(Carregando Índice)... (Carregando Índice)... |
Última revisão: 13/08/2015
Comentários de assinantes: 0
Alexei A. Grom, MD
Associate Professor of Pediatrics, Division of Rheumatology, Cincinnati Children's Hospital Medical Center, University of Cincinnati College of Medicine, Cincinnati, OH
Michael B. Jordan, MD, PhD
Assistant Professor of Pediatrics, Divisions of Immunobiology and Bone Marrow Transplantation and Immunodeficiency, Cincinnati Children's Hospital Medical Center, Cincinnati, OH
Jun Qin Mo, MD
Associate Professor of Pediatrics, Division of Pathology, Cincinnati Children's Hospital Medical Center, Cincinnati, OH
Artigo original: Grom AA, MD, Jordan MB, MD, PhD, Qin Mo J, MD. Disorders of Macrophages and Dendritic. ACP Medicine. 2012.
[The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2015 Decker Intellectual Properties Inc. All Rights Reserved.]
Tradução: Paulo Henrique Machado.
Revisão técnica: Dr.Lucas Santos Zambon
O sistema fagocítico mononuclear resulta de células dendríticas (CDs) e de monócitos/macrófagos historicamente conhecidos como histiócitos. O acúmulo e o comportamento anormal dessas células podem produzir um espectro de doenças conhecidas coletivamente como histiocitoses. Sob o ponto de vista clínico, os distúrbios histiocíticos abrangem uma ampla variedade de condições que afetam tanto crianças como adultos e variam desde lesões cutâneas benignas até distúrbios sistêmicos com progressão rápida que ameaçam a vida humana. A distinção entre essas condições se baseia no fenótipo das células histiocíticas que são a grande maioria em uma determinada lesão.1
Para compreender melhor a natureza das várias histiocitoses foi proposta uma estrutura conceitual que vincula o fenótipo dos histiócitos expandidos em um distúrbio histiocítico específico à respectiva ontogênese.2-5 Embora a origem exata de várias células histiocíticas ainda seja uma área de pesquisa ativa, aparentemente os histiócitos surgem de células precursoras hematopoiéticas CD34+. As progenitoras hematopoiéticas CD34+ amadurecem e formam histiócitos CD14+ ou CD14-. As células CD14- se diferenciam em dendrócitos ou monócitos dérmicos e macrófagos de acordo com o ambiente local. Inicialmente acreditava-se que os precursores da célula de Langerhans (CL) poderiam fazer a distinção entre CD14-. Entretanto, estudos mais recentes sugerem que pelo menos no contexto inflamatório a evolução das CLs ocorre a partir de inúmeros precursores diferentes, incluindo os monócitos.3
Usualmente, distinguem-se as várias populações de CDs e macrófagos de acordo com a morfologia e os padrões característicos da expressão de superfícies celulares e marcadores intracelulares específicos.
O compartimento das CDs inclui: (1) as células dendríticas foliculares (CDFs) do centro germinal dos linfonodos; (2) a CL da epiderme cutânea, cérvice, vagina, estômago e esôfago; (3) a CD intersticial, que representa o equivalente das CLs na derme (ou CD dérmica); (4) a CD indeterminada derivada de CLs ou de CDs intersticiais que migram para o tecido linfoide local após a captura de antígenos e (5) a célula dendrítica interdigital (CDI) da zona T de linfonodos.6,7 Tipicamente, todas essas células têm morfologia dendrítica e sua atividade fagocítica é limitada. Em vez disso, elas desempenham um papel importante na apresentação de antígenos aos linfócitos. As CLs são CDs imaturas encontradas principalmente na epiderme. As CLs, através de seus dendritos estendidos, formam uma rede contínua da epiderme sob vigilância para evitar a presença de antígenos estranhos, criando, consequentemente, uma linha importante de defesa contra o ambiente externo.3,6 Sob a ótica histopatológica, elas podem ser distinguidas com base em um alto nível de expressão de CD1a (membro do grupo CD1 de proteínas que podem apresentar antígenos lipídicos microbianos às células T) e de langerina (ou CD207, um receptor de lectina que tem alguma ação na absorção de diversos patógenos).8 Outra característica exclusiva das CLs é a presença de organelas intracitoplásmicas conhecidas por grânulos de Birbeck.3,8 O papel desses grânulos permanece obscuro. O envolvimento da langerina da superfície celular por anticorpos específicos induz a internalização e o movimento da langerina para os grânulos de Birbeck. Esta observação sugere que a formação dos grânulos de Birbeck provavelmente seja uma consequência da função de captura de antígenos pela langerina.8-10
Os dendrócitos dérmicos também possuem morfologia dendrítica característica e expressam fascina, fator XIIIa, proteínas S-100 e CD68. Ao contrário das CLs, não expressam CD1a ou langerina e não contêm grânulos de Birbeck.
Os macrófagos apresentam vários graus de atividade fagocítica. Eles derivam de promonócitos que penetram no sangue e migram para os tecidos para se diferenciarem em macrófagos. Alguns fagócitos permanecem móveis ao passo que outros residem nos tecidos, como as células de Kupffer no fígado. Os macrófagos são ricos em lisossomos contendo enzimas como a lisozima e a alfa2-antitripsina. Os estudos imunohistoquímicos dessas células revelam alta expressão de enzimas lisossômicas, assim como CD68, CD11c, CD14 e CD163. Eles não expressam CD1a ou langerina e não contêm grânulos de Birbeck.
As evidências clínicas e experimentais sugerem que as CLs desempenham papel relevante no desenvolvimento de histiocitose da célula de Langerhans (HCL), sendo que os dendrócitos dérmicos formam a população predominante de células nas lesões causadas por xantogranulomas juvenis (XGJ) e os macrófagos são os focos da patogênese de linfohistiocitose hemofagocítica (LHH) e distúrbios relacionados.12
A classificação contemporânea dos distúrbios histiocíticos faz também a distinção entre distúrbios histiocíticos benignos com vários comportamentos biológicos e distúrbios malignos autênticos [ver a Tabela 1].1,7 O foco deste capítulo são as histiocitoses não malignas.
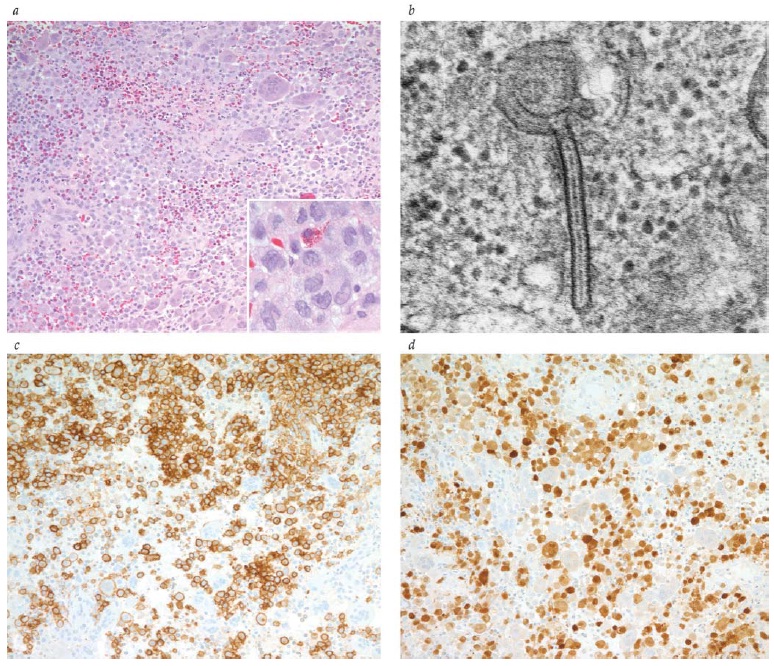
A característica histopatológica patognomônica da HCL (anteriormente conhecida por “histiocitose X”) é a presença excessiva de histiócitos lesionais do fenótipo da célula de Langerhans (i.e., CD1a, langerina positiva e presença de grânulos de Birbeck na microscopia eletrônica) com proporções variadas de macrófagos, linfócitos T, eosinófilos e células gigantes multinucleadas [ver a Figura 1].
