(Carregando Índice)... (Carregando Índice)... |
Você está em:
Inicial  acp-medicine Dermatologia
acp-medicine Dermatologia
Última revisão: 25/04/2012
Comentários de assinantes: 2
Mark Lebwohl, M.D.
Sol and Clara Kest Professor and Chairman, Department of Dermatology, Mount Sinai Medical Center
Artigo original: Lebwohl M. Cutaneous manifestations os systemic diseases. ACP Medicine. 2008;1-15.
[The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2011 Decker Intellectual Properties Inc. All Rights Reserved.]
Tradução: Soraya Imon de Oliveira
Revisão técnica: Dr. Euclides Furtado de Albuquerque Cavalcanti
As manifestações cutâneas das doenças sistêmicas são tão numerosas e variadas que um único capítulo seria insuficiente para abranger todas elas, mesmo de uma forma superficial. Em vez de fazer isso, o presente capítulo revisa as manifestações cutâneas mais importantes das doenças sistêmicas, a ser identificadas pela maioria dos médicos, e destaca os últimos avanços ocorridos no diagnóstico e na supervisão dessas patologias. Para ter acesso a discussões mais aprofundadas sobre doenças específicas, incluindo suas respectivas manifestações cutâneas, os leitores devem consultar os capítulos dedicados a cada uma dessas condições.
Em muitos dos distúrbios apresentados neste capítulo, um resultado favorável depende essencialmente do exame minucioso e do tratamento da condição sistêmica subjacente. Um achado de sarcoidose cutânea, por exemplo, pode mobilizar uma investigação para detecção de sarcoidose sistêmica. Em outras condições (p. ex., epidermólise bolhosa distrófica recessiva), o tratamento do distúrbio que afeta a pele é fundamental à supervisão da doença sistêmica.
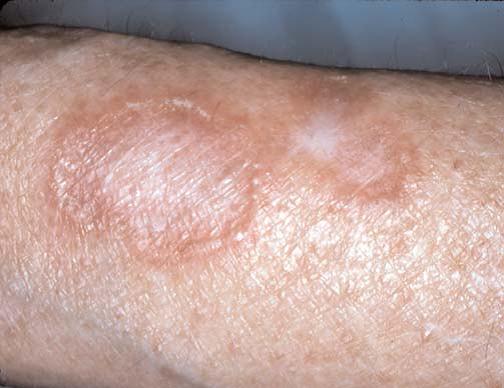
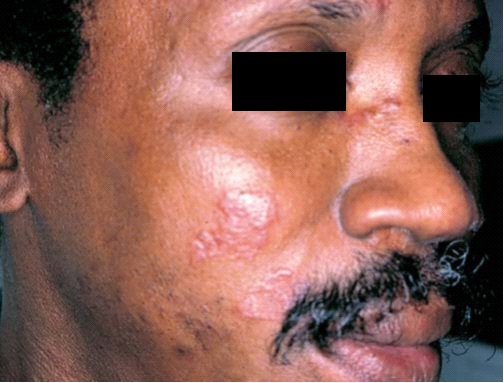
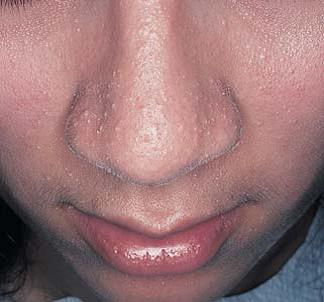
As manifestações cutâneas da sarcoidose são tão variadas quanto suas manifestações sistêmicas [ver Doença pulmonar infiltrativa difusa crônica]. As lesões que surgem na pele podem ser manchas, pápulas, nódulos ou placas; podem ser hipo ou hiperpigmentadas, eritematosas ou anulares [Figura 1]. Pápulas em torno dos olhos ou do nariz são mais características. O termo lúpus pérnio se refere aos granulomas não caseosos que resultam na formação de placas translúcidas e violáceas nas orelhas, bochechas e nariz [Figura 2]. Pode haver envolvimento de ossos subjacentes. O diagnóstico é feito por meio de biópsia de pele. O tratamento tradicional é à base de corticosteroides intralesionais, sendo que os agentes antimaláricos e o metotrexato também se mostraram úteis. Mais recentemente, o infliximabe foi utilizado com sucesso no tratamento da sarcoidose.1 Outros bloqueadores de fator de necrose tumoral alfa (TNF-alfa), como o etanercepte, foram empregados no tratamento da artrite e das lesões cutâneas associadas à sarcoidose.2 O infliximabe também foi utilizado no tratamento do lúpus pérnio.3

Figura 1. Pacientes com lesões sarcoides podem apresentar lesões anulares eritematosas na pele.
Figura 2. Lesões faciais características da sarcoidose, denominadas lúpus pérnio.
Enquanto o interferon tem sido cada mais utilizado no tratamento da hepatite C e da esclerose múltipla, há relatos de casos de sarcoidose atribuídos a esse tratamento. Foi demonstrado, nesses casos, que a terapia com infliximabe afeta a resposta.4
Alguns pacientes com sarcoidose apresentam eritema nodoso nos membros inferiores, caracterizado pelo aparecimento de nódulos eritematosos sensíveis e profundos. O lúpus pérnio está associado a um curso mais crônico da sarcoidose, enquanto o eritema nodoso indica uma doença mais aguda e benigna.5
A granulomatose de Wegener está associada a sinais mucocutâneos que podem ser distintivos ou inespecíficos. A púrpura palpável é um dos achados cutâneos mais comuns, porém úlceras, pápulas, nódulos e bolhas também já foram descritos. Além dos sintomas envolvendo os tratos respiratórios superior e inferior [ver Doença pulmonar focal e multifocal], uma deformidade do nariz em sela, ulcerações nasais e perfuração septal são sugestivas do diagnóstico de granulomatose de Wegener. O diagnóstico definitivo é estabelecido pela demonstração de uma vasculite granulomatosa necrotizante em paciente apresentando doença nos tratos respiratórios superior e inferior e glomerulonefrite. Autoanticorpos anticitoplasma de neutrófilos citoplasmáticos (c-ANCA) são detectados com frequência. A terapia padrão consiste na administração de ciclofosfamida e corticosteroides. Os bloqueadores de TNF-alfa se mostraram efetivos no tratamento de alguns pacientes com granulomatose de Wegener refratária.6 Entretanto, a terapia à base de etanercepte combinada à ciclofosfamida pode estar associada ao aumento da incidência de malignidades entre os pacientes com granulomatose de Wegener.7 Consequentemente, ainda é controverso o uso de bloqueadores de TNF-alfa no tratamento dessa doença.8
