépticas, na avaliação pré-cirúrgica da epilepsia (com eletrodos de superfície ou invasivos) e também na quantificação de crises e detecção de crises subclínicas. Crises parciais simples podem não ter alterações ao EEG ictal.
É útil para verificar adequação do tratamento, para averiguar aderência e toxicidade e para avaliar efeitos farmacocinéticos de interações medicamentosas.
Devem ser solicitados no seguimento do paciente para monitorizar possíveis efeitos colaterais das medicações utilizadas, em especial alterações de enzimas hepáticas (ácido valpróico, carbamazepina, fenitoína), hiponatremia (carbamazepina, oxcarbazepina), leucopenia (carbamazepina), plaquetopenia (valproato).
Crises epilépticas devem ser diferenciadas de outros eventos paroxísticos, com ou sem perda de consciência, de etiologia não epiléptica. Eventos não epilépticos dividem-se em fisiológicos e psicogênicos.
Eventos fisiológicos não epilépticos constituem um importante diagnóstico diferencial com epilepsia. Nos quadros sincopais, o início do evento pode ser gradual ou súbito, e, quando súbito, é mais difícil diferenciar de crise epiléptica. A presença de sintomas premonitórios pré-sincopais é bastante sugestiva de quadro sincopal, assim como a ocorrência de sensação de irregularidade dos batimentos cardíacos, sudorese profusa, palidez e náuseas. Quadros sincopais, contudo, podem apresentar-se com perda abrupta de consciência, com fenômenos motores variados, incluindo abalos clônicos sugestivos de crise (síncope convulsígena) e com ferimentos graves, o que pode tornar difícil a diferenciação das duas entidades.
Os ataques isquêmicos transitórios (AITs) geralmente são facilmente diferenciáveis de crises epilépticas, pois estes geralmente se manifestam com sintomas negativos, ao contrário das crises, que manifestam sintomas positivos. Uma situação particularmente difícil e de ocorrência rara são os episódios de tremores de membros (limb shaking episodes), que constituem manifestação não usual de isquemia cerebral, caracterizados por abalos de membros, normalmente associados a estenose crítica de carótida, com hipoperfusão cerebral.
Doenças psiquiátricas podem simular crises epilépticas. Crises não epiléticas psicogênicas podem ser confundidas com crises parciais complexas ou crises TCG. O diagnóstico pode ser difícil, sendo fundamental uma anamnese cuidadosa para levantar a suspeita diagnóstica. A monitorização por vídeo-EEG é indispensável para o diagnóstico. Crises epilépticas e crises não epilépticas psicogênicas podem coexistir em até 20% dos casos de crises não epilépticas, o que geralmente representa um desafio diagnóstico.
Outros diagnósticos diferenciais das crises epilépticas são listados na Tabela 3.
Tabela 3: Diagnósticos diferenciais das crises epilépticas
|
Consciência preservada |
Distúrbios do movimento (mioclonias não epilépticas, coréia, coreoatetose, distonia ou discinesias paroxísticas, distonia) |
|
Tiques | |
|
Distúrbios de atenção | |
|
Enxaqueca, enxaqueca basilar | |
|
Vertigem paroxística posicional benigna | |
|
Crises de pânico e de hiperventilação | |
|
Episódios isquêmicos transitórios (especialmente aqueles com fenômenos negativos – limb shaking episodes) | |
|
Amnésia global transitória | |
|
Ataxia episódica | |
|
Alterações paroxísticas na esclerose múltipla | |
|
Com perda (ou comprometimento) da consciência |
Crises não epilépticas psicogênicas |
|
Síncope e pré-síncope (vaso-vagal, reflexa, cardíaca etc.) | |
|
Quadros confusionais agudos (encefalopatias tóxico-metabólicas) | |
|
Enxaqueca basilar | |
|
Eventos durante o sono |
Mioclonias fisiológicas do sono |
|
Pesadelos | |
|
Terror noturno | |
|
Sonambulismo | |
|
Narcolepsia-cataplexia | |
|
Movimentos periódicos dos membros durante o sono | |
|
Distúrbio comportamental do sono REM |
O tratamento difere nas crises sintomáticas agudas e na epilepsia. Nas crises sintomáticas agudas, a conduta fundamental primeira é a eliminação do fator causal, o que impede a recorrência de eventos na maioria dos casos. Em lesões encefálicas agudas pode ser recomendado tratamento medicamentoso por algumas semanas (ver opções para crises focais).