A natureza exata da HCL permanece obscura.11,12 A detecção de histiócitos clonais na maior parte das formas de HCL sugere que possivelmente seja um distúrbio neoplásico com vários comportamentos biológicos.13.14 A hipótese alternativa é que a HCL seja uma doença reativa causada pela interação anormal entre a HCL e as células T.15,16 Em consonância com essa hipótese, as lesões típicas da HCL apresentam uma organização que se assemelha aos grânulos com inúmeros linfócitos ativados. Além disso, estudos recentes sobre expressão genética revelam que as células lesionais de Langerhans sobrexpressam moléculas que recrutam e ativam células T.16 Quando combinadas, essas observações dão suporte à ideia da natureza reativa da doença.
|
Tabela 1: Distúrbios Histiocíticos
|
|
Distúrbios histiocíticos benignos com vários comportamentos biológicos1 Relacionados às células dendríticas Histiocitose das células de Langerhans Xantogranulomas juvenis e distúrbios relacionados Histiocitomas solitários de vários fenótipos de células dendríticas Relacionados a macrófagos Síndromes hemofagocíticas Linfohistiocitose hemofagocítica primária Síndromes hemofagocíticas secundárias Associadas a infecções Associadas a malignidades Associadas a doenças autoimunes (síndrome da ativação de macrófagos) Outras condições Doença de Rosai-Dorfman (histiocitose sinusal com linfadenopatia maciça) Histiocitoma solitário com fenótipo de macrófago
Distúrbios histiocíticos benignos Relacionados a monócitos Leucemias relacionadas a monócitos Sarcoma ou tumor monocítico extramedular Relacionados a células dendríticas Sarcoma histiocítico relacionado a células dendríticas (localizado ou disseminado) Relacionados a macrófagos Sarcoma histiocítico relacionado a macrófagos (localizado ou disseminado) |
Apresentação clínica. A histiocitose da célula de Langerhans (HCL) se apresenta em qualquer idade, desde o período pré-natal até a velhice.17,18 A HCL pode se apresentar com envolvimento de um único sistema (ossos ou linfonodos). Esta categoria se subdivide em doença de sítio único (osso unifocal, pele ou linfonodo) e de sítios múltiplos (osso multifocal ou múltiplos linfonodos). A segunda categoria de HCL inclui HCL “multissistêmica”, definida como envolvimento de dois ou mais órgãos.
De maneira geral, os pacientes com HCL de sistema único têm uma chance maior de remissão espontânea e de resultados favoráveis. O prognóstico de doenças multissistêmicas é bem pior. O envolvimento de órgãos de “risco” como fígado, pulmão, baço, ou o sistema hematopoiético, poderá estar associado a taxas mais elevadas de mortalidade nas situações em que os pacientes não responderem imediatamente às terapias convencionais.
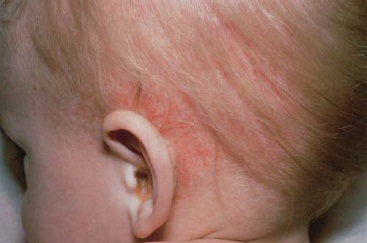
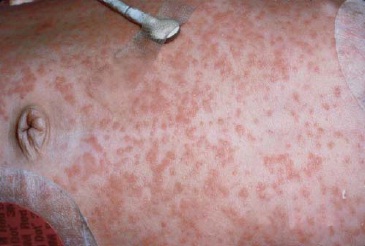
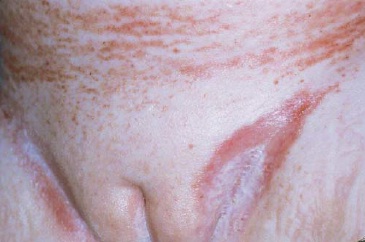
Doença de sítio único. Envolvimento da pele. Com frequência, os relatos indicam que as lesões papuloescamosas que afetam o couro cabeludo atingem 30 a 60% de pacientes. De maneira geral, em lactentes essas lesões são confundidas com seborreia. A erupção cutânea papular avermelhada que ocorre na virilha, no abdome e nas costas se assemelha às assaduras difusas causadas pelo organismo Cândida [ver as Figuras 2, 3 e 4]. Outras lesões cutâneas podem ser pustulares, purpúricas, petequiais, vesiculares ou papulonodulares. Na boca, a hipertrofia gengival ou ulcerações são muito comuns.19
Ossos. Osso é o único órgão envolvido com mais frequência.20 As lesões cranianas líticas são particularmente frequentes. Outros sítios típicos de envolvimento ósseo são os ossos vertebrais, as costelas, o fêmur e o úmero.21,22 Essas lesões ósseas podem ser sensíveis ou indolores. O envolvimento da calota craniana, da base do crânio, e dos ossos maxilofaciais pode estar associado a convulsões, perda auditiva, otite média recorrente, paralisia do nervo craniano e diabetes insípido.
Linfonodos e timo. Os linfonodos cervicais são envolvidos com mais frequência.
Apresentação de doença multissistêmica. Fígado e baço. O envolvimento hepático inclui lesões císticas ou semelhantes a tumores associadas à hepatomegalia.23,24 Esse tipo de envolvimento poderá ser acompanhado de disfunção hepática resultando em hiperbilirrubinemia, hipoalbuminemia com ascite, deficiência do fator de coagulação e, ocasionalmente, colangite esclerosante. A esplenomegalia maciça poderá produzir citopenias causadas por hiperesplenismo.
Pulmões. As características clínicas das doenças pulmonares nos casos de HCL são relativamente inespecíficas. Com frequência, a ocorrência de pneumotórax espontâneo é o primeiro sinal clínico de HCL nos pulmões. Os estudos de imagens revelam nódulos mal definidos (2 a 10 mm), opacidades reticulonodulares, cistos na zona superior ou com aspecto de favo de mel com preservação do ângulo costofrênico.25,26 A ocorrência de doença pulmonar é relativamente rara em crianças. Em adultos há uma forte conexão epidemiológica com tabagismo.
Medula óssea. O envolvimento da medula óssea em casos de HCL usualmente é observado em pacientes portadores de doença multissistêmica. Com frequência, resulta em trombocitopenia significativa, anemia e neutropenia.27,28
Sistema endócrino. Diabetes insípido é a anormalidade endócrina mais comum na HCL e, com frequência, está associado a outras anormalidades hormonais da hipófise. Em alguns pacientes o diabetes insípido pode ser a apresentação inicial da doença.29
Sistema gastrintestinal. Tipicamente, o envolvimento do sistema gastrintestinal se manifesta como diarreia e má absorção.30
Sistema nervoso central. Além do diabetes insípido secundário ao envolvimento hipofisário, uma pequena proporção de pacientes desenvolve lesões no parênquima cerebral ou no plexo coroide, com grande potencial para bloquear o fluxo do líquido espinal.31 Outra manifestação da HCL é a presença de lesões degenerativas, geralmente localizadas no núcleo dentado do cerebelo ou nos gânglios basais e podem resultar em ataxia e em disfunção comportamental e cognitiva.
Diagnóstico. A biópsia de lesões suspeitas com coloração imunohistoquímica com anticorpos específicos para DD1a e langerina é imprescindível para confirmar o diagnóstico.