A síndrome de Churg-Strauss, ou angiite granulomatosa alérgica, manifesta-se mais comumente como asma e eosinofilia. Entretanto, cerca de 40% dos pacientes desenvolvem lesões cutâneas associadas. Os achados mais comuns são petéquias e púrpura palpável simétrica nos membros inferiores. O exame da biópsia de pele revela a ocorrência de uma vasculite leucocitoclástica. Nódulos cutâneos são produzidos por granulomas necrotizantes extravasculares e também surgem pápulas nos cotovelos.9 Um dos indícios que levam ao diagnóstico desse distúrbio é a presença de anticorpos anticitoplasma de neutrófilos perinucleares (p-ANCA).10
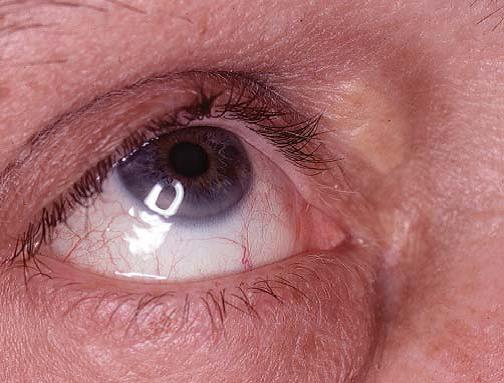
Xantomas são as manifestações cutâneas das hiperlipoproteinemias. Diversos tipos de xantomas são acompanhados por diferentes anomalias lipídicas. Os xantelasmas das pálpebras [Figura 3] constituem as manifestações mais comuns da hipercolesterolemia familiar. Entretanto, no mínimo 50% dos pacientes com lesões nas pálpebras apresentam níveis plasmáticos de lipídeos normais. Os xantomas planares são placas achatadas e amarelas que envolvem as palmas das mãos, plantas dos pés, pescoço e tórax. Esses xantomas podem ocorrer em pacientes com cirrose biliar primária ou mieloma múltiplo. Os xantomas tuberosos são grandes nódulos amarelos ou vermelhos, que aparecem nas superfícies extensoras das articulações (p. ex., cotovelos e mãos), sem se fixarem nos tendões subjacentes. Esses nódulos podem aparecer em pacientes com níveis elevados de triglicerídeos ou colesterol. Em contraste, os xantomas tendíneos, que podem surgir em pacientes com hipercolesterolemia familiar, se fixam aos tendões subjacentes dos cotovelos, tornozelos, joelhos e mãos. Os xantomas eruptivos se desenvolvem quando os níveis plasmáticos de triglicerídeos aumentam de repente. As lesões cutâneas consistem em pequenas pápulas amarelas que muitas vezes se resolvem com a diminuição dos níveis de triglicerídeos.
Figura 3. Xantelasma e arco senil em paciente com hipercolesterolemia.
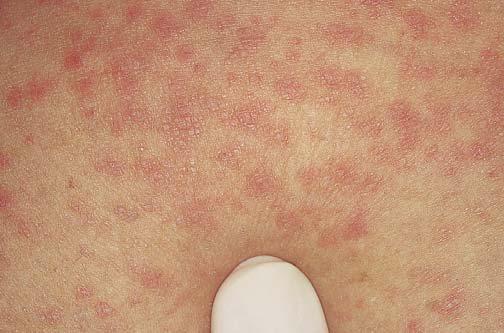
A doença de Kawasaki, também denominada síndrome do linfonodo mucocutâneo [ver Síndromes de vasculite sistêmica], é um distúrbio que acomete crianças, cujas possíveis complicações são a oclusão da artéria coronária e o infarto do miocárdio, aneurismas da artéria coronária, anormalidades eletrocardiográficas, arritmias cardíacas ou miocardite.11 Foi sugerido que uma toxina secretada por Staphylococcus aureus é responsável pela doença, contudo as evidências que comprovam sua causa precisa continuam sendo vagas.12 O diagnóstico é estabelecido com base em critérios clínicos, incluindo febre, conjuntivite, linfadenopatia e erupções cutâneas. Além das erupções cutâneas eritematosas generalizadas, o paciente pode desenvolver anomalias na mucosa oral, bem como inchaço e eritema nas mãos e nos pés. Por fim, ocorre uma notável descamação das palmas das mãos e plantas dos pés. Outras manifestações comuns são escamação e eritema nas regiões perianal e escrotal. A trombocitose constitui um achado tardio, em que as contagens de plaquetas sobem para mais de um milhão no decorrer das 2 semanas subsequentes ao início da doença. Cerca de 15 a 25% das crianças não tratadas desenvolvem aneurismas na artéria coronária, podendo morrer subitamente.13 O tratamento com administração endovenosa de imunoglobulinas reduz a frequência das anormalidades associadas à artéria coronária.14
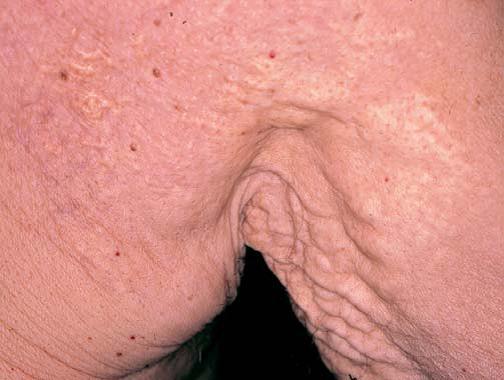
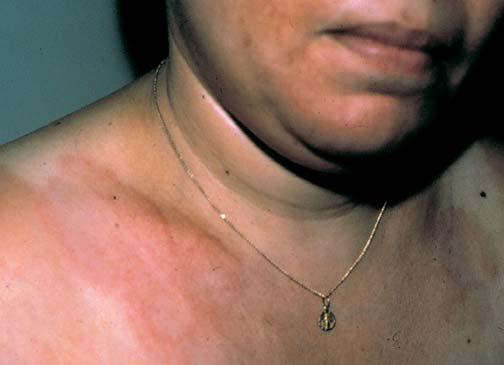
O pseudoxantoma elástico (PXE) é um distúrbio autossômico de herança recessiva, que envolve o tecido elástico e é causado por mutações associadas à proteína transportadora ABCC6.15 O PXE está associado a uma ampla variedade de manifestações sistêmicas. As estrias angioides – característica ocular da doença – são estrias que se formam na membrana de Bruch. Os pacientes comumente apresentam sangramento de retina e perda da visão. A calcificação da lâmina elástica interna das artérias pode resultar em sangramento ou oclusão desses vasos. Como consequência, os pacientes desenvolvem claudicação intermitente ao caminharem e doença arterial coronariana oclusiva em idades mais precoces. Foi descrita a ocorrência de anormalidades valvulares cardíacas.16 As lesões cutâneas consistem em manchas amarelas semelhantes aos xantomas, pápulas ou pregas cutâneas redundantes em áreas de flexão, particularmente nas regiões do pescoço e das axilas [Figura 4]. Alguns pacientes podem apresentar manifestações sistêmicas de PXE sem desenvolverem lesões cutâneas clinicamente aparentes.17 O diagnóstico é estabelecido por meio do exame de biópsia de cicatriz ou de pele aparentemente normal oriunda de regiões de flexura.18 Não existe terapia para as lesões cutâneas associadas ao PXE.
Figura 4. Pápulas xantoma-símile características do pseudoxantoma elástico. Os sítios mais comumente afetados são o pescoço e as axilas.
