(Carregando Índice)... (Carregando Índice)... |
Autor:
Victor Celso Cenciper Fiorini
Especialista em Neurologia pelo Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo (HC-FMUSP)
Última revisão: 29/08/2009
Comentários de assinantes: 0
Miopatias são doenças estruturais e/ou funcionais dos músculos, resultantes de uma variedade de etiologias. Manifestam-se fundamentalmente por fraqueza, devendo ser diferenciadas das doenças do neurônio motor, das neuropatias periféricas e das doenças da junção neuromuscular.
O primeiro passo na avaliação do paciente é identificar se a miopatia é por defeito em canais, alteração estrutural ou distúrbio metabólico. A seguir, procura-se determinar se a miopatia é hereditária ou adquirida (Tabela 1). Por último, deve-se avaliar se há disponibilidade de tratamento, mesmo que paliativo.
Tabela 1: Classificação das miopatias
|
Miopatias hereditárias |
Miopatias congênitas Distrofias musculares Canalopatias Miopatias metabólicas primárias Miopatias mitocondriais |
|
Miopatias adquiridas |
Miopatias inflamatórias Miopatias tóxicas e induzidas por drogas Miopatias metabólicas secundárias Miopatias endócrinas Miopatias infecciosas |
Este capítulo irá discorrer exclusivamente sobre as miopatias hereditárias.
As miopatias metabólicas primárias são doenças raras, causadas por defeito bioquímico dos sistemas energéticos musculares, os quais podem envolver o metabolismo de carboidratos (glicogenoses), lipídios (lipidoses), reações na mitocôndria ou no ciclo do nucleotídeo purina. Há cerca de dez tipos de defeitos do metabolismo de carboidratos associados a miopatias, sendo os mais importantes o tipo 2 (deficiência de maltase ácida ou doença de Pompe), o tipo 5 (deficiência de miofosforilase ou doença de McArdle) e o tipo 7 (deficiência de fosfofrutoquinase ou doença de Tartui). A deficiência de carnitina palmitoiltransferase é causada por um defeito no metabolismo lipídico. Como exemplos de miopatias mitocondriais, temos a deficiência do complexo IV e a deficiência de coenzima Q10. Finalmente, a deficiência de mioadenilato deaminase (MAD) é um defeito no metabolismo do nucleotídeo purina.
As miopatias congênitas são aquelas que se manifestam na infância por atraso no desenvolvimento motor ou mesmo pela síndrome do bebê hipotônico. Geralmente, são familiares e os principais exemplos são a miopatia central core, a miopatia nemalínica e a miopatia tubular (centronuclear). São essencialmente caracterizadas por alterações estruturais das fibras musculares.
As distrofias musculares são doenças hereditárias com características histológicas particulares, incluindo fibrose extensa, degeneração e regeneração de fibras e proliferação de tecido adiposo e gorduroso. Os principais representantes são a distrofia muscular de Duchenne (DMD) (recessiva ligada a X, com ausência ou diminuição importante da expressão de distrofina), a distrofia muscular de Becker (DMB) (forma mais branda da DMD), a distrofia miotônica de Steinert (herança autossômica dominante com expansão de trinucleotídeos, ou seja, instabilidade do DNA), a distrofia fáscio-escápulo-umeral (herança autossômica dominante), a distrofia cintura-membros (autossômica dominante ou recessiva, com ausência de coloração à biópsia para sarcoglicanas, calpaína, disferlina ou caveolina), a distrofia muscular de Emery-Dreifuss (recessiva ligada a X) e a distrofia muscular óculo-faríngea (autossômica dominante, com expansão curta de GCG).
As canalopatias são doenças raras causadas por distúrbios funcionais das proteínas dos canais iônicos, decorrentes de mutações específicas, incluindo as paralisias periódicas e os distúrbios com miotonia (distúrbio da contração muscular no qual o estado de contração da fibra instala-se e desaparece mais lentamente do que no estado normal).
As miopatias mitocondriais ocorrem por mutação do DNA mitocondrial (DNAmt) ou até mesmo por mutações do DNA nuclear em genes específicos para proteínas constituintes da mitocôndria. As mutações do DNA mitocondrial são inúmeras, tendo sido descritas mais de 120 delas. Manifestam-se das mais variadas formas. Os representantes mais importantes são a oftalmoplegia externa progressiva crônica (OEPC), a síndrome de Kearns-Sayre (SKS), a síndrome MELAS (encefalomiopatia mitocondrial com episódios isquêmicos e acidose láctica), a síndrome MERRF (epilepsia mioclônica com fibras vermelhas rajadas), a síndrome NARP (neuropatia, ataxia e retinite pigmentosa), a síndrome de Leigh de herança materna, além da intolerância esporádica ao exercício. As miopatias mitocondriais por alteração do DNA nuclear são muito raras e, entre elas, merece menção a síndrome de Leigh, algumas encefalomiopatias por deficiência dos complexos II ou IV da cadeia respiratória, a oftalmoplegia externa progressiva autossômica dominante e a encefalomiopatia mitocondrial neurogastrintestinal (MNGIE), por defeito no gene da timidina fosforilase.
