(Carregando Índice)... (Carregando Índice)... |
Última revisão: 17/10/2013
Comentários de assinantes: 0
Kieron Dunleavy, MD
Attending Physician/Investigator, Lymphoma Therapeutics Section, Metabolism Branch, National Cancer Institute, Bethesda, MD
Wyndham H. Wilson, MD, PhD
Senior Investigator, Chief, Lymphoma Therapeutics Section, Metabolism Branch, National Cancer Institute, Bethesda, MD
Artigo original: Dunleavy K, Wilson WH. Lymphomas. ACP Medicine. 2010;1-22.
[The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2011 Decker Intellectual Properties Inc. All Rights Reserved.]
Tradução: Soraya Imon de Oliveira.
Revisão técnica: Dr. Lucas Santos Zambon.
O linfoma está em 5º lugar entre os tipos mais comuns de câncer nos Estados Unidos. Em 2009, foram estimados 74.490 casos novos de linfoma nesse país.1 No decorrer das últimas décadas, a incidência do linfoma sofreu um aumento estável de 2 a 3% ao ano. Cerca de 85% dos casos de linfoma são do tipo não Hodgkin (LNH) e 15% são linfomas do tipo Hodgkin.
Nos Estados Unidos e no Ocidente Europeu, a incidência do linfoma de Hodgkin é maior entre os homens do que nas mulheres e segue uma distribuição bimodal entre indivíduos de diferentes idades, atingindo o maior pico na 3ª década de vida e um pico menor na 7ª década. Nos países em desenvolvimento, a doença tende a ser mais comum em homens, porém a incidência chega ao pico durante a 2ª década da vida.2 Os familiares dos pacientes com história de linfoma de Hodgkin apresentam risco aumentado de desenvolverem a doença: este risco pode ser até 10 vezes maior em irmãos do mesmo sexo e até 100 vezes mais alto em gêmeos idênticos.3,4 Existe uma concordância significativa em termos de antígeno leucocitário humano (HLA) entre os indivíduos afetados de uma mesma família. Os antígenos HLA de classe II específicos estão associados ao risco aumentado de linfoma de Hodgkin, podendo ainda estar associados a subtipos histológicos particulares da doença.5,6
A infecção pelo vírus Epstein-Barr (EBV) está associada ao linfoma de Hodgkin. Existe uma incidência aumentada deste tipo de linfoma entre indivíduos com história de mononucleose infecciosa. Um estudo prospectivo controlado encontrou títulos elevados de EBV em pacientes que posteriormente vieram a ser diagnosticados com linfoma de Hodgkin.7,8 Além disso, o genoma do EBV pode ser detectado em células malignas de aproximadamente 50% dos pacientes com linfoma de Hodgkin.9 O genoma do EBV é mais comumente detectado em amostras de tecidos de linfoma de Hodgkin obtidas de crianças pequenas, pacientes de países em desenvolvimento e em casos clássicos de linfoma de Hodgkin do subtipo de celularidade mista.
Embora uma variedade de esquemas de classificação histológica tenha sido usada em casos de linfoma de Hodgkin no passado, a atual classificação da Organização Mundial de Saúde (OMS) define 2 tipos biologicamente distintos: linfoma clássico e linfoma nodular com predominância de linfócitos.10 O linfoma de Hodgkin clássico, por sua vez, compreende 4 subtipos: esclerose nodular, celularidade mista, rico em linfócitos e depletado de linfócitos. A esclerose nodular é o subtipo mais comum, especialmente em pacientes com menos de 40 anos de idade, seguida do subtipo de celularidade mista. As denominações dos subtipos descrevem o meio histológico em que aparecem as células de Reed-Sternberg e suas variantes, bem como a extensão da infiltração linfocítica normal. As citocinas podem exercer algum papel no meio histológico do linfoma de Hodgkin, que é caracterizado pela presença de plasmócitos, eosinófilos, neutrófilos, linfócitos e, no subtipo de esclerose nodular, amplas faixas de colágeno.
A imuno-histoquímica é importante tanto para a subclassificação do linfoma de Hodgkin como para a diferenciação entre linfoma de Hodgkin e LNH. No linfoma de Hodgkin clássico, as células tumorais geralmente são positivas para CD30 (um marcador de células ativadas) e CD15 (um marcador de monócitos-macrófagos), mas são negativas para CD45 e antígenos celulares pan-B e pan-T. Em contraste, as células tumorais do subtipo de linfoma de Hodgkin nodular com predominância de linfócitos são negativas para CD30 e CD15, mas positivas para CD20, BCL6 e CD45. Contudo, a investigação molecular das células de Reed-Sternberg e suas variantes morfológicas tem sido prejudicada pelo fato de estas células representarem apenas uma pequena proporção do número total de células encontradas nos linfomas de Hodgkin; de fato, as células inflamatórias predominam.
A célula neoplásica do linfoma de Hodgkin quase sempre é uma célula B – seja a célula de Reed-Sternberg ou uma de suas variantes mononucleares [Figura 1]. Estas células malignas originam-se das células B dos centros germinativos que sofreram algum tipo de evento transformador.11 Os eventos transformadores decisivos associados ao linfoma de Hodgkin ainda precisam ser definidos. Entretanto, as mutações envolvendo o gene da I-kappa-B quinase, que promovem regulação positiva (indução) da via do fator nuclear-kappa B, foram identificadas em linfomas de Hodgkin EBV-negativos e EBV-positivos. Também foram identificadas mutações no gene FAS. Mutações no gene p53 foram encontradas em um subgrupo de casos EBV-negativos.12-14 O papel de outros genes na etiologia do linfoma de Hodgkin está sendo investigado, e o advento da tecnologia dos microarranjos (microarrays), que conseguem gerar assinaturas de expressão genética distintas para diferentes linfomas, está ampliando o conhecimento dos oncologistas sobre os eventos moleculares integrantes desta doença. Trabalhos recentes demonstraram que os miRNAs (micro-RNAs) exercem papel importante na biologia do linfoma de Hodgkin clássico.15 O aparecimento, manifestação e história natural bastante distintivos dos subtipos do linfoma de Hodgkin sugerem a existência de vias variáveis de linfomagênese, cada uma das quais envolvendo uma complexa interação entre fatores genéticos, fatores ambientais e o sistema imune dos indivíduos afetados.

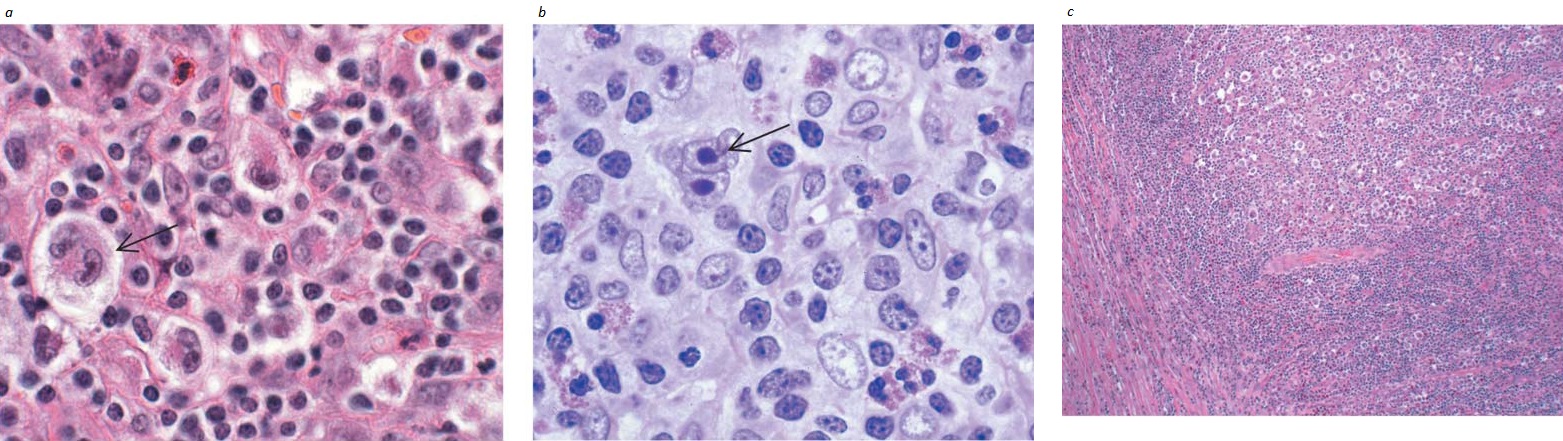
Figura 1. Amostras histológicas de subtipos de linfoma de Hodgkin. (a) Linfoma de Hodgkin do subtipo esclerose nodular; um linfonodo com células lacunares (seta) é observado em um fundo celular rico em eosinófilos. (b) Linfoma de Hodgkin clássico do subtipo de celularidade mista; nota-se um linfonodo com célula de Reed-Sternberg binucleada (seta) em um infiltrado de celularidade mista. (c) Linfoma de Hodgkin do subtipo esclerose nodular; faixas colágenas fibrosas dividem o linfonodo em nódulos.
A característica mais comum da manifestação do linfoma de Hodgkin é o aumento indolor do tamanho de 1 ou mais linfonodos, localizados em cima do diafragma. A linfadenopatia mediastinal é comum no momento da apresentação, especialmente em pacientes na faixa etária de 15 a 35 anos que apresentam esclerose nodular clássica. O envolvimento subdiafragmático é mais comum no subtipo de celularidade mista da forma clássica. O linfoma de Hodgkin nodular com predominância de linfócitos raramente envolve o mediastino ou a medula. Ocasionalmente, o linfoma de Hodgkin manifesta-se como tosse, desconforto torácico ou síndrome da veia cava superior.
O linfoma de Hodgkin é caracterizado pela disseminação ordenada a partir de uma região do linfonodo para sítios nodais contíguos. O curso do linfoma de Hodgkin não tratado é altamente variável. Em muitos pacientes jovens, a doença pode ser bastante indolente, com linfadenopatia em crescente-decrescente. Em outros pacientes, todavia, a doença pode seguir um curso mais virulento. O baço e os linfonodos do tronco celíaco frequentemente são os primeiros sítios de doença subdiafragmática, que é encontrada em 20 a 30% dos pacientes com doença confinada ao pescoço, axilas ou tórax. Tipicamente, os linfonodos para-aórticos e pélvicos são os próximos a serem envolvidos. As doenças hepática e medular estão associadas ao envolvimento esplênico. A doença intratorácica de continuidade muitas vezes resulta da disseminação para o parênquima pulmonar, pericárdio e parede torácica, a partir de uma linfadenopatia mediastínica volumosa. As lesões ósseas, que frequentemente tem característica osteoblásticas à radiografia, podem desenvolver-se por disseminação contígua a partir de linfonodos aumentados ou por disseminação hematogênica. A compressão da medula espinal pode ser causada pela extensão direta a partir de linfonodos aumentados ou pela doença vertebral.
Alguns pacientes apresentam sintomas sistêmicos proeminentes, conhecidos como sintomas B, que incluem intensa sudorese noturna, febre e perda de peso inexplicável. Ocasionalmente, os pacientes apresentam febre de Pel-Ebstein, que consiste em episódios intermitentes de febre noturna que duram vários dias e se alternam com períodos afebris. Um prurido no corpo inteiro, que costuma ser bastante incômodo e refratário aos agentes antihistamínicos, pode acompanhar o linfoma de Hodgkin. Uma característica exclusiva é a dor em sítios de linfadenopatia imediatamente após a ingesta de bebida alcoólica.
O curso clínico do linfoma de Hodgkin varia de acordo com o subtipo histológico, idade do paciente, região geográfica populacional e integridade do sistema imune. O linfoma de Hodgkin nodular com predominância de linfócitos segue um curso particularmente indolente: os sintomas constitutivos são raros, e as recidivas são frequentes, mas em geral respondem à terapia.
De uma forma geral, os pacientes idosos, que vivem em países em desenvolvimento e estão infectados pelo HIV são mais propensos a terem uma doença amplamente disseminada, com manifestação de sintomas sistêmicos no momento do diagnóstico. O subtipo de celularidade mista clássico é a variante mais comumente observada neste grupo. Nos Estados Unidos e no ocidente da Europa, os pacientes adultos jovens são os que mais frequentemente apresentam doença limitada, do subtipo esclerose nodular.
A história de doença em curso concentra-se nos sintomas sistêmicos, quando existentes. A obtenção da história deve incluir perguntas sobre a ocorrência prévia de malignidades, tratamentos quimio ou radioterápicos, doença imunossupressora ou terapia de imunossupressão. Qualquer história familiar de distúrbio linfoproliferativo é relevante e deve ser obtida.
Um exame físico detalhado deve incluir uma avaliação cuidadosa de todas as regiões de linfonodos, para determinar a extensão da linfadenopatia periférica, bem como uma avaliação da parede torácica. O exame abdominal é realizado para detectar hepatoesplenomegalia.
O propósito da avaliação diagnóstica é avaliar a extensão (estágio) do linfoma de Hodgkin, que, por sua vez, determina o prognóstico e o tratamento. Para tanto, o exame diagnóstico de maior utilidade em casos de pacientes com suspeita de linfoma é uma biópsia excisional de linfonodo bem avaliada e obtida de modo tecnicamente adequado. A avaliação anatomopatológica ideal geralmente requer um linfonodo inteiro.
Entre os exames laboratoriais adicionais, estão o hemograma completo, velocidade de sedimentação eritrocitária (VHS) e exames de bioquímica sérica, incluindo a determinação dos níveis de lactato desidrogenase (DHL). A anormalidade hematológica mais comum é a anemia normocítica normocrômica, que é encontrada com frequência em pacientes com doença extensiva e sintomática. As citopenias secundárias ao hiperesplenismo ou à doença medular são raras. Elevações modestas dos níveis de fosfatase alcalina podem ocorrer em pacientes com doença limitada, sendo que níveis mais altos estão associados ao envolvimento hepático, ósseo ou medular. A hipercalcemia é uma manifestação paraneoplásica rara. O VHS em geral está elevado na doença de Hodgkin, e o grau de elevação possui valor prognóstico em casos de pacientes com doença em estágio inicial.
Os exames de imagem para avaliação inicial do linfoma de Hodgkin devem incluir uma avaliação por radiografia torácica e exame de tomografia computadorizada (TC) do tórax, abdome e pelve (com contraste endovenoso, se possível). A TC é o padrão-ouro da modalidade de exames de imagem. A tomografia por emissão de pósitron (PET) é importante como técnica auxiliar da TC para estadiamento inicial. Seu papel é mais importante na avaliação da resposta à terapia, ajudando a distinguir entre linfoma ativo e tecido benigno nas massas residuais. Neste contexto, um PET-scan positivo é altamente preditivo de falha terapêutica.16-18 Os achados do PET, porém, requerem interpretação cautelosa, em particular diante de valores de captação específicos limítrofes. Os pacientes com resultado positivo de PET-scan após o tratamento geralmente devem ser submetidos ao exame de biópsia, para confirmar a presença de um linfoma ativo.
Um inconveniente da TC e de outras modalidades de exame de imagem é a falta de confiabilidade para uso no diagnóstico da doença esplênica e nodal oculta. Em um estudo, a acurácia geral da TC em termos de detecção da doença esplênica foi de 58%.19 A laparotomia de estadiamento, que constitui a única modalidade definitiva para detecção da doença esplênica oculta, antigamente era incluída com frequência na avaliação inicial do linfoma de Hodgkin. Hoje, seu uso é controverso e indicado apenas em casos de pacientes cujo tratamento ideal exija a avaliação da doença esplênica oculta (isto é, pacientes para os quais tenha sido planejada uma radioterapia definitiva). Como a quimioterapia é muito comumente incorporada ao tratamento inicial do linfoma de Hodgkin, a laparotomia agora é realizada apenas em casos raros. Em um estudo prospectivo, a laparotomia não exerceu impacto significativo nas taxas de sobrevida ou recidiva de pacientes com prognóstico favorável que foram randomizados para passarem pelo estadiamento laparoscópico ou clínico.20 O PET-scan, que pode detectar com maior acurácia a doença esplênica oculta no linfoma de Hodgkin, é bastante útil. O exame de ressonância nuclear magnética (RNM) e a cintilografia óssea com isótopo radioativo são rotineiramente indicados para avaliação inicial do linfoma de Hodgkin, mas podem ser úteis em casos de pacientes com doença óssea.
A incidência da infiltração medular no linfoma de Hodgkin varia de uma série para outra, estando na faixa de 2 a 32% entre os indivíduos adultos.23 Em uma revisão de 955 casos, a incidência do envolvimento medular observada no momento do diagnóstico foi de 5,2%, e este envolvimento é mais comum no subtipo de celularidade mista.22 Apesar do rendimento baixo, a biópsia de medula continua sendo recomendada para todos os pacientes recém-diagnosticados com linfoma de Hodgkin.
