(Carregando Índice)... (Carregando Índice)... |
Última revisão: 13/02/2014
Comentários de assinantes: 0
Charles M. Mansbach II, MD
Professor of Medicine and Physiology, Division of Gastroenterology, Department of Medicine, University of Tennessee Health Science Center, College of Medicine, Memphis, TN
Artigo original: Mansbach II CM. Diseases producing malabsorption and maldigestion. ACP Medicine. 2009;1-15.
[The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2011 Decker Intellectual Properties Inc. All Rights Reserved.]
Agradecimento: Figura 1 – Janet Betries. Figura 2 – Dana Burns-Pizer.
Tradução: Soraya Imon de Oliveira.
Revisão técnica: Dr. Euclides Furtado de Albuquerque Cavalcanti.
A má absorção refere-se ao comprometimento da absorção intestinal de nutrientes. Pode ser resultante de defeitos congênitos envolvendo o transporte de nutrientes, do comprometimento dos processos digestivos junto ao lúmen intestinal, ou de defeitos adquiridos nas células absortivas intestinais que revestem a superfície do intestino. A má digestão, outro fator relacionado à absorção de nutrientes, refere-se ao comprometimento da digestão de nutrientes junto ao lúmen intestinal. Embora estes dois processos sejam distintos em termos de fisiopatologia, são também interdependentes, e, na prática clínica, a má absorção é considerada o desarranjo destes processos.
A má absorção é clinicamente definida como sendo o comprometimento da absorção de gorduras (esteatorreia), uma vez que a medida da absorção de gorduras é o melhor indicador da normalidade do processo geral de absorção de nutrientes. Sob certas condições, todavia, a absorção de gorduras pode estar normal enquanto outras substâncias podem estar sendo mal absorvidas, como o ferro, vitamina B12, cálcio, sais biliares ou, em certas condições hereditárias, aminoácidos específicos, dissacarídeos e monossacarídeos.
Em geral, existem 3 causas possíveis de má absorção de gorduras: doença do intestino delgado, doença do trato biliar ou hepático e insuficiência pancreática exócrina [Tabela 1].
Tabela 1. Causas das síndromes de má absorção
|
Doenças do intestino delgado |
|
EGS |
|
Espru tropical |
|
Espru colágeno |
|
Enterite eosinofílica |
|
Enterite por radiação |
|
Amiloidose |
|
Mastocitose |
|
Abetalipoproteinemia |
|
Doença de Whipple |
|
Linfangiectasia intestinal |
|
Doença linfoproliferativa do intestino delgado |
|
Enteropatia isquêmica |
|
Infecção por Giardia lamblia |
|
SIDA |
|
Síndrome do intestino curto |
|
Ressecção ileal |
|
Ileíte (p. ex., doença de Crohn) |
|
Doenças do trato hepático e biliar |
|
Cirrose/doença hepática parenquimal |
|
Síndrome da colestase intra-hepática |
|
Colestase por obstrução extra-hepática |
|
Doenças do pâncreas |
|
Pancreatite crônica |
|
Fibrose cística |
|
Câncer |
|
Defeitos combinados ou múltiplos de digestão e absorção |
|
Hipertireoidismo |
|
Diabetes melito |
|
Síndrome carcinoide |
|
Síndrome de Zollinger-Ellison |
|
Pós-gastrectomia (tipo Billroth II) |
EGS = enteropatia glúten-sensível.
A doença do intestino delgado pode resultar na presença de quantidades moderadas de gordura nas fezes (7 a 30 g/dia com uma dieta contendo 100 g de gorduras). Os pacientes com esta condição podem perder proteína (enteropatia perdedora de proteína) através da mucosa intestinal adoecida, com consequente diminuição da concentração sérica de albumina. O paciente pode apresentar deficiências de vitaminas lipossolúveis (isto é, vitaminas A, D, E e K). Pode haver má absorção de vitamina B12 em decorrência de um íleo terminal muito adoecido ou previamente ressectado (habitualmente em mais de 60 cm). O ácido fólico também pode estar sendo mal absorvido e também pode haver hipocalcemia e hipomagnesemia.
É comum os pacientes com doença no trato biliar ou hepático apresentarem apenas pequenos aumentos no conteúdo de gordura presente nas fezes (7 a 15 g/dia) e também podem apresentar absorção precária de vitaminas lipossolúveis. A associação existente entre a doença hepática colestática (em especial a cirrose biliar primária) e a osteoporose está comprovada. A osteoporose pode ser a manifestação sintomática da doença hepática. A deficiência de vitamina K, demonstrada por um tempo de protrombina prolongado, também pode estar presente. A administração de vitamina K corrige o defeito de coagulação nos casos em que a extensão da doença hepática não é severa o bastante para impedir a síntese de fator de coagulação. É importante notar que estas deficiências vitamínicas podem ocorrer na ausência de uma esteatorreia clinicamente evidente.
Os pacientes com insuficiência pancreática exócrina podem apresentar até 80 g de gordura/dia nas fezes. Nestes indivíduos, a absorção de gorduras em geral resulta da ação da lipase gástrica. Esta enzima é encontrada nas células principais do lúmen do estômago1 e parece contribuir para qualquer absorção de lipídios que venha a ocorrer no contexto da pancreatite crônica, a exemplo do observado na fibrose cística. De fato, na fibrose cística, foram relatados níveis aumentados de lipase gástrica.2
Os sintomas da má absorção são proteicos. Na maioria dos casos evidentes, o paciente queixa-se de perda de peso, embora apresente apetite normal. Nestes casos, observa-se uma nítida alteração da qualidade das fezes e, na maioria dos casos, um aumento quantitativo de fezes. A consistência das fezes é mais mole e, na presença de excesso de gordura, as fezes passam a apresentar um odor mais fétido, flutuam na água do vaso sanitário e são mais difíceis de eliminar quando a descarga do vaso é acionada. Gotas de óleo ou o brilho dos lipídios presentes nas fezes podem aparecer na água do vaso. O excesso de gases (e não o conteúdo de gordura) faz as fezes flutuarem.3
Dependendo dos demais constituintes mal absorvidos, os pacientes podem apresentar distensão abdominal, borborigmo, cólicas abdominais (intolerância à lactose), aquisição de contusões com facilidade (deficiência de vitamina K), osteopenia ou tetania (deficiência de vitamina D e má absorção de cálcio), deficiência de ferro ou cegueira noturna (deficiência de vitamina A). Os casos mais desafiadores são aqueles em que a questão da má absorção não é abordada, devido à ausência de alterações na qualidade das fezes.
A diarreia da má absorção é classificada como diarreia osmótica e em geral cessa durante o jejum. Na má absorção de gordura, a diarreia é causada não só pelo excesso de partículas osmoticamente ativas, mas também pela presença de ácidos graxos, que estimulam a secreção de Cl- dependente de monofosfato de adenosina cíclico (cAMP) e a conversão do ácido oleico em seu produto 10-hidroxi, o 10-hidroxiesterato, que mimetiza o ingrediente ativo do óleo de rícino, o ácido ricinoleico.
Os achados físicos específicos de várias doenças podem acompanhar o estado de má absorção e auxiliar no estabelecimento do diagnóstico. Exemplificando, as alterações cutâneas associadas ao escleroderma ou à dermatite herpetiforme podem estar presentes. Os sinais de neuropatia diabética podem ser revelados. Embora a tireotoxicose possa estar associada ao excesso de gordura presente nas fezes, os pacientes com tireotoxicose geralmente comem com gula, porém absorvem um percentual normal da gordura contida nos alimentos ingeridos (95%) e, assim, não apresentam má absorção no sentido real do termo.
Nota do editor: Outro achado cutâneo Possível são os xantomas eruptivos, que são associados a hipertrigliceridemia , esta por sua vez podem causar pancreatopatia crônica e má-absorção.
Nos exames usados na investigação da má absorção, é determinado se o paciente apresenta excreção fecal excessiva de gordura [Tabela 2]. Uma grande concentração de proteínas é produzida pelo trato digestivo, especialmente pelo pâncreas, dificultando a interpretação da esteatorreia. Os carboidratos mal absorvidos que seguem para o cólon podem ser metabolizados pelas bactérias locais em ácidos graxos de cadeia curta, que, então, são parcialmente absorvidos no próprio cólon. Desta forma, a quantificação da absorção de carboidratos é inacurada, ainda que ocorra uma queda do pH fecal indicativa da presença de excesso dos ácidos graxos de cadeia curta excretados nestas condições.
Tabela 2. Testes de função de absorção intestinal
|
Teste |
Características |
Uso clínico |
|
Análise qualitativa de gordura fecal |
Exame microscópico simples, para detecção de aumento do conteúdo de glóbulos de gordura |
Avaliação eficiente para casos de aumento moderado do conteúdo de gordura fecal, ainda que a análise quantitativa de gordura fecal seja preferível |
|
Quantitativo |
Análise bioquímica da excreção de gordura durante um período de 72 horas, por titulação com NaOH. É um teste mais sensível para má absorção de gordura. Níveis normais: < 6 g/dia. não distingue entre anormalidades do intestino delgado, pâncreas ou luminais |
É o teste mais importante para identificar a má digestão ou a má absorção. É indicado para todos os pacientes com suspeita de má absorção |
|
Absorção de xilose |
Por ser uma pentose que dispensa a digestão no lúmen ou na superfície intestinal, a xilose permite avaliar a função do intestino delgado. Normalmente, > 4 g/5 h são excretadas na urina após a ingesta de 25 g. Os níveis plasmáticos de xilose devem ser de 10 a 20 mg/dL/1,73 m2 de área de superfície corporal em 60 a 75 min |
Teste indicado para todos os casos em que o conteúdo de gordura fecal estiver quantitativamente anormal. Não é tão sensível quanto a análise de gorduras, mas localiza a anomalia no intestino delgado. É um indicador da área de superfície intestinal |
|
Radiografias do intestino delgado |
Permite analisar a continuidade do intestino delgado e identificar os divertículos ou alterações da mucosa. O pâncreas acometido pode prejudicar a avaliação do duodeno |
Teste indicado para os casos de aumento quantitativo da excreção de gordura fecal |
|
Biópsia de intestino delgado obtida por via oral |
Possibilita o exame histológico direto da mucosa. Alterações características ocorrem em várias doenças produtoras de má absorção |
Indicado para os casos de aumento da excreção de gordura fecal, em particular quando o teste de xilose ou a radiografia do intestino delgado são anormais. Uma parte da amostra de biópsia pode ser testada para dissacarídeos |
|
Teste respiratório de ácido biliares |
Quando há supercrescimento bacteriano no intestino delgado ou doença ileal produtora de má absorção, o ácido 14C-glicocólico (5 mcCi) é desconjugado, metabolizado e excretado pelos pulmões como 14CO2 |
Indicado para pacientes com esteatorreia comprovada, causada por um possível supercrescimento bacteriano ou disfunção ileal |
|
Teste da bentiromida |
O peptídeo ligado a esta arilamina não absorvível é clivado especificamente pela quimiotripsina intraluminal, dando origem ao PABA que, então, é prontamente absorvido e excretado na urina |
Indicado para os casos de excreção de gordura fecal aumentada. É um teste menos sensível do que a análise quantitativa de gordura fecal. Entretanto, quando resulta positivo, estabelece o diagnóstico de níveis intraduodenais insuficientes de enzimas digestivas pancreáticas |
PABA = ácido para-aminobenzoico.
A gordura fecal pode ser analisada qualitativa e quantitativamente. A medida qualitativa da gordura fecal com coloração por Sudan III tem apresentado uma acurácia surpreendente,4 sobretudo quando quantidades clinicamente significativas de gordura são excretadas. Um grupo relatou que a contagem e medida do tamanho dos glóbulos de gordura presentes nas fezes melhorou significativamente a sensibilidade e especificidade do ensaio com Sudan.4 Contudo, como se observa em muitos testes qualitativos, a acurácia varia de acordo com o desempenho e a interpretação do teste, tornando a habilidade do observador um aspecto essencial para o sucesso.
A medida quantitativa da gordura fecal é o referencial para classificação de todos os outros testes. é importante lembrar que o teste não pode ser realizado se o paciente não puder comer pelo menos 80 g (de preferência 100 g) de gordura/dia. Entretanto, o teste requer armazenamento de fezes na geladeira por 3 dias, e isto impossibilita o equilíbrio bioquímico das gorduras em muitos contextos domiciliares ou hospitalares.