Tabela 2: Etiologia das miopatias hereditárias
|
Tipo de miopatia |
Características |
Principais causas |
|
Distrofias musculares |
Doenças hereditárias Fibrose e degeneração de fibras musculares |
Distrofia muscular de Duchenne Distrofia muscular de Becker Distrofia miotônica de Steinert Distrofia fáscio-escápulo-umeral Distrofia cintura-membros Distrofia muscular de Emery-Dreifuss Distrofia muscular óculo-faríngea |
|
Congênitas |
Familiares Manifestação na infância Atraso de desenvolvimento motor ou síndrome do bebê hipotônico Alteração da estrutura da fibra muscular |
Miopatia central core Miopatia nemalínica Miopatia tubular (centronuclear) |
|
Metabólicas primárias |
Raras Defeito bioquímico dos sistemas energéticos musculares |
Tipo 2: deficiência de maltase ácida ou doença de Pompe Tipo 5: deficiência de miofosforilase ou doença de McArdle Tipo 7: deficiência de fosfofrutoquinase ou doença de Tartui |
|
Canalopatias |
Raras Distúrbios funcionais das proteínas dos canais iônicos |
Paralisias periódicas Miotonias |
|
Mitocondriais |
Mutação do DNA mitocondrial |
Oftalmoplegia externa progressiva crônica (OEPC) Síndrome de Kearnes-Sayre (SKS) Síndrome MELAS Síndrome MERRF Síndrome NARP Síndrome de Leigh de herança materna Intolerância esporádica ao exercício |
O elemento mais importante na avaliação das miopatias hereditárias é a obtenção de um histórico detalhado.
Obviamente, é fundamental a determinação do início, da duração e da evolução dos sintomas. Por exemplo, pacientes com DMD são habitualmente identificados aos 3 anos de idade, enquanto as distrofias cintura-membros e fáscio-escápulo-umeral iniciam-se na adolescência ou após. Em relação à evolução, as miopatias podem se apresentar com fraqueza muscular constante, como nas distrofias, ou episódica, como na paralisia periódica hipocalêmica. Neste último caso, o início é agudo e a força muscular pode se normalizar após horas ou dias. Além disso, os sintomas costumam ser recorrentes.
A investigação de casos na família é de suma importância para o diagnóstico de miopatias hereditárias. O heredograma ajuda no diagnóstico de distúrbios dominantes, recessivos, autossômicos ou ligados a X. Perguntar se alguém na família precisou usar bengala ou cadeira de rodas, se havia deformidades esqueléticas ou limitações funcionais costuma ser mais útil do que a indagação sobre o fato de haver mais alguém na família com a mesma doença.
A história de fatores precipitantes que poderiam deflagrar ou exacerbar os sintomas de fraqueza ou miotonia deve ser explorada, perguntando-se não só sobre o consumo de substâncias lícitas e ilícitas, mas também sobre exercícios (glicogenoses), febre (carnitina palmitoiltransferase) e ingestão de carboidratos (paralisia periódica). Os pacientes com miotonia congênita queixam-se de piora com o frio.
Deve-se perguntar sobre sintomas cardíacos ou respiratórios, distúrbios visuais (catarata está presente na distrofia miotônica de Steinert), calvície, (idem) e sintomas gerais (para diferencial com outras doenças).
Os sintomas são divididos em positivos e negativos. Fraqueza, atrofia, intolerância ao exercício e fadiga são sintomas negativos. Os sintomas positivos são mialgias, cãibras, contraturas, miotonia e mioglobinúria (Tabela 3).
Fraqueza é o sintoma mais comum. Quando ocorre nas extremidades inferiores, o paciente costuma queixar-se de dificuldade para subir escadas, levantar-se da cadeira ou do vaso sanitário, ou ficar em pé a partir da posição agachada. Em caso de acometimento dos membros superiores, as queixas costumam ser de dificuldade para levantar cargas acima de sua cabeça ou para pentear os cabelos. Esses sintomas indicam acometimento proximal. Este padrão é o mais comum em miopatias. Mesmo assim, alguns pacientes podem se queixar sintomas de fraqueza distal, ou mesmo de acometimento de músculos inervados por pares cranianos, com disartria, diplopia, disfagia e ptose palpebral.
Fadiga é um sintoma inespecífico e de menor utilidade. Pode refletir o estado cardiopulmonar, nível de condicionamento, saúde geral, hábitos de sono ou estado emocional. Muitos pacientes com essa queixa não têm miopatia, sobretudo diante de exame neurológico normal. Entretanto, fatigabilidade pode resultar de miopatias metabólicas ou mitocondriais, sendo importante definir a duração e a intensidade do esforço desencadeante.
Mialgias, assim como a fadiga, são sintomas inespecíficos. Podem ser episódicas (miopatias metabólicas) ou praticamente constantes (miopatias inflamatórias). Entretanto, a dor é incomum e mais provavelmente se deve a distúrbio ortopédico ou reumatológico.
Cãibras são um tipo específico e involuntário de dor muscular. Podem durar de segundos a minutos e costumam localizar-se numa determinada região. Acontecem com desidratação, hiponatremia, uremia e mixedema, além de distúrbios do neurônio motor, principalmente na esclerose lateral amiotrófica (ELA).