O sistema de estadiamento de Ann Arbor para o linfoma de Hodgkin, desenvolvido há mais de 30 anos, continua sendo usado no estadiamento do LNH. Uma modificação de 4 estágios – a modificação de Cotswold – que considera fatores como a doença mediastínica volumosa, atualmente é usado para estadiar o linfoma de Hodgkin [Tabela 1].23
Tabela 1. Classificação de estadiamento do linfoma
|
Estágio* |
Classificação Ann Arbor |
Modificação de Cotswold |
|
I |
Envolvimento de uma única região de linfonodo (I) ou de um único órgão ou sítio extralinfático (IE) |
Envolvimento de uma única região de linfonodo ou estrutura linfoide |
|
II |
Envolvimento de 2 ou mais regiões de linfonodo apenas no mesmo lado do diafragma (II) ou envolvimento de um órgão ou tecido extralinfático contíguo (IIE) |
Envolvimento de 2 ou mais regiões de linfonodo apenas no mesmo lado do diafragma (o mediastino é considerado um sítio único, enquanto os linfonodos hilares são considerados bilateralmente); o número de sítios anatômicos deve ser indicado em subscrito (p. ex., II3) |
|
III |
Envolvimento de regiões de linfonodo em ambos os lados do diafragma (III), podendo incluir o baço (IIIB); um órgão ou sítio extralinfático contíguo limitado (IIIE); os ambos (IIIBE) |
Envolvimento de regiões de linfonodo em ambos os lados do diafragma: III1 (com ou sem envolvimento de linfonodos do hilo esplênico, celíacos ou portais) e III2 (com envolvimento de linfonodos para-aórticos, ilíacos e mesentéricos) |
|
IV |
Focos de envolvimento múltiplos ou disseminados de 1 ou mais órgãos ou tecidos extralinfáticos, com ou sem envolvimento linfático |
Envolvimento de 1 ou mais sítios extranodais, além do sítio para o qual é usada a designação E |
*Todos os casos são subclassificados para indicar a ausência ou presença de sintomas sistêmicos de febre significativa (> 38°C), sudorese noturna e perda de peso inexplicável que exceda 10% do peso corporal normal, nos últimos 6 meses. O estágio clínico denota o estágio determinado por todos os exames diagnósticos e por um único exame de biópsia diagnóstico. Na classificação de Ann Arbor, o termo “estágio patológico” é usado quando uma 2ª biópsia de qualquer tipo é obtida, seja negativa ou positiva. Na modificação de Cotswold, o estágio patológico é determinado pela laparotomia; “X” designa uma doença volumosa (ampliação do mediastino em mais de 1/3 ou presença de uma massa nodal > 10 cm); e “E” designa o envolvimento de um único sítio extranodal, que é contíguo ou proximal ao sítio nodal conhecido.
A análise de um amplo conjunto de dados sobre pacientes com linfoma de Hodgkin em estágio avançado identificou 7 fatores clínicos, além do estágio, que eram preditivos do desfecho final do paciente.24 Estes fatores eram a idade superior a 45 anos, doença em estágio IV, sexo masculino, anemia, hipoalbuminemia, linfopenia e neutrofilia. O International Prognostic Score (IPS) incorpora estes fatores desfavoráveis e pode ser usado para identificar pacientes com piores prognósticos que necessitem de terapias mais agressivas [Tabela 2].
Tabela 2. Escores prognósticos internacionais para o linfoma de Hodgkin em estágio avançado94
|
Escore prognóstico* |
Frequência (%) |
Sobrevida de 5 anos livre de progressão (%)† |
Sobrevida de 5 anos (%)† |
|
0 |
7 |
84 |
89 |
|
1 |
22 |
77 |
90 |
|
2 |
29 |
67 |
81 |
|
3 |
23 |
60 |
78 |
|
4 |
12 |
51 |
61 |
|
= 5 |
7 |
42 |
56 |
*Atribui-se 1 ponto pela presença de cada um dos seguintes fatores prognósticos: idade > 45 anos, doença em estágio IV, sexo masculino, níveis de albumina < 4 g/dL, níveis de hemoglobina < 10,5 g/dL, contagem de leucócitos sanguíneos = 15.000/mcL e contagem de linfócitos < 600/mcL ou < 8% do total de leucócitos.
†Baseado em dados obtidos de 5.141 pacientes.24
Uma classificação histopatológica melhorada, estadiamento acurado, radioterapia melhorada e agentes quimioterápicos efetivos contribuem para um alto índice de cura do linfoma de Hodgkin. Para o sucesso da terapia, é preciso que o tratamento seja realizado por uma equipe multidisciplinar com experiência em histopatologia, diagnóstico radiológico, oncologia médica e radioterapia.
Historicamente, a radioterapia era o tratamento preferido para linfoma de Hodgkin, enquanto a quimioterapia era reservada para casos de doença avançada ou falha da radioterapia. Entretanto, a avaliação dos efeitos tardios da radioterapia, inclusive o desenvolvimento de tumores malignos secundários e cardiopatia, levou à adoção de novas abordagens com o objetivo de alcançar índices de cura elevados com toxicidades mínimas a longo prazo.
Embora o estágio da doença seja o determinante primário do prognóstico e da terapia do linfoma de Hodgkin, a European Organization for Research and Treatment of Cancer (EORTC) subdividiu os pacientes com doença em estágio inicial (I e II) em grupos com prognósticos favorável e desfavorável, com base na presença (de pelo menos 1) ou ausência dos seguintes fatores de mau prognóstico: massa mediastinal ampla, idade igual ou superior a 50 anos, VHS alto e envolvimento de no mínimo 4 regiões de linfonodos.25
Doença favorável em estágio inicial. Durante várias décadas, a radioterapia de campo estendido sem quimioterapia foi o tratamento de escolha para os casos de doença favorável em estágio inicial. Embora as taxas de resposta completa fossem altas, 25 a 30% dos pacientes apresentavam recidiva e necessitavam de quimioterapia. O uso combinado de quimio e radioterapia agora é defendido pela maioria dos grupos, resultando em índices de cura superiores, com toxicidade a longo prazo potencialmente menor graças ao uso de doses menores de radiação.16,26,27 Uma combinação recomendada consiste na administração de uma quimioterapia de curta duração (p. ex., 2 ciclos de doxorrubicina [Adriamicina], bleomicina, vimblastina e dacarbazina [ABVD]) aliada à irradiação do campo envolvido [20 a 30 Gy]).28 Alguns grupos defendem o uso apenas da quimioterapia no tratamento da doença favorável em estágio inicial. Um recente estudo conduzido nos Estados Unidos e Canadá randomizou pacientes para receberem apenas ABVD ou ABVD com radiação e não encontrou nenhuma diferença entre ambos os grupos em termos de sobrevida geral. Embora tenha sido constatado que a sobrevida de 5 anos sem progressão da doença foi maior entre os pacientes submetidos à radioterapia, esta vantagem foi compensada pelas mortes atribuíveis a outras causas, que não a doença progressiva nem a toxicidade aguda relacionada ao tratamento neste grupo.29,30 Outro grupo relatou um estudo conduzido por uma única instituição, que comparou o uso de ABVD vs. ABVD + radioterapia do campo envolvido no tratamento de 152 pacientes com linfoma de Hodgkin em estágios I-II ou IIIA. Neste estudo, ambos os grupos não diferiram em termos de sobrevida livre de falhas terapêuticas e sobrevida geral.31
Doença desfavorável em estágio inicial. A terapia de modalidade combinada (tipicamente, 4 ciclos de ABVD com aplicação de 20 a 30 Gy de radiação no campo envolvido) é um tratamento aceito para a doença desfavorável em estágio inicial. Entretanto, dada a ocorrência de recidivas iniciais em até 15% dos pacientes tratados com ABVD, estão sendo estudados regimes quimioterápicos mais eficientes. Contudo, até agora, não há evidências que justifiquem o uso de outro esquema de quimioterapia. Assim como no caso da doença favorável em estágio inicial, o uso apenas de quimioterapia tem aumentado e foi justificado em vários estudos.16,30 No momento, estão sendo conduzidos diversos estudos que incorporam a terapia resposta-adaptada com fluorodesoxiglicose (FDG)-PET ao delineamento experimental.
A quimioterapia combinada é o tratamento de escolha para pacientes com doença em estágio avançado. Embora a combinação MOPP (mecloretamina, vincristina [Oncovin], procarbazina e prednisona) tenha sido o 1º regime multifármacos a alcançar um alto índice de cura neste grupo, o ABVD atualmente é considerado o tratamento-padrão. Em estudos comparativos randomizados, o ABVD foi mais efetivo do que o MOPP e menos tóxico do que os híbridos de MOPP.32,33 Os pacientes geralmente recebem 2 ciclos de terapia após a remissão completa, com o mínimo de 6 e o máximo de 8 ciclos.
Pacientes com linfoma de Hodgkin em estágio avançado tratados com ABVD apresentam uma sobrevida livre de eventos próxima de 70%. Mesmo assim, os pesquisadores estão tentando melhorar o resultado final com regimes mais eficientes, muitos dos quais contêm etoposídeo – um agente bastante ativo contra o linfoma de Hodgkin. O regime Standford V, que é um regime quimioterápico intensivo de curta duração com 7 fármacos (mecloretamina, doxorubicina, vimblastina, vincristina, bleomicina, etoposídeo e prednisona), foi combinado à irradiação consolidativa de sítios de linfonodos volumosos e promoveu uma sobrevida livre de progressão de 89% com uma sobrevida geral de 96% em um período de seguimento médio de 5,4 anos.34 Contudo, um estudo randomizado sugeriu que este regime não é superior ao ABVD.35 O German Hodgkin Study Group (GHSG) comparou o regime BEACOPP (bleomicina, etoposídeo, doxorubicina [Adriamicina], ciclofosfamida, vincristina [Oncovin], procarbazina e prednisona) e uma dose aumentada de BEACOPP à combinação COPP (ciclofosfamida, vincristina [Oncovin], procarbazida e prednisona) + ABVD no tratamento de pacientes com linfoma de Hodgkin em estágio avançado.36 A radioterapia, usada no tratamento da doença volumosa ou da doença residual após 8 ciclos de quimioterapia, foi administrada a 2/3 dos pacientes. Apesar da maior toxicidade, a dose aumentada de BEACOPP produziu menos falhas de indução e uma taxa maior de ausência de falha terapêutica em 5 anos, além de uma sobrevida geral melhor, quando comparada à combinação COPP/ABVD. O regime com dose aumentada de BEACOPP foi mais vantajoso para os pacientes com prognóstico ruim, definido pelo IPS. O GHSG recentemente investigou um regime de BEACOPP basal tempo-intensificado, administrado a intervalos de 14 dias, em uma tentativa de alcançar a mesma eficácia do regime BEACOPP de dose escalonada, porém com uma toxicidade menor. Em um estudo piloto, esta abordagem foi bastante efetiva37 e, atualmente, está sendo comparada ao BEACOPP de dose escalonada em um estudo randomizado. Desta forma, embora o ABVD continue sendo o tratamento-padrão para linfoma de Hodgkin em estágio avançado, regimes mais agressivos podem vir a se tornar a terapia-padrão para pacientes com prognósticos piores.
Este é um subtipo raro do linfoma de Hodgkin, que se comporta de modo bastante diferente do linfoma de Hodgkin clássico e é biologicamente similar ao linfoma de células B indolente. A sobrevida de 10 anos para os pacientes com doença em estágio inicial ultrapassa 80%, e o papel da terapia em casos de doença favorável em estágio inicial é obscuro. O EORTIC recomenda a irradiação do campo envolvido.15 Os pacientes com doença em estágio avançado apresentam prognóstico desfavorável e podem ser beneficiados por abordagens iniciais agressivas.38 Em um estudo de fase II, o anticorpo anti-CD20 rituximabe produziu uma taxa de resposta completa de 46%, porém o tempo de seguimento foi curto. O impacto deste tratamento a longo prazo requer estudos adicionais.39
Em pacientes com doença em estágio terminal que apresentam recidiva após o tratamento apenas com radiação, um tratamento subsequente com regimes como ABVD resulta em sobrevida livre de recidivas de até 70% em 10 anos.40 As recidivas que ocorrem após a quimioterapia combinada ou após a terapia de modalidade combinada são mais desafiadoras. Se a duração da remissão inicial for superior a 12 meses, o retratamento com quimioterapia (com a mesma combinação ou com uma combinação diferente) resulta em até 50% de sobrevida livre de recidivas em 5 anos.41-43
Os pacientes que falham em alcançar remissão completa com a quimioterapia inicial ou que sofrem recaída dentro de 1 ano após a quimioterapia inicial apresentam um prognóstico menos favorável e chances mais precárias de sobrevida a longo prazo se forem tratados apenas com altas doses-padrão de quimioterapia.41,44,45 Para ambos os grupos, uma dose alta de quimioterapia com ou sem radioterapia, seguida de transplante de células-tronco hematopoiéticas autólogas constitui a abordagem-padrão. Embora existam poucos dados disponíveis sobre o resultado alcançado a longo prazo, os resultados iniciais demonstraram uma sobrevida de 5 anos livre de progressão de doença de até 61%.46
Existem alguns regimes de resgate que são ativos contra o linfoma de Hodgkin recidivante e podem ser usados para controlar a doença antes da realização de um transplante autólogo. Entre estes regimes, estão o ESHAP (etoposídeo, metilprednisolona [Solu-Medrol], dose alta de citarabina [ara-C] e cisplatina [platina]), ICE (ifosfamida, carboplatina e etoposídeo) e EPOCH (etoposídeo, prednisona, vincristina [Oncovin], ciclofosfamida e doxorubicina [hidroxidaunorubicina]).47-49 A recidiva após o transplante constitui um desafio. O tratamento prolongado com doses baixas de agentes como a vimblastina ou a gencitabina pode ser administrado a estes pacientes.50,51
Diversas terapias experimentais para o linfoma de Hodgkin estão sendo investigadas. Entre estas, estão os anticorpos monoclonais, radioimunoconjugados, imunoterapia, vacinas antitumorais,52,53 e transplante de células-tronco alogênicas.54
Devido ao alto índice de cura do linfoma de Hodgkin, a toxicidade a longo prazo da terapia está se tornando um aspecto cada vez mais importante, à medida que o tempo de seguimento dos pacientes aumenta. O perfil completo de toxicidade da radiação está se tornando evidente. O risco de desenvolvimento de vários tipos de tumores sólidos aumentou dramaticamente entre os pacientes submetidos à radioterapia para tratamento do linfoma de Hodgkin.55,56 Os pacientes que foram tratados enquanto eram jovens são aqueles que apresentam maior risco. Entre os sobreviventes a longo prazo que foram submetidos à irradiação do mediastino, também foram observadas anormalidades cardiovasculares clinicamente significativas.57 Além disso, o hipotireoidismo é uma complicação tardia frequente da radioterapia.58 Embora atualmente ocorra um emprego de campos limitados, doses menores e técnicas aprimoradas de radioterapia que tendem a melhorar estas toxicidades, o acompanhamento a longo prazo se faz necessário para confirmar tais benefícios.
Os efeitos tardios da quimioterapia incluem o desenvolvimento de leucemias e mielodisplasia em pacientes tratados com agentes alquilantes, como mostarda nitrogenada e procarbazina, no regime MOPP.59 A esterilidade ocorre em cerca de 90% dos homens e em 80% das mulheres com idade acima de 25 anos, após o tratamento com um curso completo de MOPP, sendo que a incidência é menor entre as mulheres jovens.60,61 Em contraste, a leucemia aguda e a mielodisplasia raramente são relatadas em pacientes submetidos ao tratamento com ABVD, que está associado a uma incidência de infertilidade de 10 a 20%.62 A bleomicina contida no ABVD pode produzir toxicidade pulmonar a longo prazo.63
O LNH, com uma estimativa de 65.980 casos novos/ano, representa 4% de todos os casos novos de câncer registrados nos Estados Unidos.1 A incidência de LNH é maior entre os homens e com o avanço da idade. O LNH é mais frequente nos países ocidentais, e seus diferentes subtipos exibem predileções geográficas particulares. No decorrer dos últimos 20 anos, a incidência do LNH dobrou nos Estados Unidos.64 Entre os fatores que contribuíram para esta tendência estão a ampliação da população de risco de desenvolvimento de linfoma associado à AIDS e o aumento dos relatos e da detecção dos linfomas. Entretanto, estes fatores diversos são responsáveis apenas por 50% do aumento observado. Os outros motivos da elevação da incidência do LNH são desconhecidos e pouco compreendidos.64,65 As toxinas ambientais provavelmente são fatores etiológicos importantes.