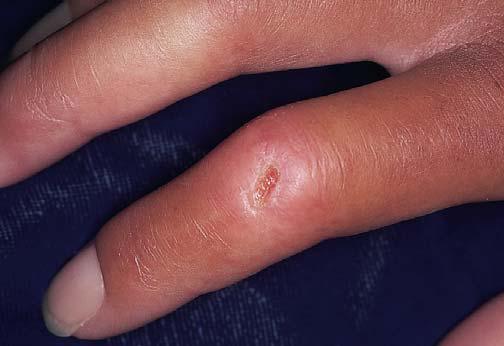
As duas manifestações cutâneas da febre reumática são o eritema marginado e os nódulos subcutâneos. O eritema marginado é uma erupção cutânea eritematosa anular, fraca e transitória que com frequência se desenvolve sobre as articulações [Figura 5]. Os nódulos subcutâneos observados no paciente com febre reumática são insensíveis, movem-se livremente e possuem diâmetro aproximado de 1 cm. Esses nódulos aparecem nas superfícies extensoras dos cotovelos, mãos ou pés.
Figura 5. Erupções cutâneas eritematosas anulares transitórias (eritema marginado) de ocorrência típica em pacientes com febre reumática.
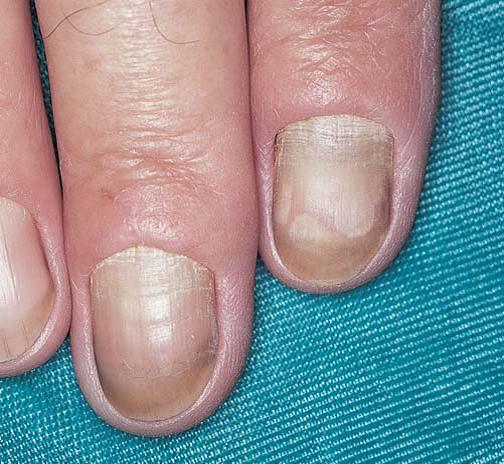
A síndrome da unha amarela é causada por uma anomalia linfática [Figura 6]. Em geral, pacientes afetados desenvolvem linfedema nas pernas, bem como efusões pleurais. Sintomas pulmonares, como bronquite recorrente, são igualmente comuns. O diagnóstico é estabelecido evidenciando a função linfática anormal associada às unhas amarelas sem outra possível causa da patologia das unhas. A permeabilidade microvascular aumentada com consequente vazamento de proteínas pode exercer papel importante no desenvolvimento da síndrome da unha amarela.19
Figura 6. Unhas amareladas sinalizam a ocorrência de uma doença linfática subjacente em pacientes com síndrome da unha amarela.
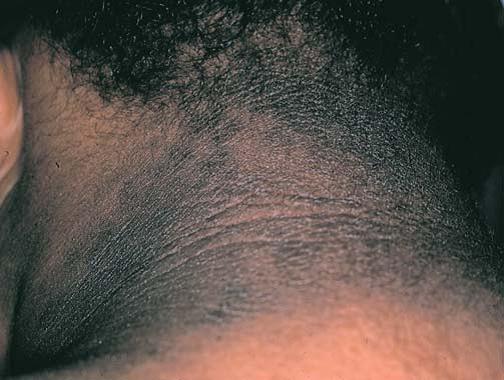
O diabetes melito está associado a numerosas manifestações cutâneas [ver Diabetes melito de tipo 1 e Diabetes melito de tipo 2]. A acantose nigricans pode ocorrer em pacientes diabéticos e com outras endocrinopatias, tais como síndrome de Cushing, acromegalia, síndrome dos ovários policísticos e doença da tireoide. A resistência à insulina constitui um fator subjacente em diversas das endocrinopatias mencionadas, além de atuar também no desenvolvimento da acantose nigricans. As lesões cutâneas consistem em manchas de aspecto aveludado e cor marrom, que aparecem nas áreas intertriginosas, sobretudo no pescoço e nas axilas [Figura 7], sendo mais comuns em indivíduos obesos com diabetes.20 A acantose nigricans também foi associada a malignidades internas, em particular ao adenocarcinoma gástrico ou outros adenocarcinomas gastrintestinais.
Figura 7. A acantose nigricans, uma acantose escura e de aspecto aveludado que ocorre em pacientes com diabetes melito e outros distúrbios endócrinos, aparece com frequência na região cervical.
A necrobiose lipoide é uma manifestação cutânea específica do diabetes. As lesões consistem em manchas atróficas crônicas com bordas eritematosas em expansão. As pernas são a região mais comumente afetada. O centro dessas lesões são amarelos por causa da gordura subcutânea visível através da derme e da epiderme atróficas. Ocasionalmente, as lesões podem apresentar ulceração. A necrobiose lipoide muitas vezes está associada a nefropatia ou retinopatia do diabetes.21
O escleroderma, outra manifestação do diabetes, consiste no aparecimento de endurações cutâneas nas costas e na parte posterior da região cervical em indivíduos obesos com diabetes de tipo 2. O escleroderma pode melhorar se o diabetes for controlado.22 Em casos menos comuns, o escleroderma ocorre em pacientes não diabéticos, após uma faringite estreptocócica. Nesses pacientes, a doença é autolimitada e se resolve em 2 anos após o início de sua manifestação. Altas doses de corticosteroides,23 radiação,24 e radiação ultravioleta A1 (UVA1)25 são empregadas no tratamento do escleroderma.
As bolhas, as úlceras neuropáticas e a pele pálida com rigidez articular do diabetes são sinais observados em pacientes diabéticos. Quando o paciente apresenta pele pálida e rigidez articular, a enduração cutânea escleroderma-símile sobre o aspecto dorsal das mãos impede a flexão ou extensão total das articulações interfalângicas proximais.
Pacientes diabéticos são propensos a contrair algumas infecções, dentre as quais o eritrasma – uma infecção por corinebactérias que produz manchas assintomáticas de cor castanho-avermelhada em sítios intertriginosos, especialmente virilha e axilas. Esses pacientes também apresentam propensão ao desenvolvimento de infecções estafilocócicas e com frequência desenvolvem furúnculos e carbúnculos. Infecções por cândida representam outro risco, em particular quando a glicemia não é controlada.
A doença de Graves consiste na tríade exoftalmo, hipertireoidismo e mixedema pré-tibial [ver Tireoide]. O mixedema pré-tibial apresenta-se como nódulos cor de pele e placas que se estendem desde a área pré-tibial até o dorso dos pés. As lesões com frequência se desenvolvem após o tratamento do hipertireoidismo, embora possam ocorrer em qualquer estágio durante a evolução da doença de Graves. Outras doenças autoimunes cutâneas, como vitiligo e alopecia em áreas (circunscrita), são mais frequentes em pacientes com doença de Graves.
A onicólise, que é separação da placa ungueal do leito ungueal, ocorre em muitos pacientes com hipertireoidismo.
O acúmulo de mucopolissacarídeos na pele de pacientes com hipotireoidismo produz espessamento e aspecto áspero, edema não depressível e aumento do tamanho do nariz, da língua e dos lábios. Os pacientes podem desenvolver uma forma específica de alopecia, perdendo o terço lateral das sobrancelhas. Em pacientes com hipotireoidismo, a diminuição do metabolismo do betacaroteno confere uma cor amarelada à pele. A elevação dos níveis de lipídeos pode causar o aparecimento de xantomas, enquanto o ressecamento da pele é uma ocorrência comum, porém inespecífica.