O tratamento crônico com drogas antiepilépticas está indicado após a ocorrência de uma segunda crise epiléptica espontânea. O tratamento deve ser iniciado com droga única (monoterapia), escolhida de acordo com a eficácia no tipo de crise ou síndrome epiléptica, tolerabilidade e particularidades específicas do paciente (Tabelas 4 a 6). A dose da medicação deve ser ajustada lentamente, até que se obtenha o controle completo de crises. Na falha da monoterapia em dose máxima tolerada, deve ser tentada uma segunda droga em monoterapia. Nas epilepsias de difícil controle, após duas tentativas sem sucesso de monoterapia em dose máxima tolerada, pode ser tentada associação de drogas antiepilépticas, empregando benzodiazepínicos ou drogas de nova geração.
Embora os estudos comparativos entre as drogas tradicionais e novas seja ainda limitado, considera-se que as novas drogas sejam mais bem toleradas, tenham menos efeitos colaterais e tenham menor potencial de interação medicamentosa. O custo de tratamento com as novas drogas ainda é consideravelmente maior que com as drogas tradicionais.
A possibilidade de piora de crises epilépticas com drogas antiepilépticas deve ser considerada, especialmente com o uso de carbamazepina, oxcarbazepina, fenitoína, vigabatrina e gabapentina em epilepsias generalizadas.
Interações farmacocinéticas e farmacodinâmicas entre as drogas antiepilépticas e outras drogas podem influenciar a ocorrência de efeitos colaterais ou modificar a eficácia do tratamento. Particularmente importante são as interações entre as drogas antiepiléticas de primeira geração com outras drogas antiepilépticas e com drogas de metabolização pelo sistema microsomal hepático. Além da importância da interação das drogas antiepilépticas entre si, deve-se atentar para interações medicamentosas das drogas antiepilépticas com os anticoagulantes orais e com os anticoncepcionais orais. Todas as drogas antiepilépticas, com exceção de ácido valpróico, lamotrigina, gabapentina e vigabatrina, diminuem a eficácia dos anticoncepcionais orais com baixa dosagem de hormônios. Nesses casos, quando é desejada anticoncepção, é preferível o emprego de métodos de barreira, DIU ou anticoncepcionais injetáveis, com menor espaçamento de doses.
O tratamento com drogas antiepilépticas aumenta em duas a três vezes o risco de malformações fetais (maiores ou menores). O uso do valproato na gestação determina ainda maior risco de malformações, com aumento da incidência de malformações do tubo neural. Embora ainda não exista evidência científica confirmando-se a eficácia no contexto de uso de drogas antiepilépticas, recomenda-se o emprego de ácido fólico em toda a mulher em idade fértil em uso de drogas antiepilépticas.
Em mulheres epilépticas que desejem engravidar, é recomendável o melhor ajuste do tratamento antes da gestação, visando-se ao melhor controle de crises, com o menor número de drogas antiepilépticas, na menor dose eficaz. A suplementação de folato deve ser iniciada antes do início da gestação, pois o maior risco de malformações ocorre nas fases iniciais da gestação. Durante a gestação não são recomendáveis mudanças abruptas no esquema medicamentoso, pelo risco de desencadeamento de crises generalizadas durante a gestação, com risco de complicações para o feto e para a gestante.
O tratamento cirúrgico da epilepsia apresenta altos índices de sucesso, com resolução completa das crises em até 80% dos casos de epilepsia focal sintomática. Ainda é subutilizado, mesmo em países do primeiro mundo, em parte devido à exigüidade de recursos especializados, mas também por noção infundada de que o procedimento é reservado para casos desesperadores, em que tudo mais foi tentado, sem sucesso ou por temor, também infundado, de que o tratamento cirúrgico tenha alto risco de seqüelas neurológicas e cognitivas.
Em pacientes com epilepsia focal sintomática refratária ao tratamento medicamentoso (como esclerose de hipocampo unilateral, displasias corticais focais, tumores de baixo grau, lesões seqüelares focais) e em pacientes com síndromes hemisféricas, a cirurgia pode apresentar elevadas taxas de sucesso, e mínimas possibilidades de seqüela (ou previsíveis, no caso das síndromes hemisféricas), especialmente se avaliados em centros especializados no tratamento cirúrgico da epilepsia.
Em outros casos, como nas epilepsias generalizadas sintomáticas ou criptogênicas o tratamento cirúrgico é limitado. Em casos que apresentem crises de queda (tônicas ou atônicas), a calosotomia pode ser um recurso paliativo.
