(Carregando Índice)... (Carregando Índice)... |
Autores:
Fabrício de Souza Neves
Médico Especialista em Reumatologia pelo Serviço de Reumatologia do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo – HC-FMUSP. Doutor em Reumatologia pela Disciplina de Reumatologia da Faculdade de Medicina da Universidade de São Paulo – FMUSP.
Célio Roberto Gonçalves
Professor Colaborador da Disciplina de Reumatologia da Faculdade de Medicina da Universidade de São Paulo – FMUSP. Médico Assistente Doutor do Serviço de Reumatologia do Hospital das Clínicas da FMUSP.
Última revisão: 11/01/2010
Comentários de assinantes: 0
A doença de Behçet (DB) é uma afecção inflamatória multissistêmica de natureza autoimune caracterizada por lesões vasculíticas que se manifestam basicamente por meio de úlceras orais recorrentes, úlceras genitais, lesões cutâneas, artrite e uveíte, além de um estado de hipercoagulabilidade responsável pela formação de trombos venosos e arteriais. É descrita em todos os continentes, sendo uma condição rara na maioria dos países. Possui, no entanto, uma distribuição geográfica peculiar, concentrando-se em regiões de alta prevalência na faixa que se estende da bacia do Mediterrâneo ao extremo Oriente asiático. Essa faixa corresponde à antiga rota comercial e migratória que unia a Ásia à Europa antes da descoberta do caminho marítimo para as Índias; disto deriva uma das denominações da DB: a “doença da rota da seda”. Na Turquia, encontra-se o maior número de casos (alcançando a prevalência de 370 por 100 mil habitantes, em algumas regiões). As prevalências são intermediárias em outros países da rota da seda (
A DB acomete indivíduos jovens (a idade média de início da doença é entre 25 e 30 anos), com incidência semelhante entre os sexos. Não se trata de doença com atividade inflamatória crônica persistente; ela cursa, mais frequentemente, com surtos de ativação intercalados por períodos mais ou menos longos de remissão. Apesar da maior parte de suas manifestações serem de caráter benigno, surtos repetidos de inflamação ocular podem levar à amaurose, que representa a principal morbidade da DB.
A curiosa distribuição geográfica da DB, acompanhando grupos étnicos e correntes migratórias, sempre sugeriu sua associação com fatores genéticos predisponentes. Em 1982, no Japão, foi descrita pela primeira vez a associação entre a DB e o alelo HLA B-51 - um dos diversos alelos responsáveis pela codificação do MCH classe I, que é responsável pela apresentação de antígenos intracelulares aos linfócitos T. Naquele país, este alelo é encontrado em 55% dos portadores da DB (a associação é menos significativa nos países ocidentais).
Quanto à fisiopatologia, uma das características mais marcantes da DB é a hiper-reatividade neutrofílica: suas lesões vasculíticas caracterizam-se por um infiltrado rico em neutrófilos (em diferentes estágios de ativação) e a quimiotaxia exacerbada dos neutrófilos da DB já havia sido descrita por investigadores também japoneses em 1975. Interessantemente, camundongos transgênicos portadores do HLA-B51 demonstraram hiper-reatividade neutrofílica (com relação ao estresse oxidativo), mas nenhuma manifestação clínica semelhante à DB.
Por outro lado, a história natural característica da DB (evoluindo em surtos recorrentes de atividade inflamatória) sugere a associação da DB com a exposição a fatores ambientais desencadeadores. Vários agentes microbianos demonstraram evidências de atuar neste processo, mas o maior destaque cabe às bactérias do gênero Streptococcus. Sorotipos incomuns de Streptococcus são encontrados na flora oral destes pacientes, sendo que este gênero de bactérias Gram positivas expressa a proteína de choque térmico HSP (Heat Shock Protein) 65 kDa. Em um importante experimento em modelo animal, a administração do peptídeo p336-351 derivado da HSP 65 kDa por via oral a ratos Lewis induziu a ocorrência de uveíte. Em pacientes com DB, tais peptídeos são capazes de estimular especificamente clones de linfócitos T.
Interessantemente, já se descreveu também a associação da DB com alelos do grupo MICA (MHC class I related gene A, gene A relacionado ao MHC classe I). Estes genes são transcritos por células humanas em situações de hipertermia, anóxia ou metabólitos tóxicos, gerando produtos do grupo das proteínas de choque térmico (HSP). A HSP humana de 60 kDa, em particular, guarda importante homologia com a HSP 65 kDa bacteriana, e é expressa em quantidade aumentada em células epiteliais humanas das lesões cutâneas da DB.