O LNH também é comumente observado em pacientes que apresentam condições de imunodeficiência ou doenças autoimunes, bem como em pacientes submetidos à terapia imunossupressora prolongada.64,66 A infecção pelo EBV está envolvida na maioria das doenças linfoproliferativas que ocorrem nestes contextos. Outros vírus ou patógenos associados, além do HIV, são o vírus linfotrópico de células T humanas de tipo I (HTLV-I), vírus da hepatite C, Helicobacter pylori, herpes vírus humano de tipo 8, Borrelia burgdorferi,67-71 Chlamydia psittaci e Campylobacter jejuni.72,73
Vários sistemas de classificação são adotados para o LNH, tais como Rappaport, Kiel, Working Formulation e Revised European-American Classification of Lymphoid Neoplasms (REAL). A atual classificação estabelecida pela OMS [Tabela 3] resultou do esforço conjunto internacional de patologistas e clínicos e é baseada em critérios da patogênese molecular do linfoma que incluem as conhecidas anormalidades genéticas características. As malignidades hematológicas são estratificadas de acordo com a linhagem ou a origem celular (células B ou T/célula natural killer [NK]).10 Além disso, junto a cada linhagem ou categoria de origem celular, as malignidades são descritas e definidas de acordo com os aspectos morfológicos, imunofenótipo, genótipo e comportamento clínico.
Tabela 3. Classificação de linfomas da OMS10
|
Neoplasias de célula B |
|
LDGCB |
|
Linfoma folicular |
|
LLC/LLP |
|
Linfoma de células de revestimento |
|
Linfoma de grandes células B (tímico) mediastínico primário |
|
Linfoma de zona marginal extranodal (linfoma MALT) |
|
Linfoma de zona marginal esplênico |
|
Linfoma de zona marginal nodal |
|
Linfoma linfoplasmocítico |
|
LB |
|
Linfoma de grandes células B intravascular |
|
Linfoma de efusão primária |
|
Linfoma plasmoblástico |
|
Granulomatose linfomatoide |
|
Linfoma de centro folicular cutâneo primário |
|
Linfoma de Hodgkin |
|
Linfoma de Hodgkin clássico |
|
Esclerose nodular |
|
Celularidade mista |
|
Rico em linfócitos |
|
Linfócito-depletado |
|
Linfoma de Hodgkin nodular com predominância de linfócitos |
|
Neoplasias de células NK e de células T maduras |
|
Linfoma de células T periférico NE |
|
LGCA |
|
Linfoma de células T angioimunoblástico |
|
Linfomas de células T periféricos cutâneos primários |
|
Micose fungoide |
|
Síndrome de Sézary |
|
Linfoma de célula T hepatoesplênico |
|
Linfoma de células T associado à enteropatia |
|
Linfoma de células T paniculite-símile subcutâneo |
|
Linfoma de célula NK/T extranodal, tipo nasal |
|
Linfoma/leucemia de células T do adulto |
|
Distúrbios linfoproliferativos associados à imunodeficiência |
|
Linfomas associados à infecção pelo HIV |
|
DLPT |
|
Outros distúrbios linfoproliferativos associados à imunodeficiência iatrogênicos |
DLPT = distúrbios linfoproliferativos pós-transplante; LB = linfoma de Burkitt; LDGCB = linfoma difuso de grandes células B ; LGCA = linfoma de grandes células anaplásicas; LLC = leucemia linfocítica crônica; LLP = linfoma linfocítico de pequenas células; MALT = tecido linfoide associado à mucosa; NK = natural killer; NE = não especificado; OMS = Organização Mundial da Saúde.
A avaliação patológica do linfoma baseia-se nas características histológicas da neoplasia e no imunofenótipo [Figura 2]. Em muitos casos, a morfologia, isoladamente, é inadequada como parâmetro de classificação, pois LNH com heterogeneidade imunofenotípica e molecular pode exibir uma aparência morfológica similar. A imunofenotipagem fornece informações valiosas para a classificação dos linfomas. As células T e B expressam diferentes antígenos de agrupamento de diferenciação (CD – cluster of differentiation) que, por sua vez, mudam conforme o grau de maturação. Usando anticorpos monoclonais dirigidos contra estes antígenos de superfície celular, a análise imunofenotípica consegue definir a célula de origem de um linfoma – célula B ou célula T – e o estágio de diferenciação. Além disso, a imuno-histoquímica pode detectar produtos genéticos que são característicos de subtipos de linfoma específicos, como a bcl-2 (uma proteína inibidora da apoptose) e a bcl-1 (uma proteína promotora da progressão do ciclo celular) [Figura 3]. Muitos destes marcadores de superfície celular estão correlacionados ao resultado e prognóstico.74-76
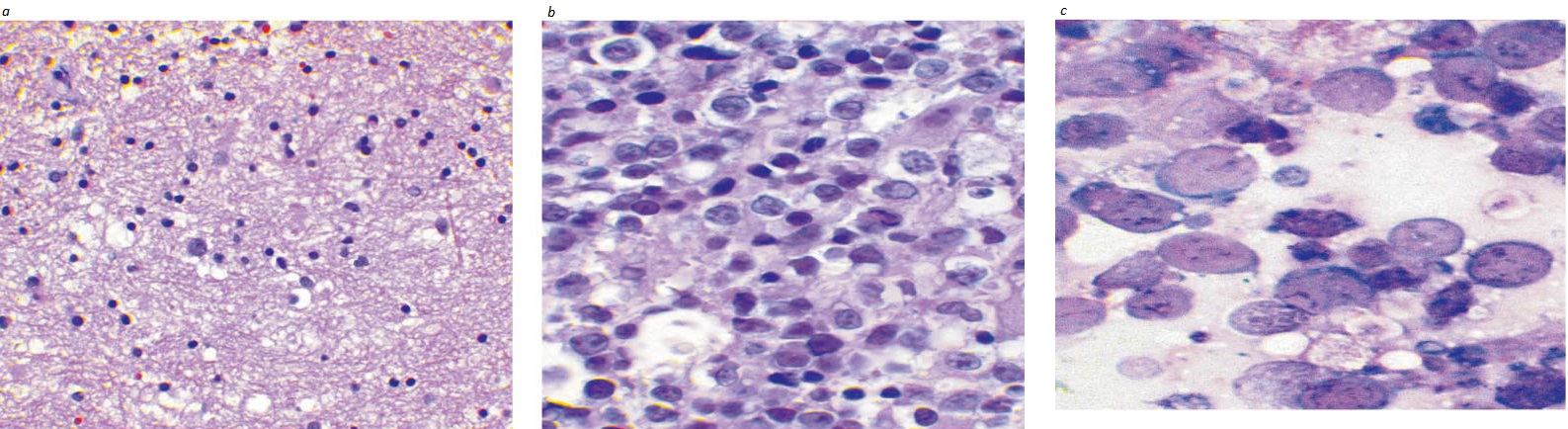
Figura 2. Imagens em (a) menor aumento; (b) aumento médio; e (c) maior aumento de uma amostra de biópsia de linfonodo de paciente com linfoma difuso de grandes células B (LDGCB), mostrando a presença de células linfoides de tamanho médio a grande, contendo núcleos vesiculares, cromatina fina e citoplasma escasso.
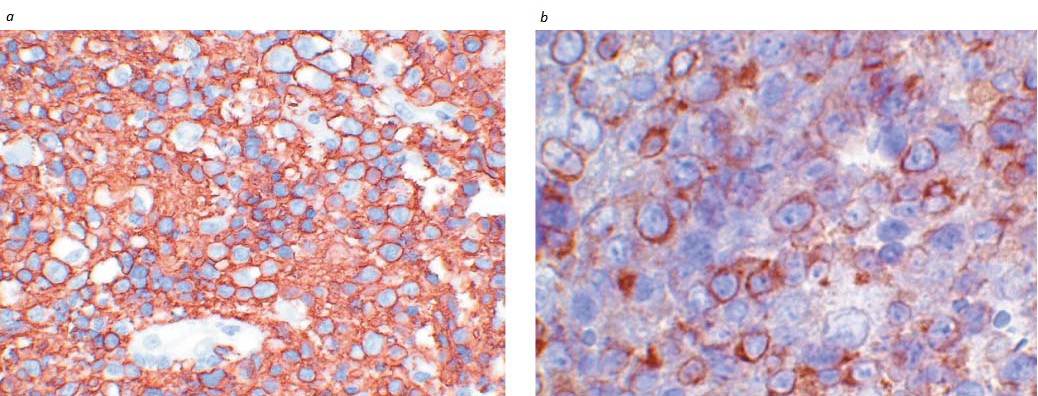
Figura 3. Análise imunofenotípica de uma amostra de biópsia de linfonodo obtida de paciente com linfoma difuso de grandes células B (LDGCB), mostrando que (a) as células tumorais expressam CD20 e (b) a coloração para detecção da proteína inibidora de apoptose bcl-2 é positiva.
O diagnóstico molecular é bastante útil quando a morfologia e a imuno-histoquímica falham em estabelecer um diagnóstico definitivo.77 Todos os linfomas possuem genes anormais, e vários destes genes estão associados à proliferação ou apoptose celular. As anormalidades genéticas (geralmente translocações) interferem no crescimento e diferenciação celular linfoide, causando assim a transformação maligna. As anormalidades costumam envolver os genes codificadores das cadeias das imunoglobulinas. Desta forma, o rearranjo do gene da imunoglobulina de cadeia leve de superfície e do gene do receptor de célula T, que pode ser detectado por imuno-histoquímica ou hibridização do DNA, pode ser útil na avaliação e diagnóstico de malignidades linfoides.
Os linfomas derivam de células B, células T ou células NK que estão em diferentes estágios de diferenciação. Exemplificando, os linfomas linfoblásticos de precursor de células B e de precursor de células T são neoplasias de linfócitos em estágio de diferenciação inicial (linfoblastos comprometidos com as linhagens de células B e T, respectivamente). Em contraste, o linfoma folicular é uma neoplasia de linfócitos mais maduros (células B do centro germinativo), assim como o linfoma de células T periféricas.
As anormalidades cariotípicas caracterizam a maioria dos LNH de células B [Tabela 4]. Entre estas, estão a translocação t(14;18)(q32;q21) com rearranjo do gene bcl-2, que está presente em 80 a 90% dos linfomas foliculares.78,79 A inibição da apoptose por bcl-2 parece exercer papel importante na patogênese dos linfomas.79 A superexpressão de bcl-2 também ocorre no LDGCB, porém com menor frequência (24 a 55% em 2 estudos), e geralmente não está associada à translocação de bcl-2.74,80 Quase todos os casos de linfoma de células de revestimento contêm a translocação t(11;14)(q13;q32), que leva à superexpressão do gene da ciclina D1, promotor da progressão dos estágios G1 a S do ciclo celular.81 O LB está associado à translocação de MYC, que mais comumente resulta de uma translocação t(8;14).10 Um subgrupo do LDGCB, caracterizado por translocações de MYC, está associado a um resultado mais desfavorável com o uso dos tratamentos-padrão para LDGCB.82,83
Tabela 4. Translocações cromossômicas no LNH74-81,180-185
|
Subtipo de linfoma* |
Translocação |
Percentual de casos afetados |
Proto-oncogene |
Função putativa |
Correlação clínica |
|
Linfoma de célula de centro folicular |
t(14;18)(q32;q21) |
80 a 90 |
bcl-2 |
Inibe a apoptose |
Sem associação com o prognóstico |
|
17p13 |
|
p53 |
Confere resistência à quimioterapia |
Transformação da célula grande e tempo menor de progressão da doença | |
|
LLP (LLC) |
Trissomia 12 |
10 a 30 |
— |
Perda da função supressora tumoral; associação com o tipo Ig-não mutante |
Doença avançada, morfologia atípica e doença resistente |
|
13q14 |
50 |
— |
Gene supressor tumoral putativo desconhecido |
Melhor sobrevida | |
|
11q22–23 |
10 a 20 |
— |
Desconhecida |
Doença agressiva e prognóstico pior | |
|
17p13 |
6 |
p53 |
Confere resistência à quimioterapia |
Menor sobrevida | |
|
LDGCB |
del(3)(q27) |
35 |
bcl-6 |
Fator de transcrição para formação do centro germinativo |
LDGCB extranodal; o impacto sobre o resultado do tratamento é controverso |
|
t(14;18)(q32;q21) |
24 a 55 |
bcl-2 |
Inibe a apoptose |
Prognóstico ruim | |
|
17p13 |
20 |
p53 |
Confere resistência à quimioterapia |
Prognóstico ruim | |
|
Linfoma de células de revestimento |
t(11;14)(q13;q32) |
— |
bcl-1/ ciclina D1 |
Regulador do ciclo celular; promove a progressão de G1 para S |
Detectada em 70% dos casos por citogenética e em quase 100% por FISH |
|
17p13 |
8 |
p53 |
Confere resistência à quimioterapia |
Prognóstico pior e morfologia blástica | |
|
LB |
t(8;14)(q24;q32) |
100 |
c-myc |
Ativador transcricional que regula a proliferação e o crescimento celular |
Presente em 100% dos casos; não totalmente específico do LB |
|
Linfoma de zona marginal |
t(11;18)(q21;q21) |
60 |
MLT-1 |
Confere resistência à terapia via inibição da apoptose |
Resistente à terapia de erradicação de Helicobacter |
|
LGCA |
t(2;5)(p23;35) |
50 a 80 |
NPM/ALK |
Ativa a tirosina quinase |
Associada a um prognóstico melhor |
LB = linfoma de Burkitt; LDGCB = linfoma difuso de grandes células B ; LGCA = linfoma de grandes células anaplásicas; LLC = leucemia linfocítica crônica; LLP = linfoma linfocítico de pequenas células; LNH = linfoma não Hodgkin; FISH = hibridização in situ fluorescente.
Apesar da maior precisão diagnóstica possibilitada pelos avanços ocorridos com as técnicas moleculares, a resposta à terapia varia amplamente entre os subtipos de linfoma específicos. No LDGCB, por exemplo, apenas 1/3 dos pacientes são curados com a terapia CHOP (ciclofosfamida, hidroxidaunorubicina, vincristina [Oncovin], prednisona), e 50% são curados com tratamento rituximabe-CHOP.84,85 Esta heterogeneidade marcante das respostas terapêuticas implica na existência de doenças distintas junto a uma mesma categoria morfológica. A determinação do perfil molecular resultou em maior discernimento acerca da diversidade biológica existente em cada uma das categorias morfologicamente definidas do linfoma.86-88 O RNA oriundo de uma amostra tumoral pode ser analisado pelo método de microarranjos de DNA, que consegue medir a expressão de milhares de genes e, desta forma, gerar uma impressão distintiva da expressão genética.
Um aspecto comumente observado no LNH é a linfadenopatia periférica indolor. Em contraste com o linfoma de Hodgkin, a linfadenopatia mediastínica é incomum na manifestação do LNH. Contudo, o envolvimento dos linfonodos abdominais e pélvicos, bem como dos sítios extranodais é mais frequente do que no linfoma de Hodgkin. A doença extranodal pode envolver qualquer órgão, contudo as localizações mais frequentes são o trato gastrintestinal (GI), pele e ossos. O linfoma indolente é tipicamente caracterizado por uma linfadenopatia em crescente-decrescente, que pode permanecer clinicamente quiescente durante vários anos, na ausência de terapia. Em contraste, o linfoma agressivo costuma ser caracterizado por uma linfadenopatia de progressão rápida, que requer terapia imediata. Mais de 1/3 dos pacientes com LNH apresentam manifestações constitutivas (conhecidas como sintomas B), que incluem a temperatura acima de 38°C, a perda inexplicável de mais de 10% do peso corporal e sudorese noturna. Os sintomas B são mais comuns nos subtipos agressivos. O envolvimento medular é significativamente mais comum no linfoma indolente do que nos subtipos agressivos, como o LDGCB.85,89
O curso clínico do LNH depende do subtipo histológico, bem como da idade e da condição do sistema imune do paciente. Alguns subtipos histológicos são bastante agressivos, como o LB e o linfoma linfoblástico. O linfoma folicular, porém, é tipicamente indolente. Os linfomas associados à AIDS, que quase sempre são agressivos, caracteristicamente estão em estágio mais avançado no momento da manifestação, com alta incidência de doença em estágio IV e maior frequência de envolvimento do sistema nervoso central (SNC) do que nos linfomas similares de pacientes HIV-negativos.90
É necessário interrogar os pacientes sobre os sintomas sistêmicos, bem como sobre qualquer história antiga de malignidade, quimioterapia ou radioterapia e doença autoimune ou imunossupressora e sobre qualquer tipo de história familiar de malignidade. Deve-se perquisar uma história de infecção com ou sem exposição a patógenos, como HIV, vírus da hepatite C ou HTLV-I. Um exame físico detalhado deve atentar especialmente para as regiões de linfonodo.