Tabela 4: Eficácia das drogas antiepilépticas sobre os diferentes tipos de crise
|
Droga |
TCG |
Focal |
Ausência |
Mioclonias |
|
Tradicionais | ||||
|
Carbamazepina* |
Sim |
Sim |
Pode piorar |
Pode piorar |
|
Fenitoína* |
Sim |
Sim |
Pode piorar |
Pode piorar |
|
Fenobarbital* |
Sim |
Variável |
Não |
Variável |
|
Valproato de sódio* |
Sim |
Sim |
Sim |
Sim |
|
Primidona* |
Sim |
Sim |
Não |
Variável |
|
Clonazepam |
Sim |
Sim |
Sim |
Sim |
|
Clobazam |
Sim |
Sim |
Variável |
Sim |
|
Nitrazepam |
Sim |
Sim |
Variável |
Sim |
|
Etossuximida*# |
Não |
Não |
Sim |
Variável |
|
Novas drogas | ||||
|
Oxcarbamazepina* |
Sim |
Sim |
Pode piorar |
Pode piorar |
|
Lamotrigina* |
Sim |
Sim |
Sim |
Variável |
|
Topiramato* |
Sim |
Sim |
Variável |
Sim |
|
Vigabatrina& |
Sim |
Sim |
Pode piorar |
Pode piorar |
|
Gabapentina |
Não? |
Sim |
Pode piorar |
Pode piorar |
|
Felbamato*# |
Sim |
Sim |
Sim |
Sim |
|
Levetiracetam# |
Sim |
Sim |
Pode piorar? |
Sim |
|
Zonisamida# |
Sim |
Sim |
Sim |
Sim |
|
Pregabalina# |
Sim? |
Sim |
Desconhecido |
Desconhecido |
* Aprovados para uso em monoterapia; # Disponíveis no Brasil apenas por importação; & Eficaz nos espasmos epilépticos
Tabela 5: Posologia, níveis séricos terapêuticos e potencial de interação medicamentosa para as principais drogas antiepilépticas
|
Drogas |
Doses (mg/dia) |
Posologia (tomadas diárias) |
Nível sérico (mg/dL) |
Interação medicamentosa |
|
Tradicionais | ||||
|
Carbamazepina* |
600-1.600 |
2-3** |
6-12 |
+++ |
|
Fenitoína* |
200-300 |
2-3 |
10-20 |
+++ |
|
Fenobarbital* |
100-200 |
1 |
15-40 |
+++ |
|
Valproato de sódio* |
500-2.500 |
2-3*** |
70-100 |
+++ |
|
Primidona* |
500-2.000 |
2-3 |
|
++ |
|
Clonazepam |
1-8 |
2-3 |
|
+ |
|
Clobazam |
10-30 |
1-3 |
|
+ |
|
Nitrazepam |
5-15 |
1-3 |
|
+ |
|
Etossuximida*# |
500-1.000 |
2-3 |
|
+ |
|
Novas drogas | ||||
|
Oxcarbamazepina* |
900-2.400 |
2-3 |
15-35 |
+ |
|
Lamotrigina*@ |
100-500 |
1-2 |
3-14 |
+ |
|
Topiramato* |
100-400 |
2-3 |
|
+ |
|
Gabapentina |
900-3.600 |
3-4 |
|
0 |
|
Vigabatrina& |
500-4.500 |
2-3 |
|
0 |
|
Felbamato |
1.200-3.600 |
2-3 |
|
+ |
|
Levetiracetam# |
1.000-3.000 |
2-3 |
|
0 |
* Aprovados para uso em monoterapia; # Disponíveis no Brasil apenas por importação; ** Duas vezes ao dia na formulação de ação prolongada; *** Uma vez ao dia na formulação de ação prolongada; @ Em associação com ácido valpróico, usar 50-200; em associação com carbamazepina, fenitoína, fenobarbital e primidona, usar 200-600; & Eficaz nos espasmos epilépticos
Tabela 6: Principais efeitos colaterais das drogas antiepilépticas
|
Droga |
Efeitos adversos dose dependentes |
Efeitos adversos idiossincráticos |
Efeitos adversos graves |
|
Tradicionais | |||
|
Carbamazepina* |
Ataxia, diplopia, tontura |
Intolerância gastrointestinal, hiponatremia, hepatotoxicidade |
Erupção cutânea, supressão de medula óssea |
|
Fenitoína* |
Ataxia |
Hiperplasia gengival, embrutecimento facial, osteomalácia |
Erupção cutânea |
|
Fenobarbital* |
Sonolência, lentificação |
Hiperatividade (crianças) |
Erupção cutânea |
|
Ácido valpróico* |
Intolerância gastrointestinal, tremor |
Hepatotoxicidade, ganho de peso, anovulação, plaquetopenia, queda de cabelo |
Encefalopatia, hiperamonemia, pancreatite, malformações fetais |
|
Primidona* |
Sonolência, lentificação |
Hiperatividade (crianças) |
Erupção cutânea |
|
Clonazepam |
Sonolência, lentificação |
Hipersecreção de vias aéreas (crianças) |
|
|
Clobazam |
Sonolência, lentificação |
|
|
|
Nitrazepam |
Sonolência, lentificação |
|
|
|
Etossuximida*# |
Sintomas gastrointestinais, soluços |
Hepatotoxicidade |
Supressão de medula óssea |
|
Novas drogas | |||
|
Oxcarbamazepina* |
Ataxia, diplopia |