Contraturas musculares são raras e são desencadeadas por exercícios em pacientes com defeito na via glicolítica.
Miotonia é um fenômeno caracterizado por ausência de relaxamento muscular após contração forçada. É mais comum nas mãos e nas pálpebras. Tipicamente, o paciente queixa-se de dificuldade para abrir a mão após um aperto de mãos cordial, ou para abrir os olhos após fechar as pálpebras com força. A exposição ao frio tipicamente piora esse quadro.
Mioglobinúria recorrente é relativamente rara e causada por excesso na liberação de mioglobina pelo músculo durante períodos rápidos de rabdomiólise. Deve-se perguntar ao paciente que se queixa de fraqueza ou mialgia após o esforço se também observa que sua urina fica escurecida.
Tabela 3: Sintomas das miopatias
|
Sintomas negativos |
Fraqueza |
Sintoma mais comum Acometimento proximal: padrão mais comum |
|
Atrofia |
Mais na musculatura proximal | |
|
Intolerância ao exercício |
Fatigabilidade: miopatias metabólicas ou mitocondriais Típica da miastenia gravis | |
|
Fadiga |
Inespecífico, pouca utilidade | |
|
Sintomas positivos |
Mialgia |
Inespecífico Episódica: miopatias metabólicas Constantes: miopatias inflamatórias |
|
Cãibras |
Causas: desidratação, hiponatremia, uremia e mixedema Diferenciar com síndrome neurônio motor (por exemplo: ELA) | |
|
Contraturas |
Raras Em pacientes com defeito na via glicolítica | |
|
Miotonia |
Ausência de relaxamento muscular após contração forçada Mais em mãos e pálpebras Piora com o frio | |
|
Mioglobinúria |
Rara Por liberação de mioglobina por rabdomiólise |
O exame físico deve ser detalhado, incluindo o exame físico geral e principalmente o neurológico. Deve-se determinar a distribuição da fraqueza, testando e graduando a força em todos os grupos musculares de forma padronizada e comparativa, simétrica, preferencialmente contra a gravidade. Não se deve esquecer-se dos músculos cranianos, como os orbiculares dos olhos e lábios, pálpebras, musculatura extra-ocular, língua e palato. Além do exame da força, é importante a determinação de atrofia ou hipertrofia. A atrofia dos músculos periescapulares associada à escápula alada sugere distrofia fáscio-escápulo-umeral. Na miotonia congênita, os músculos podem demonstrar sinais de hipertrofia. Nas distrofias de Becker e Duchenne, há pseudo-hipertrofia das panturrilhas, devido à substituição do músculo por gordura e tecido conjuntivo.
Há seis padrões de apresentação dos quadros de miopatias (Tabela 4).
Tabela 4: Padrões de apresentação dos quadros de miopatias
|
Fraqueza proximal de distribuição cintura-membros |
Padrão mais comum Simétrico, com envolvimento distal brando Envolvimento da musculatura flexora e extensora do pescoço |
|
Fraqueza distal |
Típico: envolve músculos distais dos membros superiores ou inferiores, dos compartimentos anteriores ou posteriores Em formas graves, também acomete músculos proximais O acometimento não é necessariamente simétrico Diagnóstico diferencial com neuropatias periféricas |
|
Fraqueza proximal nos membros superiores e distal nos inferiores |
Altamente sugestivo de distrofia fáscio-escápulo-umeral (principalmente se houver fraqueza de musculatura de face) Distrofia escápulo-peroneal: afeta os músculos periescapulares e o compartimento anterior dos músculos distais do membro inferior Outras doenças associadas: distrofia de Emery-Dreifuss, miopatias congênitas e deficiência de maltase ácida (doença de Pompe) |
|
Fraqueza distal nos membros superiores e proximal nos inferiores |
Envolve os flexores do punho e dos dedos e o quadríceps da coxa Assimétrica, e não envolve a face Praticamente exclusivo da miosite por corpos de inclusão Na distrofia miotônica de Steinert, o quadro é simétrico (incomum) |
|
Ptose palpebral com ou sem oftalmoplegia |
Associado ou não a distúrbio da musculatura faríngea Costuma ocorrer sem diplopia. Pode surgir fraqueza facial O acometimento de musculatura das extremidades é variável Ptose, oftalmoplegia sem diplopia e disfagia: distrofia óculo-faríngea (sobretudo quando surge a partir da meia idade) Ptose e oftalmoplegia sem disfagia: padrão de mitocondriopatias Ptose e fraqueza facial, sem oftalmoplegia: padrão da distrofia miotônica. |
|
Fraqueza importante da musculatura extensora do pescoço |
Ocorre fraqueza grave da extensão cervical: “pescoço caído” O envolvimento dos flexores é variável A fraqueza dos membros segue os padrões já descritos Quadro puro: miopatia extensora isolada do pescoço Quadro acentuado: na ELA e na miastenia gravis |
O exame isoladamente mais útil no estudo de pacientes com suspeita de miopatia é a dosagem sangüínea de CK (creatinofosfoquinase). Está elevada na maioria dos casos, mas pode estar normal nos casos de miopatias progressivas. O grau de alteração do exame também é útil na avaliação do tipo de miopatia. Por exemplo, na DMD, a CK está pelo menos dez vezes acima do normal, sendo mais freqüente o aumento acima de 100 vezes, enquanto na maioria das outras miopatias o aumento é mais discreto. Exceções a esse aumento mais discreto são na caveolinopatia, disferlinopatia e calpainopatia, nas quais a CK está muito elevada. A CK pode não estar elevada ou até mesmo diminuída em alguns casos, como perda muscular acentuada, uso de corticóide, colagenoses, alcoolismo e hipertiroidismo.