A área de superfície de absorção intestinal é medida pela capacidade do paciente de absorver xilose, um açúcar contendo 3 carbonos. Diferente da glicose, a xilose não é ativamente absorvida pelo intestino, e sim através de um processo mais lento de difusão facilitada. No teste de absorção da xilose, o paciente recebe 25 g de xilose por via oral e amostras de urina são coletadas durante 5 horas. A excreção urinária normal de xilose ultrapassa 4 g no decorrer das 5 horas. Para garantir um fluxo urinário adequado, o paciente deve beber 500 mL de água após ter ingerido a xilose. Esta ingesta deve resultar em um volume de urina de pelo menos 300 mL durante o período de coleta. A excreção de xilose pode estar falsamente baixa em pacientes com função renal diminuída ou em indivíduos com ascite, nos quais a xilose é diluída no líquido ascítico. Para evitar os resultados falso negativos, é recomendável medir a concentração de xilose no sangue [Tabela 2]. A xilose má absorvida atinge o cólon e pode ser metabolizada em hidrogênio pelas bactérias resistentes. O hidrogênio pode ser quantificado na respiração, e este teste foi descrito como sendo uma medida tão acurada quanto a medida da xilose no soro ou na urina.
Uma radiografia plana ou ultrassonografia abdominal não costuma ter utilidade na maioria dos casos de má absorção. Entretanto, 30% dos casos de pancreatite crônica apresentam calcificações visíveis à radiografia plana do abdome. A detecção da calcificação pancreática pode ser realizada pelo uso de tomografia computadorizada (TC) ou ultrassonografia. A TC e a ultrassonografia também podem identificar os ductos pancreáticos dilatados, que constituem outro sinal característico da pancreatite crônica. A pancreatografia retrógrada endoscópica também pode ser útil diante da observação de alterações ductais indicativas de pancreatite crônica [ver 4:V Doenças do pâncreas].
Os exames radiográficos do intestino delgado realizados após a ingesta de bário por via oral podem auxiliar o diagnóstico de várias anormalidades. A presença de divertículos no intestino delgado ou o comprometimento do peristaltismo, como se observa no escleroderma ou na dismotilidade intestinal idiopática, podem ser indicadores de supercrescimento bacteriano. Um exame minucioso do íleo terminal pode identificar a doença de Crohn. Em alguns pacientes com lesão produzida por radiação ou anti-inflamatórios não hormonais (AINH), é possível identificar estreitamentos. A hipoalbuminemia que afeta o intestino delgado pode produzir o sinal conhecido como “empilhamento de moedas” observado à radiografia com contraste do intestino delgado.
Um patologista experiente pode ajudar a sustentar o diagnóstico de enteropatia glúten-sensível (EGS – com ou sem dermatite herpetiforme), espru associado a hipogamaglobulinemia, doença de Whipple, doença do complexo Mycobacterium avium, síndrome de estase, amiloidose e linfangiectasia intestinal.
Mais de 90% da função pancreática exócrina deve ser destruída para que um indivíduo desenvolva má absorção sintomática [ver 4:V Doenças do pâncreas].5 O teste mais sensível de função pancreática exócrina requer a passagem de um tubo de lúmen duplo pelo interior do duodeno.6 Colecistoquinina (CCK) ou secretina são administradas por via endovenosa (IV), e as secreções gástrica e duodenal são coletadas separadamente. A secretina foi indisponibilizada para uso nos Estados Unidos quando seu fabricante descontinuou a produção, em 1999. Porém, uma nova formulação foi aprovada para uso pelo Food and Drug Administration (FDA), em 2004. Se a CCK é administrada, a atividade de lipase ou tripsina é determinada utilizando-se os substratos apropriados. Quando a secretina é administrada, são quantificados a concentração de bicarbonato e o volume de líquido no duodeno.
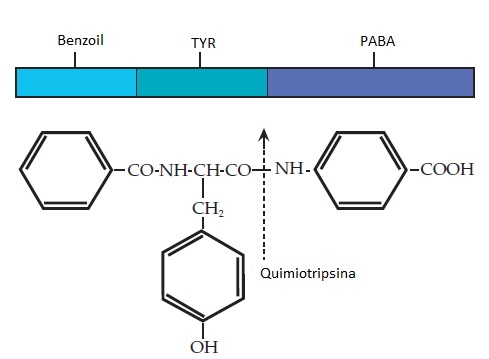
O teste de bentiromida não invasivo baseia-se na ação da tripsina sobre a bentiromida para produção de ácido para-aminobenzoico (PABA) e benzoil-tirosina [Figura 1]. O PABA é prontamente absorvido pelo intestino e excretado na urina. Em indivíduos saudáveis, quando 500 mg de bentiromida são ingeridos, pelo menos 57% do PABA aparece na urina dentro de 6 horas. Em pacientes com pancreatite crônica, a quantidade de PABA excretado é significativamente menor (em média, igual a 42%). Considerando a excreção de 57% como valor de corte, o teste de bentiromida apresenta sensibilidade de 67 a 80% e especificidade de 95%.7 O PABA também pode ser quantificado no plasma em 120 minutos após a ingesta de bentiromida, e isto pode aumentar a sensibilidade do teste.8 As determinações feitas no plasma são úteis em casos de comprometimento da excreção renal, que pode ser encontrada em idosos. O PABA é identificado por métodos colorimétricos. Desta forma, outras arilaminas podem interferir em sua determinação (p. ex., acetaminofeno, lidocaína, procainamida, sulfonamidas e diuréticos tiazida).9 Nos casos em que a absorção intestinal está comprometida, como no espru celíaco, a absorção do PABA liberado pode estar reduzida e levar a uma recuperação urinária falsamente baixa. Contudo, o teste da bentiromida resulta positivo somente quando a glândula pancreática sofre uma destruição superior a 90%. Mesmo assim, ao considerar o workup de um paciente com esteatorreia, o teste pode ser útil por usar o mesmo nível de destruição glandular para gerar esteatorreia.

Figura 1. A clivagem da molécula de bentiromida pela enzima intraduodenal quimiotripsina rende 2 fragmentos: benzoil-tirosina e ácido para-aminobenzoico (PABA). A bentiromida é composta por benzoil (azul-claro), tirosina (TYR; azul) e PABA (azul-escuro). O PABA liberado é absorvido através do intestino e excretado em quantidades significativas na urina. A ausência da quimiotripsina, como resultado de doença pancreática ou obstrução ductal, resulta na falha de liberação, absorção e excreção urinaria de PABA.
Embora a vasta maioria da proteases e lipases pancreáticas seja armazenada nos grânulos de zimogênio e liberada a partir da porção apical da célula pancreática exócrina dentro do ducto pancreático, um pequeno percentual vaza dentro do interstício glandular, é levado pela circulação e pode ser quantificado pelo ensaio do tripsinogênio sérico. Como o peptídeo ativador de tripsina ainda não foi liberado e toda a tripsina ativa é rapidamente ligada pela alfa-1-antitripsina, a forma livre circulante da tripsina é o tripsinogênio. Em pacientes com pancreatite crônica que sofrem de insuficiência exócrina, a concentração sérica de tripsinogênio é menor do que aquela encontrada em indivíduos saudáveis (2 a 18 ng/mL, em comparação aos 29 a 79 ng/mL em indivíduos sadios).10 Baixos níveis séricos de tripsinogênio parecem ter alto grau de especificidade para pancreatite crônica, porém sensibilidade apenas modesta.
Os ácidos biliares são sintetizados a partir do colesterol no fígado e devem ser conjugados pela glicina ou taurina antes de serem excretados no intestino via ducto colédoco comum. Os conjugados do ácido biliar solubilizam os produtos da hidrólise do triacilglicerol em micelas complexas e isto facilita a rápida absorção dos lipídios da dieta. Os ácidos biliares não são absorvidos com os lipídios da dieta ao nível do intestino proximal, e sim junto ao íleo distal. O pool de ácido biliar recircula 6 vezes/dia. Cerca de 95% dos ácidos biliares são reabsorvidos e recirculam na circulação entero-hepática diariamente. Em torno de 0,5 g de ácidos biliares aparecem nas fezes a cada dia e isto corresponde à taxa de síntese hepática sob condições estáveis. Se os ácidos biliares não forem adequadamente absorvidos, o indivíduo tem diarreia (enteropatia biliar). Na completa ausência de sais biliares, os ácidos graxos são absorvidos de forma menos eficiente, com até 25 a 50% dos lipídios ingeridos aparecendo nas fezes. Em pacientes com diarreia idiopática ou que apresentam diarreia pós-ressecção do íleo (= 30 cm), a má absorção dos ácidos biliares é uma possibilidade etiológica. Do mesmo modo, crianças com diarreia inexplicável podem ter um defeito congênito do transportador de sais biliares sódio-dependente presente no íleo terminal.11
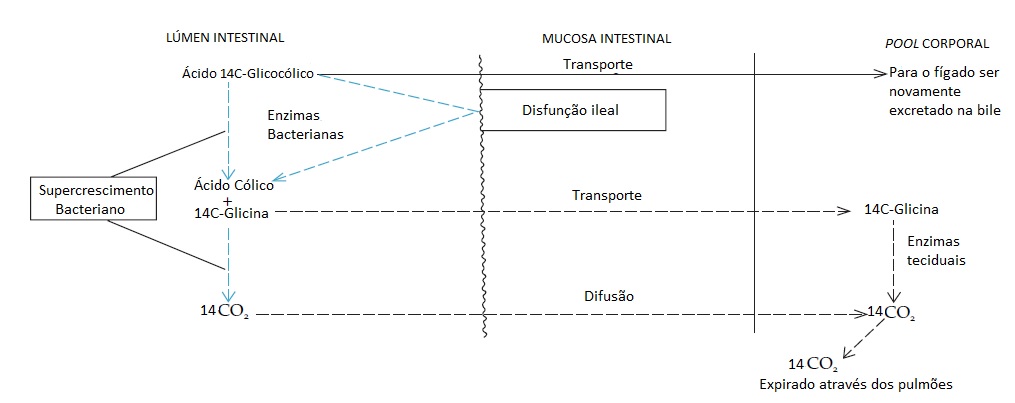
Existem 2 métodos para testar a ocorrência de má absorção de ácidos biliares, embora não sejam amplamente usados. O 1º método é o teste da respiração com 14C-ácido glicólico, e o 2º método é o teste de absorção de taurina-ácido homocólico marcado com selênio 75 (75SeHCAT). No 1º teste [Figura 2], o 14C-ácido glicólico é fornecido em quantidade mínima por via oral. Muitas bactérias são capazes de hidrolisar a amida ligada e liberar 14C-glicina. Esta pode ser absorvida e o 14CO2 é produzido no fígado, ou a 14C-glicina pode ser adicionalmente metabolizada a 14CO2 no lúmen intestinal. Em qualquer um destes eventos, o 14CO2 aparece na respiração em quantidades mensuráveis. O percentual da dose ingerida que é excretado na respiração aumenta, se no lúmen intestinal houver uma concentração de bactérias acima do normal ou se os ácidos biliares seguirem para o cólon (disfunção do íleo). Um fármaco antissecreção gástrica também pode aumentar a população bacteriana residente no intestino a um nível que resulta em um teste respiratório anormal.12 A utilidade deste teste como indicador de má absorção de ácidos biliares é, portanto, limitada. O teste de 75SeHCAT tem maior utilidade clínica em potencial, por apresentar uma correlação mais forte com a excreção de colato e devido à simplicidade da medida da retenção do 75Se com auxílio de uma câmera gama de corpo inteiro. Indivíduos normais retêm mais de 19% de uma dose de 75Se decorridos 7 dias da administração por via oral, enquanto os pacientes com disfunção significativa ou ressecção ileal retêm menos de 12%.13
Figura 2. No teste respiratório de ácidos biliares, uma pequena dose de ácido 14C-glicocólico é ingerida, e seu destino é determinado medindo-se a excreção de 14CO2na respiração. Em um indivíduo normal, pouco ácido 14C-glicocólico é metabolizado para ser excretado na respiração, porque passa direto para o íleo para ser absorvido e voltar à circulação êntero-hepática. Havendo supercrescimento bacteriano intestinal ou disfunção ileal, todavia, as enzimas bacterianas desconjugam o ácido biliar (linhas tracejadas em azul), liberando ácido cólico e 14C-glicina. A glicina radioativa pode ser transportada através da mucosa intestinal (linha tracejada em cinza, superior) e subsequentemente degrada em 14CO2 pelas enzimas teciduais. Como alternativa, a 14C-glicina pode ser metabolizada em junto ao lúmen intestinal em 14CO2 que, por sua vez, difunde-se (linha tracejada em cinza, inferior) para a circulação e é transportado até os pulmões. Em consequência, a excreção de 14CO2 é 10 vezes maior em casos de supercrescimento bacteriano ou disfunção ileal do que no estado normal.