Pacientes que apresentam qualquer uma das numerosas doenças gastrintestinais existentes podem apresentar manifestações cutâneas. De modo semelhante, pacientes com certas doenças cutâneas podem desenvolver complicações gastrintestinais.
A síndrome carcinoide caracteriza-se por um rubor episódico que pode estar associado à dor abdominal, diarreia e sibilos. Noventa por cento dos tumores carcinoides originam-se no trato gastrintestinal, embora os carcinoides bronquiais sejam ocasionais. As manifestações cutâneas menos comuns dos tumores carcinoides incluem alterações esclerodermatosas. As metástases cutâneas ocorrem como nódulos profundos e os pacientes podem desenvolver hiperceratose ou alterações pigmentosas similares àquelas observadas na pelagra.
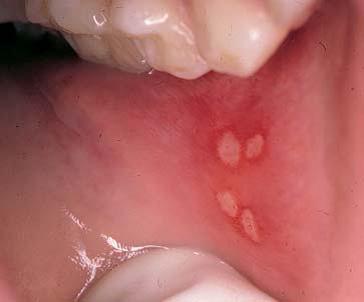
Existem diversas manifestações cutâneas específicas e inespecíficas associadas à doença inflamatória intestinal [ver Doenças inflamatórias intestinais]. Tanto a doença de Crohn como a colite ulcerativa podem evoluir para um estágio de hipercoagulação, causando trombose venosa e arterial com consequente perda de dedos e membros. A estomatite aftosa constitui outra manifestação inespecífica da doença inflamatória intestinal [Figura 8]. Em pacientes com doença de Crohn, as lesões podem assumir a forma de granulomas não caseosos, enquanto naqueles com colite ulcerativa, as lesões podem ser idênticas a aftas típicas.
Figura 8. A estomatite aftosa é um achado comum em pacientes com colite ulcerativa.
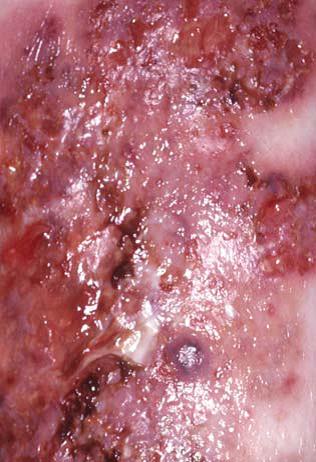
O pioderma gangrenoso ocorre em pacientes com doença de Crohn e colite ulcerativa, sendo descrita também em pacientes com hepatite crônica ativa, artrite reumatoide e numerosos distúrbios mieloproliferativos. As lesões podem ser distinguidas das demais úlceras pela observação das bordas desgastadas de cor violeta-escuro, orifícios em cratera, pústulas e drenagem purulenta [Figura 9]. O pioderma gangrenoso pode ocorrer em sítios traumatizados. O tratamento talvez requeira administração intralesional ou sistêmica de corticosteroides. Os agentes imunossupressores, como a ciclosporina, se mostraram dramaticamente efetivos. Em alguns casos, foi demonstrado que a talidomida pode ser altamente benéfica.26 O uso de infliximabe mostrou-se altamente efetivo no tratamento do pioderma gangrenoso refratário.27,28
Figura 9. O pioderma gangrenoso é caracterizado por úlceras que surgem como orifícios em forma de cratera contendo pus.
Eritema nodoso é uma paniculite septal que está associada a certo número de condições, tais como doença de Crohn, colite ulcerativa, síndrome de Behçet, sarcoidose, infecções e ingestão de estrogênios e outros fármacos. Outras manifestações da doença de Crohn incluem abscessos inguinais, além de fístulas anais e sinusais.
A designação “doença de Crohn metastática” refere-se aos granulomas não caseosos histologicamente demonstrados e localizados fora do trato gastrintestinal de pacientes com doença de Crohn. Sua apresentação clínica pode ser bastante variável e seu diagnóstico com frequência é equivocado. Em alguns casos, os pacientes apresentam escroto ou vulva acentuadamente inchados.
A doença de Cowden é um distúrbio autossômico dominante, em que o paciente desenvolve pólipos gastrintestinais hamartomatosos e numerosas lesões na pele. A doença foi atribuída à ocorrência de mutações no gene supressor tumoral PTEN.29 Pápulas semelhantes a verrugas, conhecidas como triquilemomas, parecem particularmente em torno do nariz, boca e orelhas, bem como nas mãos e nos pés [Figura 10]. Pequenas pápulas também podem se desenvolver na mucosa gengival, conferindo-lhe o aspecto de “pavimento de paralelepípedos”. Pode haver desenvolvimento de hemangiomas e lipomas.30 Foi descrito o aparecimento de um nódulo distintivo no couro cabeludo, conhecido como fibroma de Cowden. Até 50% das mulheres com doença de Cowden desenvolvem câncer de mama – uma descoberta que foi independentemente associada à mutação do gene PTEN.31 O paciente também pode desenvolver carcinomas de tireoide, adenomas de tireoide e bócios na tireoide.
Figura 10. O nariz e as bochechas do paciente estão cobertos com pequenas pápulas denominadas triquilenomas, que constituem uma característica marcante da doença de Cowden.
A dermatite herpetiforme é uma doença imune bolhosa, que está associada a uma enteropatia glúten-sensível [ver Doenças vesiculobolhosas]. As lesões cutâneas surgem como vesículas que, por serem tão pruriginosas, são rapidamente rompidas quando o indivíduo as coça, deixando apenas escoriações e crostas [Figura 11]. Essas lesões ocorrem mais comumente em cotovelos, joelhos, nádegas, costas e em pontos de pressão simétrica, contudo outros sítios também podem ser afetados. Assim como os pacientes com doença celíaca sob dieta contendo glúten, os pacientes com dermatite herpetiforme apresentam risco aumentado de desenvolver linfoma gastrintestinal.32
Figura 11. As lesões primárias da dermatite herpetiforme são vesículas que se rompem rapidamente, formando crostas e erosões.