O exame diagnóstico mais importante é uma biópsia excisional de linfonodo bem avaliada e obtida de modo tecnicamente adequado. Os exames laboratoriais devem incluir um hemograma completo, bioquímica do soro (incluindo níveis de DHL) e sorologias para HIV e hepatite. Níveis altos de DHL têm implicações prognósticas importantes para muitos subtipos de linfoma.91 Devem ser investigados os potenciais patogênicos ou vírus etiológicos suspeitos com base na avaliação clínica. Os casos de pacientes com leucemia linfocítica crônica (LLC) ou linfoma linfocítico de pequenas células (LLP) requerem a realização de eletroforese de proteínas séricas com quantificação de imunoglobulinas. Os linfomas linfoplasmacíticos, como a macroglobulinemia de Waldenström, frequentemente estão associados à paraproteinemia. É possível monitorar paraproteínas monoclonais. A deficiência de imunoglobulinas, que frequentemente acompanha estas doenças, pode predispor à aquisição de certas infecções oportunistas.
Os exames de imagem devem incluir uma radiografia torácica e uma TC de tórax, abdome e pelve, se possível com contraste endovenoso [Figuras 4 e 5]. A necessidade de realização de exames de imagem adicionais, incluindo RNM e PET-scan, depende da manifestação clínica e dos sítios afetados pela doença. A TC ou RNM do crânio são realizadas quando há forte suspeita de envolvimento do SNC [Figura 6]. Certos subtipos histológicos agressivos e contextos clínicos estão associados a um risco maior de doença no SNC.92,93 Os pacientes que apresentam maior risco de doença no SNC devem ser submetidos a uma punção lombar no momento do diagnóstico, com análise do líquido cerebrospinal por citometria de fluxo, além da avaliação citológica.93
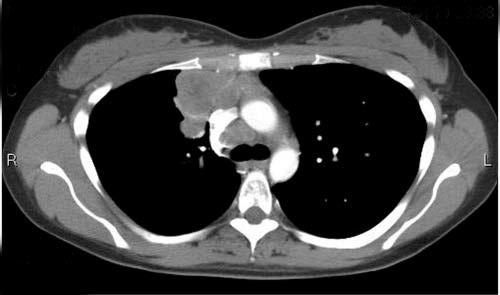
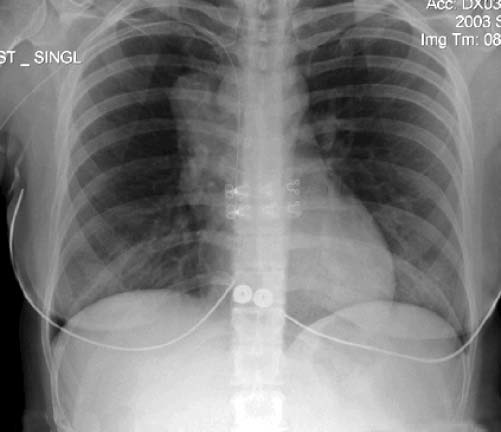
Figura 4. Varredura de tomografia computadorizada (TC) do tórax, mostrando uma massa mediastínica anterior à direita e linfadenopatia paratraqueal. Uma biópsia da massa foi consistente com um linfoma de grandes células B (tímico) mediastínico.
Figura 5. Radiografia plana do tórax, revelando o mediastino ampliado de um paciente com linfoma de grandes células B mediastínico primário.
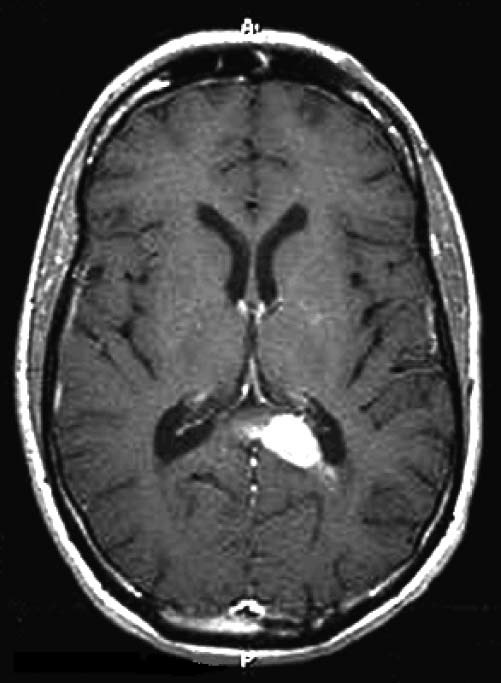
Figura 6. Varredura por imagem de ressonância magnética (RNM) do crânio, com contraste de gálio, mostrando uma massa infiltrante intensificada junto ao fórceps maior do corpo caloso. Uma biópsia revelou um linfoma difuso de grandes células B (LDGCB).
No LNH, a incidência do envolvimento medular varia de acordo com a histologia, mas pode aproximar-se de 70% em casos de linfoma de células B indolente.89 Consequentemente, a biópsia de medula óssea é considerada o exame-padrão para diagnóstico.
O sistema de estadiamento Ann Arbor, originalmente projetado para o linfoma de Hodgkin, agora tem ampla utilidade no estadiamento do LNH [Tabela 1]. Entretanto, dada a tremenda heterogeneidade do LNH e como não há disseminação ordenadamente contígua de uma região de linfonodo para outra nestas doenças, o sistema de estadiamento Ann Arbor tem valor prognóstico limitado.
Para identificar os fatores prognósticos no LNH, um projeto internacional correlacionou variáveis clínicas e resultado em 2.031 pacientes com linfoma agressivo não tratado.94 Os aspectos descritos a seguir estão associados a um pior prognóstico: idade superior a 60 anos, estágios III ou IV de Ann Arbor, níveis altos de DHL, condição funcional igual ou maior que 2 pela classificação do Eastern Cooperative Oncology Group [Tabela 5], e envolvimento de pelos menos 2 sítios extranodais. Os membros do projeto criaram o International Prognostic Index (IPI), para ser usado como ferramenta de predição de resultados. No IPI, atribui-se 1 ponto a cada aspecto encontrado. Na coorte do estudo, constatou-se que a sobrevida de 5 anos estava diretamente relacionada aos escores de IPI (sobrevida de 73%, 51%, 43% e 26%, respectivamente, para os escores 0, 1, 2, 3 e 4-5) [Tabela 6].94 Assim, o IPI é útil para predizer o resultado final alcançado por pacientes com linfoma agressivo.
Tabela 5. Eastern Cooperative Oncology Group Performance Scale
|
Condição funcional |
Definição |
|
0 |
Assintomático |
|
1 |
Sintomático e totalmente deambulante |
|
2 |
Sintomático e permanece no leito em < 50% do dia |
|
3 |
Sintomático e permanece no leito em > 50% do dia |
|
4 |
Confinado ao leito |
Tabela 6. Índice prognóstico internacional para o LNH agressivo94
|
Escore IPI* |
Grupo de risco |
Taxa de RC (%) |
Sobrevida geral de 5 anos (%) |
|
0, 1 |
Baixo |
87 |
73 |
|
2 |
Baixo intermediário |
67 |
51 |
|
3 |
Alto intermediário |
55 |
43 |
|
4, 5 |
Alto |
44 |
26 |
IPI = índice prognóstico internacional; LNH = linfoma não Hodgkin; RC = resposta completa.
* Atribui-se 1 ponto pela presença de cada uma das seguintes características: idade > 60 anos, níveis altos de lactato desidrogenase (DHL) sérico, condição funcional pelo Eastern Cooperative Oncology Group = 2, doença em estágio III ou IV de Ann Arbor, e mais de 2 sítios extranodais.
Atualmente, também há um índice prognóstico clínico disponível para pacientes com linfoma folicular.95 O Follicular Lymphoma International Prognostic Index (FLIPI), que se baseia na idade do paciente, estágio da doença, níveis séricos de DHL, níveis de hemoglobina e número de áreas nodais, é uma forma comprovada e segura de predizer a sobrevida [Tabela 7].
Tabela 7. Índice prognóstico internacional para o linfoma folicular95
|
Grupo de risco |
Número de fatores* |
Distribuição dos pacientes (%) |
SG de 5 anos (EP) |
SG de 10 anos (EP) |
RR (IC 95%) |
|
Baixo |
0 a 1 |
36 |
90,6 (1,2) |
70,7 (2,7) |
1,0 (NA) |
|
Intermediário |
2 |
37 |
77,6 (1,6) |
50,9 (2,7) |
2,3 (1,9 a 2,8) |
|
Alto |
= 3 |
27 |
52,5 (2,3) |
35,5 (2,8) |
4,3 (3,5 a 5,3) |
IC = intervalo de confiança; NA = não aplicável; SG = percentual de sobrevida geral; RR = risco relativo de morte; EP = erro padrão.
*Os fatores que produzem efeitos adversos sobre a sobrevida são: idade > 60 anos, estágios de Ann Arbor III ou IV, > 4 sítios nodais, níveis séricos de lactato desidrogenase (DHL) acima do limite normal superior e hemoglobina < 12 g/dL.
É provável que as características clínicas incluídas no IPI e no FLIPI sejam substitutas das características biológicas (ver anteriormente). As proteínas bcl-2, bcl-6 e MUM-1 tiveram suas expressões avaliadas e correlacionadas ao prognóstico, em particular no LDGCB.75,90,96-98 Vários estudos identificaram a positividade para bcl-2 como marcador de um resultado ruim. Um estudo randomizado, conduzido por um grupo francês, validou a observação de que o anticorpo monoclonal rituximabe pode vencer a resistência à quimioterapia associada à bcl-2 no LDGCB não tratado.99,100
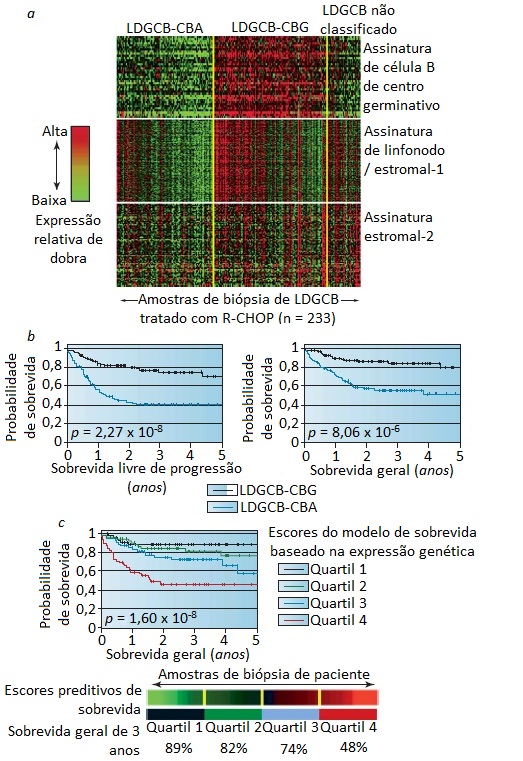
O perfil de expressão genética está emergindo como importante ferramenta prognóstica. No LDGCB, por exemplo, tumores morfologicamente idênticos podem apresentar acentuada heterogeneidade em termos de expressão genética. Estes genes podem ser classificados em assinaturas associadas aos estágios de diferenciação.101 Com base nestas assinaturas, o LDGCB pode ser classificado em 2 subtipos: LDGCB derivado de célula B do centro germinativo (denominado LDGCB de célula B germinativa-símile [CBG]); e LDGCB derivado de célula B pós-centro germinativo ou ativada (denominado LDGCB de célula B ativada-símile [CBA]) [Figura 7].102 A sobrevida geral mostrou-se significativamente melhor entre os pacientes com subtipo CBG do que entre aqueles com subtipo CBA, tanto na era pré-rituximabe como na era pós-rituximabe. A sobrevida é influenciada de modo independente pela assinatura “estromal”, que, por sua vez, é determinada pelas diferenças existentes entre as células imunes, fibrose e angiogênese observadas no microambiente tumoral [Figura 7].86,103
Figura 7. Expressão genética e expressão genética preditiva da sobrevida entre pacientes com linfoma difuso de grandes células B (LDGCB) tratados com R-CHOP. (a) Expressão genética de assinatura em célula de centro germinativo-símile (CBG), estromal-1 e estromal-2 em amostras de biópsia oriundas de pacientes com LDGCB. Os níveis relativos de expressão genética em cada amostra são representados de acordo com a escala colorida mostrada. Cada linha representa uma assinatura genética diferente, e cada coluna, uma amostra de biópsia distinta. As médias das assinaturas estromal-1 e estromal-2 são indicadas para cada paciente. O escore estromal é o componente do modelo de sobrevida multivariado contribuído pela diferença existente entre as médias das assinaturas estromal-1 e estromal-2. As amostras foram classificadas em postos em cada subtipo de LDGCB, de acordo com o escore preditivo de sobrevida, que foi gerado pelo modelo de sobrevida e incorpora tanto o escore estromal como a assinatura CBG. As assinaturas genéticas representativas são mostradas. R-CHOP denota rituximabe + quimioterapia combinada com ciclofosfamida, doxorubicina, vincristina e prednisona. (b) As estimativas de Kaplan-Meier da sobrevida livre de progressão e da sobrevida geral mostram que os pacientes com LDGCB CBG apresentaram maior probabilidade de sobrevida livre de progressão (esquerda) e de sobrevida geral (direita) do que os pacientes com LDGCB de células B ativadas (CBA). (c) As estimativas de Kaplan-Meier da sobrevida livre de progressão e da sobrevida geral mostram a existência de um precursor de sobrevida baseado na expressão genética em pacientes com LDGCB tratados com R-CHOP. As estimativas de Kaplan-Meier da sobrevida geral baseiam-se no modelo multivariado derivado das assinaturas genéticas de CBG, estromal-1 e estromal-2. Os escores preditivos de sobrevida derivados deste modelo foram usados para classificar em postos os casos de linfoma, os quais, então, foram divididos em grupos de quartis, conforme indicado. R-CHOP denota rituximabe + quimioterapia combinada com ciclofosfamida, doxorubicina, vincristina e prednisona.
A determinação do perfil de expressão genética também gerou fatores preditores de sobrevida em outros tipos de LNH, incluindo o linfoma folicular, LLC, linfoma de células B mediastínico primário e linfoma de células de revestimento.87,88,104-106 Além disso, determinar o perfil de expressão genética é útil para identificar os casos cuja aparência histológica é de LDGCB, mas que possuem perfil molecular, de modo consistente com um LB. Isto é potencialmente importante porque os pacientes com LB tratados com regimes-padrão para LDGCB alcançam um resultado final precário.107 Embora a determinação do perfil molecular seja em grande parte uma técnica experimental e sem ampla disponibilidade, promete melhorar a acurácia do diagnóstico patológico, predizer o resultado final com maior precisão e elucidar as vias de linfogênese, identificando assim alvos celulares pertinentes e abrindo caminho para o desenvolvimento de terapias mais individualizadas. A integração da imunocoloração e da técnica de microarranjos genéticos em estudos clínicos de grande porte, bem como a avaliação de seu valor prognóstico devem facilitar o desenvolvimento de modelos prognósticos novos e bastante úteis, capazes de orientar as escolhas terapêuticas.
Os linfomas de células B indolentes abrangem vários subtipos distintos, dentre os quais o linfoma folicular é o mais comum.108 Embora as terapias novas e as terapias experimentais possam oferecer futuras possibilidades de cura, estes linfomas são considerados incuráveis pela terapia convencional.109 Como estas doenças são caracterizadas por um curso crescente-decrescente e os pacientes podem viver por vários anos, até mesmo na ausência de terapia, seu manejo muitas vezes emprega uma abordagem de “espera assistida”.110 As indicações para o tratamento incluem a doença sintomática ou agressiva, uma linfadenopatia volumosa ou citopenias decorrentes do envolvimento da medula, ou ainda hiperesplenismo.
Linfoma folicular. Depois do LDGCB, o linfoma folicular ocupa o 2º lugar entre os subtipos histológicos mais prevalentes, constituindo 35% dos casos de LNH em adultos, nos Estados Unidos.10 Os pacientes tipicamente são idosos e apresentam doença disseminada no momento do diagnóstico. Durante o curso da doença, a transformação do linfoma folicular em linfoma de grandes células é observada em até 35% dos pacientes.10 A sobrevida média é de aproximadamente 10 anos, e o curso clínico é caracterizado pela resposta ao tratamento seguida de progressão da doença.
Histologicamente, o linfoma folicular é classificado em graus 1, 2 e 3, de acordo com o número de centroblastos por campo de maior aumento. O grau histológico está correlacionado com o prognóstico: os graus 1 e 2 são indolentes, enquanto o grau 3ª é agressivo e pode ser curado com quimioterapia sistêmica. O grau 3B é considerado uma variante do LDGCB, para fins de tratamento, e é potencialmente curável.