Este conjunto de informações permitiu a Lehner elaborar um modelo imunopatogenético da DB: a reação imune cruzada contra antígenos semelhantes, expressos por agentes microbianos e pelas células de indivíduos geneticamente predispostos (quando em situações de estresse infeccioso), seria a responsável por orquestrar a resposta inflamatória característica da DB. Esta resposta é caracterizada, além da hiper-reatividade neutrofílica, por elevação de citocinas promotoras de atividade Th-1 (IFNg, IL-2 e IL-12) e a presença de células NK (natural killer), sugerindo a presença predominante de um padrão de resposta imune do tipo “celular”. As citocinas pró-inflamatórias IL-1 e TNFa também são encontradas com produção aumentada.
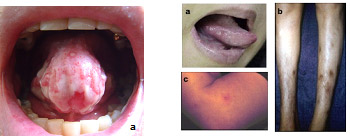
As úlceras aftosas orais recorrentes são as manifestações iniciais em até 86% dos casos, virtualmente afetando todos os pacientes ao longo da evolução da doença. Em geral, precedem as demais manifestações da DB em alguns anos; são lesões dolorosas, de fundo branco, com bordas hiperemiadas e bem definidas, medindo até
As lesões cutâneas características (80% dos casos) são o eritema nodoso (nódulo hiperemiado, doloroso, profundo, mais comum na face anterior das pernas) (Figura 1B) e a pseudofoliculite (lesões acneiformes, pustulares, em localizações não típicas de acne vulgar). Os pacientes com DB podem apresentar um quadro de hiper-reatividade cutânea responsável pelo aparecimento de pseudofoliculite associada aos atos de barbear ou depilar. Esta hiper-reatividade também é a base do teste da patergia: introduz-se obliquamente uma agulha estéril de calibre grosso (20 Gauge) na pele do antebraço até o subcutâneo. O aparecimento de um nódulo eritematoso ou pustular (não apenas hiperemia ou equimose) após
Figura 1: Manifestações clínicas da doença de Behçet. A) Úlcera aftosa oral. B) Eritema nodoso. C) Lesão pustular resultante do teste de patergia.

Artralgias e artrites (50% dos casos) são geralmente transitórias e não destrutivas, sendo mais comuns em grandes articulações dos membros inferiores.
O acometimento ocular (50% dos casos) mais característico é a uveíte, que pode ser anterior (iridociclite, associada a dor e hiperemia ocular) ou posterior (coriorretinite, que pode ser indolor e associada apenas a turvação visual). Panuveíte também pode ocorrer. Surtos repetidos de uveíte podem levar a deformidade da íris (anterior) ou oclusão vasculítica de vasos da retina (posterior), sendo frequente, no segundo caso, a perda da acuidade visual.
O comprometimento de grandes vasos (25% dos casos), venosos ou arteriais, ocorre em consequência de vaculite de vasa vasorum e do estado de hipercoagulabilidade associado à atividade inflamatória, culminando em tromboses (venosas ou arteriais, em vasos de qualquer calibre) e na formação de aneurismas arteriais. A DB não poupa a circulação pulmonar e o aneurisma de artéria pulmonar é uma complicação com elevada letalidade.
O sistema nervoso central (SNC, acometido em até 25% dos casos) pode ser afetado por tromboses dos seios venosos encefálicos (levando a cefaleia e sinais de hipertensão intracraniana), mas também de forma intraparenquimatosa com déficits neurológicos localizados associados à vasculite de pequenos vasos cerebrais ou do tronco. Meningite asséptica também ocorre e, às vezes, é manifestação inicial da DB.
Além da cavidade oral, o trato gastrintestinal (TGI) pode ser acometido (em até 25% dos casos) por úlceras aftosas ao longo de toda a extensão do tubo digestivo, causando dor abdominal, hemorragia digestiva, diarreia ou perfuração. No trato geniturinário, orquiepididimite é a manifestação mais comum (8% dos casos) e, ocasionalmente, também é manifestação de abertura da doença.