Na síndrome de Peutz-Jegher, os pacientes desenvolvem pólipos hamartomatosos no intestino delgado que estão associados ao aparecimento de manchas pigmentadas nos lábios e mucosa oral [ver Tumores cutâneos malignos]. Máculas pigmentadas também podem se desenvolver em palmas e dedos das mãos, plantas e dedos dos pés, bem como nas áreas perioral, perinasal e reto. Trata-se de uma doença herdada como caráter autossômico dominante, sendo que uma proporção significativa dos casos está associada à ocorrência de mutações no gene codificador da serina/treonina proteinacinase I1/LKB1 (STKI1/LKB1), embora tais mutações não sejam responsáveis por todos os casos.33
A epidermólise bolhosa distrófica recessiva é uma doença bolhosa congênita, com formação recorrente de bolhas e cicatrizes sobretudo nas mãos e pés [ver Doenças vesiculobolhosas]. A formação de cicatrizes resulta em pseudossindactilia, dando origem às “mãos em mitene”. A disfagia é uma queixa constante. A ingestão de alimentos ásperos pode acarretar a formação de bolhas mucosas esofágicas que, quando curadas, resultam na formação de cicatrizes e estenoses. A formação de cicatrizes esofágicas pode levar ao desenvolvimento de carcinoma de células escamosas, que é a principal causa de morte de pacientes com essa doença.34 O diagnóstico pré-natal pode ser feito em amostras de DNA proveniente das vilosidades coriônicas.35 Todas as formas de epidermólise bolhosa distrófica foram atribuídas à ocorrência de mutações no gene do colágeno de tipo VII.36
A epidermólise bolhosa simples é caracterizada pela formação de bolhas mais superficiais do aquelas observadas na epidermólise bolhosa distrófica. Além disso, é mesmo provável que essas bolhas originem cicatrizes, não ameacem a vida do paciente e não estejam associadas à doença gastrintestinal.
A epidermólise bolhosa juncional, caracterizada pela formação de bolhas disseminadas na pele e nas membranas mucosas, pode ser fatal. Esse distúrbio está associado à atresia pilórica.
Existem várias formas de amiloidose local e sistêmica [ver Leucemias linfoides crônicas e distúrbios de plasmócitos]. De forma associada ao mieloma múltiplo, fibrilas amiloides constituídas por cadeias leves de imunoglobulinas depositam-se na pele. Pápulas translúcidas e brilhantes se desenvolvem em particular nas pálpebras. Por causa da deposição de amiloide nos vasos sanguíneos, o paciente apresenta sangramento espontâneo. Um traumatismo mínimo resulta em petéquias e púrpura. Também pode haver macroglossia em alguns pacientes com amiloidose associada ao mieloma e em certos casos de amiloidose sistêmica primária. As manifestações sistêmicas da amiloidose associada ao mieloma e da amiloidose sistêmica primária são bastante variadas; 50% dos pacientes desenvolvem hepatomegalia. A amiloidose pode afetar o coração, resultando em insuficiência cardíaca ou infarto do miocárdio. A sobrevida dos pacientes submetidos ao transplante cardíaco em decorrência da amiloidose cardíaca é menor do que a sobrevida dos pacientes submetidos ao transplante de coração por outras indicações.37 A amiloidose do trato gastrintestinal pode resultar em má absorção e enteropatia perdedora de proteína. O tratamento com talidomida (até 400 mg/dia) e dexametasona intermitente é rapidamente efetivo para alguns pacientes, porém é comum produzir efeitos adversos.38
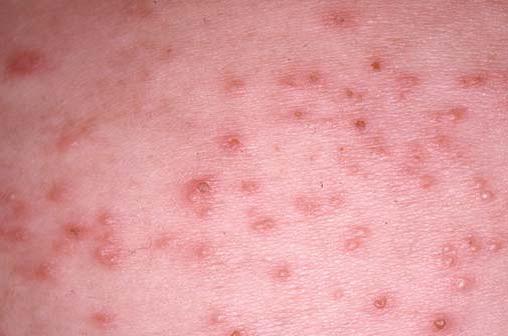
A mastocitose é causada pela infiltração de mastócitos na pele e em outros órgãos [ver Doenças produtoras de má absorção e má digestão]. A urticária pigmentosa se refere às lesões cutâneas que aparecem na maioria dos pacientes com mastocitose. Manchas castanho-avermelhadas e pápulas semelhantes a nevos são achados característicos [Figura 12]. O toque das lesões individuais resulta em urticação – um fenômeno conhecido como sinal de Darier. Prurido, rubor, dor abdominal, náusea, vômitos e diarreia são queixas comuns.
Figura 12. Múltiplas manchas de cor marrom, parecidas com nevos, são observadas em pacientes com urticária pigmentosa.
A maioria dos pacientes com mastocitose desenvolve a forma indolor da doença, mesmo quando os mastócitos infiltram a medula óssea.39 A doença de mastócitos maligna ou sistêmica e agressiva pode afetar baço, fígado, linfonodos, pele e medula óssea. Histologicamente, os infiltrados contêm mastócitos não metacromáticos atípicos que são monoclonais em alguns pacientes.40 Crianças com urticária pigmentosa costumam apresentar melhor prognóstico do que adultos com a doença.41
O diagnóstico da mastocitose é estabelecido pela demonstração da presença dos mastócitos em uma biópsia de pele. Como os mastócitos degranulam facilmente, dificultando sua identificação, as biópsias com frequência geram resultados falso-negativos.
As porfirias resultam da síntese de hemoglobina defeituosa, que conduz ao excesso de porfirinas no sangue e nos tecidos corpóreos [ver Porfirias]. A porfiria cutânea tardia caracteriza-se por fotossensibilidade, formação de vesículas (em especial no dorso das mãos) [Figura 13] e hipertricose. A condição pode estar associada à ingestão de álcool ou medicações, como os estrogênios. O diagnóstico das porfirias pode ser estabelecido pela detecção de níveis elevados de porfirina na urina. O exame de urina com auxílio de uma lâmpada de Wood muitas vezes revela uma fluorescência rosa-avermelhada atribuível ao alto nível de porfirinas presente na urina. A sobrecarga de ferro associada à hemocromatose hereditária pode promover o desenvolvimento de porfiria cutânea tardia. A infecção pelo vírus da hepatite C também foi associada a esse tipo de porfiria. A flebotomia constitui uma terapia efetiva.
Figura 13. Formação de crosta e cicatriz após o aparecimento de vesículas e bolhas na porfiria cutânea tardia.
A Aids pode resultar em infecções e neoplasias cutâneas - cuja extensão e gravidade frequentemente são dramáticas. Graças ao desenvolvimento de terapias antirretrovirais efetivas, a frequência das infecções oportunistas em pacientes infectados pelo HIV diminuiu de forma acentuada. A presente seção enfoca algumas manifestações cutâneas seletas de infecções e outras doenças associadas à Aids (para ter acesso a uma discussão mais abrangente dos distúrbios associados à infecção pelo HIV, ver HIV e Aids, além dos demais capítulos que abordam condições específicas).
Infecções virais. Infecções virais banais, como o molusco contagioso, em geral autolimitadas e facilmente curáveis, transformam-se em doenças disseminadas e crônicas que representam um enorme problema para o paciente com Aids. Pápulas umbilicais brancas, normalmente medindo poucos milímetros de diâmetro, podem atingir 1 a 2 cm de diâmetro num paciente com Aids. De modo semelhante, o condiloma acuminado, produzido pela infecção com papilomavírus humano, muitas vezes é de difícil tratamento em pacientes aidéticos.