Há algumas doenças não musculares que também aumentam dos níveis de CK, mas não acima de dez vezes. Entre elas, destacam-se a doença do neurônio motor, a síndrome de Guillain-Barré e a polirradiculoneurite desmielinizante inflamatória crônica. Condições que cursam com aumentos transitórios de CK incluem hipotiroidismo, realização de eletroneuromiografia (ENMG), hipoparatiroidismo, além de trauma, injeções subcutâneas e intramusculares, viroses, crises epilépticas e exercício vigoroso, nunca chegando a aumentos acima de cinco vezes o normal.
Os níveis de CK podem aumentar em decorrência de medicações, como estatinas, fibratos, niacina, zidovudina, colchicina, cloroquina e ciclosporina. A dosagem de CK também pode ser influenciada por aspectos raciais e de gênero. Afrodescendentes e indivíduos com massa muscular elevada podem ter níveis elevados de CK. Há casos de elevações transitórias de CK de caráter benigno e hereditário (Tabela 5).
Finalmente, é extremamente improvável que o aumento de CK menor de três vezes o limite superior da normalidade, associado à ausência de fraqueza muscular ou dor, signifique miopatia.
Tabela 5: Diferenciais para variações nos níveis de CK
|
Diagnóstico |
Padrão da CK |
|
Distrofia muscular de Duchenne |
Aumento de 10x a > 100x |
|
Perda muscular acentuada, uso de corticóide, colagenoses, alcoolismo e hipertiroidismo |
Elevada, diminuída ou normal |
|
Doença do Neurônio Motor, síndrome de Guillain-Barré, polirradiculoneurite desmielinizante inflamatória crônica |
Aumento < 10x |
|
Hipotiroidismo, eletroneuromiografia, hipoparatiroidismo, trauma, injeções subcutâneas e intramusculares, viroses, crises epilépticas e exercício vigoroso |
Aumento transitório até 5x |
Outros testes para miopatias, como dosagens séricas de aldolase, aspartato e alanina aminotransferases (AST e ALT), desidrogenase láctica (DHL) podem estar alterados. AST, ALT e DHL podem estar alteradas em outras condições que não miopatias. No caso de hepatotoxicidade, por exemplo, o aumento da dosagem de gamaglutamil transferase (GGT) irá esclarecer a dúvida.
A dosagem de isoenzimas de CK não é útil nas miopatias. A fração MM elevada é típica de miopatia, mas a fração MB, miocárdica, também está elevada nas miopatias e não necessariamente significa acometimento cardíaco.
A eletroneuromiografia deve fazer parte da rotina da investigação do paciente com miopatia, auxiliando na definição de qual é o músculo doente e excluindo a presença de neuropatia motora ou doença da junção neuromuscular. O estudo da condução nervosa é tipicamente normal nos pacientes com miopatia. O exame de eletromiografia por agulha mostra evidência de unidades motoras de pequena amplitude, curta duração, com recrutamento aumentado, extremamente importante para o diagnóstico. Além disso, por mostrar qual músculo está acometido, pode servir de guia para a biópsia muscular. O exame pode ser normal em pacientes com miopatia, e a decisão deve ser tomada em um contexto clínico amplo, com base em história, exame físico e laboratório.
Em casos sugestivos de miopatia, a biópsia muscular deve ser realizada para a confirmação diagnóstica. No entanto, muitas formas de miopatias hereditárias podem ser diagnosticadas mediante testes genéticos moleculares, sendo possível dispensar a realização da biópsia. O procedimento pode ser aberto ou por meio de agulhamento, minimamente invasivo. A escolha do local a ser biopsiado é importante, não devendo ser feito o exame em locais em que a força muscular esteja menor ou igual a 3 (vence a gravidade, mas não a resistência). Nesses locais, a biópsia só mostrará miopatia terminal. De modo geral, nos membros superiores, o músculo de eleição é o bíceps e nos membros inferiores, o vasto lateral. Eventualmente, a biópsia pode ser guiada por algum método auxiliar de imagem (ultra-sonografia, tomografia ou ressonância magnética). Os fragmentos biopsiados são submetidos à análise por meio de microscopia óptica, eletrônica, testes bioquímicos e imuno-histoquímicos. Na maioria das vezes, apenas a microscopia óptica já é suficiente para o diagnóstico. No entanto, em distrofias musculares, por exemplo, para a etiologia, é necessário o estudo imuno-histoquímico, como a coloração para distrofina nas distrofias de Duchenne e Becker. A microscopia eletrônica é útil na investigação de algumas miopatias congênitas e nas doenças mitocondriais. O fragmento muscular pode ser processado para análise bioquímica com a finalidade de determinar uma anormalidade enzimática específica, por exemplo.