Como os linfomas foliculares de graus 1 e 2 são considerados incuráveis, existem várias abordagens terapêuticas razoáveis. Para pacientes selecionados, com doenças em estágios I e II de Ann Arbor, o adiamento da terapia constitui uma opção aceitável. Em um estudo sobre o linfoma folicular em estágio inicial, mais da metade dos pacientes dispensaram tratamento durante um período de 6 anos de seguimento, e a sobrevida dos pacientes não tratados foi comparável à sobrevida dos pacientes tratados de outra série.110
O tratamento apenas com radiação foi avaliado no linfoma folicular em estágio inicial. Nenhum estudo prospectivo randomizado comparou a radioterapia a outra modalidade terapêutica, contudo o uso apenas da radiação no tratamento de pacientes seletos produziu taxas de sobrevida de 10 anos livre de doença superiores a 60%.111 A fludarabina é um fármaco bastante efetivo no linfoma folicular, apresentando altas taxas de resposta tanto na doença não tratada como na doença recidivante.112 Os agentes alquilantes, como a ciclofosfamida e a prednisona, também promovem altas taxas de resposta e podem ser combinados à fludarabina.113 A terapia de modalidade combinada também produz taxas de resposta satisfatórias em casos de doença em estágio inicial.
O rituximabe, um anticorpo monoclonal dirigido contra o CD20, é bastante efetivo no tratamento do linfoma folicular. Este fármaco promove taxas de resposta de até 73% em pacientes não tratados e de 43% em pacientes com doença recidivante ou refratária.114,115 Em pacientes previamente responsivos ao rituximabe, a taxa de resposta ao retratamento é de 40%.116 Os regimes quimioterápicos, como CHOP ou fludarabina com rituximabe, são altamente ativos e podem produzir respostas duradouras.117,118 Em um estudo randomizado sobre o uso de CHOP com ou sem rituximabe no tratamento do linfoma folicular, o rituximabe foi associado a uma taxa de resposta significativamente maior e um benefício terapêutico mais duradouro.119 O rituximabe também exerce um papel em potencial na terapia de manutenção, em que pode melhorar a sobrevida livre de progressão, e também na terapia combinada à quimioterapia sistêmica no tratamento inicial do linfoma folicular.117,120 Estes resultados sugerem que o rituximabe tende a tornar-se um componente-padrão da quimioterapia inicial.
Anticorpos anti-CD20 de última geração, projetados com melhorias adicionais em relação ao rituximabe, estão sendo desenvolvidos. Entre estes agentes, estão o ofatumumabe e o veltuzumabe, que apresentam atividade modesta em pacientes com linfoma folicular recidivante.121,122 Recentemente, os anticorpos monoclonais dirigidos contra antígenos diferentes do CD20, como o galiximabe, que tem como alvo o CD80, e o epratuzumabe, que constitui um anticorpo humanizado anti-CD22, têm apresentado atividade promissora no linfoma folicular.123,124
A radioimunoterapia do linfoma folicular envolve a distribuição de radioterapia dirigida ao tecido tumoral, por meio da conjugação de um anticorpo anti-CD20 ao ítrio-90 ou ao iodo-131. Os radioimunoconjugados anti-CD20 ibritumomabe tiuxetana e tositumomabe são aprovados para uso no tratamento do linfoma folicular recidivante ou refratário. Em um estudo randomizado sobre linfoma folicular recidivante ou refratário ou linfoma transformado, a taxa de resposta geral foi melhor com o uso de ibritumomabe tiuxetana do que com o uso de rituximabe (80% vs. 56%).125 A radioimunoterapia combinada à quimioterapia sistêmica também está sendo avaliada.126 Em um estudo que usou tositumomabe como tratamento inicial do linfoma folicular, 75% dos pacientes apresentaram resposta integral, e a média da sobrevida livre de progressão foi de 6,1 anos.127 O papel exato da radioimunoterapia no tratamento do linfoma folicular ainda precisa ser definido, mas os resultados obtidos até agora são bastante promissores.
O transplante de células-tronco, tanto autólogo como alogênico, pode induzir remissões duráveis no linfoma folicular. O alotransplante pode ser curativo em casos de pacientes seletos.128 Outra estratégia imune inclui a vacinação idiotípica, que envolve o isolamento de antígeno tumor-específico – a proteína idiotípica – a partir da neoplasia, seguido de conjugação e manipulação imunológica in vitro. Embora esta estratégia seja efetiva para muitos tipos de tumor, os resultados até então alcançados no linfoma folicular são conflitantes.129,130 Recentemente, um inibidor de alta afinidade das proteínas da família bcl-2 apresentou resultados promissores.131 Muitos outros agentes apresentaram atividade no linfoma folicular recidivante ou refratário, como o inibidor de proteassoma bortezomibe e também a lenalidomida, que é um análogo estrutural e funcional da talidomida.132,133
Linfoma linfocítico de pequenas células (LLP) e leucemia linfocítica crônica (LLC). O LLP representa o tumor sólido constituinte da LLC, e as 2 entidades são consideradas biologicamente relacionadas. A maioria dos pacientes é idosa. Todos os pacientes com LLC apresentam envolvimento do sangue periférico e da medula óssea no momento do diagnóstico, enquanto o LLP pode ser diagnóstico na ausência destes achados. O tratamento da LLC e do LLP é semelhante ao tratamento do linfoma folicular, em que a fludarabina e o rituximabe exercem papéis terapêuticos importantes.
Linfoma da zona marginal. Os linfomas de células B da zona marginal extranodal do tecido linfoide associado à mucosa (linfomas MALT) constituem 7 a 8% de todos os linfomas de célula B e uma ampla proporção dos linfomas gástricos primários.10 Estes linfomas tendem a ser encontrados no trato GI, glândulas salivares, pulmões, acessórios oculares, mama e outros sítios extranodais. A maioria dos pacientes apresenta doença em estágio inicial. Uma história de distúrbio inflamatório crônico, como gastrite crônica associada a Helicobacter ou síndrome de Sjögren, é comum. Em um pequeno percentual de pacientes com linfomas MALT gástricos, linfodo-negativos e superficiais, é possível haver remissões sustentadas após a erradicação de Helicobacter com terapia antibiótica.134 Contudo, estes tumores são sensíveis à quimioterapia, rituximabe ou radioterapia, usados de forma isolada ou combinados. O tratamento da doença em estágio inicial pode resultar em remissões duradouras.135 Os linfomas MALT tendem a seguir um curso indolente, permanecendo localizados por longos períodos.
O linfoma de zona marginal esplênico, considerado uma entidade à parte na classificação da OMS, envolve o baço, linfonodos hilares esplênicos, medula e sangue periférico.10 Este linfoma segue um curso indolente característico e é classicamente tratado com esplenectomia. No entanto, o tratamento com rituximabe pode enfim substituir esta abordagem. O rituximabe tem sido usado no tratamento de linfomas de zona marginal extranodais.136 O linfoma de zona marginal nodal é uma rara neoplasia de células B nodal primária, considerada a contraparte nodal do linfoma MALT.
Entre os linfomas de células B agressivos estão o linfoma de células de revestimento, LDGCB, LB e linfoma linfoblástico. O LDGCB é o mais comum e corresponde a cerca de 30 a 40% de todos os LNH que ocorrem em pacientes adultos no países ocidentais.10 Tanto o LDGCB como o LB ocorrem no contexto da infecção pelo HIV. Diferente do que se dá com os linfomas de célula B indolentes, os subtipos agressivos são (como indica o nome) mais rapidamente progressivos. Com exceção do linfoma de células de revestimento, estes subtipos são potencialmente curáveis. Desta forma, estes tumores exigem uma avaliação diligente, bem como tratamento adequado imediato.
Linfoma de células de revestimento. O linfoma de células de revestimento é uma neoplasia de células B, que é composta por células linfoides de tamanho pequeno a médio e contornos nucleares irregulares. Ocorre, em média, ao redor dos 60 anos de idade e predomina na população masculina.10 A sobrevida média dos pacientes é de 3 anos.137 As variantes morfológicas do linfoma de células revestimento incluem 2 variantes blastoides, que estão associadas a um prognóstico pior. A translocação t(11;14) está presente na maioria dos casos. Quase todos estes tumores expressam ciclina D1 (bcl-1), e, assim, um anticorpo monoclonal dirigido contra esta proteína é utilizado na rotina de confirmação do diagnóstico. Os linfomas de células de revestimento podem responder à quimioterapia agressiva, mas as respostas em geral são breves, e a doença é considerada incurável. Dependendo das circunstâncias clínicas, as estratégias de tratamento variam da observação ao tratamento agressivo.
No linfoma de células de revestimento, embora a taxa de resposta geral seja de 89% com CHOP, a sobrevida geral média é de meros 37 meses.138 Contudo, o regime hiper-CVAD (hiperfracionamento de ciclofosfamida, vincristina, doxorubicina [Adriamicina] e dexametasona, alternadas com metotrexato e citarabina) é efetivo, e a adição de rituximabe intensifica seu efeito.139 Em um estudo sobre linfoma de células de revestimento não tratado, o DA-EPOCH-R (regime dose-ajustado contendo etoposídeo, prednisona, vincristina [Oncovin], ciclofosfamida, doxorubicina-rituximabe) produziu uma taxa de resposta completa da ordem de 93%.140 Estudos sobre doses altas de quimioterapia aliadas ao transplante autólogo de células-tronco ou ao transplante alogênico estão em andamento, mas até agora têm produzido resultados variáveis.141 Sendo assim, o transplante deve ser atualmente considerado para uso experimental no tratamento do linfoma de células de revestimento. Outros agentes que demonstraram atividade contra este tipo de linfoma são o inibidor de proteassoma bortezomibe; o inibidor do alvo da rapamicina em mamíferos (mTOR) tensirolimo; e o agente imunomodulador lenalidomida.132,142-144
Linfoma de grandes células B difusos (LDGCB). O LDGCB é o subtipo histológico de LNH mais prevalente. Em termos de histologia, este linfoma consiste na proliferação difusa de grandes células B neoplásicas. Embora os pacientes sejam diagnosticados em média na 7ª década da vida, o LDGCB afeta crianças e adultos de qualquer idade. Os pacientes podem apresentar doença nodal ou extranodal, localizada ou disseminada. O sítio extranodal mais comum é o trato GI, mas qualquer tecido extranodal pode ser afetado. O LDGCB pode surgir de novo ou resultar da transformação histológica de um linfoma de células B indolente. As variantes morfológicas existentes em cada categoria de LDGCB são: centroblástica, imunoblástica, rica em células T/histiócitos e anaplásica.10 O LDGCB pode surgir em qualquer órgão. Desta forma, diferentes comportamentos clínicos e histórias naturais são encontrados em alguns subtipos, como no linfoma de células B mediastínico primário, que surge no mediastino, ou o linfoma do SNC (ver adiante).
O uso apenas de radiação é inadequado como tratamento do LDGCB e está associado a altas taxas de recorrência, tanto ao nível local como distalmente.145 Por este motivo, todos os estágios da doença requerem no mínimo quimioterapia sistêmica. No caso da doença em estágio inicial (I ou II), a questão da adequação do tratamento do paciente apenas com quimioterapia ainda é controversa. O benefício proporcionado pelo tratamento de modalidade combinada foi demonstrado em um estudo randomizado, que comparou um curso integral de CHOP a um curso limitado de CHOP aliado à irradiação do campo envolvido no tratamento do linfoma agressivo em estágio inicial.146 Neste estudo, o tratamento de modalidade combinada foi associado a taxas superiores de sobrevida geral e de sobrevida livre de doença iguais a 82% e 77%, respectivamente. Entretanto, uma análise de seguimento mostrou uma convergência das curvas de sobrevida geral, devida ao aumento da incidência de recidivas sistêmicas tardias no grupo da modalidade terapêutica combinada.147 Além disso, o estudo do Eastern Cooperative Oncology Group não demonstrou nenhum benefício promovido pelo tratamento de modalidade combinada em comparação aos benefícios alcançados apenas com a quimioterapia.148 Estes resultados sugerem que a irradiação do campo envolvido pode ser desnecessária quando são usados tratamentos sistêmicos mais efetivos, como R-CHOP ou ACVBP (doxorubicina [Adriamicina], ciclofosfamida, vindesina, bleomicina e prednisona).85,149
Alguns pacientes com LDGCB mediastínico primário em estágio inicial (LCBMP), todavia, podem necessitar de irradiação do campo envolvido após a quimioterapia. Em um estudo envolvendo 50 pacientes com LCBMP não tratado, que receberam MACOP-B (metotrexato com leucovorina de resgate, doxorubicina [Adriamicina], ciclofosfamida, vincristina [Oncovin], prednisona e bleomicina) seguido de radiação, 66% apresentaram positividade persistente na varredura com gálio após o tratamento apenas com quimioterapia, sugerindo a existência de doença ativa.150 Contudo, o tratamento do LCBMP empregando outros regimes que não incluíam a irradiação do campo envolvido, como DA-EPOCH, produziu resultados satisfatórios.75,149 Além disso, o rituximabe melhorou o resultado alcançado com o tratamento do LDGCB.85 Desta forma, é razoável limitar o uso da irradiação do campo envolvido aos pacientes que apresentam positividade persistente nas varreduras de TEP após receberem estes tratamentos.
A doença avançada é tratada com quimioterapia sistêmica. O regime CHOP, desenvolvido nos anos 1970, promove remissões prolongadas em cerca de 35% dos pacientes com LDGCB.84 Foram desenvolvidos alguns regimes mais modernos, em que foram introduzidas melhorias em relação ao CHOP.85,151-155 Em um estudo randomizado, a adição de rituximabe ao CHOP resultou em taxas de resposta e de sobrevida melhores em pacientes com mais de 60 anos de idade. Desta forma, o regime R-CHOP emergiu como padrão de fato para o tratamento do LDGCB.85 O benefício promovido pela adição do rituximabe ao CHOP foi, ainda, validado pelo estudo do US Intergroup, conduzido em uma população de pacientes similar, e também pelo estudo MabThera International Trial (MINT), que randomizou pacientes com bom prognóstico e idade superior a 60 anos para receberem CHOP ou um regime CHOP-símile, com ou sem rituximabe.156,157 Nos casos de linfoma agressivo com prognóstico ruim, o regime ACVBP melhorou as taxas de sobrevida livre de eventos e de sobrevida geral, em comparação ao uso apenas de CHOP, na população de pacientes com mais de 60 anos.152 O sucesso do regime DA-EPOCH no tratamento do LDGCB não tratado levou os pesquisadores a investigarem o regime DA-EPOCH-R. Este regime, por sua vez, promoveu taxas de sobrevida livre de progressão e de sobrevida geral aproximadas de 80% em um período médio de seguimento de 4 anos, e, atualmente, está sendo comparado ao regime R-CHOP em um estudo randomizado do Cancer and Leukemia Group B (CALGB).75,153 Diferente do CHOP, o regime DA-EPOCH parece ser efetivo no tratamento do LDGCB com um alto índice proliferativo.75 O regime CHOP dose-denso (administrado a cada 2 semanas) foi comparado ao CHOP administrado a cada 3 semanas, com ou sem adição de etoposídeo (CHOP vs. EPOCH), no tratamento de pacientes jovens e idosos.154,155 Entre os pacientes jovens com linfomas agressivos cujos prognósticos eram favoráveis, observou-se uma sobrevida melhor associada ao regime EPOCH. Os pacientes idosos apresentaram resultados melhores em resposta ao regime CHOP dose-denso, em comparação do regime EPOCH. Atualmente, o CHOP dose-denso com rituximabe está sendo investigado e comparado ao regime R-CHOP administrado a cada 3 semanas.
O tratamento inicial do LDGCB agressivo com altas doses de quimioterapia e transplante autólogo continua sendo estudado. No entanto, ainda não foi comprovado de maneira definitiva que este tratamento seja mais efetivo do que a quimioterapia convencional.156,157 Um estudo demonstrou que esta abordagem produz resultados melhores do que o CHOP, mas esta conclusão foi confundida pela inclusão de múltiplos subtipos de linfoma neste estudo.158 Além disso, o regime R-CHOP atualmente é considerado o padrão de tratamento do LDGCB, diminuindo assim o valor dos estudos que avaliam o uso isolado de CHOP.