Agora, que o transportador de ácido biliar sódio-dependente humano está clonado, estão sendo descobertos defeitos congênitos que promovem má absorção de ácido biliar e consequente diarreia.11 Estes defeitos podem ser a causa da má absorção primária de ácido biliar.
A EGS era conhecida como doença celíaca em crianças e espru idiopático ou não tropical em adultos. Em 1960, foi reconhecido que estas doenças eram a mesma entidade, causada pelo glúten (a principal proteína do trigo) e, mais especificamente, pela gliadina (o componente álcool-solúvel do glúten).14
A doença celíaca em geral é considerada menos comum nos Estados Unidos do que na Europa Ocidental. Entretanto, um amplo estudo multicentros realizado recentemente indicou que a prevalência da doença celíaca nos Estados Unidos, entre os pacientes sintomáticos (1 em 56) e entre os indivíduos fora de risco (1 em 133), é similar àquela relatada na Europa.15 Muitos pacientes atualmente são identificados como celíacos que não manifestam os sintomas clássicos de esteatorreia e perda de peso.
A EGS ou doença Celiaca está associada aos haplótipos HLA-DQ2 (DQA1*501, DQB1* 201) e HLA-DQ8 (DQA1*031, DQB1*302). Em grupos de gêmeos monozigóticos, 30% dos gêmeos opostos do caso-incidência não têm EGS. Esta condição levou à crença na existência de outro fator (não genético) desconhecido, que é importante como causa da doença. Um peptídeo contendo 33 aminoácidos componente da gliadina é comprovadamente mal digerido pelas proteases e causa ativação de células T em pacientes com EGS.16
A EGS está associada ao comprometimento da liberação de CCK. As células contendo CCK podem estar numericamente reduzidas ou defeituosas, a ponto de a quantidade de CCK presente na mucosa duodenal estar bastante diminuída.14 Esta deficiência de CCK acarreta diminuição da quantidade de lipase pancreática e ácidos biliares que chegam ao lúmen intestinal em resposta aos lipídios da dieta. As células da cripta intestinal são as principais células intestinais secretoras de líquido, via secreção de Cl- dependente de cAMP com secreção concomitante de água. Na EGS, a porção críptica do complexo viloso está amplamente expandida, com consequente aumento da secreção de água. Como as células localizadas na ponta dos vilos (que normalmente absorvem água) estão adoecidas e em número reduzido, a absorção de água e eletrólitos não é tão efetiva quanto o normal, e o intestino torna-se secretor.14 Desta forma, a concentração de ácidos biliares no lúmen intestinal cai para níveis inferiores àqueles esperados para o simples comprometimento da liberação de CCK. A capacidade dos ácidos biliares de solubilizar os produtos da lipólise depende da presença de sais biliares em concentração maior do que a concentração micelar crítica de 1,4 mM.17 Normalmente, o intestino possui uma concentração pós-prandial de sais biliares igual a 10 mM.18 Uma 3ª causa de esteatorreia em pacientes com EGS é a diminuição da área de superfície de absorção intestinal. As bordas em escova existentes na superfície dos enterócitos maduros são severamente afetadas na EGS. Além disso, as estruturas vilosas estão achatadas. Estas 2 condições resultam em uma área de superfície severamente reduzida que limita a absorção dos lipídios. A redução da área de superfície pode ser estimada pelo teste de absorção de D-xilose. Uma 4ª causa de esteatorreia deriva da natureza imatura dos enterócitos das células absortivas intestinais, que ficam voltadas para o lúmen intestinal. Estas células são precariamente equipadas para montar os quilomícrons e suas respectivas cargas de triacilglicerol, ambos processos complexos que requerem funções celulares maduras.
Manifestações clínicas. Embora a doença celíaca possa surgir na infância e ser responsável pela abstinência de glúten, as crianças com esta doença apresentam remissão logo nos primeiros anos de adolescência, mesmo que consumam uma dieta contendo glúten. Na fase adulta, estes pacientes – dos quais 25% ou mais eram sintomáticos na infância – podem apresentar diversas queixas. Em geral, a perda de peso, fadiga, cólicas abdominais, distensão abdominal e inchaço abdominal pela presença de gases e diarreia (esteatorreia) são proeminentes, embora os pacientes nem sempre apresentem perda de apetite. Em alguns pacientes, a doença inicialmente é insidiosa e os sintomas são leves. É somente depois de serem tratados que estes pacientes percebem, em retrospectiva, o quanto estavam doentes. Em estudos populacionais, nos quais a presença da doença foi determinada por biópsia intestinal, os indivíduos com biópsia consistente com EGS frequentemente eram assintomáticos, mas às vezes apresentavam estatura inferior que a de de seus irmãos não afetados. Como nenhum achado conduz especificamente ao diagnóstico, em especial na ausência de uma esteatorreia clinicamente evidente, pode demorar para se perceber que o paciente tem EGS. Este problema tende mais a ocorrer no caso de pacientes que não têm esteatorreia, mas têm osteoporose, sofrem contusões com facilidade em decorrência da deficiência de vitamina K ou têm anemia ferropriva. Esta última condição é comum em pacientes adultos com EGS.19 Um estudo sugeriu que as fraturas podem ser o único sinal de má absorção em pacientes com EGS não detectada.20
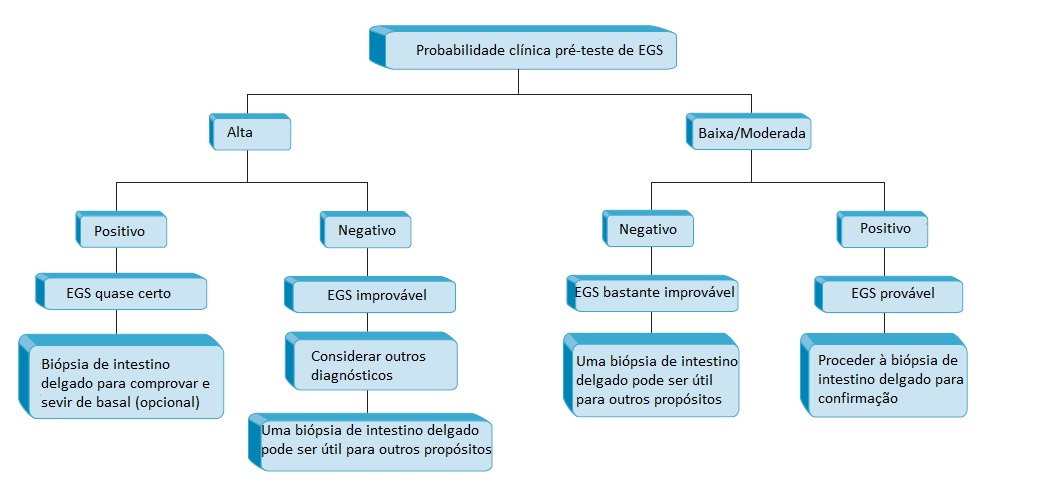
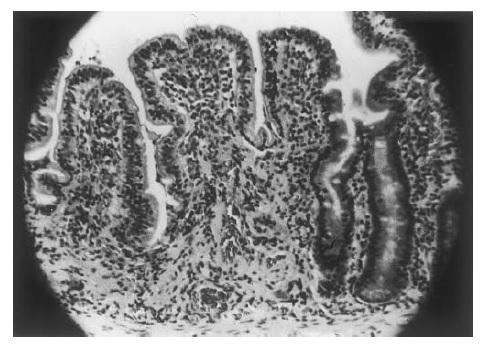
Exames laboratoriais. Em um paciente com alta suspeita de EGS (p. ex., parente em 1º grau de um paciente com EGS comprovada; paciente com história de doença causadora de diarreia na infância, que tenha sido avaliado por um especialista e tratado com dieta especial; ou paciente com má absorção que não seja alcoólatra nem apresente outras explicações evidentes para a má absorção de gorduras), um resultado positivo no teste de anticorpos antitransglutaminase tecidual é quase certamente diagnóstico [Figura 3].21 Como alternativa, o diagnóstico talvez seja estabelecido com base nos achados do exame de biópsia de intestino delgado [Figura 4]. Os achados clássicos incluem uma atrofia parcial ou total das vilosidades; enterócitos de aspecto anormal na extremidade das vilosidades; aumento dos linfócitos intraepiteliais; infiltração da lâmina própria, consistindo predominantemente de linfócitos e macrófagos; aumento do tamanho das criptas, tanto na vertical como na horizontal; e aumento do número de figuras mitóticas.14 Estes achados, embora sejam típicos, não são patognomônicos. Para que o diagnóstico seja definitivo, o paciente deve responder à terapia dietética. É possível esperar a melhora dos sintomas em 80% dos pacientes dentro de um período de 1 mês, contudo a melhora histológica é consideravelmente mais demorada. Outros 10% dos pacientes permanecem irresponsivos por um período de até 2 meses, enquanto o restante pode demorar até 2 anos para responder. Mesmo que um controle rígido da dieta seja adotado, os achados do exame de biópsia talvez não sejam normalizados. Mais frequentemente, o paciente permanece bem controlado, devido à melhora sintomática associada à dieta. Entretanto, muitos pacientes eventualmente são testados quando já estão curados ou se encontram em uma situação que os força a cometer indiscrição alimentar. Este erro inevitavelmente resulta na recorrência dos sintomas, sustentando ainda mais o diagnóstico.
Figura 3. Procedimento para diagnóstico da enteropatia glúten-sensível (EGS).
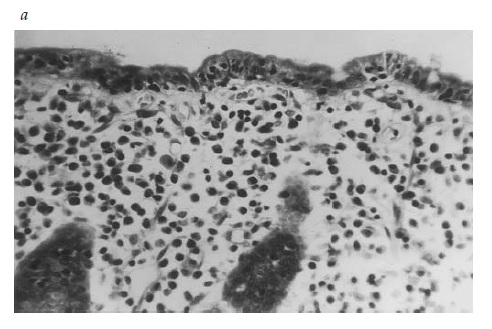
Figura 4. A amostra de biópsia de intestino delgado de um paciente com espru celíaco não tratado (a) mostra uma superfície plana contendo infiltração plasmocítica na região subepitelial e linfócitos intraepiteliais (aumento: 400x). Em contraste, uma amostra de biópsia obtida de um paciente com insuficiência pancreática exócrina (b) é indistinguível de uma amostra normal e mostra vilosidades altas e encurvadas, além de uma infiltração mononuclear mínima na região subepitelial (aumento: 100x).
Outro teste útil é a identificação de um anticorpo antiendomísio. Este anticorpo está presente em até 95% dos casos e raramente é encontrado nos indivíduos do grupo controle.22 Outros testes de detecção de má absorção, como o teste de absorção de D-xilose ou os testes de detecção de gordura nas fezes, podem resultar anormais. Níveis baixos de fatores de coagulação, anemia por deficiência de folato ou de ferro, ou, ainda, osteoporose, são condições que podem estar presentes. Entretanto, nenhuma destas condições é específica para EGS.
Nota do editor: Alem do anticorpo anti-endomísio, pode ser dosado o anticorpo anti-gliadina e anticorpo antitransglutamidase, todos sensíveis e comm alta acurácia para o diagnóstico, mas caso seja retirado o glúten da dieta, pode ocorrer que os anticorpos desapareçam ou seu títulos diminuam sensivelmente.