Hiponatremia |
|
|
Lamotrigina* |
Ataxia, diplopia |
Insônia, irritabilidade |
Erupção cutânea |
|
Topiramato* |
Comprometimento cognitivo |
Anorexia, nefrolitíase, afasia |
Glaucoma agudo, encefalopatia |
|
Vibagatrina& |
|
Psicose |
Retinopatia |
|
Gabapentina |
Sonolência |
Hiperatividade (crianças), ganho de peso, edema de tornozelo |
|
* Aprovados para uso em monoterapia; # Disponíveis no Brasil apenas por importação; & Eficaz nos espasmos epilépticos
A prioridade inicial é buscar hipoglicemia e realizar as manobras de suporte avançado de vida (garantir vias aéreas, ventilação, circulação etc.).
A conduta farmacológica com medicação antiepiléptica é reservada para casos selecionados, uma vez que a grande maioria das crises é autolimitada. Nesse caso, quando indicado, o objetivo do uso de agentes antiepilépticos é cessar uma crise prolongada e prevenir novas crises. Seu uso deve ser criterioso, norteado pelo cenário clínico em que a crise ocorre.
Em pacientes epilépticos que chegam à unidade de emergência com persistência ou nova crise, deve ser investigada, inicialmente, aderência ao tratamento ou mudanças recentes no esquema medicamentoso, além de associação de outras medicações que podem reduzir a eficácia das drogas antiepilépticas, ocorrência de problemas clínicos agudos, com alterações metabólicas, infecciosas ou febre, além de traumatismo cranioencefálico, que podem atuar como fatores desencadeantes de crises. Na ausência de fatores desencadeantes agudos, o manejo do tratamento deve ser feito, preferencialmente, pelo médico responsável pelo acompanhamento ambulatorial do paciente. Nos pacientes epilépticos com etiologia já investigada, pode-se, criteriosamente, prescindir de novo exame de imagem. A TC pode ainda ser necessária para excluir lesões secundárias à crise, como traumatismo craniano. Exame do líquido céfalo-raquidiano (LCR) deve ser realizado quando há suspeita de meningite ou encefalite.
Agentes antiepilépticos são, em geral, pouco eficazes no controle de crises epilépticas agudas sintomáticas decorrentes de distúrbios metabólicos. Nesses casos, o melhor tratamento é a correção da causa. Assim, geralmente não se inicia tratamento com agentes antiepilépticos nessa situação.
Não devem ser administrados benzodiazepínicos se a crise já tiver cessado e o doente estiver no período pós-ictal. Nesse contexto, o emprego de benzodiazepínicos não tem indicação e pode acentuar a depressão do SNC, prolongando o período de recuperação do nível de consciência ou acentuando o quadro confusional. Além disso, os benzodiazepínicos têm duração de efeito curta (não mais que 30 minutos para o diazepam), não sendo agentes eficazes para a prevenção de recorrência de crises. Esses agentes devem ser reservados para casos em que se caracterize estado de mal epiléptico, em crises com duração superior a 5 minutos.
No caso de crises agudas sintomáticas secundárias a lesões neurológicas agudas, habitualmente se empregam agentes antiepilépticos na prevenção de recorrência de crises, embora sua eficácia possa ser limitada nesse contexto.
O agente mais empregado é a fenitoína, por não ser sedativa e ser passível de administração endovenosa em dose de ataque, permitindo rápido início de ação (15 a 20 mg/kg de peso, EV, diluído em soro fisiológico, com monitorização cardíaca, taxa máxima de 50 mg/min). Recomenda-se a manutenção da medicação antiepiléptica durante toda a fase aguda e habitualmente se procede sua retirada a partir da 12ª. semana.
A maior parte dos doentes com crise única na emergência não apresentará recorrência de crises. Logo, a introdução de agentes antiepilépticos não está indicada na maioria dos casos.