Estudos genéticos moleculares já estão disponíveis comercialmente e podem ser feitos com análise do sangue periférico. Esses testes podem substituir a realização da biópsia muscular. Têm importância não só no diagnóstico, mas também na identificação de portadores assintomáticos e aconselhamento genético.
Alguns outros testes específicos podem ser úteis na avaliação de determinadas doenças musculares. No caso das miopatias hereditárias, exame de urina com pesquisa de mioglobinúria e o teste de McArdle, além dos testes para diagnóstico de causas de miopatias adquiridas.
O teste de McArdle é feito com exercícios isométricos do antebraço com auxílio de um dinamômetro. Colhem-se amostras de sangue para dosagem de amônia e lactato antes do início do teste, para a determinação basal dessas substâncias. Há quem o faça com auxílio de manguito de pressão, insuflando-o com pressão 20 mmHg acima da pressão sistólica, para promover isquemia. O paciente, então, inicia rapidamente exercícios com esse membro, como apertar uma bola de borracha, até a exaustão. O manguito é esvaziado e o sangue é colhido após 1, 3, 5, 10 e 15 minutos do teste. O aumento de três vezes no nível de lactato representa o normal. O teste é normal nas lipidoses, em alguns distúrbios glicolíticos com fraqueza muscular fixa, como na doença de Pompe. O nível de lactato aumenta patologicamente nos casos de deficiência de fosfofrutoquinase e miofosforilase, e é reduzido na de fosfoglicerato mutase.
As miopatias metabólicas são caracterizadas por intolerância ao exercício, fraqueza muscular, fatigabilidade, mialgias, cãibras musculares e mioglobinúria. Manifestam-se durante atividades que necessitam de aumento do consumo de energia. São raras e já foram mencionadas.
As canalopatias também são doenças raras e devem ser consideradas em pacientes com episódios recorrentes de fraqueza ou miotonia. Tipicamente, há um fator precipitante. A paralisia periódica hipocalêmica é um distúrbio autossômico dominante dos canais de cálcio (tipo 1) ou dos canais de sódio (tipo 2). Manifesta-se da puberdade à terceira década de vida, com ataques episódicos de fraqueza muscular com discreta elevação de CK e alterações à ENMG e anatomopatológicas detectáveis apenas durante as crises. Os eventos costumam ser desencadeados por ingestão de carboidratos (tipicamente aos domingos e segundas-feiras, após o consumo de massas), após exercício, gravidez, estresse emocional e frio. Os músculos da face, respiração e esfíncteres são geralmente poupados. Os primeiros músculos a ficarem fracos são geralmente os que melhoram primeiro também. Os níveis de K+ costumam estar abaixo de 3,0. O fósforo também costuma estar diminuído. Outras canalopatias são bem mais raras e incluem a paralisia hipercalêmica, a paramiotonia congênita, a miotonia congênita de Thomsen e a miotonia generalizada de Becker.
A hipertermia maligna é uma condição rara, mas grave, autossômica dominante, por alteração dos canais de cálcio, com aumento importante de CK, desencadeada por procedimentos anestésicos, sobretudo agentes inalatórios, como o halotano, sevoflurano e fenflurano, além de relaxantes musculares como a succinilcolina. Ocorre entre 1:5.000 a 1:100.000 anestesias, mas a prevalência da mutação genética é maior que 1:3.000 indivíduos. Os sinais clássicos são hipertermia grave, taquicardia, taquipnéia, hipercapnia, aumento no consumo de oxigênio, acidemia, rigidez muscular e rabdomiólise, todos decorrentes de hipermetabolismo. Em virtude de um defeito nos receptores de rianodina, há aumento incontrolável do cálcio mioplasmático, o qual ativa processos bioquímicos relacionados à ativação muscular. É fatal se não for tratada. Testes in vitro da resposta do músculo biopsiado ao halotano, cafeína e outras drogas podem ser feitos para o diagnóstico. A mortalidade há 30 anos era de 80%; atualmente, caiu para 5%.
As distrofias musculares congênitas devem ser separadas em dois grupos: associadas ou não a retardo mental. As que se não se associam a retardo mental são a deficiência de merosina, a proteinopatia relacionada a fukutina e a doença de Ullrich. A doença de Fukuyama, a distrofia músculo-olho-cérebro e a síndrome de Walker-Warburg são associadas a desenvolvimento cerebral anormal e retardo mental.
As distrofinopatias (Duchenne e Becker) são miopatias progressivas, recessivas ligadas ao X. A DMD é a forma mais comum de distrofia muscular. Ocorre em 1:3.300 nascidos do sexo masculino e tem prevalência de 3 em 100.000 habitantes. A DMB ocorre em 1:18.