O tratamento da EGS é uma dieta estritamente livre de glúten. Como a dieta isenta de glúten é um compromisso para toda a vida, é mais onerosa do que uma dieta normal e pode impor limitações sociais, não deve ser recomendada sem que o diagnóstico de EGS tenha sido firmemente estabelecido, seja por histologia ou por exames sorológicos confiáveis. Uma dieta livre de glúten proíbe a ingesta de trigo, centeio e cevada. A aveia é considerada um alimento seguro, mas costuma ser evitada na fase inicial, quando a resposta clínica à dieta está sendo avaliada. Manter o paciente na dieta é difícil em alguns casos, pois muitos alimentos contêm glúten escondido. A manutenção de uma dieta isenta de glúten é importante, porque os linfomas intestinais tendem mais a se desenvolver em pacientes que não seguem este tipo de dieta.23
Os grupos de apoio, como aqueles organizados pela Celiac Society of America, nos Estados Unidos, podem ser úteis, especialmente logo após o diagnóstico da doença. Informações como o que procurar nos rótulos das embalagens e receitas interessantes podem contribuir bastante no sentido de manter o paciente na dieta livre de glúten. Durante o período de avaliação, o paciente não deve consumir cervejas e uísques, que podem conter glúten em quantidade suficiente para sensibilizá-lo. Depois que a resposta à dieta estiver nitidamente estabelecida, estas bebidas podem ser experimentadas para determinar se o paciente é sensível. Outros produtos geralmente considerados como isentos glúten e que na verdade o contêm são os sorvetes, a óstia sagrada e até mesmo alguns fármacos (como preenchedores). Apesar das restrições, existem muitas opções dietéticas permitidas ao paciente, tais como alguns cereais matinais, leite, queijo, ovos, carne vermelha, aves, peixe, chocolate e produtos feitos com farinha de milho, arroz ou batata.
Se o paciente for irresponsivo, a explicação mais provável é que a dieta não está sendo fielmente seguida. Nesses casos, é útil contar com um nutricionista que acompanhe o histórico alimentar do paciente.14 Menos frequentemente, o paciente desenvolve síndrome da estase intestinal ou insuficiência pancreática. Quando estes problemas subsidiários são diagnosticados e tratados com sucesso, o paciente geralmente apresenta resposta à dieta. Entretanto, uma minoria dos pacientes que não responde à dieta livre de glúten sofre de uma condição conhecida como espru refratário.24 Os pacientes com espru refratário podem necessitar de tratamento com corticosteroides e outros agentes imunossupressores, incluindo a azatioprina.25
Dermatite herpetiforme. Muitos pacientes com dermatite herpetiforme têm EGS.21 As lesões vesiculares e intensamente pruriginosas surgem nos joelhos, cotovelos, ombros e nádegas [ver 2:I Manifestações cutâneas de doenças sistêmicas]. As biópsias cutâneas das lesões da dermatite herpetiforme apresentam os característicos depósitos de imunoglobulina A (IgA). Com a adoção de uma dieta livre de glúten, as lesões dermatológicas e intestinais melhoram, indicando a existência de uma ligação entre ambas. Entretanto, as lesões cutâneas respondem ao tratamento com dapsona, ao contrário das lesões intestinais, e isto indica que também existem diferenças entre as 2 condições.
Diabetes melito de tipo 1. Os pacientes com diabetes de tipo 1 apresentam risco aumentado de desenvolvimento de doença celíaca. Quando os anticorpos antiendomísio foram usados em um estudo, a EGS foi encontrada em 47 pacientes diabéticos (6%) – uma incidência significativamente maior do que se esperaria encontrar por acaso.26 Mesmo quando não mostram sinais evidentes de má absorção, os pacientes com diabetes melito de tipo 1 apresentam risco de desenvolvimento de doença celíaca. Por este motivo, a avaliação da doença celíaca pode ser recomendável para estes pacientes.27 Um estudo relatou uma prevalência de 5,7% do espru celíaco entre pacientes com diabetes de tipo 1, além do achado de aumento da incidência de doenças autoimunes entre estes pacientes.28 A prevalência dos distúrbios autoimunes entre os pacientes com doença celíaca parece estar relacionada à duração da exposição do glúten, que justifica ainda mais o diagnóstico e tratamento antecipados da EGS.29
O espru tropical é uma doença de má absorção que aparece em certas áreas do mundo, sobretudo nos trópicos, e afeta tanto os povos indígenas quanto os turistas. Em 2 populações minuciosamente estudadas, 5 a 13% dos norte-americanos que viviam em Porto Rico por pelo menos 6 meses apresentaram sintomas de espru tropical. Os indivíduos exilados dos Estados Unidos que voltam dos trópicos e outras áreas endêmicas de espru tropical podem apresentar sintomas desta doença passados mais de 10 anos do retorno.30 Os voluntários das Forças de Paz norte-americanas que passaram algum tempo no Paquistão desenvolveram lesões demonstráveis no intestino delgado e anormalidades funcionais que foram revertidas ao normal no decorrer de vários meses, após a volta para casa.31 Os indianos e paquistaneses que vivem nos Estados Unidos podem demorar mais (até 4 anos) para excretar quantidades normais de D-xilose.32 A causa exata das alterações observadas no intestino delgado são obscuras, porém a síndrome do espru tropical parece ser causada por 1 ou mais espécies de bactérias coliformes, como as espécies de Klebsiella,33 que colonizam o trato intestinal superior.
Diagnóstico. Os sintomas do espru tropical diferem dos sintomas da EGS. A perda de peso, causada principalmente pela anorexia, é bastante proeminente, assim como a diarreia. Língua dolorida (70% dos pacientes), edema de extremidades de membros inferiores (25%), deficiência de folato e vitamina B12 (75 a 100%) ou resultado anormal no teste de Schilling (96 a 100%) são achados significativamente mais comuns no espru tropical do que no espru não tropical.33 Os sintomas podem ser bastante severos, por vezes levando à morte do paciente nas áreas endêmicas. Contudo, com o tratamento adequado, o prognóstico em geral é excelente tanto para os pacientes que permanecem nos trópicos como para aqueles que voltam para os Estados Unidos.
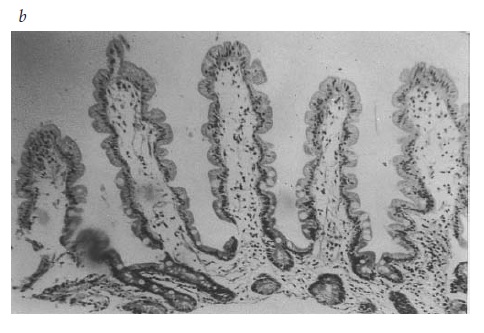
O diagnóstico de espru tropical é estabelecido por meio de um exame de biópsia do intestino delgado de pacientes com manifestação clínica compatível e história de viagem. Os vilos, que são semelhantes a folhas ou agulhas rombudas, e a lâmina própria estão lotados de células inflamatórias [Figura 5]. Em norte-americanos e europeus normais, é possível observar as estruturas vilosas [Figura 4]. Ao considerar esta doença, é preciso notar que os resultados da biópsia intestinal de residentes de áreas endêmicas ou de turistas que não se hospedam nos hotéis mais conhecidos ao viajarem para áreas endêmicas seriam classificados como anormais se fossem obtidos de residentes dos Estados Unidos ou da Europa.
Figura 5. Em uma amostra de biópsia de intestino delgado de um paciente com espru tropical, as vilosidades estão mais amplas e curtas, enquanto as criptas estão mais profundas. Estas alterações resultam em uma proporção vilos:criptas igual a 1:1 (aumento: 100x).
Tratamento. O tratamento do espru tropical deve começar com o uso de acido fólico (5 mg/dia).33 Esta terapia está associada à melhora rápida do apetite e elimina a maioria dos sintomas clínicos. Em pacientes com sintomas de curta duração (< 4 meses), a administração de folato por 6 meses a 1 ano pode ser suficiente. Para os pacientes com sintomas de maior duração (> 4 meses), recomenda-se a adição de antibióticos (p. ex., tetraciclina, 2 g/dia, por 1 ano). A maioria dos pacientes que voltam para os Estados Unidos ganham peso rapidamente, mesmo que os resultados dos testes de absorção ou dos exames de biópsia intestinal não sejam normalizados.
O espru colagenoso é uma doença rara e devastadora, em que há formação de camada de colágeno sob os enterócitos do intestino delgado. A relação existente entre o espru colagenoso e a colite colagenosa é obscura, contudo o aspecto histológico básico da deposição subepitelial de colágeno é o mesmo em ambas as condições. A origem da colite colagenosa é desconhecida, mas a condição se desenvolve em cerca de metade dos pacientes com doença celíaca refratária (indivíduos irresponsivos à dieta de exclusão do glúten).34 Embora esteja comprovado que o colágeno de tipo 6 é depositado na colite colagenosa mais comumente diagnosticada, o tipo de colágeno da camada que forma no intestino delgado de pacientes com espru celíaco é desconhecido. Na colite colagenosa, os sintomas (principalmente a diarreia) costumam ser modestos. No espru colagenoso, porém, os sintomas são mais fortes e incluem evidente má absorção. Esta gravidade dos sintomas deve-se provavelmente à atuação do colágeno como barreira à difusão, que impede a difusão dos nutrientes para dentro dos capilares porta ou dos linfáticos.
Diagnóstico. O diagnóstico do espru colagenoso é estabelecido pelo clássico quadro histológico de atrofia das vilosidades e deposição subepitelial de colágeno. Entretanto, se houver erro diagnóstico e o paciente for considerado como tendo EGS com base no achatamento estrutural das vilosidades, este paciente em geral não responderá à dieta livre de glúten.
Tratamento. A terapia para o espru colagenoso é incerta. O problema mais comum é a diarreia osmótica produzida pelo estado geral de má absorção induzida pelo processo patológico. Nesta situação, o paciente é tratado como se tivesse síndrome do intestino curto. Alguns pacientes respondem à terapia com esteroides. Poucos pacientes respondem aos esteroides e a uma dieta de exclusão de glúten, sendo que nestes casos a melhora da condição do paciente eventualmente possibilita afunilar a dosagem de esteroide.35
O trato gastrintestinal (GI) é o maior órgão linfoide do corpo. O ambiente ao qual este sistema imune é exposto é repleto de antígenos estranhos que devem ser sorteados, identificados e, se necessário, atacados. Assim, não surpreende que possa haver desenvolvimento de disfunção intestinal em pacientes imunodeficientes, em particular naqueles com deficiência de IgA, pois esta é a imunoglobulina mais importante do intestino. Alguns pacientes que apresentam uma das síndromes hipogamaglobulinêmicas podem desenvolver má absorção.36 Os pacientes com deficiência de IgA em geral também têm história de infecções respiratórias recorrentes,36 e isto os distingue ainda mais dos pacientes com EGS. A causa mais comum de má absorção encontrada nesta condição é a giardíase.
Diagnóstico. A suspeita diagnóstica de espru associado a hipergamaglobulinemia é considerada quando o paciente apresenta sinais e sintomas de má absorção e baixos níveis séricos de imunoglobulinas, em especial de IgA. As amostras de biópsia intestinal não mostram plasmócitos e, assim, podem ser distinguidas com facilidade das biópsias obtidas de pacientes com EGS, em que os plasmócitos são abundantes. Os plasmócitos também são prontamente visualizados nas amostras de biópsias normais. Os organismos de Giardia lamblia também podem ser encontrados no espru associado a hipergamaglobulinemia .
Tratamento. Frequentemente, os sintomas intestinais associados ao espru hipergamaglobulinêmico melhoram com a administração de 750 mg de metronidazol/dia, durante 10 dias, para tratamento de giardíase.
Nota do editor: As principais imunodeficiências associadas com este tipo de espru são a imunodeficiência comum variada a deficiência de IgA.
A ressecção em massa do intestino delgado é empregada no tratamento de várias doenças, incluindo a isquemia mesentérica, volvo e doença de Crohn. Como o intestino necessita de uma determinada área de superfície sobre a qual a absorção possa ocorrer, a diminuição desta área a uma extensão inferior à área considerada essencial resulta em má absorção. Dependendo da quantidade de intestino extirpada, os resultados podem variar de levemente inconvenientes a catastróficos. A preservação da valva ileocecal ameniza os sintomas. O íleo responde à ressecção jejunal sofrendo hiperplasia, e de forma bem mais efetiva do que o jejuno responde a uma ressecção ileal. Do mesmo modo, existem mecanismos especializados que atuam no íleo e estão ausentes no jejuno, como os transportadores de sais biliares e de vitamina B12. A manutenção de um pool adequado de ácidos biliares é importante para a absorção de gorduras, pois a área reduzida da superfície de absorção dos pacientes submetidos à ressecção intestinal torna necessário que a absorção de gorduras seja o mais eficiente possível. Como alternativa, o íleo pode realizar a maioria das funções do jejuno, com exceção da absorção de ácido fólico, Ca2+ e Fe2+. Entretanto, estas moléculas podem ser substituídas por uma medicação apropriada.