Tratamento. Baseando-se em diversos testes clínicos, a Histiocyte Society estratificou os pacientes com HCL em grupos de “baixo risco” e de “alto risco” de acordo com o envolvimento de sistemas únicos ou múltiplos e com o tipo de órgão envolvido.32 Ver no site http://www.histiocytesociety.org as orientações atuais para os tratamentos de pacientes com HCL multissistêmica.

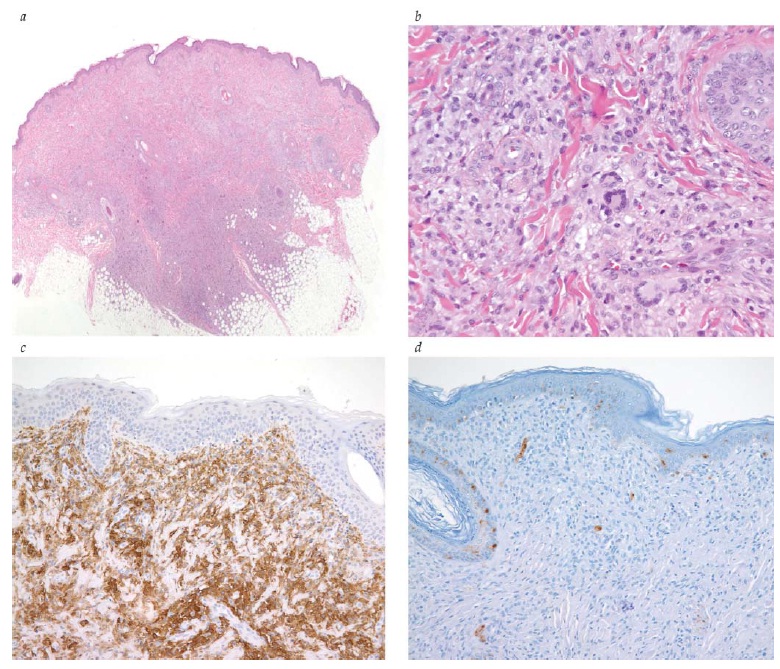
Figura 1 - Histiocitose da célula de Langerhans: lesão craniana lítica em um garoto de 10 anos de idade. (a) A lesão consiste predominantemente de histiócitos acompanhados de eosinofilia abundante inter-relacionada com algumas células gigantes multinucleadas semelhantes aos osteoclastos (coloração com hematoxilina-eosina; ampliação de 200 vezes em relação ao original). Os histiócitos são grandes e apresentam características recortadas e sulcadas, ou com núcleos com a forma de um grão de café e cromatina vesicular (coloração com hematoxilina-eosina) (inset). (b) Grânulo de Birbeck (inclusão citoplásmica na forma de raquete de tênis) na microscopia eletrônica. (c e d) As células de Langerhans mostram uma forte coloração imunohistoquímica positiva para CD1a (c, ampliação de 200 vezes em relação ao original) e S-100 (d, ampliação de 200 vezes em relação ao original).
Tipicamente, os pacientes com doença cutânea limitada não precisam fazer terapia sistêmica, embora seja comum o uso de esteroides tópicos.32 A mostarda de azoto para uso tópico e o psoraleno com raios ultravioleta A (PUVA) de ondas longas são opções de segunda linha. De maneira geral, a curetagem é suficiente para fins diagnósticos e terapêuticos em pacientes com lesões ósseas localizadas, embora os esteroides intralesionais ou baixas doses de radiação também sejam opções válidas.32 Os bifosfonatos como pamidronato e clodronato são utilizados para controlar a dor e, possivelmente, para controlar a progressão da histiocitose da célula de Langerhans (HCL).33,34 Entretanto, esses pacientes devem ser observados de perto para permitir a busca de evidências da presença de alguma doença em outros órgãos, tendo em vista que muitos desses indivíduos desenvolvem algum tipo de envolvimento multissistêmico. A radioterapia é especialmente apropriada nos casos de lesões localizadas com risco de colapso na coluna ou no fêmur. O tratamento com prednisona e vimblastina é a melhor opção nas situações em que as lesões se localizarem nos ossos orbitais, mastoides, temporais ou esfenoides.
Os pacientes com envolvimento multissistêmico, principalmente indivíduos com órgãos de “risco” envolvidos (i.e., fígado, baço, pulmão, medula óssea), exigem tratamento sistêmico mais agressivo, incluindo prednisona, vimblastina, 6-mercaptopurina e metotrexato.35 Vários estudos colaborativos de grande porte demonstraram que a quimioterapia com múltiplos agentes mantida durante períodos mais longos de tempo resultou em taxas mais elevadas de resposta com um número menor de recorrências.
Figura 2 - Esta criança de três anos desenvolveu manchas com crosta hemorrágica no couro cabeludo, atrás das orelhas e nas dobras da virilha. A biópsia da pele revelou a presença de infiltrados histiocíticos típicos da histiocitose da célula de Langerhans.
Atualmente, a Histiocyte Society disponibiliza orientações padrões de tratamento médico para pacientes que não tenham sido tratados em testes clínicos. O teste internacional seguinte (LCH IV) foi iniciado em 2011 e inclui estratos para quase todos os pacientes diagnosticados com HCL. Os pacientes que não responderem adequadamente às terapias padrões atuais apresentarão maus prognósticos, sendo, portanto imprescindível a aplicação de terapia agressiva de emergência (como a inclusão no LCHJ-S 2005).
As células da histiocitose da célula de Langerhans (HCL) expressam CD1a e CD52,36 dois alvos potenciais para intervenções terapêuticas com anticorpos monoclonais. Os pacientes com respostas insuficientes à terapia de recuperação são candidatos ao transplante de células-tronco hematopoiéticas.
As células lesionais da maior parte dos distúrbios não relacionados à HCL têm imunofenótipos idênticos: fator XIIIa+, CD163+, CD14+, S-100-, CD1a-, langerina negativa e grânulo de Birbeck negativo [ver a Figura 5]. Este grupo inclui XGJ, reticulohistiociose multicêntrica e doença de Erdheim-Chester.
Figura 3 - Placas abdominais violáceas endurecidas e manchas eczematosas maceradas nas dobras da virilha que aumentam e diminuem nesta menina com 9 meses de idade com pancitopenia e histiocitose da célula de Langerhans.
Esta condição é muito rara. A maior parte das crianças com Xantugranuloma Juvenil (XGJ) se apresentam durante os primeiros 2 anos de vida com lesões maculopapulares de cor marrom amarelada na pele. Ocasionalmente essas lesões podem se estender até os tecidos subcutâneos. O envolvimento extracutâneo ocorre em torno de 5 a 10% de pacientes. Existem relatos de lesões nodulares nos pulmões, fígado, baço, pâncreas, glândulas adrenais, intestinos, rins, linfonodos, medula óssea e sistema nervoso central (SNC).37-39 Usualmente o XGJ cutâneo segue um curso benigno, com involução gradual durante períodos de tempo variando de alguns meses a vários anos. As lesões podem desaparecer completamente ou deixar cicatrizes atróficas. Ao final, a maioria das lesões sistêmicas também passa por um processo involutivo, embora as lesões oculares e no SNC possam deixar danos residuais. O XGJ multissistêmico pode ser tratado com regimes que se baseiam na histiocitose da célula de Langerhans, incluindo corticosteroides e alcaloides da vinca.40
Figura 4 - As pápulas vermelhas com disseminação generalizada e placas no tórax e no abdome desta criança com histiocitose da célula de Langerhans foram associadas à hepatoesplenomegalia, à linfadenopatia generalizada e à pneumonite.