O diagnóstico da DB é exclusivamente clínico. Em 1990, foram publicados os critérios diagnósticos do International Study Group for Behçet’s Disease, que logo se converteram em padrão para estudos clínicos mundialmente (Tabela 1). Porém, embora tenham sido publicados sob a denominação “critérios diagnósticos”, os próprios autores destacaram que a maior utilidade do conjunto de critérios é assegurar a uniformidade de pacientes incluídos em estudos, e não o diagnóstico do caso individual (um paciente com úlceras orais e eritema nodoso recorrentes que desenvolve trombose venosa na ausência de um estado pró-trombótico identificável, p. ex., provavelmente tem a melhor conduta ao ser diagnosticado com DB, embora sem preencher os critérios necessários, e acompanhado seguidamente para o surgimento de outros sinais ou sintomas característicos da doença – em particular, o acometimento ocular).
Tabela 1: Critérios de classificação da doença de Behçet
|
1. Ulcerações orais recorrentes (ao menos 3 vezes num período de 12 meses) |
|
2. Ulceração genital recorrente |
|
3. Lesões oculares (uveíte anterior, uveíte posterior, células no vítreo ou vasculite retiniana) |
|
4. Lesões cutâneas (eritema nodoso, pseudofoliculite, lesões papulopustulares ou acneiformes) |
|
5. Teste de patergia positivo |
Um paciente pode ser classificado como portador da DB quando o critério 1 está presente associado a quaisquer dois dos demais critérios, não necessariamente de forma simultânea.
Também não existem marcadores laboratoriais associados à atividade da DB em toda a sua gama de possíveis manifestações. A atividade da DB pode ser melhor avaliada clinicamente por meio da aplicação de um questionário padronizado denominado Behçet’s Disease Current Activity Form (BDCAF), recentemente validado para uso no Brasil.
As manifestações mais frequentes da DB são avaliadas apenas clinicamente: úlceras orais, genitais, lesões cutâneas e articulares são contempladas pela anamnese e pelo exame físico, e a atividade mucocutânea e articular da DB pode ser quantificada pelo questionário BDCAF. Provas tradicionais de atividade inflamatória (particularmente a PCR) podem estar elevadas durante os períodos de exacerbação em algumas manifestações específicas (como nas artrites), mas não em outras (como nas manifestações mucocutâneas). A avaliação de sistemas orgânicos viscerais apenas em bases clínicas, no entanto, não demonstra suficiente confiabilidade e exige a realização de avaliações complementares. A avaliação oftalmológica para a pesquisa de uveíte/vasculite retiniana, p. ex., é mandatória no momento da suspeita diagnóstica da DB e deve ser repetida, ao menos, sempre que sinais ou sintomas sugerirem a possibilidade de novo acometimento ocular. Endoscopia digestiva alta e colonoscopia são indicados na presença de sintomas gastrintestinais. Exames de imagem do encéfalo (preferencialmente a ressonância nuclear magnética), acompanhados de avaliação liquórica, estão indicados na suspeita de acometimento do SNC. Angiotomografia ou angiorressonância são os exames indicados para a avaliação de grandes vasos torácicos e abdominais; a ultrassonografia com Doppler pode ser suficiente para os vasos periféricos. É interessante lembrar que a elevação da PCR, na ausência de atividade da doença clinicamente evidente, pode sugerir inflamação em grandes vasos profundos, cursando com pouca sintomatologia.
Pacientes com úlceras orais e genitais recorrentes, mas que não desenvolvem alterações em outros sistemas orgânicos mesmo após vários anos de evolução, são portadores de aftose complexa. Ao menos parte dos pacientes com DB iniciam sua história desta forma, e, portanto, o diagnóstico diferencial nas fases iniciais da doença é virtualmente impossível. Eventualmente, a biópsia de uma lesão evidenciando vasculite com infiltrado neutrofílico pode indicar o diagnóstico da DB de forma precoce, mas a ausência deste achado não exclui de todo essa possibilidade. Desse modo, muitos autores recomendam, como melhor conduta, o acompanhamento periódico de pacientes com aftose complexa a fim de detectar precocemente possíveis manifestações da DB, particularmente oftalmológicas.
Duas entidades relacionadas ao grupo das espondiloartropatias compartilham muitos achados com a DB: a artrite reativa, que comumente cursa com úlceras orais, lesões cutâneas e genitais (psoriasiformes, diferentes das lesões aftosas da DB), inflamação ocular (tipicamente conjuntivite) e oligoartrite de membros inferiores; e a doença inflamatória intestinal, que além de alterações digestivas (também comuns na DB) pode cursar com uveíte, artrite e lesões cutâneas (do tipo pioderma gangrenoso, também descrito na DB). O achado de sacroiliite e a presença do HLA-B27 favorecem o diagnóstico de espondiloartropatia, mas suas ausências não o excluem de todo. Na ausência de marcadores patognomônicos, muitas vezes é a relativa predominância de achados clínicos mais característicos de uma entidade em particular que define o melhor diagnóstico.