Diagnóstico. O diagnóstico de síndrome do intestino curto é estabelecido por uma história de ressecção intestinal combinada às manifestações clínicas da síndrome, como diarreia, esteatorreia, perda de peso, deficiências de oligoelementos, hiponatremia e hipocalemia.
Tratamento. O tratamento de pacientes com síndrome do intestino curto depende da localização e da quantidade de intestino extirpado. As proteínas requerem a maior área de superfície para absorção.37 Desta forma, alcançar um nível de assimilação adequado pode tornar-se problemático, apesar da hidrossolubilidade das proteínas e de seus produtos hidrolíticos. As vitaminas e minerais também precisam ser adicionados a qualquer regime terapêutico, dependendo da parte do intestino que estiver faltando. O tratamento pode incluir múltiplas refeições pequenas diárias, consumo de alimentos de rápida absorção (p. ex., suplementos calóricos enlatados), picar ou moer os alimentos, e consumo de alimentos contendo triglicerídeos de cadeia intermediária, que podem ser absorvidos na ausência de sais biliares.37 Os alimentos ricos em ácidos graxos poli-insaturados (p. ex., óleos vegetais) são mais facilmente absorvidos do que as carnes, que contêm mais gordura saturada. Por fim, os suplementos dietéticos totalmente hidrolisados são absorvidos de forma rápida. Para retardar o trânsito intestinal, a difenoxilato-atropina, loperamida ou tintura de ópio desodorizada podem ser usadas de maneira efetiva. Um método alternativo consiste em fazer o paciente beber uma pequena quantidade de óleo de girassol antes da refeição. O lipídio segue rápido para o íleo (se este não tiver sido removido), cólon ou ambos38 e elabora o peptídeo YY (PYY).39 O PYY constitui a pausa ileal putativa, que retarda o esvaziamento gástrico. Fazer os pacientes provarem dietas diferentes muitas vezes os capacita a ingerir alimentos por via oral, em vez de fazê-los receber nutrição parenteral total, que é menos desejada.
A lesão intestinal, de modo geral, é uma consequência comum da exposição à radiação ionizante durante o tratamento oncológico. A lesão no intestino delgado é mais comum em pacientes previamente submetidos à cirurgia abdominal, que pode restringir a movimentação do intestino delgado. O íleo terminal pode ser envolvido durante a irradiação pélvica. Para evitar a lesão por radiação, é recomendável irradiar os pacientes sobre uma plataforma giratória, para que a quantidade máxima de radiação incida sobre mais de uma parte do intestino.
A doença de Whipple é uma rara doença multissistêmica causada pela bactéria Tropheryma whippelii.40 O bacilo pode disseminar-se amplamente por todo o corpo, porém os sítios de invasão apresentam poucos sinais de inflamação, sugerindo que uma deficiência autoimune pode levar à predisposição à doença.41 O diagnóstico correto é imperativo, pois a taxa de mortalidade é de quase 100% entre os pacientes não tratados com antibiótico.
Manifestações clínicas. Classicamente, a doença de Whipple surge em homens de meia-idade com um caso de artrite não deformante que geralmente se desenvolveu vários anos antes da manifestação dos sintomas intestinais. As artralgias, diarreia, dor abdominal e perda de peso são as manifestações cardinais da doença de Whipple. Outras queixas incluem febre, distensão abdominal, linfadenopatia, hiperpigmentação da pele e esteatorreia.42 Muitos pacientes expressam o isótipo HLA-B27. Ocasionalmente, os sintomas intestinais estão ausentes, mesmo em alguns pacientes que apresentam envolvimento do sistema nervoso central (SNC).43 Em um caso bem comprovado, embora incomum, o envolvimento intestinal não foi identificado nem mesmo após o exame de extensivas biópsias em 2 laboratórios, apesar do fato de o paciente apresentar os demais sintomas típicos da doença.44 O envolvimento cardíaco e pulmonar também pode estar presente.45
Exames laboratoriais. O reconhecimento da doença de Whipple em pacientes sem sintomas nem envolvimento intestinal patológico tem se tornado mais frequente desde o advento das técnicas de reação em cadeia da polimerase (PCR), que identificam o exclusivo RNA ribossômico 16S de T. whippelii.46
O diagnóstico baseia-se na identificação dos clássicos macrófagos positivos para ácido periódico de Schiff (PAS), que contêm partículas falciformes.42 Sem dúvida, o sítio de biópsia que mais comumente fornece resultados positivos é o intestino. A lesão histológica mostra as vilosidades distendidas (vilosidades deformadas) repletas de macrófagos PAS-positivos espumosos e dilatação linfática. Em casos extremos, é possível observar uma superfície vilosa plana. Estes achados devem ser diferenciados, no contexto clínico apropriado, dos achados associados à doença causada pelo complexo M. avium, em que os macrófagos PAS-positivos também estão presentes. Uma coloração para bacilos acidorresistentes deve permitir esta diferenciação. O envolvimento do SNC – ocasionalmente, associado à presença de macrófagos típicos no líquido cerebrospinal (LCS), sustentada pela técnica mais sensível de PCR – pode ser observado na presença ou ausência de sintomas neurológicos,47 enquanto um resultado de PCR negativo pode predizer uma baixa probabilidade de recidiva clínica.48 Em alguns casos, torna-se necessário obter uma biópsia cerebral, que pode ser orientada por imagem de ressonância magnética (IRM).
Tratamento. Como a doença de Whipple é bastante incomum, torna-se difícil estabelecer um plano terapêutico bem definido. O tratamento originalmente proposto consistia na administração de penicilina (250 mg/dia) e estreptomicina (1 g; via intramuscular [IM]) por um período de 2 semanas, seguida da administração de tetraciclina (1 g) por 1 ano. Hoje em dia, o trimetoprima-sulfametoxazol (1 comprimido de potência dobrada, 2 vezes/dia) é tipicamente administrado durante 1 ano. Todos os agentes antimicrobianos são usados nas doses habituais.
Embora os sintomas intestinais e sistêmicos respondam prontamente aos tratamentos, o aspecto mais preocupante é o tratamento das manifestações do SNC. Nos pacientes que a princípio não apresentam envolvimento do SNC, a manifestação inicial dos sintomas de envolvimento do SNC costuma ocorrer pelo menos 1 ano após o tratamento dos sintomas sistêmicos e intestinais, em especial quando o antibiótico administrado não atravessa a barreira hematoencefálica. É possível que um desenvolvimento progressivo de demência seja observado, contudo os sinais patognomônicos de doença do SNC, quando presentes, são as miorritmias óculo-mastigatória e óculo-facial-esquelética.49 Portanto, os anticorpos que atravessam a barreira hematoencefálica são necessários no tratamento da doença de Whipple. É interessante notar que o curto período de administração de penicilina-estreptomicina é suficiente para bloquear os sintomas do SNC, ao passo que até mesmo a terapia prolongada com trimetoprima-sulfametoxazol ocasionalmente não evita as manifestações do SNC associadas à doença de Whipple.50 A tetraciclina isoladamente não erradica a doença do SNC e não deve ser fornecida por si só, ainda que seja considerada efetiva no tratamento dos sintomas intestinais e sistêmicos. Um aspecto importante a ser lembrado é que, em 50% dos pacientes, o LCS pode conter os macrófagos da doença de Whipple ou material PCR-positivo, mesmo na ausência de sintomas do SNC.46 Uma vez alcançada a melhora do envolvimento do SNC, o tratamento em geral é inútil, apesar de ser possível observar alguma melhora e a não progressão da doença com o tratamento.
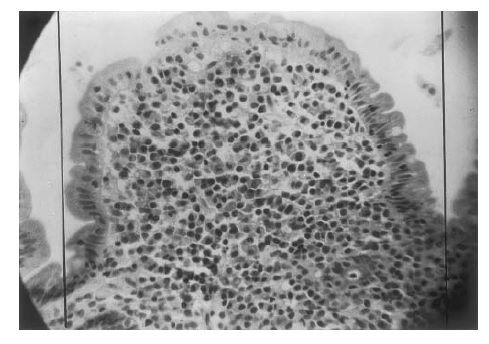
A doença imunoproliferativa do intestino delgado (DIID), antigamente conhecida como linfoma intestinal primário, é uma condição em que a lâmina própria do intestino delgado é intensamente infiltrada por linfócitos, enquanto os enterócitos sobrejacentes permanecem morfologicamente normais [Figura 6]. Trata-se de um distúrbio de células B envolvendo o tecido linfoide associado à mucosa (MALT). A doença é rara nos países industrializados e mais comum nas populações menos favorecidas, principalmente entre os indivíduos que estão na 2ª ou 3ª décadas da vida, com predominância masculina. Em uma série de pacientes chineses, 6 dos 45 indivíduos que apresentavam linfoma intestinal tinham DIID.51 Estes pacientes apresentavam má absorção severa. Entre os pacientes com linfoma sem DIID, 65% apresentavam dores abdominais, perda de peso, massas abdominais, obstrução e perfuração. A DIID está associada às cadeias pesadas (de IgA), com paraproteínas presentes no soro, urina ou líquido jejunal. A duodenografia mostra pregas espessadas e muitas elevações nodulares na ausência de ulceração. O diagnóstico pode ser estabelecido por meio do exame de biópsia de intestino delgado em 85% dos casos.52 No início de seu curso, a doença parece ser tratável com antibióticos. Entretanto, se não for contida, a doença acaba evoluindo para formas mais agressivas de linfoma.53
Figura 6. Amostra de biópsia de intestino delgado obtida de um paciente com linfoma intestinal primário, mostrando uma única vilosidade ampliada (aumento: 400x). O epitélio é constituído por células colunares normais, porém a lâmina própria está repleta de plasmócitos e outras células mononucleares. As biópsias cirúrgicas obtidas deste paciente revelaram evidências de um linfoma histiocítico subepitelial generalizado.
A linfangiectasia intestinal costuma ser uma condição congênita, em que os vasos linfáticos deformados comprometem o transporte de quilomícrons dos enterócitos para o ducto linfático mesentérico. Um quadro fisiopatológico semelhante é observado em certos casos de DIID, enterite granulomatosa, enterite tuberculosa ou doença de Whipple, em que a drenagem linfática normal é bloqueada.
O bloqueio da drenagem linfática pode resultar em ascite quilosa, quilúria ou quilometrorreia.54 A enteropatia perdedora de proteína e a linfopenia são aspectos proeminentes. Uma modesta esteatorreia também está presente, com uma excreção de gorduras que comumente chega a 20 g/dia. Na forma congênita da doença, é possível observar o linfedema das pernas ou de uma perna e um braço. Ao exame endoscópico, podem ser observadas vilosidades brancas, nódulos brancos e elevações na submucosa.55 A aparência esbranquiçada da mucosa é sem dúvida produzida pela retenção de triacilglicerol do quilomícron. A radiografia com duplo contraste de bário mostra protrusões nodulares regulares e pregas mucosas espessas na ausência de ulceração.56 Ao exame histológico, são observados linfáticos dilatados com vilosidades em forma de clava, por vezes em pacientes assintomáticos, cujo resultado final é benigno.
O tratamento é voltado para a identificação de qualquer processo causal. Para os pacientes que apresentam a condição congênita, nos quais a melhora do envolvimento linfático não é esperada, uma dieta com baixo teor de gordura suplementada com triglicerídeos de cadeia intermediária costuma ser útil. A cirurgia pode ser realizada para remover áreas isoladas de disfunção linfática, caso estas áreas possam ser identificadas, ou, ainda, para anastomosar um ducto linfático ao sistema venoso. Em alguns casos, um desvio peritônio-venoso (LeVeen) é útil.