Sua utilização em doente com crise isolada é restrita àqueles casos em que há alto risco de recorrência. Para uma conduta adequada, portanto, deve ser feita uma estratificação do risco de recorrência das crises. Para isso são necessários os resultados de dois exames:
Neuroimagem (RM de crânio);
EEG.
Caso ambos os exames sejam normais, o risco de recorrência de crises é menor que 30%, sendo maior nos primeiros meses após a crise inicial, declinando progressivamente. Quando ambos os exames são anormais, o risco de recorrência chega a 70%.
A decisão de se iniciar tratamento crônico com medicação antiepiléptica deve ser discutida com o doente, ponderando o risco de recorrência e o impacto de uma nova crise na vida do doente.
Como dito anteriormente, doentes com crise única de etiologia não esclarecida idealmente não devem receber alta hospitalar até que se tenham dados completos de investigação que forneçam subsídios para decidir sobre a introdução ou não de medicação antiepiléptica.
Independentemente da etiologia da crise epiléptica, o doente pode apresentar-se na emergência em situação que caracterize o chamado estado de mal epiléptico (Tabela 7).
O conceito de estado de mal epiléptico está intrinsecamente ligado ao fato de crises prolongadas potencialmente causarem dano ao SNC. Estudos experimentais e clínicos demonstram que crises não controladas predispõem o cérebro a crises de mais difícil controle. O tratamento precoce, portanto, se justifica. Uma vez que a maior parte das crises epilépticas cessa espontaneamente em poucos minutos, alguns autores recomendam operacionalmente que condutas para estado de mal epiléptico sejam adotadas após 5 minutos contínuos de crise ou a ocorrência de duas ou mais crises sem que seja recuperada a consciência entre os ataques.
Essa situação caracteriza emergência médica que requer tratamento imediato e adequado. Segundo alguns estudos, sua mortalidade pode chegar a 20%, não sendo justificável nenhum atraso no tratamento. Portanto, qualquer crise que dure 5 minutos ou mais deve ser tratada agressivamente.
Crises não epilépticas psicogênicas podem ser diagnosticadas erroneamente como estado de mal epiléptico. Em alguns estudos, de 20% a 40% dos doentes com suposto estado de mal epiléptico apresentavam na realidade crises não epilépticas de origem psicogênica. Esse diagnóstico deve ser reconhecido prontamente para tratamento adequado.
Em doentes com história prévia de epilepsia, algumas etiologias são mais freqüentes e devem ser investigadas, como a suspensão ou retirada abrupta de agentes, mudança no esquema medicamentoso, especialmente de benzodiazepínicos e barbitúricos (por vezes iatrogênica) e traumatismo cranioencefálico. Doentes epilépticos têm maior risco de desenvolver traumatismo craniano devido a crises.
O protocolo de tratamento farmacológico no estado de mal epiléptico varia de serviço para serviço, baseado na experiência dos médicos assistentes, assim como na disponibilidade de agentes para serem utilizados.
A conduta medicamentosa inclui:
infusão endovenosa de benzodiazepínicos. O lorazepam (0,1 a 0,15 mg/kg de peso, EV, em 1 a 2 minutos) é a droga de escolha pra abortar a crise. A dose pode ser repetida após 5 minutos. O diazepam é uma alternativa (dose de 5 a 10 mg, EV, em 1 a 2 minutos). As desvantagens do diazepam incluem a curta duração do efeito (apenas cerca de 30 minutos), em razão de sua alta lipossolubilidade, com conseqüente recirculação, baixa ligação aos receptores de benzodiazepínicos no SNC e menor eficácia que o lorazepam;
utilização de benzodiazepínicos, que, no estado de mal epiléptico, deve ser seguida de administração de agentes antiepilépticos com duração de ação mais prolongada no SNC, como fosfenitoína (20 mg/kg, EV, com taxa máxima de 150 mg/min), não disponível facilmente no Brasil, ou fenitoína endovenosa. A dose da fenitoína é de 15 a 20 mg/kg de peso, devendo ser diluída em 250 a 500 mL de soro fisiológico (não pode ser diluída em soro glicosado) e infundida à velocidade máxima de 50 mg/min. O doente deve ser observado durante a infusão, de preferência com monitorização eletrocardiográfica. Podem ocorrer hipotensão e arritmias durante a infusão;