As opções de tratamento para o LDGCB recidivante incluem a quimioterapia de resgate e a quimioterapia com doses altas aliada ao transplante autólogo ou alogênico. Para os pacientes que apresentam recidiva quimiossensível na ausência de envolvimento medular, a terapia com doses altas e transplante autólogo geralmente oferece chances melhores de resposta e remissão.156 Com um percentual maior dos atuais pacientes recebendo regimes mais efetivos do que o CHOP, o benefício relativo proporcionado pela terapia com doses altas diminuirá, pois os pacientes de hoje apresentarão doença mais resistente. Benefícios limitados podem ser alcançados com o uso dos regimes de resgate convencionais. E a escolha entre a quimioterapia de resgate ou a quimioterapia de doses altas com transplante autólogo é influenciada por diversos fatores. Os pacientes que apresentam recidiva após receberem transplante autólogo podem ser beneficiados por um transplante alogênico.159
Linfoma primário do sistema nervoso central (LPSNC). O linfoma primário do sistema nervoso central (LPSNC) é um linfoma raro, que permanece confinado no SNC. Do ponto de vista histológico, em geral, trata-se de um LDGCB. O tratamento do LPSNC difere do tratamento do LNH sistêmico, pois muitos dos agentes quimioterápicos usados no tratamento deste último não atravessam a barreira hematoencefálica. Entretanto, pode haver uma justificativa lógica para o uso destes agentes no tratamento de pacientes em que o LPSNC rompe esta barreira. O LPSNC é responsivo à radiação, mas as respostas produzidas tendem a ser de curta duração, e as taxas de recidiva são altas. O metotrexato é bastante efetivo sobre o LPSNC, mas quando usado de forma isolada produz baixa taxa de sobrevida média livre de progressão (apenas 12,8 meses).160 O metotrexato combinado a outros agentes quimioterápicos é potencialmente curativo, em particular em casos de pacientes com menos de 60 anos de idade, com prognóstico geral de sobrevida de 34% em 5 anos.161
O papel da radioterapia de consolidação subsequente à quimioterapia no tratamento do LPSNC é controverso. A radioterapia craniana total está associada a uma significativa neurotoxicidade, em particular em pacientes idosos. Por este motivo, se os resultados alcançados forem equivalentes àqueles produzidos pela quimioterapia isolada, a radioterapia deve ser adiada até a recidiva da doença. Em um estudo alemão, o grupo de Bonn et al. relatou resultados satisfatórios com o uso da quimioterapia e adiou a radioterapia.148 Esta pode vir a tornar-se a abordagem-padrão para o tratamento do LPSNC, no futuro.160,162 O papel do rituximabe no tratamento deste linfoma ainda precisa ser mais bem definido. No entanto, como estas doenças são positivas para CD20, assim como o LDGCB sistêmico, o rituximabe pode ser benéfico quando adicionado à quimioterapia.
Linfoma de Burkitt (LB). O LB é um linfoma altamente agressivo que ocorre em 3 contextos clínicos: endêmico, esporádico e associado à imunodeficiência. O LB endêmico ocorre em crianças pequenas que vivem na região equatorial da África. Clinicamente, estes pacientes costumam apresentar envolvimento dos ossos da mandíbula e da face. O LB esporádico, que é mais comum em crianças e adultos jovens, frequentemente se manifesta como doença abdominal ou envolvimento de outro sítio extranodal. O LB associado à imunodeficiência geralmente é observado em pacientes infectados pelo HIV.
Existem graus variáveis de positividade para a infecção pelo EBV com LB. Todos os pacientes apresentam a translocação de MYC.
O LB é prontamente curável com quimioterapia sistêmica. Regimes bastante intensivos, que incluem terapia intratecal profilática, são tradicionalmente empregados e continuam sendo considerados o padrão de tratamento, apesar das toxicidades significativas.163,164
Por diversos motivos, entre os quais a desorganização da imunovigilância e a estimulação antigênica crônica, a incidência do linfoma é significativamente aumentada entre os pacientes infectados pelo HIV. Cerca de 4% dos pacientes com AIDS apresentam LNH no momento do diagnóstico da AIDS. Embora a incidência de todos os subtipos de linfoma seja maior entre os pacientes infectados pelo HIV, o risco é 1.000 vezes maior para o LB e até 400 vezes maior para o linfoma agressivo.149,165 O linfoma de efusão primária e o linfoma plasmablástico são quase exclusivamente encontrados em pacientes infectados pelo HIV, porém ambos estão associados a um prognóstico bastante ruim. Os linfomas também são observados com maior frequência após o transplante de órgãos sólidos; em algumas síndromes autoimunes e síndromes de imunodeficiência hereditária; e associado a certas terapias, como o metotrexato.
As estratégias de tratamento com doses baixas eram consideradas padrão nos primeiros anos do tratamento dos linfomas associados à AIDS. Entretanto, as terapias antirretrovirais altamente ativas (HAART) melhoraram a sobrevida e, assim, permitiram tratar os linfomas de maneira mais agressiva, com intensão curativa, e não apenas paliativa.166 As abordagens quimioterápicas sistêmicas para LB e LDGCB associado à AIDS incluem o regime CHOP, contudo a eficácia deste regime é menor, e sua toxicidade é maior do que em pacientes HIV-negativos com linfomas similares.167 Um dos principais aspectos controversos do tratamento do linfoma associado à AIDS tem sido a suspensão da HAART durante a quimioterapia. A HAART pode intensificar a toxicidade dos fármacos quimioterápicos, e isto pode levar à necessidade de se reduzir a dose de quimioterapia, com consequente comprometimento da eficácia terapêutica. Uma abordagem consiste em suspender a HAART durante a quimioterapia, com imediata retomada após a conclusão de todos os ciclos. O regime infusional de EPOCH, com suspensão de HAART, produziu uma taxa de resposta completa de 74% e uma taxa de sobrevida geral de 72% em um período de seguimento médio de 53 meses.95 Estes resultados foram significativamente superiores àqueles obtidos com o regime CHOP. O AIDS Malignancy Consortium atualmente está testando o regime EPOCH combinado ao rituximabe em um estudo randomizado.
Vários distúrbios linfoproliferativos estão associados à infecção pelo EBV. O distúrbio linfoproliferativo pós-transplante (DLPT), que frequentemente ocorre após o transplante de um órgão sólido, engloba um amplo espectro de doenças com graus variáveis de agressividade clínica. Este distúrbio geralmente é tratado com a retirada da imunossupressão crônica, mas pode haver necessidade de quimioterapia nos casos em que a DLPT se comporta como um linfoma agressivo ou é resistente à descontinuação da imunossupressão. Os distúrbios linfoproliferativos associados ao metotrexato também podem estar associados à infecção pelo EBV. Por fim, a granulomatose linfomatoide, um distúrbio linfoproliferativo envolvendo sítios extranodais, é dirigida pela infecção por EBV e tratada com interferon ou quimioterapia, dependendo do grau de doença.168
Os linfomas de precursores linfoblásticos são citologicamente idênticos à leucemia linfoblástica aguda. De uma forma geral, são linfomas de células T e ocorrem mais comumente em meninos adolescentes ou homens jovens. Os pacientes costumam apresentar contagem elevada de leucócitos e uma ampla massa mediastínica. O tratamento é feito à base de regimes apropriados para a leucemia aguda e que incluam profilaxia contra o envolvimento do SNC.
Os linfomas derivados de células T maduras e de células NK são incomuns, correspondendo a cerca de 12% de todos os LNH.10 Sua incidência varia de forma significativa, conforme a localização geográfica, sendo maior na Ásia. Nas classificações de linfoma anteriores à atual classificação da OMS, alguns destes linfomas eram considerados não classificáveis ou não eram reconhecidos. A história natural de linfoma de células T maduras ou de linfoma de células NK varia de indolente a agressiva. Contudo, diferente dos linfomas de células B agressivos, a maioria dos linfomas agressivos de células T e NK é incurável. As exceções importantes, todavia, incluem o linfoma de grandes células anaplásicas (LGCA) sistêmico, que é altamente curável quando expressa a proteína de fusão quinase do linfoma anaplásico (ALK), e o linfoma angiocêntrico nasal (LAN) NK e T em estágio inicial, que é curável com altas doses de radioterapia.169,170 Embora uma descrição na íntegra destas doenças relativamente raras esteja além do escopo deste capítulo, vários subtipos importantes são destacados adiante.
Os linfomas cutâneos de células T incluem a micose fungoide, síndrome de Sézary, LGCA cutâneo primário e papulose linfomatoide. A micose fungoide manifesta-se na pele, sob a forma de manchas e placas. A doença segue um curso indolente e possui uma longa história natural. Pode haver desenvolvimento de eritroderma geral. A disseminação generalizada da doença é em geral um evento tardiamente observado na micose fungoide. Algumas terapias tópicas e locais, como a radiação ultravioleta e a terapia cutânea com feixe de elétrons, são bastante efetivas no tratamento da micose fungoide. Entre os agentes sistêmicos estão o bexatoteno (um novo retinoide sintético seletivo para receptor X retinoico), a denileucina diftitox (uma toxina de fusão) e os inibidores de histona desacetilase.171,172 O transplante alogênico foi investigado como tratamento potencialmente curativo para a micose fungoide em estágio avançado.173
A síndrome de Sézary consiste na presença de células T neoplásicas, eritroderma e linfadenopatia. Diferente da micose fungoide, a síndrome de Sézary é bastante agressiva.
O LGCA afeta a pele e difere do LGCA sistêmico. Nos casos de envolvimento cutâneo localizado, as terapias dirigidas ao tratamento da pele são apropriadas. A doença extracutânea requer terapias mais agressivas. A papulose linfomatoide geralmente segue um curso clínico benigno e às vezes requer terapia, como doses baixas de metotrexato.
O LGCA, um raro linfoma de células T que corresponde a 3% de todos os LNH em indivíduos adultos, ocorre tipicamente em homens. O LGCA é bastante curável, e os pacientes apresentam taxa de sobrevida a longo prazo de 70%, após o tratamento quimioterápico sistêmico. A principal característica histológica do LGCA é a positividade para CD30. Além disso, a maioria destes tumores expressa antígeno epitelial de membrana.174 O LGCA está associado à translocação t(2;5)(p23;q35), que resulta na expressão da nucleofosmina ALK. A positividade para ALK, observada em mais de 50% dos LGCALGCA, está associada a um prognóstico melhor do que a doença ALK-negativa.169 O LGCA ALK-positivo ocorre com frequência em crianças e adultos jovens. A maioria dos pacientes apresenta doença em estágio avançado e pode ter envolvimento de sítios extranodais. O LGCA responde muito bem à quimioterapia sistêmica, em particular quando a doença é ALK-positiva.
Este tipo de linfoma é um tipo distinto de linfoma de células T periférico, que está associado à presença de células B EBV-positivas dispersas em amostras de biópsia. Clinicamente, esta doença é caracterizada pela manifestação de sintomas sistêmicos, erupção cutânea, organomegalia, hipergamaglobulinemia e anemia hemolítica. Os pacientes exibem imunodeficiência e são particularmente suscetíveis a infecções. O linfoma de células T angioimunoblástico geralmente segue um curso clínico agressivo e costuma ser incurável por quimioterapia convencional.175
Um número significativo de linfomas de células T predominantemente nodais são excluídos das demais categorias do linfoma de células T da classificação da OMS. Este grupo de linfomas é bastante diversificado, tanto em termos de morfologia como clinicamente. Os pacientes em geral apresentam resposta precária a longo prazo à quimioterapia convencional, com alta taxa de recidiva e taxas baixas de sobrevida geral.176 A quimioterapia com doses altas seguida de transplante de células-tronco autólogo tem sido empregada com algum sucesso no tratamento de pacientes com doença recidivante, proporcionando alguma chance de remissão prolongada.177,178 Os resultados iniciais obtidos com o transplante alogênico no tratamento do linfoma de células T periférico recidivante são promissores e estão associados a algum efeito evidente de enxerto vs. linfoma.179 Recentemente, o pralatrexato, um inibidor metabólico semelhante ao ácido fólico, apresentou certo grau de atividade no linfoma de células T periférico (e há pouco foi aprovado para uso pelo Food and Drug Administration).180 Outros agentes, como os anticorpos monoclonais dirigidos contra antígenos da célula T (CD30 e CD52) e imunotoxinas (p. ex., denileucina difitox), também estão sendo investigados.
Os autores não possuem relações comerciais com os fabricantes de produtos e prestadores de serviços mencionados neste capítulo.
1. Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2009. CA Cancer J Clin 2009;59:225–49.
2. Correa P, O’Conor GT. Epidemiologic patterns of Hodgkin disease. Int J Cancer 1971;8:192.
3. Grufferman S, Cole P, Smith PG, et al. Hodgkin disease in siblings. N Engl J Med 1977;296:248.
4. Mack TM, Cozen W, Shibata DK, et al. Concordance for Hodgkin disease in identical twins suggesting genetic susceptibility to the young-adult form of the disease. N Engl J Med 1995;332:413.
5. Klitz W, Aldrich CL, Fildes N, et al. Localization of predisposition to Hodgkin disease in the HLA class II region. Am J Hum Genet 1994;54:497.
6. Harty LC, Lin AY, Goldstein AM, et al. HLA-DR, HLADQ, and TAP genes in familial Hodgkin disease. Blood 2002;99:690.
7. Mueller N, Evans A, Harris NL, et al. Hodgkin disease and Epstein-Barr virus: altered antibody pattern before diagnosis. N Engl J Med 1989;320:689.
8. Alexander FE, Jarrett RF, Lawrence D, et al. Risk factors for Hodgkin disease by Epstein-Barr virus (EBV) status: prior infection by EBV and other agents. Br J Cancer 2000;82:1117.
9. Weiss LM, Chen YY, Liu XF, et al. Epstein-Barr virus and Hodgkin disease: a correlative in situ hybridization and polymerase chain reaction study. Am J Pathol 1991; 139:1259.
10. Swerdlow SH, Campo E, Harris NL, editors. World Health Organization classification of tumors of haematopoietic and lymphoid tissues. Lyon: IARC; 2008.
11. Kuppers R, Rajewsky K. The origin of Hodgkin and Reed/ Sternberg cells in Hodgkin disease. Annu Rev Immunol 1998;16:471.
12. Emmerich F, Meiser M, Hummel M, et al. Overexpression of I kappa B alpha without inhibition of NF-kappaB activity and mutations in the I kappa B alpha gene in Reed-Sternberg cells. Blood 1999;94:3129.
13. Jungnickel B, Staratschek-Jox A, Brauninger A, et al. Clonal deleterious mutations in the IkappaBalpha gene in the malignant cells in Hodgkin lymphoma. J Exp Med 2000; 191:395.
14. Thomas RK, Re D, Wolf J, et al. Part I: Hodgkin lymphoma: molecular biology of Hodgkin and Reed-Sternberg cells. Lancet Oncol 2004;5:11.
15. Navarro A, Gaya A, Martinez A, et al. MicroRNA expression profiling in classic Hodgkin lymphoma. Blood 2008;111:2825–32.
16. Diehl V, Thomas RK, Re D. Part II: Hodgkin lymphoma: diagnosis and treatment. Lancet Oncol 2004;5:19.
17. Staak JO, Dietlein M, Engert A, et al. [Hodgkin lymphoma in nuclear medicine: diagnostic and therapeutic aspects]. Nuklearmedizin 2003;42:19.
18. Zinzani PL, Fanti S, Battista G, et al. Predictive role of positron emission tomography (PET) in the outcome of lymphoma patients. Br J Cancer 2004;91:850.
19. Castellino RA, Hoppe RT, Blank N, et al. Computed tomography, lymphography, and staging laparotomy: correlations in initial staging of Hodgkin disease. AJR Am J Roentgenol 1984;143:37.
20. Carde P, Hagenbeek A, Hayat M, et al. Clinical staging versus laparotomy and combined modality with MOPP versus ABVD in early-stage Hodgkin disease: the H6 twin randomized trials from the European Organization for Research and Treatment of Cancer Lymphoma Cooperative Group. J Clin Oncol 1993;11:2258.
21. Franco V, Tripodo C, Rizzo A, et al. Bone marrow biopsy in Hodgkin lymphoma. Eur J Haematol 2004;73:149.
22. Howell SJ, Grey M, Chang J, et al. The value of bone marrow examination in the staging of Hodgkin lymphoma: a review of 955 cases seen in a regional cancer centre. Br J Haematol 2002;119:408.
23. Lister TA, Crowther D, Sutcliffe SB, et al. Report of a committee convened to discuss the evaluation and staging of patients with Hodgkin disease: Cotswolds meeting. J Clin Oncol 1989;7:1630.
24. Hasenclever D, Diehl V. A prognostic score for advanced Hodgkin disease. International Prognostic Factors Project on Advanced Hodgkin Disease. N Engl J Med 1998; 339:1506.
25. Cosset JM, Henry-Amar M, Meerwaldt JH, et al. The EORTC trials for limited stage Hodgkin’s disease. The EORTC Lymphoma Cooperative Group. Eur J Cancer 1992;28A:1847.
26. Karmiris TD, Grigoriou E, Tsantekidou M, et al. Treatment of early clinically staged Hodgkin disease with a combination of ABVD chemotherapy plus limited field radiotherapy. Leuk Lymphoma 2003;44:1523.