Na rara condição congênita conhecida como abetalipoproteinemia, os pacientes não desenvolvem quilomicronemia pós-prandial porque são incapazes de acoplar corretamente a apolipoproteína B ao quilomícron em desenvolvimento. Como os lipídios e as vitaminas lipossolúveis são transportadas do intestino nos quilomícrons, a consequente diminuição da absorção destas moléculas resulta em esteatorreia sintomática, anormalidades neurológicas, uma variante de retinite pigmentosa e hemácias espiculadas. Contrastando com as teorias antigas acerca da etiologia da abetalipoproteinemia, os pacientes que sofrem desta condição transcrevem normalmente o RNA mensageiro (mRNA) da apolipoproteína a partir da qual a proteína é traduzida de maneira correta. Mesmo assim, a apolipoproteína B não é secretada pela célula intestinal. O defeito associado a esta condição reside nas várias mutações ocorridas no gene codificador do componente M da proteína de transporte de triglicerídeos microssomal.57 Este complexo proteico que consiste no componente M de 97 kDa e no componente P (proteína dissulfeto isomerase) de 55 kDa, ajuda a translocar a apolipoproteína através da membrana do retículo endoplasmático.58 Se esta etapa não ocorrer, a apolipoproteína é degrada pelas peptidases citosólicas e microssomais. O resultado deste defeito é que tanto o intestino como o fígado não conseguem produzir nem secretar suas lipoproteínas ricas em triacilglicerol, quilomícrons e lipoproteínas de densidade muito baixa (VLDL), respectivamente. Como os quilomícrons não podem transportar a gordura para fora do enterócito, presume-se (embora não esteja comprovado) que os 80% dos lipídios absorvidos são absorvidos através da veia porta.59
Além de apresentarem os sintomas intestinais, os pacientes com abetalipoproteinemia desenvolvem problemas neurológicos sérios. Tais problemas neurológicos podem ser causados, em parte, por uma deficiência de ácidos graxos essenciais e, em parte, pelo comprometimento da distribuição de lipídios aos nervos ou pela interferência na síntese local de lipídios. O resultado é uma condição desmielinizante que causa ataxia sensorial em decorrência da perda das sensações de posição e vibração. Os sintomas são semelhantes, porém menos severos do que aqueles associados à ataxia de Friedreich.60 Os pacientes podem apresentar enfraquecimento muscular e movimentos atetoides. Os pacientes também apresentam retinite pigmentosa, geralmente acompanhada de uma discreta perda da acuidade visual, todavia com preservação da visão central. Além das anormalidades neurológicas, os pacientes apresentam acantócitos no sangue. Os acantócitos são hemácias espiculadas que, apesar da expectativa de vida quase normal, são mais suscetíveis ao traumatismo mecânico nos ensaios in vitro.
Estes pacientes apresentam baixos níveis plasmáticos de triacilglicerol e colesterol. Ao exame histológico, observa-se que os enterócitos estão carregados de gordura. Apesar deste fenótipo, a quantidade de esteatorreia é modesta (cerca de 20 g/dia).
A abetalipoproteinemia geralmente é descoberta durante a infância, pois os pacientes afetados pela doença apresentam falha de crescimento e esteatorreia. Em adultos, a doença pode ser identificada pela observação de uma combinação de achados neurológicos e oftalmológicos, pela morfologia das hemácias, níveis plasmáticos baixíssimos de lipídios e esteatorreia moderada. Ao exame da biópsia de intestino delgado, observa-se que os enterócitos estão abarrotados de lipídios, mesmo após o jejum de um dia para outro, indicando que os lipídios absorvidos estão impossibilitados de sair dos enterócitos.61
O tratamento deve incluir vitamina E, além de outras vitaminas lipossolúveis e triglicerídeos de cadeia intermediária, para diminuir a esteatorreia (se necessário).
A gastrenterite eosinofílica é uma doença rara que se caracteriza pela presença de infiltração eosinofílica de 1 ou mais partes do trato GI, em qualquer ponto desde o esôfago até o cólon, aliada à manifestação de sintomas GI. O infiltrado eosinofílico não apresenta nenhuma causa evidente, como uma infestação parasitária, por exemplo. Muitos pacientes apresentam diátese alérgica subjacente (p. ex., febre do feno, asma, dermatite atópica ou alergias farmacológicas).
Não se sabe o que provoca a agregação eosinofílica no trato GI observada na enterite eosinofílica. Entretanto, evidências sugerem que os eosinófilos, uma vez ativados, podem produzir citocinas que autoperpetuam o acúmulo de eosinófilos adicionais. Estas citocinas são a interleucina (IL)-3, IL-5 e fator estimulador de colônias de granulócitos-macrófagos (CSF-GM), que foram identificados em eosinófilos de pacientes, mas não foram detectados nos indivíduos do grupo controle que apresentavam síndrome do intestino irritável. A produção local destas citocinas é sugerida pelo achado de que os níveis séricos de IL-5 estão normais em pacientes com gastrenterite eosinofílica, em contraste com o observado nos pacientes com síndrome hipereosinofílica, que possuem níveis sanguíneos elevados de IL-5.62
Embora os eosinófilos sejam um constituinte normal do trato GI, na gastrenterite eosinofílica, os eosinófilos parecem ser mais numerosos e mais invasivos do que o normal. Exemplificando, a invasão eosinofílica das criptas do intestino delgado é a principal característica da gastrenterite eosinofílica. Uma eosinofilia periférica é observada com frequência, ainda que nem sempre esteja presente.
A gastrenterite eosinofílica pode ser dividida em 2 formas básicas: uma massa tumoral de eosinófilos produtores de uma lesão do tipo granulomatosa; e uma forma mais difusamente infiltrativa. No 1º caso, as lesões são frequentes na porção distal do estômago e podem produzir sintomas obstrutivos. Como alternativa, as massas podem ser encontradas em uma região mais proximal do estômago, intestino delgado ou cólon. Quando as lesões ocorrem no intestino delgado ou no cólon, a condição deve ser diferenciada de um linfoma ou da doença de Crohn.63 No caso da doença difusa envolvendo o intestino delgado, a infiltração pode ser mucosa, com sintomas de enteropatia perdedora de proteínas ou má absorção. Quando a localização primária da infiltração são as camadas musculares do intestino, os sintomas obstrutivos são comuns. Por fim, a doença pode ser encontrada junto à área subserosa do intestino, com uma resultante ascite eosinofílica.64
A maioria dos pacientes responde a medidas conservativas e esteroides. A cirurgia deve ser evitada, a menos que seja necessária para aliviar uma obstrução persistente ou do intestino delgado.
Um regime de 40 mg de prednisona administrada por via oral todas as manhãs e afunilada lentamente, ao longo de 2 semanas, é a terapia mais efetiva para pacientes que apresentam sintomas obstrutivos e ascite. Caso haja necessidade de administração de doses altas de esteroides para se manter a remissão, a azatioprina pode ser adicionada, por apresentar efeito poupador de esteroides.
A terapia de eliminação dietética pode ser benéfica para pacientes com envolvimento da camada mucosa.
Na América do Norte, a prevalência da doença de Crohn varia de 26 a 198,5 casos em cada 100.000 indivíduos, com uma incidência máxima entre os descendentes de judeus.65,66
A doença de Crohn, que consiste em uma doença intestinal estenosante e fistulizante, pode comprometer a absorção intestinal atuando através de pelo menos 2 mecanismos: disfunção ileal e síndrome da estase [ver Síndrome de supercrescimento bacteriano, adiante]. No caso da ressecção ileal ou do envolvimento ileal severo pela doença de Crohn, o íleo fica impedido de absorver normalmente os sais biliares. Nesta situação, a deficiência de sais biliares pós-prandial ocorre na porção superior do intestino. Esta condição pode piorar nas fases mais tardias do dia, com a ingesta de uma refeição, uma vez que o pool de ácidos biliares vai sendo progressivamente depletado a cada refeição.67 A deficiência pós-prandial de sais biliares instala-se mesmo que o fígado responda à perda de ácidos biliares a partir da circulação êntero-hepática, aumentando a síntese destes ácidos. O aumento da síntese de sais biliares é inadequado, pois toda vez que a vesícula biliar se contrai em resposta à refeição, a maioria do pool de sais biliares é perdida para o cólon,68 se uma parte significativa do íleo tiver sido extirpada. Desta forma, o fígado não tempo de promover uma reposição de sais biliares suficiente para a absorção completa da refeição que acabou de ser ingerida ou da próxima refeição. Esta condição é denominada enteropatia biliar e pode ocorrer diante da ressecção de mais de 30 cm do íleo terminal. O excesso de líquido presente no cólon é causado pela secreção de Cl- dirigida por cAMP, especificamente pelos ácidos biliares di-hidroxilados, quenodesoxicolato e desoxicolato, em vez do ácido cólico tri-hidroxilado.69
A doença de Crohn com má absorção é sugerida pela história (p. ex., ressecção ileal prévia); pela percepção de um intestino sensível e espessado ao exame físico; ou por exames de imagem (p. ex., exame do intestino delgado com bário mostrando a ausência do íleo terminal e a presença de estreitamentos, úlceras estreladas ou revestimento de mucosa pedregoso). A disfunção ileal secundária à ressecção ileal é sugerida pela presença de diarreia sem esteatorreia, mas pode ser difícil diferenciar entre esta manifestação e a síndrome da estase. A má absorção de lipídios seria sugerida por um aumento do conteúdo fecal de gordura.
A perda de ácidos biliares para o cólon e, portanto, para a circulação êntero-hepática pode estar associada a uma esteatorreia mínima ou nula.70
Diante de uma ressecção ileal mais extensa (= 100 cm), todavia, a diarreia é causada não só pelos ácidos biliares, como também pelos ácidos graxos precariamente absorvidos (esteatorreia).71 Assim, a diarreia associada à doença de Crohn pode ser causada não pela doença ativa, e sim pelas consequências da ressecção ileal. Este contexto é sugerido pelo achado de uma diarreia que ocorre quando o paciente se alimenta pela 1ª vez no pós-cirúrgico, um momento em que a atividade da doença pode ser de certo modo secundária à ressecção de áreas de atividade de doença, ou pelo fato de o paciente apresentar diarreia mínima ou nenhuma diarreia antes da cirurgia e diarreia proeminente no pós-cirúrgico.
Devido à estenose presente em alguns pacientes com doença de Crohn, é possível que haja desenvolvimento da síndrome da estase [ver Síndrome da estase (supercrescimento bacteriano), adiante].
Quando a diarreia é causada pela perda de ácidos biliares, o tratamento consiste na administração de colestiramina (4 g antes das refeições e na hora de dormir).71 Esta resina liga-se preferencialmente aos ácidos biliares di-hidroxilados, diminuindo sua concentração aquosa e sua proporção no pool de ácidos biliares totais. Ambos os efeitos são benéficos. No caso das ressecções ileais mais amplas com esteatorreia proeminente, a colestiramina na verdade pode intensificar a diarreia e a má absorção, porque diminui a concentração aquosa de ácidos biliares na porção superior do intestino ao ser tomada antes das refeições. Quando isto ocorre, os ácidos graxos de cadeia intermediária são usados como substitutos dos ácidos graxos de cadeias longas. O resultado desta estratégia frequentemente deixa a desejar. A absorção da vitamina B12 também deve ser avaliada em todos os pacientes submetidos à ressecção ileal e, caso esteja anormal, requer a administração de vitamina B12 por via parenteral.
Alguns pacientes com doença de Crohn severa são submetidos a uma extensa ressecção intestinal, que resulta efetivamente no desenvolvimento da síndrome do intestino curto. De modo semelhante, os pacientes que possuem numerosas fístulas enteroentéricas também apresentam sintomas destas síndrome, pois as fístulas podem fazer o quimo se desviar de grandes seções de intestino delgado. Estes 2 tipos de pacientes devem ser tratados como se tivessem síndrome do intestino curto [ver Síndrome do intestino curto, anteriormente].
A síndrome de supercrescimento bacteria ocorre quando a estase intestinal cria uma oportunidade para as bactérias proliferarem localmente. Esta condição tem múltiplas causas, dentre as quais as mais proeminentes são o diabetes, escleroderma, diverticulose intestinal, alça aferente subsequente a uma gastrojejunostomia de Billroth II, e obstrução intestinal decorrente de estreitamentos, aderências ou câncer. Estes distúrbios podem estar presentes por vários anos, antes do desenvolvimento dos sintomas. Estes podem surgir em um paciente aparentemente estável, em consequência da administração de um inibidor de bomba de prótons (que diminui a produção de ácido gástrico e permite o supercrescimento bacteriano junto ao estômago e intestino delgado) ou de um opiáceo (que diminui ainda mais a motilidade intestinal).