administração de dose adicional de 7 a 10 mg/kg, caso as crises epilépticas persistam e não ocorra controle completo dessas crises (no caso da fosfenitoína, a dose adicional também é de 7 a 10 mg/kg). A maior vantagem da fosfenitoína é a possibilidade de administração com velocidade 3 vezes maior que a fenitoína;
prescrição de fenobarbital, caso o paciente ainda tenha crises epilépticas (clinicamente ou pela monitorização de EEG). É importante lembrar que se o doente em questão encontra-se em abstinência de fenobarbital (ele é epiléptico, usa fenobarbital, mas parou nos últimos dias por conta própria), esse é o agente de escolha (antes da fenitoína). A dose é de 10 a 20 mg/kg de peso, intravenosa, a uma velocidade de 50 a 75 mg/min. Recomenda-se iniciar 10 mg/kg e repetir se necessário. O fenobarbital tem a vantagem de apresentar meia-vida longa, porém pode deprimir intensamente o nível de consciência, sendo por vezes necessário suporte ventilatório;
administração de valproato endovenoso (uma excelente alternativa antes do fenobarbital), na dose de 25 mg/kg de peso, embora seja raramente disponível no Brasil;
intubação orotraqueal (se já não realizada pelo nível de consciência), caso o paciente ainda persista em estado de mal epiléptico após o uso de benzodiazepínico, fenitoína (ou fosfenitoína), valproato e/ou fenobarbital. Deve-se colocar o paciente em um ventilador mecânico e imediatamente prescrever um dos três agentes: midazolam, propofol ou pentobarbital. É imprescindível a monitorização por EEG (preferencialmente de forma contínua), visando-se especialmente à identificação do estado de mal subclínico (ou ocorrência de crises sutis), orientando-se assim o ajuste de agentes. Ainda não existem estudos comparativos da eficácia relativa desses agentes no estado de mal epiléptico;
midazolam: bastante usado e disponível, porém a ocorrência de taquifilaxia pode ser problemática, requerendo doses progressivamente maiores para obter o mesmo efeito terapêutico. A dose inicial é de 0,2 mg/kg, IV, lentamente e manutenção de de 1 a 10 µg/kg/min;
propofol: anestésico geral, de ação curta, de alto custo e pode causar hipotensão. A dose inicial é de 1 a 2 mg/kg, IV, seguida de manutenção de 1 a 15 mg/kg/h;
pentobarbital: disponível, de baixo custo, muito longa ação (vários dias); pode causar hipotensão, por vezes requerendo o emprego de agentes vasoativos. A dose é de 10 a 15 mg/kg, IV, durante uma hora, com manutenção de 0,5 a 1,0 mg/kg/h.
É importante que sejam introduzidos, além de agentes para combater o estado de mal, agentes antiepilépticos para o tratamento crônico, antes de se proceder ao desmame dos agentes empregados no tratamento do estado de mal.
Tabela 7: Causas mais freqüentes de estado de mal epiléptico
|
Exacerbação de crises em doente epiléptico: deve-se suspeitar de uso irregular de medicação, suspensão abrupta ou troca intempestiva de medicação, por vezes iatrogênica |
|
Traumatismo cranioencefálico |
|
Processos infecciosos do SNC, como meningites e encefalites (principalmente a meningoencefalite herpética) |
|
Lesões agudas vasculares do SNC (AVCH, hemorragia subaracnóidea) |
|
Neoplasias do SNC |
|
Abstinência de drogas sedativas do SNC |
|
Intoxicação exógena (álcool, cocaína, anfetaminas, lítio, teofilina, antidepressivos tricíclicos, isoniazida, antiepilépticos) |
A epilepsia é caracterizada por ocorrência de crises espontâneas recorrentes (duas ou mais).
Crise epiléptica é a expressão clínica decorrente de descarga anormal, excessiva e sustentada do tecido cerebral.
Crise aguda sintomática (ou crise provocada) é decorrente de uma causa imediata identificada, como distúrbio metabólico-eletrolítico, intoxicação exógena, abstinência de drogas sedativas ou insulto neurológico agudo.
Crise única (isolada): uma ou mais crises que recorrem no período de 24 horas. Pode corresponder a uma crise aguda sintomática ou a primeira manifestação de epilepsia.
A epilepsia é uma das doenças neurológicas mais freqüentes; pode se manifestar em qualquer fase da vida, com dois picos de incidência, no primeiro ano de vida e após a sétima década.
Na suspeita de uma crise epiléptica, deve-se detalhar o evento, caracterizando os sintomas iniciais, a seqüência em que eles ocorreram, o contexto em que a crise ocorreu, comprometimento do nível de consciência, fenomenologia clínica, duração e alterações pós-ictais.
A anamnese cuidadosa geralmente permite diferenciação entre crises focais e primariamente generalizadas.