Outra situação particular é o diagnóstico da DB feito a partir da manifestação de trombose e aneurismas arteriais, com manifestações apenas frustras em outros sistemas. Nesses casos em que predomina o componente vascular da DB, o quadro pode ser semelhante à arterite de Takayasu (que também cursa com retinite). O acometimento do leito venoso sistêmico e a presença de lesões mucosas (ainda que brandas) podem reforçar a possibilidade de DB.
O tratamento da DB deve ser direcionado às manifestações presentes em cada caso individual.
Preparados com corticosteroides em base emoliente para uso tópico podem ser usados no tratamento das úlceras orais. Algumas apresentações comerciais de corticosteroides tópicos têm associações com anestésicos locais que melhoram o controle sintomático da dor. Parar de fumar e tratar adequadamente afecções dentárias, periodontites e gengivites são medidas provavelmente benéficas a estes pacientes. Para reduzir a intensidade e a frequência dos episódios de exacerbação, a primeira escolha é a colchicina por via oral, iniciando com 0,5 mg/dia, podendo ser aumentada a dose até o melhor controle dos sintomas ou surgimento de efeitos colaterais. Talidomida (evitar em mulheres), dapsona e pentoxifilina podem ser usadas com a mesma finalidade, como drogas de segunda linha ou em associação à colchicina. O uso da penicilina benzatina como profilaxia de infecções estreptocócicas (um dos principais suspeitos como agente desencadeador de surtos da DB) revelou-se eficaz na redução da frequência de episódios de exacerbação quando associado à colchicina.
Artralgias e artrites, normalmente transitórias (acompanhando os períodos de exacerbação), não têm potencial destrutivo. São em geral adequadamente controladas com antiinflamatórios não-hormonais, e a frequência dos episódios pode ser diminuída pelos mesmos agentes sugeridos para o controle das exacerbações mucocutâneas (inclusive a penicilina benzatina). Se a intensidade ou a frequência dos episódios articulares justificarem seu uso, drogas anti-reumáticas tradicionais podem ser utilizadas (sulfassalazina e metotrexato são os agentes de primeira escolha). Porém, nos casos de artrite persistente, recomenda-se reavaliar o diagnóstico diferencial (P. ex.: considerar avaliação intestinal para pesquisa de doença inflamatória intestinal; avaliação radiológica das articulações sacroilíacas e pesquisa do HLA-B27 para investigação de espondiloartropatia).
Os casos de DB com acometimento ocular devem ser acompanhados por oftalmologista com regularidade. Episódios agudos de uveíte anterior podem ser manejados com colírios de corticosteroides e midriáticos, enquanto na uveíte posterior está indicado o uso de corticosteroides sistêmicos (prednisona 0,5 mg/kg/dia), tendo como opção a injeção subcapsular de corticosteroide. Além do tratamento do episódio agudo, devido ao risco de perda visual com surtos sucessivos de inflamação ocular, está indicado o uso de imunossupressores, para redução da frequência e intensidade das exacerbações (o risco de perda da visão útil é estimado em 30% no sexo masculino e 17% no sexo feminino, nos casos de DB ocular). Azatioprina e ciclosporina são os agentes de primeira escolha, por terem maior respaldo na literatura. Azatioprina demonstrou reduzir efetivamente o número de episódios de inflamação ocular em ensaio clínico randomizado, duplo-cego, placebo-controlado (além de reduzir a ocorrência de manifestações mucocutâneas e articulares). Além disso, demonstrou melhorar o prognóstico visual em longo prazo: quando reavaliados após 7 anos, o grupo tratado com azatioprina tinha melhor função ocular que o grupo controle. A ciclosporina também demonstrou eficácia em ensaio controlado, enquanto a colchicina de forma isolada não pareceu ser eficaz na prevenção de ataques de inflamação ocular. Dentre os agentes biológicos, existem relatos e séries de casos demonstrando a eficácia do intererferon-a (possivelmente, por sua ação reguladora na resposta imune celular) e do infliximabe (pelo bloqueio do TNFa, citocina importante na resposta inflamatória efetora) no controle da inflamação ocular (além das manifestações mucocutâneas e articulares). São opções nos casos refratários aos imunossupressores tradicionais.