A disfunção intestinal observada na síndrome da estase é provavelmente causada pelas glicosidases bacterianas, que hidrolisam os grupos carboidrato promotores da extensiva glicosilação encontrada nas proteínas da borda em escova apical.72 Apesar de os ácidos biliares serem desconjugados na síndrome da estase – e isto teoricamente poder levar ao comprometimento da solubilização dos produtos da hidrólise dos triglicerídeos – estudos mostraram que a concentração de ácidos graxos na fase aquosa do conteúdo intestinal pós-prandial na verdade é normal.73 A micrografias eletrônicas, porém, mostram que os enterócitos são danificados, com os lipídios absorvidos acumulando-se no retículo endoplasmático e sem seguirem normalmente para o aparelho de Golgi.73
Manifestações clínicas. Os sintomas da síndrome de supercrescimento bacteriano são semelhantes aos sintomas associados a outros estados de má absorção e incluem esteatorreia e anemia. O paciente pode ter deficiência de vitamina B12, a qual pode ter causas diversas, como a ligação da vitamina a bactérias,74,75 metabolização bacteriana da vitamina a metabólitos sem atividade metabólica e algum grau de internalização da vitamina B12 pelas bactérias para uso no próprio metabolismo bacteriano. Os níveis de ácido fólico geralmente elevados são secundários à produção bacteriana de folato.76 Os níveis séricos de albumina podem estar baixos em consequência de uma enteropatia perdedora de proteínas, permanecendo reduzidos por vários meses após o tratamento adequado do paciente. O diagnóstico geralmente é estabelecido em indivíduos que apresentam má absorção, em um contexto clínico consistente. A diverticulose intestinal (em geral, do jejuno) mais frequentemente permanece sem ser notada até que uma radiografia do intestino delgado é solicitada.
Exames laboratoriais. Estabelecer o diagnóstico de síndrome de supercrescimento bacteriano é complicado. Para tanto, a forma mais correta envolve a inserção de um tubo de aspiração dentro do intestino. O líquido deve ser quantitativamente cultivado, tanto de forma aeróbica como de forma anaeróbica. Na maioria dos casos, mais de 105 organismos anaeróbicos são encontrados. Como alternativa, pode ser empregado o teste de respiração do hidrogênio não invasivo. Para este teste, pode ser usado um alto nível de hidrogênio em repouso ou um aumento rápido dos níveis de hidrogênio na respiração em resposta a um substrato fermentável, como a glicose ou a lactulose. Outro teste respiratório é o teste da 1 g de (14C)-D-xilose, que quantifica os níveis de 14CO2 na respiração.
O tratamento de 1ª escolha para a síndrome da estase é a correção cirúrgica dos defeitos, como uma alça aferente que abriga bactérias ou uma fístula jejunocólica. Se esta opção cirúrgica estiver indisponível, como ocorre em casos de diverticulose do intestino delgado, então a dosagem recorrente de um antibiótico se torna necessária. Um curso de tetraciclina com doses de 1 a 2 g/dia administradas por 7 a 10 dias produz resultados satisfatórios. Como alternativa, pode ser usado outro antibiótico que seja ativo contra bactérias anaeróbicas (p. ex., 1 comprimido de potência dupla de trimetoprima-sulfametoxazol, 2 vezes/dia). O paciente terá de ser tratado novamente, caso os sintomas clínicos reapareçam, ou então poderá receber tratamento por 1 semana a cada mês.
O envolvimento intestinal ocorre com frequência em pacientes com amiloidose sistêmica, em especial naqueles com polineuropatia. Entre os pacientes com mais de 85 anos de idade, 36% apresentam envolvimento intestinal com amiloidose,77 embora a maioria seja assintomática. Ao exame de endoscopia, é possível observar erosão mucosa, friabilidade ou protrusões polipoides.78 O diagnóstico é estabelecido com base no exame de biópsias intestinais de espessura integral ou obtidas pela boca. Quando uma biópsia de intestino é obtida pela boca, sua profundidade deve ser suficiente para que as arteríolas estejam visíveis, de modo a possibilitar a demonstração de um amiloide eventualmente presente. As arteríolas coradas com vermelho do Congo que exibem uma tonalidade esverdeada sob luz polarizada confirmam o diagnóstico. O seguimento com radiografia do intestino delgado pode mostrar a existência de pregas intestinais inchadas, possivelmente com alças intestinais separadas. Se houver esteatorreia, esta pode ser resultante de supercrescimento bacteriano decorrente de desmotilidade intestinal ou comprometimento da absorção de ácidos biliares.79 Não há uma terapia específica disponível. Se houver supercrescimento bacteriano, então os antibióticos apropriados devem ser administrados.
Nesta rara condição, a pele (99% dos casos), os ossos (9%), fígado (12%), baço (11%), linfonodos e trato GI apresentam envolvimento com presença de mastócitos proliferantes. Diarreia, dor abdominal ou ambos (23% dos casos), úlceras pépticas (4%) e prurido com eritema (36%) podem ser observados. Cefaleia, fadiga e mal-estar são observados em 12% dos casos. Pode haver, ainda, disfunção cognitiva. A eosinofilia é observada em 12 a 50% dos casos.69 Muitas destas manifestações da doença são secundárias à ação da histamina liberada pelos mastócitos. A liberação de histamina pode ser precipitada por álcool, aspirina, narcóticos e AINH, acarretando distúrbios episódicos de eritema, diarreia, dor abdominal e hipotensão, que podem progredir para síncope.80
O excesso de histamina é excretado na urina em cerca de 75% dos pacientes, e isto torna o teste de excreção urinária útil para fins diagnósticos. A excreção urinária de um metabólito da prostaglandina D2 oriundo dos mastócitos pode ser um teste ainda mais útil.81 Radiografias do intestino delgado podem mostrar a existência de nodulação ou pregas espessadas. Estes achados não são diagnósticos, mas podem indicar a ocorrência de doença no intestino delgado.
A superprodução de ácido gástrico mediada pela histamina pode causar ulceração péptica. Quando isto ocorre, os inibidores de bomba de prótons são efetivos no controle dos sintomas. Na pele, a urticária pigmentosa pode ser tratada de maneira efetiva com antagonistas de receptor H1, como a difenidramina (25 mg a cada 6 a 8 horas). Se a diarreia persistir, é possível administrar 100 mg de cromolina de sódio por via oral, 4 vezes/dia.
A maioria dos casos de pancreatite crônica é causada por alcoolismo. A doença hereditária é rara. Os pacientes afetados sofrem perda de peso decorrente da má absorção dos alimentos. A má absorção produzida pela pancreatite é discutida em outro capítulo [ver 4:5 Doenças do pâncreas].
Uma das consequências da cirurgia gástrica é a esteatorreia, principalmente em pacientes submetidos à ressecção gástrica de Billroth II com gastrojejunostomia. Nesta operação, o antro e uma porção variável do corpo do estômago são extirpados, o estômago é fechado com suturas, e uma gastrojejunostomia é criada. Desta forma, os alimentos desviam do duodeno e da porção mais proximal do jejuno, que são os sítios de concentração máxima de células secretoras de secretina e CCK, bem como de absorção ativa de folato, cálcio e ferro. Cerca de metade dos pacientes submetidos ao procedimento de Billroth II desenvolvem esteatorreia (10 a 15 g de gordura/dia). Esta condição é considerada resultante da entrada dos alimentos no jejuno, na ausência de sítios hormônio-sensíveis no duodeno, que continua recebendo sinais apropriados para liberação de hormônio. Assim, a contração da vesícula biliar é fraca, e há diminuição da liberação de enzimas pancreáticas no intestino, com consequente mistura precária do quimo às enzimas pancreáticas e sais biliares. A alça aferente, que drena o duodeno e o jejuno proximal, pode estar bloqueada ou atônica e abrigar bactérias. O paciente pode desenvolver síndrome da estase, diante da presença de bactérias em número suficiente. Em consequência do estômago reduzido, os pacientes afetados não conseguem comer tanto quanto comiam no passado. Esta diminuição do consumo de alimentos, combinada à esteatorreia, faz que muitos pacientes submetidos ao procedimento de Billroth II mantenham um peso estável abaixo do que tinham antes da cirurgia. A osteopenia e a anemia ferropriva também são observadas. Pequenas perdas de sangue constantes a partir do sítio de osteotomia gástrica combinadas ao comprometimento da absorção de ferro contribuem para a condição de deficiência de ferro, que é a forma mais comum de anemia. A deficiência de folato secundária à incapacidade de gerar folato monoglutâmico absorvível a partir do folato heptaglutâmico não absorvível (a forma de folato comumente encontrada na dieta) também está presente.82 Uma condição menos comumente observada é a deficiência de vitamina B12 decorrente de hipocloridria e ressecção das células parietais gástricas contendo fator intrínseco. O tratamento da esteatorreia em geral é desnecessário, pois a condição é clinicamente insignificante. O ferro, o cálcio ou a vitamina B12 e o ácido fólico devem ser repostos, conforme a indicação. Para o paciente com saciedade precoce, uma medida eficaz pode ser o consumo de múltiplas refeições pequenas.
Os pacientes podem manifestar sintomas de EGS após a cirurgia gástrica.83 É provável que estes pacientes tenham EGS clinicamente silenciosa anterior à cirurgia. A operação em si produz uma esteatorreia modesta (10 a 15 g de gordura/dia) em 50% dos casos, até mesmo em pacientes com intestino aparentemente normal. Em um paciente com EGS previamente compensada, todavia, a cirurgia é um estímulo suficiente para acarretar a manifestação da sintomatologia clínica. Sendo assim, os pacientes pós-gastrectomizados que apresentam uma esteatorreia excessiva devem ser avaliados quanto à hipótese de EGS. A enteropatia inflamatória que se desenvolve nos pacientes após a gastrectomia também pode ser indicação da existência de uma EGS até então silenciosa.84
O autor não mantém relações comerciais com os fabricantes dos produtos ou prestadores de serviços mencionados neste capítulo.
1. Moreau H, Gargouri Y, Bernadal A, et al. Etude biochemique et physiologique des lipases préduodéales d’origines animale et humaine. Rev Fr Corps Gras 1988;35:169.
2. Roulet M, Weber A, Roy C. Perspectives in cystic f brosis. Toronto: Cystic Fibrosis Foundation; 1980.
3. Levitt MD, Duane WC. Floating stools: flatus versus fat. N Engl J Med 1972;286:973.
4. Fine KD, Ogunji F. A new method of quantitative fecal fat microscopy and its correlation with chemically measured fecal fat output. Am J Clin Pathol 2000;113:528.
5. DiMagno EP, Go VLW, Summerskill WHJ. Relation between pancreatic enzyme outputs and malabsorption in severe pancreatic insuff ciency. N Engl J Med 1973;288:813.
6. Chowdhury RS, Forsmark CE. Pancreatic function testing. Aliment Pharmacol Ther 2003;17:733.
7. Kato H, Nakao A, Kishimoto W, et al. 13C-labeled trioctanoin breath test for exocrine pancreatic function test in patients after pancreatoduodenectomy. Am J Gastroenterol 1993;88:64.
8. Lang C. Value of serum PABA as a pancreatic function test. Gut 1984;25:508.
9. Bando N, Ogawa T, Tsuji H. Enzymatic method for selective determination of 4 aminobenzoic acid in urine. Clin Chem 1990;36:1937.
10. Jacobson DG, Curlington C, Connery K, et al. Trypsin-like immunoreactivity as a test for pancreatic insufficiency. N Engl J Med 1984;310:1307.
11. Oelkers P, Kirby LC, Heubi JE, et al. Primary bile acid malabsorption caused by mutations in the ileal sodium-dependent bile acid transporter gene. J Clin Invest 1997; 99:1880.
12. Shindo K, Yamazaki R, Koide K, et al. Alteration of bile acid metabolism by cimetidine in healthy humans. J Investig Med 1996;44:462.
13. Nyhlin H, Merrick MV, Eastwood MA, et al. Evaluation of ileal function using 23-selena-25-homotaurocholate, a g-labeled conjugated bile acid. Gastroenterology 1983;84: 63.
14. Maki M, Collin P. Coeliac disease. Lancet 1997;349:1755.
15. Fasano A, Berti I, Gerarduzzi T, et al. Prevalenc of celiac disease in at-risk and not-at-risk groups in the United States: a large multicenter study. Arch Intern Med 2003;163:286.