Atenção especial deve ser dada ao primeiro episódio, pois a crise pode representar apenas uma manifestação de um insulto clínico ou neurológico agudo. Tratar apenas a crise e ignorar as outras possibilidades seria por analogia prescrever antitérmico e antitussígeno para todos pacientes com tais sintomas, ignorando a possibilidade de diagnósticos de maior gravidade e com implicações terapêuticas como uma pneumonia bacteriana.
O primeiro passo é a avaliação clínica completa, que inclui a atenção a vias aéreas, respiração, sistema cardiovascular e realização de glicemia capilar (dextro).
A crise epiléptica pode indicar um problema clínico ou neurológico subjacente e se manifestar com sinais e sintomas da doença de base, como:
- quadro de febre, rigidez de nuca, confusão e convulsões: podem indicar uma meningite com vasculite, encefalite herpética, meningoencefalite tuberculosa ou fúngica etc.;
- história de traumatismo craniano (hemorragia do SNC);
- distúrbios metabólicos: hipoglicemia, hiperglicemia, distúrbios eletrolíticos, hipóxia etc.
- história de tentativa de suicídio com ingestão de tóxicos: pode indicar antidepressivos, isoniazida, lítio, teofilina, anticolinérgicos, organofosforados etc.
- história de doença ou lesão neurológica prévia: acidente vascular cerebral isquêmico (AVCI) ou hemorrágico (AVCH), neurocisticercose, neurocirurgia prévia etc.
- lesões neurológicas agudas concomitantes com a crise epiléptica: trauma cranioencefálico, hemorragia subaracnóidea, hemorragia intraparenquimatosa, metástases tumorais para o SNC, tumores primários do SNC etc.
- história de etilismo importante, crônico, com redução ou ausência da ingesta de álcool nas últimas horas, sugerindo abstinência alcoólica.
A investigação de uma primeira crise difere daquela para pacientes com epilepsia. Na primeira crise, normalmente abordada na emergência, devem-se excluir causas de crises sintomáticas agudas (distúrbios eletrolíticos, ácido-básicos, uremia, hipo ou hiperglicemia, lesões do SNC, encefalite etc.).
Crises epiléticas são manifestações comuns tanto no pronto-socorro quanto no ambulatório. A abordagem sistematizada permite controle completo das crises na maioria dos pacientes. A abordagem da primeira crise no setor de emergência deve enfatizar a conduta diagnóstica, visando-se a identificar doenças neurológicas agudas e excluir causas potencialmente fatais, como síncope cardíaca. No contexto ambulatorial, a abordagem é direcionada a confirmar o diagnóstico e instituir tratamento adequado e individualizado. A referência para especialistas é recomendada sempre que possível, em especial nos casos em que houver dúvida e maior dificuldade no controle das crises.
O EEG é o exame mais importante na abordagem de pacientes com epilepsia.
A RM de crânio é o exame de escolha na investigação de epilepsia.
A TC de crânio tem menor valor que a RM, podendo ser útil para descartar doenças neurológicas na emergência.
A monitorização por vídeo-EEG está indicada em pacientes em que exista dúvida diagnóstica, para caracterização do tipo de crise, avaliação da terapêutica, pré-cirúrgica e em casos de epilepsia refratária ao tratamento medicamentoso.
Crises epilépticas devem ser diferenciadas de outros eventos paroxísticos, com ou sem perda de consciência, de etiologia não epiléptica.
Eventos fisiológicos não epilépticos constituem um importante diagnóstico diferencial com epilepsia (por exemplo, síncope, AIT etc.).
Crises epilépticas e crises não epilépticas psicogênicas podem coexistir em até 20% dos casos de crises não epilépticas, o que geralmente representa um desafio diagnóstico.
Nas crises sintomáticas agudas, a conduta fundamental primeira é a eliminação do fator causal (investigar e tratar/remover a causa), o que impede a recorrência de eventos na maioria dos casos. Em casos especiais, tratar com droga antiepiléptica por curto período.
Crise única: investigar, avaliar risco de recorrência e tratar em situações particulares.
Objetivo do tratamento da epilepsia: controle de crises, tratamento de co-morbidades, especialmente a depressão. Minimizar efeitos colaterais de drogas antiepilépticas, minimizar o impacto psicossocial da epilepsia e permitir ao paciente um estilo de vida com o mínimo de limitações sob o ponto de vista familiar, de trabalho e social.
Epilepsia: determinar o(s) tipo(s) de crise(s) e síndrome epiléptica. Instituir tratamento com droga adequada, monitorar resposta clínica, efeitos colaterais clínicos e laboratoriais. Ajustar doses para controle completo de crises. Após dois anos de controle de crises, avaliar risco de recorrência para cada caso e considerar retirada da medicação.