27. Bonadonna G, Bonfante V, Viviani S, et al. ABVD plus subtotal nodal versus involved-field radiotherapy in early-stage Hodgkin disease: long-term results. J Clin Oncol 2004;22:2835.
28. Bessell EM, MacLennan KA, Toghill PJ, et al. Suprahyoid Hodgkin’s disease stage IA. Radiother Oncol 1991;22:190.
29. Straus DJ, Portlock CS, Qin J, et al. Results of a prospective randomized clinical trial of doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD) followed by radiation therapy (RT) versus ABVD alone for stages I, II, and IIIA nonbulky Hodgkin disease. Blood 2004;104:3483.
30. Meyer RM, Gospodarowicz MK, Connors JM, et al. Randomized comparison of ABVD chemotherapy with a strategy that includes radiation therapy in patients with limited-stage Hodgkin’s lymphoma: National Cancer Institute of Canada Clinical Trials Group and the Eastern Cooperative Oncology Group. J Clin Oncol 2005;23: 4634–42.
31. Strauss DJ, Portlock CS, Qin J, et al. Results of a prospective randomized clinical trial of doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD) followed by radiation therapy (RT) versus ABVD alone for stages I, II, and IIIA nonbulky Hodgkin disease. Blood 2004;104:3483–9.
32. Canellos GP, Niedzwiecki D. Long-term follow-up of Hodgkin disease trial. N Engl J Med 2002;346:1417.
33. Duggan DB, Petroni GR, Johnson JL, et al. Randomized comparison of ABVD and MOPP/ABV hybrid for the treatment of advanced Hodgkin disease: report of an Intergroup trial. J Clin Oncol 2003;21:607.
34. Horning SJ, Hoppe RT, Breslin S, et al. Stanford V and radiotherapy for locally extensive and advanced Hodgkin disease: mature results of a prospective clinical trial. J Clin Oncol 2002;20:630.
35. Chisesi T, Federico M, Levis A, et al. ABVD versus Stanford V versus MEC in unfavourable Hodgkin’s lymphoma: results of a randomised trial. Ann Oncol 2002;13 Suppl 1:102.
36. Diehl V, Franklin J, Pfreundschuh M, et al. Standard and increased-dose BEACOPP chemotherapy compared with COPP-ABVD for advanced Hodgkin disease. N Engl J Med 2003;348:2386.
37. Sieber M, Bredenfeld H, Josting A, et al. 14-Day variant of the bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, and prednisone regimen in advanced-stage Hodgkin’s lymphoma: results of a pilot study of the German Hodgkin’s Lymphoma Study Group. J Clin Oncol 2003;21:1734–9.
38. Diehl V, Sextro M, Franklin J, et al. Clinical presentation, course, and prognostic factors in lymphocyte-predominant Hodgkin disease and lymphocyte-rich classical Hodgkin disease: report from the European Task Force on Lymphoma Project on Lymphocyte-Predominant Hodgkin Disease. J Clin Oncol 1999;17:776.
39. Ekstrand BC, Lucas JB, Horwitz SM, et al. Rituximab in lymphocyte-predominant Hodgkin disease: results of a phase 2 trial. Blood 2003;101:4285.
40. Ng AK, Li S, Neuberg D, et al. Comparison of MOPP versus ABVD as salvage therapy in patients who relapse after radiation therapy alone for Hodgkin disease. Ann Oncol 2004;15:270.
41. Lohri A, Barnett M, Fairey RN, et al. Outcome of treatment of first relapse of Hodgkin disease after primary chemotherapy: identification of risk factors from the British Columbia experience 1970 to 1988. Blood 1991;77:2292.
42. Viviani S, Santoro A, Negretti E, et al. Salvage chemotherapy in Hodgkin disease: results in patients relapsing more than twelve months after first complete remission. Ann Oncol 1990;1:123.
43. Vassilakopoulos TP, Angelopoulou MK, Siakantaris MP, et al. Hodgkin lymphoma in first relapse following chemotherapy or combined modality therapy: analysis of outcome and prognostic factors after conventional salvage therapy. Eur J Haematol 2002;68:289.
44. Yuen AR, Rosenberg SA, Hoppe RT, et al. Comparison between conventional salvage therapy and high-dose therapy with autografting for recurrent or refractory Hodgkin disease. Blood 1997;89:814.
45. Bonfante V, Santoro A, Viviani S, et al. Outcome of patients with Hodgkin disease failing after primary MOPP-ABVD. J Clin Oncol 1997;15:528.
46. Diehl V, Stein H, Hummel M, et al. Hodgkin’s lymphoma: biology and treatment strategies for primary, refractory, and relapsed disease. Hematol-Am Soc Hematol Educ Program 2003;225–47.
47. Aparicio J, Segura A, Garcera S, et al. ESHAP is an active regimen for relapsing Hodgkin’s disease. Ann Oncol 1999;10:593.
48. Moskowitz CH, Bertino JR, Glassman JR, et al. Ifosfamide, carboplatin, and etoposide: a highly effective cytoreduction and peripheral-blood progenitor-cell mobilization regimen for transplant-eligible patients with nonHodgkin’s lymphoma. J Clin Oncol 1999;17:3776.
49. Dunleavy K, Butrynski J, Steinberg S, et al. Phase II study of EPOCH infusional chemotherapy in relapsed or refractory Hodgkin’s lymphoma (HL): a report on toxicity, efficacy, and prognostic indicators of outcome. J Clin Oncol 2004;22:6598.
50. Little R, Wittes RE, Longo DL, et al. Vinblastine for recurrent Hodgkin’s disease following autologous bone marrow transplant. J Clin Oncol 1998;16:584.
51. Santoro A, Bredenfeld H, Devizzi L, et al. Gemcitabine in the treatment of refractory Hodgkin’s disease: results of a multicenter phase II study. J Clin Oncol 2000;18:2615.
52. Schnell R, Borchmann P, Schulz H, et al. Current strategies of antibody-based treatment in Hodgkin disease. Ann Oncol 2002;13 Suppl 1:57.
53. Landgren O, Bjorkholm M, Konradsen HB, et al. A prospective study on antibody response to repeated vaccinations with pneumococcal capsular polysaccharide in splenectomized individuals with special reference to Hodgkin lymphoma. J Intern Med 2004;255:664.
54. Dean RM, Bishop MR. Allogeneic hematopoietic stem cell transplantation for lymphoma. Clin Lymphoma 2004;4: 238.
55. Gustavsson A, Osterman B, Cavallin-Stahl E. A systematic overview of radiation therapy effects in Hodgkin lymphoma. Acta Oncol 2003;42:589.
56. Chronowski GM, Wilder RB, Levy LB, et al. Second malignancies after chemotherapy and radiotherapy for Hodgkin disease. Am J Clin Oncol 2004;27:73.
57. Adams MJ, Lipsitz SR, Colan SD, et al. Cardiovascular status in long-term survivors of Hodgkin disease treated with chest radiotherapy. J Clin Oncol 2004;22:3139.
58. Illes A, Biro E, Miltenyi Z, et al. Hypothyroidism and thyroiditis after therapy for Hodgkin’s disease. Acta Haematol 2003;109:11.
59. Kaldor JM, Day NE, Clarke EA, et al. Leukemia following Hodgkin disease. N Engl J Med 1990;322:7.
60. Horning SJ, Hoppe RT, Kaplan HS, et al. Female reproductive potential after treatment for Hodgkin disease. N Engl J Med 1981;304:1377.
61. Chapman RM, Sutcliffe SB, Malpas JS. Male gonadal dysfunction in Hodgkin disease: a prospective study. JAMA 1981;245:1323.
62. Anselmo AP, Cartoni C, Bellantuono P, et al. Risk of infertility in patients with Hodgkin disease treated with ABVD vs MOPP vs ABVD/MOPP. Haematologica 1990;75:155.
63. Putterman C, Polliack A. Late cardiovascular and pulmonary complications of therapy in Hodgkin disease: report of three unusual cases, with a review of relevant literature. Leuk Lymphoma 1992;7:109.
64. Fisher SG, Fisher RI. The epidemiology of non-Hodgkin lymphoma. Oncogene 2004;23:6524.
65. Chiu BC, Weisenburger DD. An update of the epidemiology of non-Hodgkin lymphoma. Clin Lymphoma 2003; 4:161.
66. Loren AW, Porter DL, Stadtmauer EA, et al. Post-transplant lymphoproliferative disorder: a review. Bone Marrow Transplant 2003;31:145.
67. Poiesz BJ, Papsidero LD, Ehrlich G, et al. Prevalence of HTLV-I–associated T-cell lymphoma. Am J Hematol 2001;66:32.
68. Engels EA, Chatterjee N, Cerhan JR, et al. Hepatitis C virus infection and non-Hodgkin lymphoma: results of the NCI-SEER multi-center case-control study. Int J Cancer 2004;111:76.
69. Wotherspoon AC. Gastric lymphoma of mucosaassociated lymphoid tissue and Helicobacter pylori. Annu Rev Med 1998;49:289.
70. Staudt MR, Kanan Y, Jeong JH, et al. The tumor microenvironment controls primary effusion lymphoma growth in vivo. Cancer Res 2004;64:4790.
71. Cerroni L, Zochling N, Putz B, et al. Infection by Borrelia burgdorferi and cutaneous B-cell lymphoma. J Cutan Pathol 1997;24:457.
72. Ferreri AJ, Guidoboni M, Ponzoni M, et al. Evidence for an association between Chlamydia psittaci and ocular adnexal lymphomas. J Natl Cancer Inst 2004;96:586.
73. Lecuit M, Abachin E, Martin A, et al. Immunoproliferative small intestinal disease associated with Campylobacter jejuni. N Engl J Med 2004;350:239.
74. Gascoyne RD, Adomat SA, Krajewski S, et al. Prognostic significance of Bcl-2 protein expression and Bcl-2 gene rearrangement in diffuse aggressive non-Hodgkin lymphoma. Blood 1997;90:244.
75. Wilson WH, Grossbard ML, Pittaluga S, et al. Dose-adjusted EPOCH chemotherapy for untreated large B-cell lymphomas: a pharmacodynamic approach with high efficacy. Blood 2002;99:2685.
76. Ichikawa A, Kinoshita T, Watanabe T, et al. Mutations of the p53 gene as a prognostic factor in aggressive B-cell lymphoma. N Engl J Med 1997;337:529.
77. Krause JR, Shahidi-Asl M. Molecular pathology in the diagnosis and treatment of non-Hodgkin lymphomas. J Cell Mol Med 2003;7:494.
78. Weiss LM, Warnke RA, Sklar J, et al. Molecular analysis of the t(14;18) chromosomal translocation in malignant lymphomas. N Engl J Med 1987;317:1185.
79. Cleary ML, Sklar J. Nucleotide sequence of a t(14;18) chromosomal breakpoint in follicular lymphoma and demonstration of a breakpoint-cluster region near a transcriptionally active locus on chromosome 18. Proc Natl Acad Sci U S A 1985;82:7439.
80. Hill ME, MacLennan KA, Cunningham DC, et al. Prognostic significance of BCL-2 expression and bcl-2 major breakpoint region rearrangement in diffuse large cell non-Hodgkin lymphoma: a British National Lymphoma Investigation study. Blood 1996;88:1046.
81. Bosch F, Jares P, Campo E, et al. PRAD-1/cyclin D1 gene overexpression in chronic lymphoproliferative disorders: a highly specific marker of mantle cell lymphoma. Blood 1994;84:2726.
82. Klapper W, Stoecklein H, Zeynalova S, et al. Structural aberrations affecting the MYC locus indicate a poor prognosis independent of clinical risk factors in diffuse large B-cell lymphomas treated within randomized trials of the German High-Grade Non-Hodgkin’s Lymphoma Study Group (DSHNHL). Leukemia 2008;22:2226–9.
83. Savage KJ, Johnson NA, Ben-Neriah S, et al. MYC gene rearrangements are associated with a poor prognosis in diffuse large B-cell lymphoma patients treated with R-CHOP chemotherapy. Blood 2009;114:3533–7.
84. Fisher RI, Gaynor ER, Dahlberg S, et al. Comparison of a standard regimen (CHOP) with three intensive chemotherapy regimens for advanced non-Hodgkin lymphoma. N Engl J Med 1993;328:1002.
85. Coiffier B, Lepage E, Briere J, et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med 2002;346:235.
86. Rosenwald A, Wright G, Chan WC, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med 2002; 346:1937.
87. Rosenwald A, Wright G, Leroy K, et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J Exp Med 2003;198:851.
88. Rosenwald A, Wright G, Wiestner A, et al. The proliferation gene expression signature is a quantitative integrator of oncogenic events that predicts survival in mantle cell lymphoma. Cancer Cell 2003;3:185.
89. Canioni D, Brice P, Lepage E, et al. Bone marrow histological patterns can predict survival of patients with grade 1 or 2 follicular lymphoma: a study from the Groupe d’Etude des Lymphomes Folliculaires. Br J Haematol 2004;126:364.
90. Little RF, Pittaluga S, Grant N, et al. Highly effective treatment of acquired immunodef ciency syndrome–related lymphoma with dose-adjusted EPOCH: impact of antiretroviral therapy suspension and tumor biology. Blood 2003;101:4653.
91. Dumontet C, Drai J, Bienvenu J, et al. Prof les and prognostic values of LDH isoenzymes in patients with non-Hodgkin lymphoma. Leukemia 1999;13:811.
92. van Besien K, Ha CS, Murphy S, et al. Risk factors, treatment, and outcome of central nervous system recurrence in adults with intermediate-grade and immunoblastic lymphoma. Blood 1998;91:1178.
93. Hegde U, Filie A, Little RF, et al. High incidence of occult leptomeningeal disease detected by flow cytometry in newly diagnosed aggressive B-cell lymphomas at risk of central nervous system involvement: the role of flow cytometry versus cytology. Blood 2005;105:496.
94. A predictive model for aggressive non-Hodgkin lymphoma. The International Non-Hodgkin Lymphoma Prognostic Factors Project. N Engl J Med 1993;329:987.
95. Solal-Celigny P, Roy P, Colombat P, et al. Follicular lymphoma international prognostic index. Blood 2004;104: 1258.
96. Hans CP, Weisenburger DD, Greiner TC, et al. Conf rmation of the molecular classif cation of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood 2004;103:275.
97. Sohn SK, Jung JT, Kim DH, et al. Prognostic signif cance of bcl-2, bax, and p53 expression in diffuse large B-cell lymphoma. Am J Hematol 2003;73:101.
98. Jerkeman M, Anderson H, Dictor M, et al. Assessment of biological prognostic factors provides clinically relevant information in patients with diffuse large B-cell lymphoma: a Nordic Lymphoma Group study. Ann Hematol 2004; 83:414.
99. Wilson W, Pittaluga S, O’Connor P. Rituximab may overcome BCL-2–associated chemotherapy resistance in untreated diffuse large B-cell lymphomas [abstract]. Blood 2001;98:A-1447.
100.Mounier N, Briere J, Gisselbrecht C, et al. Rituximab plus CHOP (R-CHOP) overcomes bcl-2–associated resistance to chemotherapy in elderly patients with diffuse large B-cell lymphoma (DLBCL). Blood 2003;101:4279.
101.Staudt LM. Molecular diagnosis of the hematologic cancers. N Engl J Med 2003;348:1777.
102.Alizadeh AA, Eisen MB, Davis RE, et al. Distinct types of diffuse large B-cell lymphoma identif ed by gene expression prof ling. Nature 2000;403:503.
103.Lenz G, Wright G, Dave SS, et al. Stromal gene signatures in large-B-cell lymphomas. N Engl J Med 2008;359: 2313–23.
104.Dave S, Wright G, Tan B. A molecular predictor of survival following diagnosis of follicular lymphoma. Blood 2003; 102:617a.
105.Rosenwald A, Alizadeh AA, Widhopf G, et al. Relation of gene expression phenotype to immunoglobulin mutation genotype in B cell chronic lymphocytic leukemia. J Exp Med 2001;194:1639.
106.Wiestner A, Rosenwald A, Barry TS, et al. ZAP-70 expression identif es a chronic lymphocytic leukemia subtype with unmutated immunoglobulin genes, inferior clinical outcome, and distinct gene expression prof le. Blood 2003;101:4944.
107.Dave SS, Fu K, Wright GW, et al. Molecular diagnosis of Burkitt’s lymphoma N Engl J Med 2006;354:2431–42.
108.A clinical evaluation of the International Lymphoma Study Group classif cation of non-Hodgkin’s lymphoma. The Non-Hodgkin’s Lymphoma Classif cation Project. Blood 1997;89:3909.
109.Seymour JF. New treatment approaches to indolent non-Hodgkin lymphoma. Semin Oncol 2004;31:27.