A DMD começa a se manifestar por volta de 2 a 3 anos de idade, com quase todos os pacientes sintomáticos por volta dos 5 anos. A criança começa inicialmente com quadro de dificuldade para correr, pular e subir escadas. Nota-se marcha característica, anserina, com lordose lombar e hipertrofia das panturrilhas. A fraqueza muscular é simétrica e afeta inicialmente os músculos proximais e dos membros inferiores. A corrida e os saltos vão ficando cada vez mais difíceis e os meninos começam a usar as mãos para se levantarem do chão (sinal de Gowers). Há acometimento cardíaco e cognitivo (memória de trabalho e disfunção executiva). Ao exame, observa-se hipertrofia das panturrilhas, lordose lombar acentuada, marcha em gingado, encurtamento do tendão de Aquiles e hipo ou arreflexia. Entre os
A idade de início dos sintomas na DMB é entre 5 e 15 anos (média aos 12). O acometimento é mais brando, o comprometimento cardíaco e cognitivo está ausente ou é bem mais leve do que na DMD e não há manifestações gastrintestinais. Os pacientes perdem a capacidade de deambulação a cerca dos 16 anos e vivem além dos 30 anos, com óbito de insuficiência ventilatória/cor pulmonale aos 30-60 anos. O primeiro sintoma costuma ser fraqueza nos membros inferiores, e dor na panturrilha é o sintoma inicial em 25%. Os níveis de CK não ultrapassam cinco vezes o limite superior da normalidade.
A distrofia miotônica de Steinert é a forma mais prevalente de distrofia muscular em adultos. Existe correlação direta entre o número de repetições de trinucleotídeos com a idade de início e a gravidade da doença. A doença pode se iniciar desde o período neonatal até a vida adulta. Ocorre fraqueza lenta e progressiva da musculatura facial e do esternocleidomastóideo, associando-se a calvície frontal e ptose, com as feições acentuadas. Fraqueza bulbar e cervical está presente com freqüência. Os pacientes desenvolvem tipicamente fraqueza nas mãos e pé caído. O fenômeno miotônico é característico e pode ser observado durante a percussão dos músculos tenares ou da língua. Observa-se uma contração involuntária sustentada. A maioria dos pacientes tem envolvimento cardíaco (bloqueio de ramo e miocardiopatia), o que eleva o risco de morte súbita. Pode ocorrer fraqueza diafragmática e intercostal, resistência à insulina, catarata e hipogonadismo. É comum haver sonolência excessiva. Como resultado de esvaziamento gástrico precoce, podem ocorrer náuseas, vômitos e saciedade precoce. Do ponto de vista laboratorial, observam-se aumento de CK, alterações ao ECG, eletromiografia com descargas miotônicas e padrão miopático, hiperglicemia e catarata ao exame de “lâmpada de fenda”.
A distrofia fáscio-escápulo-umeral é uma afecção lentamente progressiva e de gravidade variável. Inicia-se na infância ou adolescência, mas pode surgir até na quinta década. A fraqueza tem predomínio facial e da cintura escapular. Os doentes são incapazes de assoviar, sorrir ou mesmo fechar completamente os olhos, podendo inclusive adormecer com os olhos abertos. Não costuma acometer a musculatura extra-ocular. É comum haver escápula alada, a qual fica mais evidente quando o paciente eleva os membros superiores lateralmente. Em 20% dos casos, há acometimento da cintura pélvica, sobretudo do compartimento anterior (variedade escápulo-peroneal). Os pacientes costumam ter expectativa de vida normal, uma vez que é raro que ocorra insuficiência respiratória. O diagnóstico é feito por teste genético.
O termo distrofia cintura-membros é reservado para distrofias musculares não-congênitas, com fraqueza proximal progressiva e ausência de deficiência de distrofina. Esses distúrbios são causados por deficiência de diferentes proteínas, como sarcoglicanas, calpaína, disferlina e caveolina. Os sintomas iniciam-se no começo da infância ou vida adulta. A progressão e a distribuição são igualmente variáveis entre os pacientes e os subtipos genéticos. O mais freqüente é o quadro progressivo de fraqueza de cinturas, às vezes associado a miocardiopatia. A hipertrofia de panturrilhas é freqüente, mas não a regra. Há considerável sobreposição com o quadro de distrofinopatias. Os níveis de CK costumam ser mais elevados nas disferlinopatias. A coloração da biópsia muscular para sarcoglicana, disferlina, calpaína e caveolina costuma ajudar na distinção entre as doenças.
É durante a infância precoce ou adolescência que tipicamente se apresenta o quadro de distrofia muscular de Emery-Dreifuss. A tríade clássica de sintomas consiste de contraturas precoces (cotovelos, joelhos, tornozelos e coluna), fraqueza e atrofia da musculatura umeral e peroneal, além de distúrbio de condução cardíaca que pode levar a síncope ou morte súbita. Os níveis de CK podem estar aumentados e a biópsia muscular mostrar alterações distróficas inespecíficas. A análise do DNA ou do produto do gene (emerina) é necessária para confirmar o diagnóstico. É freqüente que o paciente precise de marca-passo cardíaco.
A distrofia muscular óculo-faríngea manifesta-se na quarta a sexta décadas de vida. Caracteriza-se clinicamente por ptose, disfagia e fraqueza muscular proximal leve. De modo variável, pode ocorrer acometimento da musculatura extra-ocular, devendo-se distinguir a doença da miastenia grave e da oftalmoplegia externa progressiva. A CK pode estar normal ou levemente aumentada. A eletromiografia mostra padrão miopático. A biópsia mostra vacúolos cercados e filamentos tubulares anucleados.