O tratamento crônico com drogas antiepilépticas está indicado após a ocorrência de uma segunda crise epiléptica espontânea.
O tratamento deve ser iniciado com droga única (monoterapia), escolhida de acordo com a eficácia no tipo de crise ou síndrome epiléptica, tolerabilidade e particularidades específicas do paciente.
Na falha da monoterapia em dose máxima tolerada, deve ser tentada uma segunda droga em monoterapia.
Nas epilepsias de difícil controle, após duas tentativas sem sucesso de monoterapia em dose máxima tolerada, pode ser tentada associação de drogas antiepilépticas, empregando benzodiazepínicos ou drogas de nova geração.
Co-morbidade psiquiátrica: identificar e tratar co-morbidades psiquiátricas, principalmente depressão.
Epilepsia refratária (após ao menos duas monoterapias com drogas eficazes em dose máxima tolerada): certificar-se do diagnóstico de epilepsia, do(s) tipo(s) de crise(s) e síndrome epiléptica e possível associação de crises não epilépticas psicogênicas. Investigação com RM e EEG. Tentar associação de drogas antiepilépticas. Se refratário: encaminhar para avaliação pré-cirúrgica com vídeo-EEG. Considerar procedimento cirúrgico (especialmente nas epilepsias focais sintomáticas – lesionais).
Mulher em idade fértil: caso não deseje engravidar, orientar anticoncepção e impacto da medicação antiepiléptica sobre efeitos da pílula. Caso deseje engravidar, orientar potencial teratogênico (relativamente baixo) e importância da aderência ao tratamento durante a gestação. Orientar suplementação de ácido fólico antes do início da gestação e orientar aleitamento materno.
Idosos: doses menores, titulação lenta, monitorar efeitos colaterais, especialmente os cognitivos.
Insuficiência hepática, renal e co-morbidades clínicas: selecionar adequadamente as drogas para cada caso, considerando interações medicamentosas.
No estado de mal epiléptico, é imprescindível o manejo rápido, sistematizado e monitorização eletroencefalográfica.
Após benzodiazepínico, deve se prescrever fenitoína ou fosfenitoína. A droga seguinte é fenobarbital e/ou valproato endovenoso. Persistindo o estado de mal, deve-se realizar intubação orotraqueal e as alternativas são midazolam, propofol ou pentobarbital.
1. Bradley W, Daroff R, Fenichel G, Jankovic J. Neurology in clinical practice. 4. ed. Philadelphia: Elsevier; 2004.
2. Castro LHM. Crise epiléptica. In: Martins HS, et al. Pronto-socorro: diagnóstico e tratamento em emergências. 2. ed. Barueri: Manole; 2008. p.439-45.
3. Chang BS, Lowenstein DH. Epilepsy. N Engl J Med. 2003;349(13):1257-66. Disponível em: www.ncbi.nlm.nih.gov/pubmed/14507951.
4. Duncan JS, Sander JW, Sisodiya SM, Walker MC. Adult epilepsy. Lancet. 2006;367:1087-100. Disponível em: www.ncbi.nlm.nih.gov/pubmed/16581409.
5. Fisher RS, van Emde Boas W, Blume W, Elger C, Genton P, Lee P, et al. Epileptic seizures and epilepsy: definitions proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia 2005;46(4):470-2. Disponível em: www.ncbi.nlm.nih.gov/pubmed/15816939.
6. Guerreiro CAM, Guerreiro MM, Cendes F, Lopes-Cendes I. Epilepsia. São Paulo: Lemos Editorial; 1999.
7. Lowenstein DH, Cloyd J. Out-of-hospital treatment of status epilepticus and prolonged seizures. Epilepsia. 2007;48(Suppl 8):96-8. Disponível em: www.ncbi.nlm.nih.gov/pubmed/18330013.
8. Lowenstein DH. Seizures and epilepsy. In: Harrison’s principles of internal medicine. 17. ed. Philadelphia: McGraw-Hill; 2008. p.2498-512.
10. Nitrini R, Bacheschi A. A neurologia que todo médico deve saber. São Paulo: Atheneu; 2003.
11. Pinto LF, Castro LHM. Crise epiléptica. In: Cavalcanti EFA, Martins HS, et al. Clínica médica: dos sinais e sintomas ao diagnóstico e tratamento. Barueri: Manole; 2007. p.897-902.
12. Spencer SS. Seizures and epilepsy. In: Goldman L, et al. Cecil Medicine. 23. ed. Philadelphia: Elsevier; 2008. p.2676-87.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.