Reticulohistiocitose multicêntrica é uma doença rara que acomete adultos mais velhos, embora existam relatos de alguns casos em crianças. As lesões cutâneas típicas são pápulas firmes de cor vermelha, marrom ou amarela. As lesões patognomônicas são pápulas periungueais na forma de “contas de coral” e pápulas localizadas na orelha e lesões vermiculares nas bordas das narinas. A combinação de doença cutânea com artropatia erosiva é o padrão clínico mais característico.41 A distribuição da artropatia em pequenas articulações das mãos pode ser confundida com osteoartrite erosiva. O curso natural da doença poderá progredir para artropatia destrutiva grave e lesões cutâneas desfigurantes. Nesses casos, recomenda-se enfaticamente um tratamento agressivo com medicamentos imunossupressivos. Os agentes do fator alfa de necrose antitumoral e o alendronato também foram incluídos na lista dos medicamentos usados nos casos de reticulohistiocitose multicêntrica.
Figura 5 - Xantogranuloma juvenil: lesão cutânea no antebraço direito de uma menina de três anos . (a) A pele mostra uma lesão proliferativa na derme que se estende até o tecido adiposo subcutâneo (coloração com hematoxilina-eosina; ampliação de 20 vezes em relação ao original). (b) A lesão se compõe de histiócitos em forma de espuma com alguns linfócitos espalhados e células gigantes de Touton multinucleadas (coloração com hematoxilina-eosina; ampliação de 400 vezes em relação ao original). As células de Touton são histiócitos carregados de lipídeos onde ocorre o agrupamento de múltiplos núcleos ao redor de uma pequena ilha de citoplasma. (c e d) Os histiócitos apresentam reação positiva para o fator XIIIa (c, ampliação de 200 vezes em relação ao original) e reação negativa para S-100 (d, ampliação de 200 vezes em relação ao original) de acordo com medições da coloração imunohistoquímica.
A doença de Erdheim-Chester (ou histiocitose poliostótica esclerosante) é um distúrbio histiocítico multissistêmico raro.42 Nesse tipo de doença, a expansão histiocítica produz infiltrados xantogranulomatosos de múltiplos sistemas de órgãos, tais como pele, pulmões, retroperitôneo, tecido retro-orbital, ossos e parênquima cerebral. O envolvimento de ossos longos de uma forma simétrica é bem característico. O prognóstico da doença de Erdheim-Chester aparentemente é pior em comparação com outros distúrbios histiocíticos. Há relatos de mortes secundárias à fibrose pulmonar ou insuficiência renal como consequência de fibrose retroperitoneal. Nesse tipo de distúrbio, as células histiocíticas expandidas nas lesões podem ser diferenciadas de HCL com base na coloração positiva para o fator XIIa, CD163, CD14 e S-100, enquanto que a coloração para CD1a e langerina é negativa, e na ausência de grânulos de Birbeck. Não há tratamento definido especificamente para essa condição, embora haja relatos de respostas satisfatórias ao interferon alfa,43 ao imatinib e aos agentes inibidores da interleucina-1.
A LHH é uma constelação de distúrbios não malignos, mas geralmente com risco de vida, associados a uma lista crescente de causas genéticas primárias e de causas secundárias adquiridas. Esses distúrbios se caracterizam pelo acúmulo de células mononucleares benignas bem diferenciadas com fenótipo macrofágico.44 Considerando que esses macrófagos representam um subgrupo de histiócitos distintos de células dendríticas (CDs), esta entidade deverá ser diferenciada de HCL e de outros distúrbios de CDs. Na classificação atual de distúrbios histiocíticos, a LHH subdivide-se em primária ou LHH familiar e secundária ou LHH reativa.1 Entretanto, sob o ponto de vista clínico, é extremamente difícil distinguir uma da outra. A linfohistiocitose hemofagocítica familiar (LHHF) é uma miríade de distúrbios imunes recessivos autossômicos. Usualmente, os sintomas clínicos de LHHF se tornam evidentes nos primeiros dois meses de vida, embora haja relatos de apresentações iniciais que ocorreram aos 22 anos de idade.45 A LHH secundária tende a ocorrer em crianças mais velhas e, com frequência, está associada a um episódio infeccioso identificável, mais marcadamente as infecções pelo vírus Epstein-Barr (VEB) ou pelo citomegalovírus. O grupo de distúrbios hemofagocíticos inclui também a LHH associada a malignidades. Embora haja muitas semelhanças clínicas entre a síndrome da ativação de macrófagos (SAM) e LHH primária e secundária, ainda não há uma compreensão sobre a relação exata entre essas condições.46
Os mecanismos patológicos da LHH não são totalmente compreendidos. Na LHH primária a expansão descontrolada de células T e de macrófagos foi associada à diminuição funcional das células exterminadoras naturais (NKC, do inglês natural killer cells) e das células T citotóxicas47 em decorrência das mutações em vários genes cujos produtos estão envolvidos na via citolítica.48-51 A atividade citotóxica dessas células é mediada pela liberação de grânulos citotóxicos especializados contendo várias classes de proteínas expressas somente em células citotóxicas, incluindo a perforina e as granzimas. Após a ativação das células citotóxicas esses grânulos são liberados para a superfície das células, sendo que seu conteúdo é liberado na sinapse imunológica com a célula alvo. A perforina facilita a liberação dos grânulos no interior do citoplasma da célula alvo, enquanto que as granzimas desencadeiam apoptose depois que estiverem no citoplasma da célula alvo. A disfunção citolítica causada por mutações no gene que codifica a perforina ocorre em 15 a 40% de pacientes com LHHF.48 As mutações em outro gene – MUNC13-4 – foram associadas ao desenvolvimento de LHH em aproximadamente 10 a 30% de pacientes com herança de linfohistiocitose hemofagocítica.49 A proteína codificada pelo gene MUNC13-4 é importante para a fixação e a fusão dos grânulos citotóxicos com a membrana citoplasmática. Embora as células citolíticas dos pacientes com LHHF produzidas por mutações no MUNC13-4 produzam quantidades suficientes de perforina, a baixa capacidade para liberar o conteúdo dos grânulos citolíticos para a sinapse imunológica com a célula alvo reduz profundamente a atividade citolítica. Mais recentemente, as mutações em dois outros genes que codificam proteínas que facilitam a fusão granular em eventos do trânsito intracelular foram associadas ao desenvolvimento de LHH primária: a sintaxina 11,50 membro da família SNARE de proteínas e a proteína 2 de ligação com a sintaxina (STXBP2, também conhecida por MUNC18-2).51
Alguns defeitos nas funções citotóxicas dependentes de grânulos dos linfócitos também foram implicados em três outras doenças genéticas associadas à síndrome hemofagocítica. Consequentemente, as mutações no gene que codifica a Rab27a, uma das moléculas efetoras do MUNC18-4, foram associadas ao desenvolvimento da síndrome de Griscelli tipo 2.52 As mutações no gene Lizst foram identificadas como uma das causas da síndrome de Chediak-Higashi.53 A síndrome de Hermansky-Pudlak tipo II foi associada a mutações em AP3B.54 Todos esses distúrbios podem ser complicados pelo desenvolvimento de LHH.
Linfohistiocitose hemofagocítica após a exposição ao vírus Epstein-Barr (EB) e, ocasionalmente, a outros tipos de vírus, denominada mononucleose infecciosa fulminante, é a complicação com risco de vida mais frequente da síndrome linfoproliferativa ligada ao X (LPX). A síndrome LPX1 é causada por mutações hemizigóticas no gene SH2D1A, que codifica o SAP (proteína associada à SLAM), produzindo respostas anormais de células NK e uma deficiência de célula T NK não variante.55 A síndrome LPX2 é causada por mutações no gene BIRC4, que codifica a X1AP (proteína inibidora da apoptose ligada ao X) e foi descrita como uma forma de LHH familiar ligada ao X.56,57 Observações recentes sugerem que os linfócitos de pacientes com ambos os tipos de LPX demonstram a presença de apoptose reduzida induzida por ativação que contribui para a proliferação linfomatosa descontrolada.