16. Shan L, Molberg O, Parrot I, et al. Structural basis for gluten intolerance in celiac sprue. Science 2002;297:2218.
17. Hofmann AF. The function of bile salts in fat absorption. Biochem J 1963;89:57.
18. Mansbach CM II, Cohen RS, Leff PB. Isolation and properties of the mixed micelles present in intestinal content during fat digestion in man. J Clin Invest 1975;56:781.
19. Farrell RJ, Kelly CP. Celia sprue. N Engl J Med 2002; 346:180.
20. Vasquez H, Mazure R, Gonzalez D, et al. Risk of fractures in celiac disease patients: a cross-sectional, case-control study. Am J Gastroenterol 2000;95:183.
21. Dieterich W, Laag E, Schópper H, et al. Autoantibodies to tissue transglutaminase as predictors of coeliac disease. Gastroenterology 1998;115:1317.
22. Volta V, Molinaro N, deFranceschi L, et al. IgA antiendomysial antibodies on human umbilical cord tissue for celiac disease screening. Dig Dis Sci 1995;40:1902.
23. Holmes GKT, Prior P, Lane MR, et al. Malignancy in coeliac disease: effect of a gluten-free diet. Gut 1989;30:333.
24. Ryan BM, Kelleher D. Refractory celiac disease. Gastroenterology 2000;119:243.
25. Gawkrodger DJ, Vestey JP, O’Mahouny S. Dermatitis herpetiformis and established coeliac disease. Br J Dermatol 1993;129:694.
26. Rensch MJ, Merenich JA, Lieberman M, et al. Gluten-sensitive enteropathy in patients with insulin-dependent diabetes mellitus. Ann Intern Med 1996;124:564.
27. Holmes GK. Coeliac disease and type 1 diabetes mellitus, the case for screening. Diabet Med 2001;18:169.
28. Not T, Tommasini A, Tonini G, et al. Undiagnosed celiac disease and risk of autoimmune disorders in type 1 diabetes mellitus. Diabetologia 2001;44:151.
29. Ventura A, Magazzu G, Greco L. Duration of exposure to gluten and risk for autoimmune disorders in patients with celiac disease. SIGEP Study Group for Autoimmune Disorders in Celiac Disease. Gastroenterology 1999;117: 297.
30. Klipstein FA, Falaiye JM. Tropical sprue in expatriates from the tropics living in the continental United States. Medicine (Baltimore) 1969;48:475.
31. Lindenbaum J, Gerson CD, Kent TH. Recovery of small intestinal structure and function after residence in the tropics: I. Studies in Peace Corps volunteers. Ann Intern Med 1971;74:218.
32. Gerson CD, Kent TH, Saha JR, et al. Recovery of small intestinal structure and function after residence in the tropics: II. Studies in Indians and Pakistanis living in New York City. Ann Intern Med 1971;75:41.
33. Haghighi P, Wolf PL. Tropical sprue and subclinical enteropathy: a vision for the nineties. Crit Rev Clin Lab Sci 1997;34:313.
34. Robert ME, Ament ME, Weinstein WM. The histologic spectrum and clinical outcome of refractory and unclassified sprue. Am J Surg Pathol 2000;24:676.
35. McCashland TM, Donovan JP, Strobach RS, et al. Collagenous enterocolitis: a manifestation of gluten-sensitive enteropathy. J Clin Gastroenterol 1992;15:45.
36. Hermaszewski RA, Webster AD. Primary hypogammaglobulinaemia: a survey of clinical manifestations and complications. Q J Med 1993;86:31.
37. Ladefoged K, Hessov I, Jarnum S. Nutrition in short-bowel syndrome. Scand J Gastroenterol 1996;216 Suppl:122.
38. Lin HC, Zhao X-T, Wang L. Fat absorption is not complete by midgut but is dependent on load of fat. Am J Physiol 1996;271(1 Pt 1):G62.
39. Lin HC, Zhao X-T, Wong H. Fat-induced ileal brake in the dog depends on peptide YY. Gastroenterology 1996;110: 1491.
40. Redman DA, Schmidt TM, McDermott RP, et al. Identification of the uncultured bacillus of Whipple’s disease. N Engl J Med 1992;327:393.
41. Bentley SD, Maiwald M, Murphy LD, et al. Sequencing and analysis of the genome of the Whipple’s disease bacterium Tropheryma whipplei. Lancet 2003;361:637.
42. Durand DV, Lecomte C, Cathebras P, et al. Whipple disease: clinical review of 52 cases. The SNFMI Research Group on Whipple Disease. Medicine (Baltimore) 1997; 76:170.
43. Dobbins WO III. HLA antigens in Whipple’s disease. Arthritis Rheum 1987;30:102.
44. Mansbach CM II, Shelburne J, Stevens RD, et al. Lymph node bacilliform bodies morphologically resembling those of Whipple’s disease in a patient without intestinal involvement. Ann Intern Med 1978;89:64.
45. Kelly CA, Egan M, Rawlinson J. Whipple’s disease presenting with lung involvement. Thorax 1996;51:343.
46. Swartz MN. Whipple’s disease—past, present and future. N Engl J Med 2000;342:648.
47. von Herbay A, Ditton H-J, Schumacher F, et al. Whipple’s disease: staging and monitoring by cytology and polymerase chain reaction analysis of cerebrospinal fluid. Gastroenterology 1997;113:434.
48. Ramzan NN, Loftus E Jr, Burgart LJ, et al. Diagnosis and monitoring of Whipple disease by polymerase chain reaction. Ann Intern Med 1997;126:520.
49. Louis ED, Lynch T, Kaufmann P, et al. Diagnostic guidelines in central nervous system Whipple’s disease. J Ann Neurol 1996;40:561.
50. Feurle GE, Marth T. An evaluation of antimicrobial treatment for Whipple’s disease: tetracycline versus trimethoprim-sulfamethoxazole. Dig Dis Sci 1994;39:1642.
51. Shih LY, Liaw SJ, Dunn P, et al. Primary small-intestinal lymphomas in Taiwan: immunoproliferative small-intestinal disease and nonimmunoproliferative small-intestinal disease. J Clin Oncol 1994;12:1375.
52. Halphen M, Najjar T, Jaafoura H, et al. Diagnostic value of upper intestinal fiber endoscopy in primary small intestinal lymphoma: a prospective study by the Tunisian-French Intestinal Lymphoma Group. Cancer 1986;58:2140.
53. Khojasteh A, Haghighi P. Immunoproliferative small intestinal disease: portrait of a potentially preventable cancer from the Third World. Am J Med 1990;89:483.
54. Fox C, Lucani G. Disorders of the intestinal mesenteric lymphatic system. Lymphology 1993;26:61.
55. Aoyagi K, Iida M, Yao T, et al. Characteristic endoscopic features of intestinal lymphangiectasia: correlation with histological findings. Hepatogastroenterology 1997;44:133.
56. Aoyagi K, Iida M, Yao T, et al. Intestinal lymphangiectasia: value of double-contrast radiographic study. Clin Radiol 1994;49:814.
57. Sharp D, Blinderman L, Combs KA, et al. Cloning and gene defects in microsomal triglyceride transfer protein associated with abetalipoproteinaemia. Nature 1993;365:65.
58. Gordon DA, Jamil H, Gregg RE, et al. Inhibition of the microsomal triglyceride transfer protein blocks the step of apolipoprotein B lipoprotein assembly but not the addition of bulk core lipids in the second step. J Biol Chem 1996;271:33047.
59. Mansbach CM II, Dowell RF, Pritchett D. Portal transport of absorbed lipids in the rat. Am J Physiol 1991;261:G530.
60. Isselbacher KJ, Scheig R, Plotkin GR, et al. Congenital b-lipoprotein deficiency: an hereditary disorder involving a defect in the absorption and transport of lipids. Medicine (Baltimore) 1964;43:347.
61. Ways PO, Parmentier CM, Kayden HJ, et al. Studies on the absorptive defect for triglyceride in abetalipoproteinemia. J Clin Invest 1967;46:35.
62. Desreumaux P, Blogot F, Seguy D, et al. Interleukin 3, granulocyte-macrophage colony-stimulating factor, and interleukin 5 in eosinophilic gastroenteritis. Gastroenterology 1996;110:768.
63. Madhotra R. Eosinophilic gastroenteritis masquerading as ampullary adenoma. J Clin Gastroenterol 2002;34:240.
64. Klein NC, Hargrove RL, Sleisenger MH, et al. Eosinophilic gastroenteritis. Medicine (Baltimore) 1970;49:299.
65. Loftus EV Jr, Schoenfeld P, Sandborn WJ. The epidemiology and natural history of Crohn’s disease in population-based patient cohorts from North America: a systematic review. Aliment Pharmacol Ther 2002;16:51.
66. Sugimura K, Taylor KD, Lin YC, et al. A novel NOD2/ CARD15 haplotype conferring risk for Crohn disease in Ashkenazi Jews. Am J Hum Genet 2003;72:509.
67. Van Deest BW, Fordtran JS, Morawski SG, et al. Bile salt and micellar fat concentration in proximal small bowel contents of ileectomy patients. J Clin Invest 1968;47:1314.
68. Low-Beer TS, Wilkins RM, Lack L, et al. Effect of one meal on enterohepatic circulation of bile salts. Gastroenterology 1974;67:490.
69. Merhjian HS, Phillips SF, Hofmann AF. Colonic secretion of water and electrolytes induced by bile acids: perfusion studies in man. J Clin Invest 1971;50:1569.
70. Mansbach CM II, Newton DF, Stevens RD. Fat digestion in patients with bile acid malabsorption but minimal steatorrhea. Dig Dis Sci 1980;25:353.
71. Hofmann AF, Poley JR. Role of bile acid malabsorption in pathogenesis of diarrhea and steatorrhea in patients with ileal resection: I. Response to cholestyramine or replacement of dietary long chain triglyceride by medium chain triglyceride. Gastroenterology 1972;62:918.
72. Riepe S, Goldstein J, Alpers DH. Effect of secreted Bacteroides proteases on human intestinal brush border hydrolases. J Clin Invest 1980;66:314.
73. Ament ME, Shimoda SS, Saunders DR, et al. Pathogenesis of steatorrhea in three cases of small intestinal stasis syndrome. Gastroenterology 1972;63:728.
74. Gianella RA, Broitman SA, Zamcheck N. Vitamin B12 uptake by intestinal microorganisms: mechanisms and relevance to syndromes of bacterial overgrowth. J Clin Invest 1971; 50:1100.
75. Gregg CR. Enteric bacterial flora and bacterial overgrowth syndrome. Semin Gastrointest Dis 2002;13:2000.
76. Hoffbrand AV, Tabaqchali S, Booth CC, et al. Small intestinal bacterial flora and folate status in gastrointestinal disease. Gut 1971;12:27.
77. Rocken C, Saeger W, Linke RP. Gastrointestinal amyloid deposits in old age: report of 110 consecutive autopsical patients and 98 retrospective bioptic specimens. Pathol Res Pract 1994;190:641.
78. Tada S, Iida M, Yao KK, et al. Endoscopic features in amyloidosis of the small intestine: clinical and morphologic differences between chemical types of amyloid protein. Gastrointest Endosc 1994;40:45.
79. Suhr O, Danielsson A, Steen L. Bile acid malabsorption caused by gastrointestinal motility dysfunction? An investigation of gastrointestinal disturbances in familial amyloidosis with polyneuropathy. Scand J Gastroenterol 1992;27:201.
80. Golkar L, Bernhard JD. Mastocytosis. Lancet 1997;349:1379.
81. Morrow JD, Guzzo C, Lazarus G, et al. Improved diagnosis of mastocytosis by measurements of the major urinary metabolite of prostaglandin D2. J Invest Dermatol 1995; 104:937.
82. Rosenberg IH. Folate absorption and malabsorption. N Engl J Med 1975;293:1303.
83. Bai J, Moran C, Martinez C. Celiac sprue after surgery of the upper gastrointestinal tract: report of 10 patients with special attention to diagnosis, clinical behavior, and follow-up. J Clin Gastroenterol 1991;13:521.
84. Kitis G, Holmes GTK, Cooper BT. Association of coeliac disease and inflammatory bowel disease. Gut 1980;21:636.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.