Ptose e oftalmoparesia são aspectos centrais da SKS e da OEPC. A OEPC é uma miopatia pura com fraqueza extra-ocular que se manifesta por ptose e oftalmoparesia. Outros músculos podem estar afetados, causando fraqueza da face, orofaringe e membros. Ao contrário, a SKS é uma doença de múltiplos sistemas, de início na juventude ou idade adulta, que produz oftalmoparesia, degeneração pigmentar da retina, e combinações variáveis de bloqueio de condução cardíaca e hiperproteinorraquia (> 100 mg/dL), além de ataxia cerebelar. Esses pacientes podem ainda ter baixa estatura e diabete melito, hipoparatiroidismo, miocárdio e nefropatia. A biópsia muscular identifica fibras vermelhas rajadas e o defeito molecular.
A síndrome MELAS caracteriza-se por episódios de isquemia cortical na idade jovem (< 40 anos), encefalopatia (crises epilépticas, demência ou ambas) e disfunção mitocondrial, caracterizada por acidose láctica, fibras vermelhas rajadas ou ambas. A prevalência da mutação mais comum é de
A ocorrência de epilepsia, mioclonias, ataxia e biópsia muscular mostrando fibras vermelhas rajadas define o diagnóstico de MERRF. Também pode ocorrer perda auditiva, demência, neuropatia periférica, baixa estatura, intolerância ao exercício, lipomas e acidose láctica. O diagnóstico é confirmado pela pesquisa da mutação genética. O diagnóstico diferencial inclui outras causas de epilepsias mioclônicas progressivas, como síndrome de Unverricht-Lundborg, doença dos corpos de Lafora, lipofuscinose ceróide neuronal e sialidose.
Pacientes com mutação maior que 90% da subunidade 6 do complexo V da cadeia respiratória desenvolverão um quadro devastador de encefalomiopatia, caracterizada por regressão do desenvolvimento psicomotor, crises convulsivas, acidose láctica e lesões necrotizantes subagudas nos gânglios da base e outras estruturas da substância cinzenta, na linha média do cérebro e tronco. Isso caracteriza a síndrome de Leigh de herança materna (MILS). A ressonância magnética mostra lesões características da região periaquedutal do mesencéfalo e ponte, e no bulbo, adjacente ao quarto ventrículo. Podem afetar outras estruturas do sistema nervoso central e periférico. O diagnóstico diferencial inclui a doença de Refsum, a abetalipoproteinemia e outras doenças mitocondriais.
Entre as miopatias metabólicas, o tratamento é experimental e consiste na reposição da enzima deficiente. Ainda em fase experimental, alguns resultados são promissores.
O quadro de paralisia periódica hipocalêmica deve ser familiar ao emergencista. O tratamento é feito com a prevenção de desencadeantes, e reposição de potássio. Prescreve-se a dose de
O tratamento da hipertermia maligna é feito em quatro etapas: suspender imediatamente a administração do agente inalatório ou da succinilcolina, aumentar o volume-minuto para diminuir a ETCO2, pedir ajuda e administrar dantrolene, nessa ordem. A dose inicial é de 2,5 mg/kg, máximo sugerido de 10 mg/kg, titulando-se pela freqüência cardíaca e taxa de extração de gás carbônico (ETCO2). Depois, procede-se ao resfriamento do doente, com pacotes de gelo nas axilas e virilhas, lavado gástrico com solução gelada e medidas mais agressivas, se necessário. O objetivo é atingir a temperatura de 38,5°C. As arritmias não devem ser tratadas com bloqueadores de canais de cálcio, e os distúrbios tóxico-metabólicos investigados periodicamente e corrigidos. O dantrolene deve ser mantido a 1 mg/kg a cada 4 a 8 horas por
As distrofinopatias (DMD e DMB) devem ser tratadas com uso de corticosteróides. Prednisona 0,75 mg/kg/dia é capaz de produzir melhora significativa na força muscular e prolongar o tempo de deambulação dos pacientes em cerca de três anos. Na presença de efeitos colaterais, a dosagem pode ser diminuída para até 0,3 mg/kg/dia, ainda capaz de produzir resultado satisfatório. Com menos efeitos colaterais, o deflazacort na dose de 0,9 mg/kg/dia pode ser prescrito, com o mesmo perfil de sucesso. Além disso, estão indicadas fisioterapia, órteses, cirurgia ortopédica, dependendo do caso.
Não há tratamento específico para a distrofia miotônica. Podem ser prescritos dispositivos ortopédicos e fenitoína, 300 mg/dia, para suprimir a miotonia. A fenitoína é a droga de escolha para essa finalidade em detrimento de quinidina e procainamida por menor índice de efeitos colaterais sobre a condução cardíaca. Eventualmente, os pacientes podem necessitar do implante de um marca-passo.
O tratamento das outras distrofias e das miopatias congênitas é fisioterapia e sintomáticos, não havendo tratamento específico.