Considerados como um conjunto, esses distúrbios genéticos ainda são responsáveis por menos da metade dos casos diagnosticados de LHH em crianças, incluindo muitos casos familiares que ainda aguardam definição molecular.
A presença de defeitos na atividade citotóxica dependente de grânulos dos linfócitos em diversas doenças associadas às síndromes hemofagocíticas destaca a importância desta função para conter algumas respostas inflamatórias. Ainda não estão suficientemente claros os mecanismos exatos que fazem a ligação das funções das células NK deficientes e dos linfócitos T citotóxicos com a expansão de macrófagos ativados. Uma provável explicação está relacionada ao fato de que a atividade citolítica deficiente observada em pacientes com LHH poderá diminuir a capacidade para controlar algumas infecções. Mais especificamente, as células NK e os linfócitos T citotóxicos não conseguem destruir as células infectadas e, consequentemente, não conseguem eliminar a fonte de estimulação antigênica. Por sua vez, essa estimulação antigênica persistente produz uma ativação persistente acionada por antígenos e a proliferação de células T associadas à escalada na produção de citocinas que estimulam os macrófagos. Levantou-se também a hipótese de que as células citotóxicas anormais possivelmente não consigam gerar sinais apoptóticos apropriados para eliminar macrófagos ativados e células T durante o estágio de contração das respostas imunes, resultando na expansão persistente de células T e de macrófagos que segregam citocinas pró-inflamatórias. Os macrófagos se tornam hemofagocíticos como resultado da estimulação contínua das citocinas pró-inflamatórias (em especial o interferon gama).58
As principais características clínicas da LHH incluem febre persistente e hepatoesplenomegalia. O envolvimento do SNC é bastante comum e varia de aumento na irritabilidade até meningoencefalite manifesta. Observa-se com frequência a presença de linfadenopatia generalizada e erupções polimorfas. Esses sintomas clínicos estão associados a uma queda vertiginosa em pelo menos duas entre as três linhas de células sanguíneas (leucócitos, eritrócitos e plaquetas). Usualmente, a queda na contagem de plaquetas é um achado precoce.
A presença de hepatomegalia significativa é frequente. Alguns pacientes desenvolvem icterícia branda. Com frequência, os testes da função hepática revelam níveis elevados de transaminases séricas e níveis ligeiramente moderados de bilirrubina sérica. Existem também relatos da presença de hipoalbuminemia moderada. Tipicamente, os níveis séricos de amônia são normais ou apenas ligeiramente elevados.
As descobertas laboratoriais adicionais nos casos de LHH incluem níveis séricos altamente elevados de triglicérides e de ferritina. A elevação nos níveis de ferritina é particularmente acentuada (acima de 10.000 ng/mL na maioria dos pacientes). Usualmente obtém-se o diagnóstico de LHH com base nas orientações diagnósticas desenvolvidas pela International Histiocyte Society.1,46 Os critérios incluem (a) diagnóstico molecular com base em mutações específicas encontradas no gene PRF1 ou MUNC13-4 ou (b) diagnóstico clínico de acordo com os seguintes critérios clínicos: (1) febre persistente; (2) esplenomegalia; (3) citopenias envolvendo pelo menos duas linhas celulares; (4) hipertrigliceridemia e/ou hipofibrinogenemia; (5) hiperferritinemia e (6) hemofagocitose na medula óssea. Mais recentemente, foram incluídos dois critérios adicionais: (7) ausência ou baixa atividade citolítica da célula NK e (8) níveis de alta solubilidade do receptor alfa da interleucina-2. O diagnóstico clínico definitivo de LHH exige a presença de pelo menos cinco entre os oito critérios.
Levando-se em consideração que o curso natural da linfohistiocitose hemofagocítica poderá se tornar fatal muito rapidamente, recomenda-se iniciar o tratamento quando houver uma alta suspeita clínica, mesmo antes da conclusão da avaliação. Atualmente, a terapia inicial mais eficaz para LHH consiste de combinações de quimioterapia pró-apoptótica, como o tratamento à base de etoposida (VP-16), e medicamentos imunossupressivos (esteroides e, em alguns casos, globulina antitimócito [ATG]) com foco principal nas células T ativadas e nos histiócitos 46. O tratamento definitivo e a cura potencial da linfohistiocitose hemofagocítica familiar (LHHF) são possíveis apenas com transplante de células-tronco hematopoiéticas.
A síndrome da ativação de macrófagos (SAM) se refere às síndromes que se apresentam como complicações de doenças reumáticas. Assim como outras síndromes hemofagocíticas, a SAM é causada pela ativação e expansão excessivas de linfócitos T e de histiócitos macrofágicos com atividade hemofagocítica.59-61 Embora a característica patognomônica da SAM, isto é, os elementos hematopoiéticos normais de histiócitos fagocitantes usualmente seja observada na medula óssea, esse tipo de célula pode se infiltrar em quase todos os órgãos do corpo humano. Além disso, a expansão dessas células poderá gerar respostas inflamatórias sistêmicas maciças associadas a três características essenciais: citopenias, disfunção hepática e coagulopatia assemelhando-se à coagulação intravascular disseminada. Trata-se de uma condição com risco de vida com relatos de mortalidade variando de 20 a 30%.60-62 Embora a SAM tenha sido associada a quase todas as doenças reumáticas, é disparadamente mais comum nos casos de artrite idiopática juvenil sistêmica (AIJS). Por outro lado, cerca de 10% de pacientes com AIJS contraem SAM completamente desenvolvida,62 enquanto que a incidência de SAM “subclínica” branda ocorre em até um terço de pacientes portadores de doença sistêmica ativa.63,64 Além da AIJS, o lúpus eritematoso sistêmico (LES) e a doença de Kawasaki são outras duas condições reumatólogicas nas quais a SAM aparentemente ocorre com mais frequência do que em outros tipos de doença. Embora a maior parte dos pacientes desenvolva essa síndrome em algum momento durante o curso de uma doença reumática primária, não é comum a SAM ocorrer na apresentação inicial das enfermidades reumatológicas. Com base em alguns estudos epidemiológicos, a SAM ocorre com mais frequência em adultos em associação com a doença de Still com início na vida adulta, LES e várias síndromes vasculíticas.
Embora não haja relatos de casos familiares de SAM na artrite idiopática juvenil sistêmica, dados recentes revelam que os pacientes com condições como LHHF e AIJS/SAM apresentam defeitos funcionais na rota de desgranulação de exossomas.65 Além disso, esses defeitos estão associados a polimorfismos nucleotídeos simples nos genes MUNC13-4 e PRF1 relacionados à linfohistiocitose hemofagocítica familiar.66-69
As descobertas clínicas em casos manifestos de SAM geralmente são dramáticas.60-62 Tipicamente, os pacientes com condição crônica se tornam gravemente enfermos e se apresentam com febre persistente, alterações no estado mental, linfadenopatia, hepatoesplenomegalia e disfunção hepática. A presença da síndrome hemorrágica que se assemelha à coagulação intravascular disseminada é outra característica marcante da síndrome da ativação de macrófagos. Erupções hemorrágicas na pele causadas por petéquias leves a lesões equimóticas, epistaxe, hematêmese secundária a hemorragia na parte superior do trato gastrintestinal e hemorragia retal são condições observadas com bastante frequência nesse grupo de pacientes. Esses sintomas clínicos estão associados a quedas vertiginosas em pelo menos duas entre as três linhas de células sanguíneas (leucócitos, eritrócitos e plaquetas). Usualmente, a queda na contagem de plaquetas é um achado precoce. Levando-se em consideração que, em geral, a aspiração de medula óssea revela a presença de hipercelularidade significativa e de megacariócitos normais, aparentemente essas citopenias não são secundárias à produção celular inadequada. O aumento na destruição das células por fagocitose ou por consumo nos sítios inflamatórios é a explicação mais provável. Quedas acentuadas na velocidade de hemossedimentação (VHS), a despeito do nível elevado persistente de proteína C reativa, são mais uma característica laboratorial que provavelmente reflita o grau de hipofibrinogenemia secundária ao consumo de fibrinogênio e à disfunção hepática. Na realidade, o envolvimento hepático é comum nos casos de SAM. Alguns pacientes desenvolvem icterícia branda. Com frequência, os testes da função hepática mostram níveis séricos elevados da atividade transaminase e níveis ligeiramente elevados de bilirrubina sérica. Há também relatos da presença de hipoalbuminemia moderada. Tipicamente, os níveis séricos de amônia são normais ou apenas ligeiramente elevados.