As infecções pelo vírus do herpes simples em pacientes com Aids a princípio são crônicas e erosivas, e formam úlceras amplas que não se curam [ver Infecções por herpesvírus]. Há relatos de cepas de vírus do herpes simples resistentes ao aciclovir em pacientes com Aids.42 Tais pacientes requerem tratamento com outros agentes antivirais, como o foscarnet. Mutações nos genes codificadores da timidinacinase e da DNA polimerase dos vírus do herpes simples podem conferir resistência ao aciclovir e ao foscarnet.43 Foi demonstrado que o uso tópico de cidofovir em gel produz efeitos benéficos contra o herpes infeccioso em pacientes infectados pelo HIV.44
As infecções de herpes zóster constituem um sinal comum da infecção pelo HIV. No hospedeiro não infectado pelo HIV, o herpes zóster é caracterizado pela existência de vesículas agrupadas apresentando distribuição dermatômica. A erupção é autolimitada, resolvendo-se em 1 a 2 semanas. Em contraste, a infecção pelo herpes zóster pode se desenvolver em erupções vesiculares disseminadas em pacientes aidéticos. Alguns dos pacientes com Aids desenvolvem lesões herpéticas crônicas que persistem durante meses.
Infecções fúngicas. Pacientes infectados pelo HIV comumente apresentam infecções causadas por fungos. As infecções moniliais incluem candidíase oral e inguinal. Várias infecções fúngicas que raramente causam infecção disseminada em pacientes cujo sistema imune esteja normal (p. ex., criptococose, histoplasmose, aspergilose e esporotricose) emergem como patologias graves em pacientes aidéticos [ver Infecções micóticas em hospedeiros comprometidos].
Infecções bacterianas. As infecções causadas por bactérias são mais frequentes e severas em pacientes com Aids do que naqueles com sistema imune normal. A angiomatose bacilar, causada por Bartonella henselae, manifesta-se como pápulas e nódulos de cor púrpura, que podem ser confundidos com sarcoma de Kaposi (ver adiante). O paciente pode apresentar febre crônica e calafrios, além de lesões ósseas. Evidências epidemiológicas sugerem que gatos podem constituir uma fonte de infecção humana.45 O diagnóstico comumente é estabelecido por meio de testes sorológicos, contudo, no futuro a reação em cadeia da polimerase (polymerase chain reaction – PCR) poderá proporcionar um modo rápido e conveniente de estabelecer esse diagnóstico.46 A condição é resolvida mediante tratamento com antibióticos orais [ver Infecções causadas por Brucella, Francisella, Yersinia pestis e Bartonella].
Escabiose e outras erupções pruriginosas. A escabiose (sarna), uma erupção cutânea severamente pruriginosa, apresenta predileção por nádegas, genitais, área periumbilical e zonas interdigitais. A escabiose norueguesa, a forma parasítica da doença associada à formação de crostas espessas, foi descrita em pacientes com síndrome de Down e em indivíduos imunossuprimidos. Nos últimos anos, a escabiose norueguesa foi relatada mais frequentemente em pacientes com Aids. As crostas produzidas nessa condição contêm milhares de ácaros que são facilmente observáveis ao microscópio. As tocas (de parasitas) são lesões lineares de chegam a medir 1 cm de comprimento. O ácaro causador, Sarcoptes scabiei, pode ser identificado por exame microscópico de raspados a partir das tocas.
Folículos pustulares eosinofílicos e erupção papular da Aids são erupções cutâneas pruriginosas que acometem pacientes infectados pelo HIV. Foi sugerido que a erupção papular pruriginosa observada nos pacientes aidéticos pode representar uma reação às picadas dos artropódos.47 Tanto os folículos pustulares eosinofílicos como a erupção papular da Aids caracterizam-se por uma coceira severa. É comum observar pápulas cor de pele e escoriações em ambos os casos. Pacientes com foliculite pustular eosinofílica podem desenvolver pústulas e pápulas eritematosas. Ambas as condições respondem ao tratamento com radiação ultravioleta de tipo B.
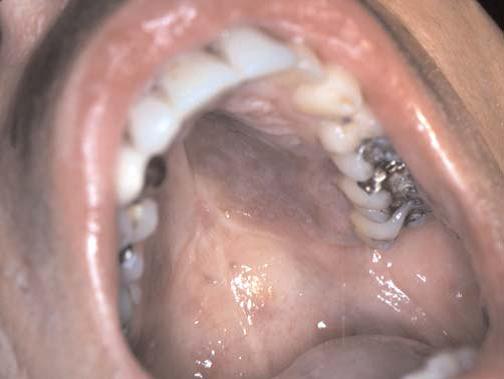
O sarcoma de Kaposi é um neoplasma vascular de progressão lenta, originalmente descrito em homens idosos italianos e judeus [ver Tumores cutâneos malignos]. Subsequentemente, uma forma de progressão mais rápida desse distúrbio foi descrita em pacientes imunossuprimidos com linfoma e também em pacientes submetidos ao transplante renal sob terapia farmacológica imunossupressora. Uma forma agressiva foi descrita em pacientes com Aids [Figura 14]. O sarcoma de Kaposi clássico costuma afetar os membros inferiores e progride somente de modo gradual para outros sítios. Em contraste, o sarcoma de Kaposi associado a Aids pode ocorrer em qualquer superfície do corpo, incluindo as membranas mucosas [Figura 15]. O herpesvírus humano do tipo 8 foi implicado tanto na forma clássica como na forma associada a Aids do sarcoma de Kaposi.48 Os tratamentos utilizados são radioterapia, crioterapia e injeção intralesional com vimblastina. A quimioterapia sistêmica também pode ser efetiva. Nos pacientes aidéticos, o sarcoma de Kaposi é mai bem tratado por meio de regimes antirretrovirais.
Figura 14. O sarcoma de Kaposi é a malignidade de ocorrência mais frequente em pacientes com Aids. Costuma se manifestar como manchas, placas ou pápulas de cor púrpura. A presença de manchas púrpuras no pé é observada em pacientes com sarcoma de Kaposi clássico, mas, nesta foto, foi detectada em paciente com sarcoma de Kaposi associado a Aids.
Figura 15. O sarcoma de Kaposi pode afetar as membranas mucosas. A foto mostra o envolvimento do palato em paciente com Aids.
A leucoplaquia pilosa oral, outra condição descrita em pacientes infectados pelo HIV, consiste no aparecimento de pápulas brancas lineares nas superfícies laterais da língua, que conferem o conhecido aspecto piloso. É possível distinguir a leucoplaquia pilosa oral da candidíase oral, pois as lesões da primeira não podem ser removidas por fricção, como ocorre na segunda.
Outros estados de imunodeficiência herdados ou adquiridos compartilham algumas características clínicas. A suscetibilidade a infecções moniliais ou bacterianas é maior diante de certos distúrbios, como doença granulomatosa crônica e neutropenia induzida por quimioterapia. De modo similar, úlceras orais ocorrem em pacientes com neutropenia cíclica e naqueles com imunossupressão induzida por quimioterapia.
Algumas drogas imunossupressoras apresentam efeitos cutâneos característicos. Os corticosteroides, quando utilizados por tempo prolongado, provocam uma fragilidade vascular que resulta em púrpura esteroide. Esses fármacos também podem causar atrofia cutânea, formação de estrias e erupções acneiformes. A ciclosporina está associada à hipertricose. A estomatite aftosa constitui um efeito característico de numerosos fármacos imunossupressores, em particular dos agentes supressores da função da medula óssea. A imunossupressão crônica pode levar ao desenvolvimento de linfoma e câncer de pele não melanoma. Evitar a excessiva exposição à luz solar pode prevenir o desenvolvimento desta última condição.