Acometimento vascular ativo, com formação de aneurismas, deve ser tratado com altas doses de corticosteroides (preferencialmente em pulsoterapia) e agentes imunossupressores (por extrapolação de outras síndromes vasculíticas, dá-se preferência à ciclofosfamida, também em pulsos). Na existência de tromboses, anticoagulação com heparina seguida por cumarínicos via oral está indicada. Após a fase aguda, a necessidade de manter a anticoagulação deve ser avaliada a cada caso individualmente: uma vez que a hipercoagulabilidade está associada à atividade da doença, pacientes que mantêm episódios de exacerbação, apesar da terapia imunossupressora, talvez possam se beneficiar com a manutenção da anticoagulação. Intervenção cirúrgica pode ser necessária para a correção definitiva de aneurismas com risco de ruptura. De forma semelhante (imunossupressão e anticoagulação), são tratadas as tromboses de seios venosos encefálicos. Também por extrapolação de outras síndromes, a vasculite do SNC é geralmente tratada com pulsos de ciclofosfamida e corticosteroides em altas doses, enquanto a inflamação do TGI é tratada com sulfassalazina e corticosteroides por via oral.
A doença de Behçet é uma entidade inflamatória que pode acometer múltiplos sistemas orgânicos.
A maioria dos pacientes tem um curso benigno da doença, mas que pode comprometer de forma importante sua qualidade de vida pelo aparecimento recorrente de lesões dolorosas orais e genitais.
Estabelecido o diagnóstico, drogas de uso comum como colchicina ou pentoxifilina podem ser suficientes no controle desses casos. Porém, em todos os casos (suspeitos ou confirmados) de DB, a avaliação oftalmológica é imprescindível, pois a importante frequência de acometimento ocular (às vezes com poucos sintomas) é a principal morbidade em médio e longo prazo, e a perda visual pode ser evitada com a imunossupressão de forma adequada.
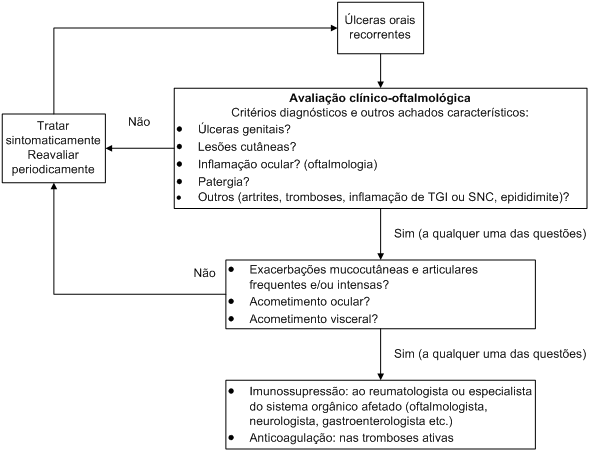
Agentes imunossupressores também podem ser indicados se a frequência ou gravidade de outras manifestações da doença justificarem seu uso e, nesses casos, o acompanhamento com reumatologista habituado no manejo de doenças autoimunes é recomendado. Particularmente, nos casos de acometimento visceral, a abordagem multidisciplinar entre diferentes especialidades pode ser necessária (Figura 2).
Figura 2: Fluxograma para condução de um caso sugestivo típico de doença de Behçet.
1. Calguneri M, et al. Effect of prophylatic benzathine penicillin on mucocutaneous symptoms of Behçet’s disease. Dermatology. 1996;192:125-128.
2. International Society of Behçet’s Disease (ISBD). Disponível em: www.behcet.ws.
3. International Study Group for Behçet’s Disease. Criteria for diagnosis of Behçet’s disease. Lancet. 1990;335:1078-1080.
4. Neves FS, et al. Síndrome de Behçet: à procura de evidências. Rev Bras Reumatol. 2006; 46(Supl 1):21-29.
5. Neves FS, et al. Cross-cultural adaptation of the Behçet’s Disease Current Activity Form (BDCAF) to Brazilian Portuguese language. Clin Rheumatol. 2007;26:1263-1267.
6. Sakane T, et al. Behçet’s disease. N Engl J Med. 1999;21:1248-1291.
7. Yazici H, et al. A controlled trial of azathioprine in Behçet’s syndrome. N Engl J Med. 1990;322:281-285.
8. Yurdakul S, et al. Behçet syndrome. Curr Opin Rheumatol. 2004;16:38-42.
9. Yurdakul S, et al. A double-blind trial of colchicine in Behçet’s syndrome. Arthritis Rheum. 2001;44:2686-2692.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.