Encefalopatia é outra característica clínica da síndrome da ativação de macrófagos documentada com bastante frequência. Alterações no estado mental, convulsões e coma são as manifestações mais comuns da doença no SNC. Observou-se em alguns estudos a presença de pleiocitose no líquido cerebroespinal com níveis proteicos ligeiramente altos. Deterioração significativa da função renal foi observada em diversas séries e em um relato foi associada a taxas de mortalidade particularmente elevadas.62 Diversos relatos fizeram referência à presença de infiltrados pulmonares e macrófagos hemofagocíticos encontrados em casos de lavagem broncoalveolar.
As descobertas laboratoriais complementares em casos de SAM incluem níveis séricos excessivamente elevados de triglicérides e ferritina. Assim como nos casos de histiose hemofagocítica familiar, a elevação nos níveis de ferritina é particularmente acentuada e, aparentemente, se iguala ao grau de ativação de macrófagos.
Não há critérios diagnósticos válidos para SAM e, na maioria das vezes, é muito difícil obter diagnósticos na fase inicial da condição. Consequentemente, os pacientes portadores de doença reumatológica subjacente persistentemente ativa e com queda na velocidade de hemossedimentação e na contagem de plaquetas, principalmente em combinação com elevação nos níveis séricos do dímero D e de ferritina, devem levantar suspeitas de síndrome de ativação de macrófagos iminente. Usualmente obtém-se o diagnóstico de SAM pela confirmação da presença de hemofagocitose na medula óssea. No entanto, a confirmação da presença de hemofagocitose na medula óssea pode se tornar extremamente difícil em decorrência de erros de amostragem, em especial nos estágios iniciais da síndrome. Além disso, acredita-se de uma forma cada vez mais convincente que os macrófagos hemofagocíticos podem infiltrar nos tecidos e não na medula óssea (i.e., fígado, linfonodos, pele e pulmões).
Infelizmente, a aplicação dos critérios diagnósticos de LHH em pacientes com AIJS com suspeita de SAM é muito problemática. Alguns marcadores de LHH, tais como linfadenopatia, esplenomegalia e hiperferritinemia, são características comuns de AIJS ativa e, consequentemente, não distinguem SAM da manifestação súbita da AIJS convencional. Outros critérios diagnósticos de LHH, incluindo citopenias e hipofibrinogenemia, se tornam evidentes somente nos estágios finais da doença. Isso se relaciona ao fato de que os pacientes com AIJS apresentam contagens elevadas de leucócitos e de plaquetas e níveis altos de fibrinogênio sérico como parte da resposta inflamatória da doença. Portanto, nas situações em que desenvolverem SAM, os pacientes atingem o grau de citopenias e hipofibrinogenemia observado nos casos de LHH apenas nos estágios finais da síndrome, quando o tratamento se transforma em um grande desafio. Esta situação é ainda mais problemática para o diagnóstico de SAM em pacientes com lúpus eritematoso sistêmico, nos quais as citopenias autoimunes são comuns e é difícil distingui-las daquelas causadas pela síndrome de ativação de macrófagos. Nesses pacientes, a presença de condições como hiperferritinemia extrema e elevação no nível da lactato desidrogenase deve levantar suspeitas sobre a presença de SAM. Recentemente foram iniciadas algumas tentativas para modificar os critérios de LHH com o objetivo de aumentar a sensibilidade e a especificidade para o diagnóstico de SAM em condições reumáticas.
A síndrome da ativação de macrófagos (SAM) é uma condição com risco de vida associada a uma taxa elevada de mortalidade. Portanto, a identificação precoce desta síndrome e a intervenção terapêutica imediata para produzir respostas rápidas são medidas extremamente importantes. Grande parte dos médicos inicia com pulsoterapia à base da aplicação intravenosa de metilprednisolona (30 mg/kg por três dias consecutivos), seguida por quatro doses divididas de 2 a 3 mg/kg/dia. Recomenda-se iniciar a administração parenteral de ciclosporina se a resposta aos esteroides não for evidente.70,71 Na maior parte dos pacientes com SAM, a adição de ciclosporina não apenas possibilita o controle rápido dos sintomas, mas também permite evitar o uso excessivo de esteroides. Os pacientes nos quais a SAM permanecer ativa, apesar do uso de corticosteroides e de ciclosporina, são um grande desafio. Nesses pacientes recomenda-se considerar a hipótese de usar o protocolo HLH-2004 desenvolvido pela International Histiocyte Society para tratamento de LHH46 [ver a Tabela 2]. Recentemente, foi apresentada a sugestão de que em pacientes que não responderem à combinação de esteroides e ciclosporina, principalmente em indivíduos com danos renais e hepáticos, a ATG possivelmente seja uma alternativa mais segura para o etoposídeo.72 A ATG destrói as células T CD4+ e CD8+ através da lise celular dependente do complemento. Alguns pacientes apresentam também uma depleção leve de monócitos. Embora nos casos documentados esse tipo de tratamento tenha sido bem tolerado, é importante ressaltar que, em geral, as reações às infusões ocorrem com a administração de ATG e, nas situações em que este for o tratamento de escolha, recursos laboratoriais e suporte médico adequados devem ser disponibilizados para uso imediato. Além disso, os pacientes tratados com ATG ou com medicamentos similares são substancialmente imunossuprimidos e necessitam de um suporte rigoroso, isto é, típico de receptores de transplante de medula ou de órgãos sólidos. Ainda não está muito clara a utilidade dos medicamentos biológicos no tratamento de SAM.73,74
Histiocitose sinusal com linfadenopatia maciça ou doença de Rosai-Dorfman é um distúrbio policlonal de etiologia incerta.75 A origem exata das células típicas da doença de Rosai-Dorfman não é muito clara, embora seu fenótipo tenha muita semelhança com macrófagos ativados. Essas células são S-100+, CD682+, CD1632+, CD1a- e langerina negativa.
|
Tabela 2: HLH-2004: Orientações Diagnósticas Revisadas para Linfohistiocitose Hemofagocítica46
|
|
O diagnóstico de LHH pode ser estabelecido se uma entre as duas condições abaixo for atendida:
Diagnóstico molecular consistente com LHH (i.e., mutações documentadas em PRF1 ou MUNC13-4) ou
Observância dos critérios diagnósticos de LHH (i.e., pelo menos na presença de cinco entre os oito critérios abaixo): Febre persistente Esplenomegalia Citopenias (afetando = 2 entre três linhas de sangue periférico): Hemoglobina < 9 g/100 mL (em lactentes < 4 semanas < 10 g/ 100 mL) Plaquetas < 100 x 103/mL Neutrófilos < 1 x 103/mL Hipertrigliceremia e/ou hipofibrinogenemia Triglicérides de jejum = 3,0 mmol/L (i.e., = 265 mg/dL) Fibrinogênio = 1,5 g/L (i.e., = 150 mg/dL) Hemofagocitose na medula óssea* ou no baço ou em linfonodos, sem evidências de malignidades Ferritina sérica = 500 µg/L Atividade baixa ou ausente da célula NK (de acordo com referência do laboratório local) Nível sérico elevado do receptor alfa da interleucina 2 solúvel em água (de acordo com referência do laboratório local).
|
HLH (hemophagocytic lymphohistiocytosis) = LHH (linfohistiocitose hemofagocítica); NK – natural killer (células exterminadoras naturais).