As manifestações cutâneas podem constituir as principais características de algumas infecções sistêmicas. Exemplificando, pacientes com uma notável septicemia podem desenvolver coagulação intravascular disseminada (CIVD), a qual resulta em infartos cutâneos e hemorragia nas camadas internas da pele. A seguir, são descritos os principais aspectos característicos de algumas infecções sistêmicas selecionadas.
As manifestações cutâneas da endocardite cutânea incluem petéquias, hemorragias ungueais (estrias lineares vermelhas sob a unha), nódulos de Osler (nódulos arroxeados e sensíveis no coxim dos dedos da mão e do pé) e lesões de Janeway (manchas arroxeadas e indolores nas palmas das mãos e plantas dos pés). As lesões cutâneas são causadas por êmbolos sépticos ou vasculite. O tratamento da infecção subjacente conduz à resolução das manifestações cutâneas [ver Endocardite infecciosa].
A síndrome do choque tóxico estafilocócico foi identificada pela primeira vez em mulheres menstruadas que usavam tampões superabsorventes [ver Infecções por cocos gram-positivos]. Essa síndrome é causada por uma exotoxina produzida por algumas cepas de S. aureus.49 Foram implicadas as infecções estafilocócicas nos ossos e tecidos moles, entre outros sítios. Os pacientes desenvolvem eritema difuso semelhante à queimadura solar, com inchaço de mãos e pés, seguido de descamação também em mãos e pés. Há também eritema de membranas mucosas, febre e hipotensão. Pode haver surgimento de sintomas gastrintestinais, comprometimento da função renal, elevação das enzimas hepáticas, trombocitopenia e miosite.
A síndrome da pele escaldada estafilocócica (SPEE) é causada por uma toxina esfoliativa circulante produzida pelo grupo de fagos 11 do S. aureus. A formação generalizada de bolhas com amplas áreas de descamação é característica do distúrbio. Além de sensibilidade, eritema e esfoliação da pele, os pacientes apresentam febre. A fonte de infecção estafilocócica nem sempre é aparente. às vezes, a infecção surge em uma ferida ou em abscessos ocultos. Como a infecção estafilocócica em geral ocorre em locais afastados da região de pele afetada, o S. aureus não cresce em culturas de pele.
A SPEE deve ser diferenciada da necrólise epidérmica tóxica, que comumente afeta adultos e envolve membranas mucosas. A SPEE costuma afetar crianças e poupa as membranas mucosas. Além disso, a necrose epidérmica tóxica pode durar várias semanas e está associada a uma elevada mortalidade, enquanto a SPEE tem duração de poucos dias e, em geral, apresenta resultado satisfatório. Em termos de histologia, a SPEE apresenta formação de bolhas na epiderme superior, sendo que a cavidade dessas bolhas contém células acantolíticas de aspecto normal flutuando livremente. Na necrólise epidérmica tóxica, as bolhas se formam na camada basal da epiderme e as células epidérmicas são necróticas. O tratamento com antibióticos efetivos contra S. aureus elimina a causa subjacente da SPEE.
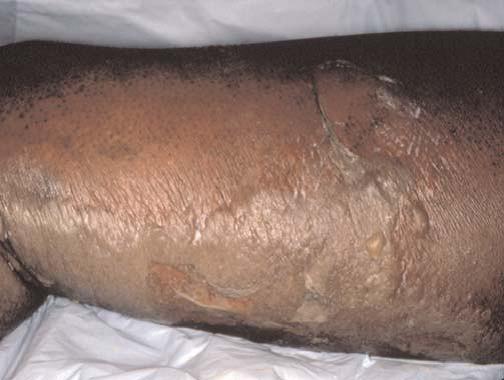
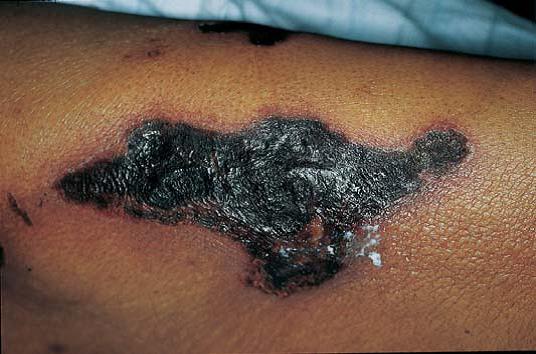
A fasciite necrotizante é causada pela infecção anaeróbia mista de uma úlcera ou ferida cirúrgica/traumática. A pele afetada é eritematosa, aquecida e sensível, e desenvolve bolhas hemorrágicas caracterizadas por amplas áreas de formação de crosta [Figura 16]. Essas bolhas progridem e originam áreas gangrenosas de rápida expansão, que se estendem para baixo da fáscia. O debridamento cirúrgico é essencial ao tratamento dessa infecção que ameaça a vida do paciente.50
Figura 16. Formação de crosta em amplas áreas de pele em paciente com fasciite necrotizante.
A meningocococemia pode ocorrer tanto em epidemias como em casos isolados [ver Infecções causadas por Neisseria]. Os pacientes apresentam febre, dor de cabeça e erupções cutâneas hemorrágicas. Se não forem tratados, desenvolvem CIVD que resulta em extensa hemorragia, hipotensão e, por fim, morte. O organismo causador, Neisseria meningitidis, em geral é detectado em amostras de líquido cerebrospinal, mas também pode ser encontrado em esfregaços, culturas de material proveniente das lesões cutâneas ou culturas sanguíneas. O tratamento à base de antibióticos e a assistência de suporte constituem aspectos essenciais da terapia.
A escarlatina inicialmente manifesta-se como faringite causada por Streptococcus de grupo A [ver Infecções por cocos gram-positivos]. O paciente desenvolve erupções cutâneas generalizadas em 1 a 2 dias após o início da faringite. As erupções são caracterizadas por pápulas eritematosas em pontos localizados, que podem ser mais facilmente palpadas do que vistas. Outras lesões características são a língua “em morango” com revestimento esbranquiçado e as manchas petequiais lineares encontradas nas regiões de pregas corpóreas (linhas de Pastia). Conforme as erupções desaparecem, as palmas das mãos e plantas dos pés começam a descamar. O tratamento com penicilina resulta na rápida resolução de todos os sintomas.
A infecção por Vibrio vulnificus tem início a partir de um traumatismo mínimo e prolongado, que é produzido quando o indivíduo nada em lagos ou no mar, ou durante a limpeza de alimentos de origem marinha. O paciente desenvolve celulite acompanhada de linfangite e bacteremia. Em pacientes com cirrose hepática, a infecção pode ocorrer após a ingestão de mariscos crus. Esses pacientes desenvolvem bolhas hemorrágicas, leucopenia e CIVD.51 A condição deve ser tratada com antibióticos e a supervisão das complicações pode requerer assistência de suporte intensiva.