110.Advani R, Rosenberg SA, Horning SJ. Stage I and II follicular non-Hodgkin lymphoma: long-term follow-up of no initial therapy. J Clin Oncol 2004;22:1454.
111.Ott OJ, Rodel C, Gramatzki M, et al. Radiotherapy for stage I–III nodal low-grade non-Hodgkin lymphoma. Strahlenther Onkol 2003;179:694.
112.Lenz G, Hiddemann W, Dreyling M. The role of fludarabine in the treatment of follicular and mantle cell lymphoma. Cancer 2004;101:883.
113.Santini G, Chisesi T, Nati S, et al. Fludarabine, cyclophosphamide and mitoxantrone for untreated follicular lymphoma: a report from the Non-Hodgkin Lymphoma Co-operative Study Group. Leuk Lymphoma 2004; 45:1141.
114.Colombat P, Salles G, Brousse N, et al. Rituximab (antiCD20 monoclonal antibody) as single f rst-line therapy for patients with follicular lymphoma with a low tumor burden: clinical and molecular evaluation. Blood 2001; 97:101.
115.Davis TA, White CA, Grillo-Lopez AJ, et al. Single-agent monoclonal antibody eff cacy in bulky non-Hodgkin lymphoma: results of a phase II trial of rituximab. J Clin Oncol 1999;17:1851.
116.Davis TA, Grillo-Lopez AJ, White CA, et al. Rituximab anti-CD20 monoclonal antibody therapy in non-Hodgkin lymphoma: safety and eff cacy of re-treatment. J Clin Oncol 2000;18:3135.
117.Czuczman MS, Weaver R, Alkuzweny B, et al. Prolonged clinical and molecular remission in patients with low-grade or follicular non-Hodgkin lymphoma treated with rituximab plus CHOP chemotherapy: 9-year follow-up. J Clin Oncol 2004;22:4659.
118.Czuczman MS, Fallon A, Mohr A, et al. Rituximab in combination with CHOP or fludarabine in low-grade lymphoma. Semin Oncol 2002;29:36.
119.Hiddemann W, Forstpointer R, Kneba M, et al. The addition of rituximab to combination chemotherapy CHOP has a long lasting impact on subsequent treatment in remission in follicular lymphoma but not in mantle cell lymphoma: results of two prospective randomized studies of the German Low Grade Lymphoma Study Group (GLSG). Blood 2004;104:161.
120.Hainsworth JD, Litchy S, Burris HA 3rd, et al. Rituximab as first-line and maintenance therapy for patients with indolent non-Hodgkin lymphoma. J Clin Oncol 2002; 20:4261.
121.Hagenbeek A, Gadeberg O, Johnson P, et al. First clinical use of ofatumumab, a novel fully human anti-CD20 monoclonal antibody in relapsed or refractory follicular lymphoma: results of a phase 1/2 trial. Blood 2008;111: 5486–95.
122.Morschhauser F, Leonard JP, Fayad L, et al. Humanized anti-CD20 antibody, veltuzumab, in refractory/recurrent non-Hodgkin’s lymphoma: phase I/II results. J Clin Oncol 2009;27:3346–53.
123.Czuczman MS, Thall A, Witzig TE, et al. Phase I/II study of galiximab, an anti-CD80 antibody, for relapsed or refractory follicular lymphoma. J Clin Oncol 2005;23: 4390–8.
124.Leonard JP, Coleman M, Ketas JC, et al. Phase I/II trial of epratuzumab (humanized anti-CD22 antibody) in indolent non-Hodgkin’s lymphoma. J Clin Oncol 2003;21:3051–9.
125.Witzig TE, Gordon LI, Cabanillas F, et al. Randomized controlled trial of yttrium-90–labeled ibritumomab tiuxetan radioimmunotherapy versus rituximab immunotherapy for patients with relapsed or refractory low-grade, follicular, or transformed B-cell non-Hodgkin lymphoma. J Clin Oncol 2002;20:2453.
126.Gordon LI, Witzig T, Molina A, et al. Yttrium 90–labeled ibritumomab tiuxetan radioimmunotherapy produces high response rates and durable remissions in patients with previously treated B-cell lymphoma. Clin Lymphoma 2004;5:98.
127.Kaminski MS, Tuck M, Estes J, et al. 131I-Tositumomab therapy as initial treatment for follicular lymphoma. N Engl J Med 2005;352:441.
128.van Besien K, Loberiza FR Jr, Bajorunaite R, et al. Comparison of autologous and allogeneic hematopoietic stem cell transplantation for follicular lymphoma. Blood 2003; 102:3521.
129.McCarthy H, Ottensmeier CH, Hamblin TJ, et al. Antiidiotype vaccines. Br J Haematol 2003;123:770.
130.Kwak LW. Translational development of active immunotherapy for hematologic malignancies. Semin Oncol 2003; 30:17.
131.Wilson WH, O’Connor OA, Czuczman MS, et al. Phase 112 a study of ABT-263 in relapsed or refractory lymphoid malignancies. Blood (ASH Annual Meeting Abstracts) Nov 2009;114:3442.
132.O’Connor OA, Wright J, Moskowitz C, et al. Phase II clinical experience with the novel proteasome inhibitor bortezomib in patients with indolent non-Hodgkin’s lymphoma and mantle cell lymphoma. J Clin Oncol 2005; 23:676.
133.Witzig TE, Wiernik PH, Moore T, et al. Lenalidomide oral monotherapy produces durable responses in relapsed or refractory indolent non-Hodgkin’s lymphoma. J Clin Oncol 2009;27:5404–9.
134.Wotherspoon AC, Doglioni C, Diss TC, et al. Regression of primary low-grade B-cell gastric lymphoma of mucosaassociated lymphoid tissue type after eradication of Helicobacter pylori. Lancet 1993;342:575.
135.Cavalli F, Isaacson PG, Gascoyne RD, et al. MALT Lymphomas. Hematol-Am Soc Hematol Educ Program 2001;241.
136.Nuckel H, Meller D, Steuhl KP, et al. Anti-CD20 monoclonal antibody therapy in relapsed MALT lymphoma of the conjunctiva. Eur J Haematol 2004;73:258.
137.Lenz G, Dreyling M, Hiddemann W. Mantle cell lymphoma: established therapeutic options and future directions. Ann Hematol 2004;83:71.
138.Meusers P, Engelhard M, Bartels H, et al. Multicentre randomized therapeutic trial for advanced centrocytic lymphoma: anthracycline does not improve the prognosis. Hematol Oncol 1989;7:365.
139.Hagemeister FB. Mantle cell lymphoma: non-myeloablative versus dose-intensive therapy. Leuk Lymphoma 2003; 44 Suppl 3:S69.
140.Wilson W, Neelapu S, Rosenwald A, et al. Idiotype vaccine and dose-adjusted EPOCH-rituximab treatment in untreated mantle cell lymphoma: preliminary report on clinical outcome and analysis of immune response. Blood 2003;102:358.
141.Jacobsen E, Freedman A. An update on the role of high-dose therapy with autologous or allogeneic stem cell transplantation in mantle cell lymphoma. Curr Opin Oncol 2004;16:106.
142.Goy A, Younes A, McLaughlin P, et al. Phase II study of proteasome inhibitor bortezomib in relapsed or refractory B-cell non-Hodgkin’s lymphoma. J Clin Oncol 2004;23: 667.
143.Witzig TE, Geyer SM, Ghobrial I, et al. Phase II trial of single-agent temsirolimus (CCI-779) for relapsed mantle cell lymphoma. J Clin Oncol 2005;23:5347–56.
144.Wiernik PH, Lossos IS, Tuscano JM, et al. Lenalidomide monotherapy in relapsed or refractory aggressive nonHodgkin’s lymphoma. J Clin Oncol 2008;26:4952–7.
145.Spicer J, Smith P, Maclennan K, et al. Long-term follow-up of patients treated with radiotherapy alone for early-stage histologically aggressive non-Hodgkin lymphoma. Br J Cancer 2004;90:1151.
146.Miller TP, Dahlberg S, Cassady JR, et al. Chemotherapy alone compared with chemotherapy plus radiotherapy for localized intermediate- and high-grade non-Hodgkin lymphoma. N Engl J Med 1998;339:21.
147.Miller T, Sahlberg S, Cassady JR, et al. CHOP alone compared to CHOP plus radiotherapy for early stage aggressive non-Hodgkin’s lymphomas: update of the Southwest Oncology Group (SWOG) randomized trial. Blood 2001;98: 724.
148.Horning SJ, Weller E, Kim K, et al. Chemotherapy with or without radiotherapy in limited-stage diffuse aggressive non-Hodgkin lymphoma: Eastern Cooperative Oncology Group study 1484. J Clin Oncol 2004;22:3032.
149.Reyes FL, Munck JN, Morel P, et al. Superiority of chemotherapy alone with the ACVBP regimen over treatment with 3 cycles of CHOP plus radiation in low risk localized aggressive lymphoma: the LNH93-1 GELA study [abstract]. Proc Am Soc Hematol 2002, Abstract 343.
150.Zinzani PL, Martelli M, Magagnoli M, et al. Treatment and clinical management of primary mediastinal large B-cell lymphoma with sclerosis: MACOP-B regimen and mediastinal radiotherapy monitored by (67)gallium scan in 50 patients. Blood 1999;94:3289.
151. Messori A, Vaiani M, Trippoli S, et al. Survival in patients with intermediate or high grade non-Hodgkin lymphoma: meta-analysis of randomized studies comparing third generation regimens with CHOP. Br J Cancer 2001;84:303.
152.Tilly H, Lepage E, Coiffier B, et al. Intensive conventional chemotherapy (ACVBP regimen) compared with standard CHOP for poor-prognosis aggressive non-Hodgkin lymphoma. Blood 2003;102:4284.
153.Wilson WH, Dunleavy K, Pittaluga S, et al. Phase II study of dose-adjusted EPOCH and rituximab in untreated diffuse large B-cell lymphoma with analysis of germinal center and post-germinal center biomarkers. J Clin Oncol 2008;26:2717–24.
154.Pfreundschuh M, Trumper L, Kloess M, et al. Two-weekly or 3-weekly CHOP chemotherapy with or without etoposide for the treatment of young patients with good-prognosis (normal LDH) aggressive lymphomas: results of the NHL-B1 trial of the DSHNHL. Blood 2004;104:626.
155.Pfreundschuh M, Trumper L, Kloess M, et al. Two-weekly or 3-weekly CHOP chemotherapy with or without etoposide for the treatment of elderly patients with aggressive lymphomas: results of the NHL-B2 trial of the DSHNHL. Blood 2004;104:634.
156.Shipp MA, Abeloff MD, Antman KH, et al. International consensus conference on high-dose therapy with hematopoietic stem cell transplantation in aggressive non-Hodgkin lymphomas: report of the jury. J Clin Oncol 1999; 17:423.
157.Gisselbrecht C, Lepage E, Molina T, et al. Shortened first-line high-dose chemotherapy for patients with poor-prognosis aggressive lymphoma. J Clin Oncol 2002;20: 2472.
158.Milpied N, Deconinck E, Gaillard F, et al. Initial treatment of aggressive lymphoma with high-dose chemotherapy and autologous stem-cell support. N Engl J Med 2004; 350:1287.
159.Seropian S, Bahceci E, Cooper DL. Allogeneic peripheral blood stem cell transplantation for high-risk non-Hodgkin lymphoma. Bone Marrow Transplant 2003;32:763.
160.Batchelor T, Carson K, O’Neill A, et al. Treatment of primary CNS lymphoma with methotrexate and deferred radiotherapy: a report of NABTT 96-07. J Clin Oncol 2003; 21:1044.
161.Sandor V, Stark-Vancs V, Pearson D, et al. Phase II trial of chemotherapy alone for primary CNS and intraocular lymphoma. J Clin Oncol 1998;16:3000.
162.Pels H, Schmidt-Wolf IG, Glasmacher A, et al. Primary central nervous system lymphoma: results of a pilot and phase II study of systemic and intraventricular chemotherapy with deferred radiotherapy. J Clin Oncol 2003; 21:4489.
163.Magrath I, Adde M, Shad A, et al. Adults and children with small non-cleaved-cell lymphoma have a similar excellent outcome when treated with the same chemotherapy regimen. J Clin Oncol 1996;14:925.
164.Reiter A, Schrappe M, Parwaresch R, et al. Non-Hodgkin lymphomas of childhood and adolescence: results of a treatment stratified for biologic subtypes and stage: a report of the Berlin-Frankfurt-Munster Group. J Clin Oncol 1995;13:359.
165.Dal Maso L, Franceschi S. Epidemiology of non-Hodgkin lymphomas and other haemolymphopoietic neoplasms in people with AIDS. Lancet Oncol 2003;4:110.
166.Little RF, Wilson WH. Update on the pathogenesis, diagnosis, and therapy of AIDS-related lymphoma. Curr Infect Dis Rep 2003;5:176.
167.Ratner L, Lee J, Tang S, et al. Chemotherapy for human immunodeficiency virus–associated non-Hodgkin lymphoma in combination with highly active antiretroviral therapy. J Clin Oncol 2001;19:2171.
168.Wilson WH, Kingma DW, Raffeld M, et al. Association of lymphomatoid granulomatosis with Epstein-Barr viral infection of B lymphocytes and response to interferon-alpha 2b. Blood 1996;87:4531.
169.Gascoyne RD, Aoun P, Wu D, et al. Prognostic significance of anaplastic lymphoma kinase (ALK) protein expression in adults with anaplastic large cell lymphoma. Blood 1999; 93:3913.
170.Ribrag V, Ell Hajj M, Janot F, et al. Early locoregional high-dose radiotherapy is associated with long-term disease control in localized primary angiocentric lymphoma of the nose and nasopharynx. Leukemia 2001;15:1123.
171.Foss F. Mycosis fungoides and the Sezary syndrome. Curr Opin Oncol 2004;16:421.
172.Piekarz RL, Fry R, Turner M, et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J Clin Oncol 2009;27:5410–7.
173.Soligo D, Ibatici A, Berti E, et al. Treatment of advanced mycosis fungoides by allogeneic stem-cell transplantation with a nonmyeloablative regimen. Bone Marrow Transplant 2003;31:663.
174.Falini B, Pileri S, Zinzani PL, et al. ALK+ lymphoma: clinico-pathological findings and outcome. Blood 1999; 93:2697.
175.Dogan A, Attygalle AD, Kyriakou C. Angioimmunoblastic T-cell lymphoma. Br J Haematol 2003;121:681.
176.Coiffier B, Brousse N, Peuchmaur M, et al. Peripheral T-cell lymphomas have a worse prognosis than B-cell lymphomas: a prospective study of 361 immunophenotyped patients treated with the LNH-84 regimen. The GELA (Groupe d’Etude des Lymphomes Agressives). Ann Oncol 1990;1:45.
177.Rodriguez J, Caballero MD, Gutierrez A, et al. High-dose chemotherapy and autologous stem cell transplantation in peripheral T-cell lymphoma: the GEL-TAMO experience. Ann Oncol 2003;14:1768.
178.Song KW, Mollee P, Keating A, et al. Autologous stem cell transplant for relapsed and refractory peripheral T-cell lymphoma: variable outcome according to pathological subtype. Br J Haematol 2003;120:978.
179.Corradini P, Dodero A, Zallio F, et al. Graft-versuslymphoma effect in relapsed peripheral T-cell non-Hodgkin lymphomas after reduced-intensity conditioning followed by allogeneic transplantation of hematopoietic cells. J Clin Oncol 2004;22:2172.
180.O’Connor OA, Horwitz S, Hamlin P, et al. Phase II-I-II study of two different doses and schedules of pralatrexate, a high-affinity substrate for the reduced folate carrier, in patients with relapsed or refractory lymphoma reveals marked activity in T-cell malignancies. J Clin Oncol 2009; 27:4357–64.
181.Tilly H, Rossi A, Stamatoullas A, et al. Prognostic value of chromosomal abnormalities in follicular lymphoma. Blood 1994;84:1043.
182.Knauf WU, Knuutila S, Zeigmeister B, et al. Trisomy 12 in B-cell chronic lymphocytic leukemia: correlation with advanced disease, atypical morphology, high levels of sCD25, and with refractoriness to treatment. Leuk Lymphoma 1995;19:289.
183.Dohner H, Stilgenbauer S, Dohner K, et al. Chromosome aberrations in B-cell chronic lymphocytic leukemia: reassessment based on molecular cytogenetic analysis. J Mol Med 1999;77:266.
184.Raty R, Franssila K, Jansson SE, et al. Predictive factors for blastoid transformation in the common variant of mantle cell lymphoma. Eur J Cancer 2003;39:321.
185.Ueda C, Akasaka T, Ohno H. Non-immunoglobulin/BCL6 gene fusion in diffuse large B-cell lymphoma: prognostic implications. Leuk Lymphoma 2002;43:1375.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.