*Se a atividade hemofagocítica não tiver sido comprovada no momento da apresentação, deve-se incentivar uma nova busca de atividade hemofagocítica. Se a amostra de medula óssea não for conclusiva é necessário colher material de outros órgãos.
De maneira geral, esses pacientes se apresentam com adenopatia cervical bilateral indolor associada a febres, sudorese noturna, indisposição e perda de peso. Outros grupos de linfonodos também poderão ser afetados. Embora não seja comum o envolvimento de tecidos extranodais como pele, trato respiratório superior, ossos, olhos e tecidos retro-orbitais, ele poderá ocorrer sem participação aparente dos linfonodos.
Sob a ótica histopatológica, os linfonodos afetados revelam a presença de infiltração sinusal maciça em histiócitos de grandes dimensões, juntamente com linfócitos e células plasmáticas. Eritrócitos, linfócitos e células plasmáticas intactas poderão ser absorvidos pelas células histiocíticas através de um processo conhecido por emperipolese. Ao contrário da hemofagocitose, a emperipolese não está associada à destruição das células absorvidas.
O tratamento é incerto e as resoluções espontâneas são comuns. Os esteroides são usados em pacientes com envolvimento sistêmico e há relatos de que a quimioterapia foi útil em algumas circunstâncias.76
__________________________________________________________
Alexei Grom, MD, recebeu honorários de consultoria da Novartis.
Michael B. Jordan, MD, PhD, e Jun Qin Mo, MD não mantêm nenhuma relação comercial com os fabricantes dos produtos ou provedores de serviços mencionados neste capítulo.
1. Favara BE, Feller AC, Pauli M, et al. Contemporary classification of histiocytic disorders. The WHO Committee On Histiocytic/Reticulum Cell Proliferations. Reclassification Working Group of the Histiocyte Society. Med Pediatr Oncol 1997;29:157–66.
2. Weitzman S, Jaffe R. Uncommon histiocytic disorders: the non-Langerhans cell histiocytoses. Pediatr Blood Cancer 2005;45:256–64.
3. Merad M, Ginhoux F, Collin M. Origin, homeostasis and function of Langerhans cells and other langerin-expressing dendritic cells. Nat Rev Immunol 2008;8:935–47.
4. Katz SI, Tamaki K, Sach DH. Epidermal Langerhans cells are derived from cells originating in bone marrow. Nature 1979;282:324–6.
5. Abla O, Egeler MR, Weitzman S. Langerhans cell histiocytosis: current concepts and treatments. Cancer Treat Rev 2010;36:354–9.
6. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature 1998;392:245–52.
7. Pileri SA, Grogan TM, Harris NL, et al. Tumours of histiocytes and accessory dendritic cells: an immunohistochemical approach to classification from the International Lymphoma Study Group based on 61 cases. Histopathology 2002;41:1–29.
8. Valladeau J, Ravel O, Dezuter-Dambuant C, et al. Langerin, a novel C-type lectin specific to Langerhans cells, is an endocytic receptor that induces the formation of Birbeck granules. Immunity 2000;12:71–81.
9. Hunger RE, Sieling PA, Ochoa MT, et al. Langerhans cells utilize CD1a and langerin to efficiently present nonpeptide antigens to T cells. J Clin Invest 2004;113:701–8.
10. McDermott R, Ziylan U, Spehner D, et al. Birbeck granules are subdomains of endosomal recycling compartment in human epidermal Langerhans cells, which form where langerin accumulates. Mol Biol Cell 2002;13:317–35.
11. Nicholson HS, Egeler RM, Nesbit ME. The epidemiology of Langerhans cell histiocytosis. Hematol Oncol Clin North Am 1998;12:379.
12. Bechan GI, Egeler RM, Arceci RJ. Biology of Langerhans cells, and Langerhans cell histiocytosis. Int Rev Cytol 2006;254:1–43.
13. Willman CL, Busque L, Griffith BB, et al. Langerhans' cell histiocytosis (histiocytosis X)—a clonal proliferative disease. N Engl J Med 1994;331:154.
14. Yu RC, Chu C, Buluwela L, Chu AC. Clonal proliferation of Langerhans cells in Langerhans' cell histiocytosis. Lancet 1994;343:767.
15. Savasan S. An enigmatic disease: childhood Langerhans cell histiocytosis in 2005. Int J Dermatol 2006;45:182–8.
16. Allen CE, Li L, Peters T, et al. Cell-specific gene expression in Langerhans cell histiocytosis lesions reveals a distinct gene expression profile compared to epidermal Langerhans cells. J Immunol 2010;184:4557–679.
17. Munn S, Chu AC. Langerhans cell histiocytosis of the skin. Hematol Oncol Clin North Am 1998;12:269.
18. Minkov M, Prosch H, Steiner M, et al. Langerhans cell histiocytosis in neonates. Pediatr Blood Cancer 2005;45:802–7.
19. Lau L, Krafchik B, Trebo MM, Weitzman S. Cutaneous Langerhans cell histiocytosis in children over one year. Pediatr Blood Cancer 2006;46:66.
20. Slater JM, Swarm OJ. Eosinophilic granuloma of the bone. Med Pediatr Oncol 1980;8:151.
21. Garg S, Mehta S, Dormans JP. Langerhans cell histiocytosis of the spine in children. Long-term follow up. J Bone Joint Surg Am 2004;86-A:1740.
22. D'Ambrosio N, Soohoo S, Warshall C, et al. Craniofacial and intracranial manifestations of Langerhans cell histiocytosis: report of findings in 100 patients. AJR Am J Roentgenol 2008;191:589.
23. Kaplan KJ, Goodman ZD, Ishak KG. Liver involvement in Langerhans cell histiocytosis: a study of nine cases. Mod Pathol 1999;12:370.
24. Jaffe R. Liver involvement in the histiocytic disorders of childhood. Pediatr Dev Pathol 2004;7:214.
25. Odame I, Li P, Lau L, et al. Pulmonary Langerhans cell histiocytosis: a variable disease in childhood. Pediatr Blood Cancer 2006;47:889.
26. Vassallo R, Ryu JH, Colby TV, et al. Pulmonary Langerhans cell histiocytosis. N Engl J Med 2000;342:1969.
27. McClain K, Ramsay NK, Robison L, et al. Bone marrow involvement in histiocytosis X. Med Pediatr Oncol 1983;11:167–71.
28. Minkov M, Potschger U, Grois N, et al. Bone marrow assessment in Langerhans cell histiocytosis. Pediatr Blood Cancer 2007;49:694.
29. Donadieu J, Rolon MA, Thomas C, et al. Endocrine involvement in pediatric-onset Langerhans cell histiocytosis: a population-based study. J Pediatr 2004;144:344.
30. Hait E, Liang M, Degar B, et al. Gastrointestinal tract involvement in Langerhans cell histiocytosis: case report and review of the literature. Pediatrics 2006;118:1593.
31. Grois N, Fahrner B, Arceci RJ, et al. Central nervous system disease in Langerhans cell histiocytosis. J Pediatr 2010;156:873.
32. Satter EK, High WA. Langerhans cell histiocytosis: a review of the current recommendations of the Histiocyte Society. Pediatr Dermatol 2008;25:291–5.
33. Farran RP, Zaretski E, Egeler RM. Treatment of Langerhans cell histiocytosis with pamidronate. J Pediatr Hematol Oncol 2001;23:54–6.