O tratamento da SKS e OECP é sintomático. Eventualmente, pode ser necessário o implante de marca-passo cardíaco. A função mitocondrial pode melhorar com o uso de coenzima Q10, na dose de
No caso da síndrome MELAS, há relatos anedóticos de que os corticóides seriam benéficos nos AVCs agudos. As crises respondem com a maioria dos anticonvulsivantes, e muitos evitam o uso de valproato pelo risco de causar deficiência de carnitina. Implantes cocleares podem ser usados para o tratamento da disfunção auditiva. Os pacientes também fazem a suplementação tradicional padrão para SKS e EOPC. O tratamento da síndrome MERRF obedece ao mesmo padrão.
Miopatias são doenças estruturais e/ou funcionais dos músculos, cuja manifestação fundamental é fraqueza muscular.
Fazem diagnóstico diferencial com doenças do neurônio motor, com neuropatias periféricas e com doenças da junção neuromuscular.
São divididas em hereditárias e adquiridas.
As formas hereditárias são divididas em: miopatias congênitas, distrofias musculares, canalopatias, miopatias metabólicas primárias e miopatias mitocondriais.
É necessário realizar uma anamnese detalhada com informações sobre o padrão evolutivo da fraqueza, seus fatores desencadeantes e quanto à presença de casos na família.
Os sintomas positivos das miopatias são mialgias, cãibras, contraturas, miotonia e mioglobinúria.
Os sintomas negativos das miopatias são fraqueza, atrofia, intolerância ao exercício (fatigabilidade) e fadiga.
No exame físico deve-se detalhar a distribuição da fraqueza, lembrando dos músculos cranianos, além de avaliar se há atrofia ou hipertrofia de músculos.
Há seis padrões de apresentação clínica dos quadros de miopatias (Tabela 4).
O padrão mais comum é a fraqueza proximal de distribuição cintura-membros.
O exame isoladamente mais útil no estudo de pacientes com suspeita de miopatia é a dosagem sangüínea de CK. A dosagem de suas isoenzimas não é útil nesses casos.
A eletroneuromiografia faz parte da abordagem diagnóstica, sendo o estudo da condução nervosa tipicamente normal nos pacientes com miopatia. É capaz de delimitar o músculo acometido, guiando biópsias.
A biópsia muscular é o exame que define o diagnóstico em boa parte dos casos. Não deve ser feito em músculos com força de grau menor ou igual a 3. De modo geral, nos membros superiores, o músculo de eleição é o bíceps, e nos membros inferiores, o vasto lateral.
Estudos genéticos moleculares (exames em sangue periférico) podem substituir a biópsia para o diagnóstico de muitas miopatias hereditárias.
O teste de McArdle é feito com exercícios isométricos do antebraço com auxílio de um dinamômetro. Colhem-se amostras de sangue para dosagem de amônia e lactato antes e após os exercícios. É útil em diagnóstico de miopatias metabólicas: o nível de lactato aumenta patologicamente nos casos de deficiência de fosfofrutoquinase e miofosforilase, e é reduzido na de fosfoglicerato mutase.
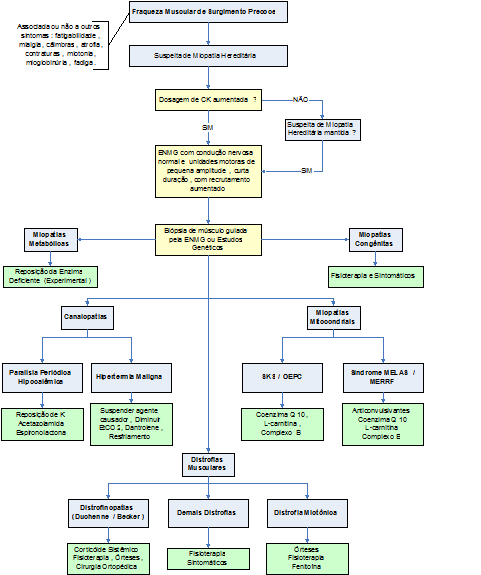
Algoritmo 1: Guia para diagnóstico e tratamento das miopatias hereditárias.

1. Angelini C. The role of corticosteroids in muscular dystrophy: a critical appraisal. Muscle Nerve 2007;36:424-435.
2. Darras B. Muscular dystrophies. Continuum Lifelong Learning in Neurology 2006;12(3).
3. Finsterer J. Primary periodic paralises. Acta Neurol Scand 2008;117:145-58.
4. Hirano M. Mitochondrial diseases. In Brust JCM. Current diagnosis and treatment in neurology. New York: McGraw Hill; 2007.
5. Jackson C. A clinical approach to the patient with suspected myopathy. Continuum Lifelong Learning in Neurology 2006;12(3).
6. Laing NG. Congenital myopathies. Curr Opin Neurol 2007;20:583-9.
7. Moxley III RT et al. Practice parameter: corticosteroid treatment of Duchenne dystrophy: report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committe of the Child Neurology Society. Neurology 2005;64:13.
8. Rosenberg H, Davis M, James D, Pollock N, Stowel K. Malignant hyperthermia. Orphanet J Rare Dis 2007;2:21.
9. Saperstein D. Muscle channelopathies. Continuum Lifelong Learning in Neurology 2006;12(3).
10. Walsh RJ. Metabolic myopathies. Continuum Lifelong Learning in Neurology 2006;12(3).
11. Williams O. Diseases of muscle. In; Brust JCM. Current diagnosis and treatment in neurology. New York: McGraw Hill; 2007.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.