34. Moromoto A, Shioda Y, Imamura T, et al. Nationwide survey of bisphosphonate therapy for children with reactivated Langerhans cell histiocytosis in Japan. Pediatr Blood Cancer 2011;56:110–5.
35. Gadner H, Grois N, Potchger U, et al. Improved outcome in multisystem Langerhans cell histiocytosis is associated with therapy intensification. Blood 2008;111:2556.
36. Jordan MB, McClain KL, Yan X, et al. Anti-CD52 antibody, alemtuzumab, binds to Langerhans cells in Langerhans cell histiocytosis. Pediatr Blood Cancer 2005;44:251.
37. Janssen D, Harms D. Juvenile xanthogranuloma in childhood and adolescence: a clinicopathologic study of 129 patients from Kiel pediatric tumor registry. Am J Surg Pathol 2005;29:21.
39. Freyer DR, Kennedy R, Bostrom BC, et al. Juvenile xanthogranuloma: forms of systemic disease and their clinical implications. J Pediatr 1996;129:227.
40. Stover DG, Alapati S, Regueira O, et al. Treatment of juvenile xanthogranuloma. Pediatr Blood Cancer 2008;51:130.
41. Trotta F, Castellino G, Monaco AL. Multicentric reticulohistiocytosis. Best Pract Res Clin Rheumatol 2004;18:759–72.
42. Veyssier-Belot C, Cacoub P, Caparros-Lefebvre D, et al. Erdheim-Chester disease. Clinical and radiologic characteristics of 59 cases. Medicine (Baltimore) 1996;75:157.
43. Braiteh F, Boxrud C, Esmaeli B, Kurzrock R. Successful treatment of Erdheim-Chester disease, a non-Langerhans cell histiocytosis with interferon-alpha. Blood 2005;106:2992.
44. Filipovich HA. Hemophagocytic lymphohistiocytosis. Immunol Allergy Clin North Am 2002;22:281–300.
45. Clementi R, Emi L, Maccario R, et al. Adult onset and atypical presentation of hemophagocytic lymphohistiocytosis in siblings carrying PRF1 mutations. Blood 2002;100:2266–7.
46. Henter JI, Horne A, Arico M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemopagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007;48:124–31.
47. Sullivan KE, Delaat CA, Douglas SD, Filipovich AH. Defective natural killer cell function in patients with hemophagocytic lymphohistiocytosis and in first degree relatives. Pediatr Res 1998;44:465–8.
48. Stepp SE, Dufourcq-Lagelouse R, Le Deist F, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science 1999;286:1957–9.
49. Feldmann J, Callebaut I, Raposo G, et al. MUNC13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell 2003;115:461–73.
50. Zur Stadt U, Schmidt S, Kasper B, et al. Linkage of familial hemophagocytic lymphohistiocytosis (FHL) type-4 to chromosome 6q24 and identification of mutations in syntaxin 11. Hum Mol Genet 2005;14:827–34.
51. Zur Stadt U, Rohr J, Seifert W, et al. Familial hemophagocytic lymphohistiocytosis type 5 (FHL-5) is caused by mutations in MUNC18-2 and impaired binding to syntaxin11. Am J Hum Genet 2009;85:482–92.
52. Menasche G, Pastural E, Feldman J, et al. Mutations in Rab27a cause Griscelli syndrome associated with haemophagocytic syndrome. Nat Genet 2000;25:173–6.
53. Barbosa MD, Nguyen QA, Tchernev VT, et al. Identification of the homologous beige and Chediak-Higashi syndrome genes (LYST). Nature 1996,382:262–5.
54. Badolato R, Parolini S. Novel insights from adaptor protein 3 complex deficiency. J Allergy Clin Immunol 2007;120:735–41.
55. Coffey AJ, Brooksbank RA, Brandau O, et al. Host response to EBV infection in X-linked lymphoproliferative disease results from mutations in an SH2-domain encoding gene. Nat Genet 1998;20:129.
56. Rigaud S, Fondaneche MC, Lambert N, et al. XIAP deficiency in humans causes an X-linked lymphoproliferative disease. Nature 2006;444:110–4.
57. Marsh RA, Madden L, Kitchen BJ, et al. XIAP deficiency: a unique primary immunodeficiency best classified as X-linked familial hemophagocytic lymphohistiocytosis and not as X-linked lymphoporliferative disease. Blood 2010;116:1079–82.
58. Jordan MB, Hildeman D, Kappler J, Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood 2004;104:735–43.
59. Athreya BH. Is macrophage activation syndrome is a new entity? Clin Exp Rheumatol 2002;20:121–3.
60. Hadchouel M, Prieur AM, Griscelli C. Acute hemorrhagic, hepatic, and neurologic manifestations in juvenile rheumatoid arthritis: possible relationship to drugs or infection. J Pediatr 1985;106:561–6.
61. Grom AA. NK dysfunction: a common pathway in systemic onset juvenile rheumatoid arthritis, macrophage activation syndrome, and hemophagocytic lymphohistiocytosis. Arthritis Rheum 2004;50:689–98.
62. Sawhney S, Woo P, Murray KJ. Macrophage activation syndrome: a potentially fatal complication of rheumatic disorders. Arch Dis Child 2001;85:421–6.
63. Bleesing J, Prada A, Villanueva J, et al. The diagnostic significance of soluble CD163 and soluble IL2Ra chains in macrophage activation syndrome and untreated new onset systemic juvenile idiopathic arthritis. Arthritis Rheum 2007;56:965–71.
64. Behrens EM, Beukelman T, Paessler M, Cron RQ. Occult macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis. J Rheumatol 2007;34:1133–8.
65. Grom AA, Villanueva J, Lee S, et al. Natural killer cell dysfunction in patients with systemic-onset juvenile rheumatoid arthritis and macrophage activation syndrome. J Pediatr 2003;142:292–6.
66. Villanueva J, Lee S, Giannini EH, et al. Natural killer cell dysfunction is a distinguishing feature of systemic onset juvenile rheumatoid arthritis and macrophage activation syndrome. Arthritis Res Ther 2005;7:R30–37.
67. Hazen MM, Woodward AL, Hofman I, et al. Mutations of the hemophagocytosis-associated gene UNC13D in a patient with systemic juvenile idiopathic arthritis. Arthritis Rheum 2008;58:567–70.
68. Zhang K, Biroscak J, Glass DN, et al. Macrophage activation syndrome in systemic juvenile idiopathic arthritis is associated with MUNC13D gene polymorphisms. Arthritis Rheum 2008;58:2892–6.
69. Vastert SJ, van Wijk R, D'Urbano LE, et al. Mutations in the perforin gene can be linked to macrophage activation syndrome in patients with systemic onset juvenile idiopathic arthritis. Rheumatology (Oxford) 2010;49:441–9.
70. Stephan JL, Kone-Paut I, Galambrun C, et al. Reactive haemophagocytic syndrome in children with inflammatory disorders. A retrospective study of 24 patients. Rheumatology (Oxford) 2001;40:1285–92.
71. Ravelli A, Magni-Manzoni S, Pistorio A, et al. Preliminary diagnostic guidelines for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. J Pediatr 2005;146:598–604.
72. Coca A, Bundy KW, Marston B, et al. Macrophage activation syndrome: serological markers and treatment with anti-thymocyte globulin. Clin Immunol 2009;132:10–8.
74. Zeft A, Hollister R, LaFleur B, et al. Anakinra for systemic juvenile arthritis: the Rocky Mountain experience. J Clin Rheumatol 2009;15:161–4.
75. Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol 1990;7:19.
76. Pulsoni A, Anghel G, Falcucci P, et al. Treatment of sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): report of a case and literature review. Am J Hematol 2002;69:67.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.