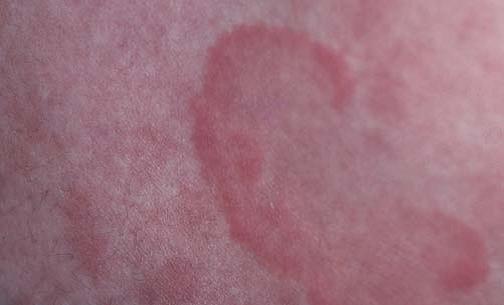
A doença de Lyme é causada pela espiroqueta Borrelia burgdorferi, sendo transmitida primariamente pelo carrapato Ixodes scapularis [ver Doenças infecciosas causadas por Rickettsia, Ehrlichia e Coxiella]. A lesão cutânea característica – o eritema crônico migratório – manifesta-se a princípio como uma mancha ou pápula eritematosa no sítio da picada do carrapato. No decorrer de alguns dias e semanas, a lesão eritematosa se expande e dá origem a um anel avermelhado com centro claro. Quando não são tratadas, as lesões duram semanas ou meses. A disseminação hematógena das espiroquetas ocorre após várias semanas, resultando em múltiplas manchas anulares de eritema crônico migratório [Figura 17]. As complicações sistêmicas incluem uma artrite aguda envolvendo uma ou mais articulações grandes, que surge algumas semanas após o início da manifestação dos sintomas. Cerca de 10% dos pacientes desenvolve uma artrite erosiva crônica. Pode haver manifestação de sintomas neurológicos, como a paralisia de Bell, bem como complicações cardíacas, incluindo insuficiência cardíaca e anomalias de condução cardíaca.
Uma doença semelhante à doença de Lyme, conhecida como doença da erupção cutânea associada ao carrapato do sul, foi atribuída à infecção por Borrelia (causada por B. lonestari) transmitida por carrapatos das espécies Amblyomma americanum e Ixodes scapularis. Embora inicialmente tenha sido relatada no sul dos Estados Unidos, alguns casos também foram registrados nas regiões nordeste e central desse mesmo país.
Figura 17. Várias semanas após a infecção primária da doença de Lyme, a disseminação hematógena das espiroquetas resulta no aparecimento de múltiplas manchas de eritema crônico migratório.
A febre maculosa das Montanhas Rochosas (FMMR) é uma doença transmitida por carrapatos e causada por Rickettsia rickettsii [ver Doenças infecciosas causadas por Rickettsia, Ehrlichia e Coxiella]. Caracteriza-se pela manifestação repentina de febres, calafrios e dor de cabeça. Decorridos cerca de 4 dias, o paciente desenvolve uma erupção cutânea eritematosa nos punhos e tornozelos que, em seguida, se torna pruriginosa. Essas erupções, então, disseminam-se centralmente e envolvem membros, tronco e face.
Como a FMMR está associada a uma elevada mortalidade, é necessário tratar os pacientes imediatamente com uma injeção endovenosa de cloranfenicol ou tetraciclina, caso haja suspeita da doença. O diagnóstico, então, pode ser estabelecido por meio de uma biópsia de pele: a imunofluorescência utilizando anticorpos contra R. rickettsii revela a presença do organismo nas paredes dos vasos sanguíneos cutâneos. Os testes sorológicos, entre eles a reação de Weil-Felix, podem confirmar o diagnóstico após a fase aguda da doença.
A síndrome do nevo de células basais é um distúrbio autossômico dominante atribuído à inativação por mutação do gene PTCH.53 Os pacientes desenvolvem carcinoma de células basais em idade precoce [ver Tumores cutâneos malignos]. Múltiplas anormalidades esqueléticas estão associadas a essa síndrome, sendo que os indivíduos afetados também podem desenvolver cistos mandibulares. Há calcificação lamelar da foice do cérebro, além de outras anomalias neurológicas, como os meduloblastomas
A síndrome do nevo epidérmico é caracterizada pela ocorrência de manifestações sistêmicas, tais como convulsões, retardo mental, cegueira e anomalias esqueléticas, associadas ao aparecimento de amplos nevos epidérmicos. Os nevos consistem em longas estrias pigmentadas, que são lineares ou em redemoinho, e envolvem amplas áreas corpóreas [Figura 18].
Figura 18. A síndrome do nevo epidérmico é caracterizada pela presença de estrias de pigmentação lineares ou em redemoinho, que envolvem amplas áreas corpóreas.
A incontinência pigmentar é uma síndrome herdada que afeta a pele e o sistema nervoso. A ocorrência de mutações no gene NEMO, que constitui um dos componentes essenciais da cascata de sinalização do fator nuclear-?B, responde por 85% dos casos.54 O padrão de herança é ligado ao cromossomo X e dominante, sendo letal para fetos do sexo masculino. As primeiras manifestações cutâneas surgem em algumas semanas após o nascimento e, ocasionalmente, ocorrem in utero. Tais manifestações constituem padrões lineares de lesões vesiculobolhosas. Em algumas semanas, essas lesões evoluem para pápulas verrucosas e, eventualmente, redemoinhos pigmentados. À parte dos sintomas neurológicos, os pacientes podem apresentar anomalias oculares, alopecia cicatricial e malformações esqueléticas.
A hipomelanose de Ito, também conhecida como incontinência pigmentar do acrômio, consiste em redemoinhos de hipopigmentação que estão associados à sintomas neurológicos em 50% dos pacientes. As lesões cutâneas já estão presentes ao nascimento ou podem se desenvolver no início da infância. Além de convulsões e retardo mental, o paciente também pode apresentar anomalias esqueléticas e oculares.
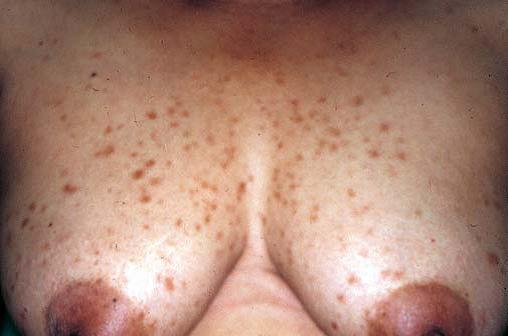
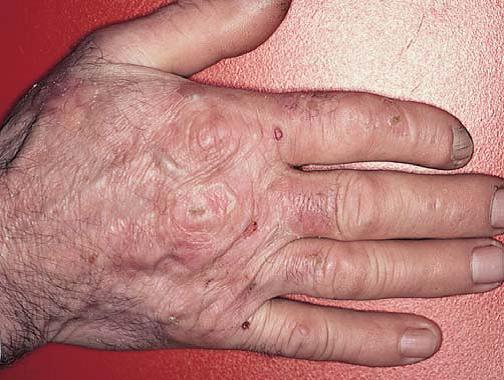
A neurofibromatose é um distúrbio autossômico dominante de ocorrência comum, que afeta a pele e o sistema nervoso [ver Tumores cutâneos benignos]. As lesões cutâneas são neurofibromas cutâneos, que constituem nódulos moles, da cor da pele e, com frequência, pedunculados [Figura 19]. As manchas cor de café com leite são manchas planas e uniformemente pigmentadas, cujo diâmetro pode medir vários centímetros. A maioria dos pacientes com neurofibromatose de tipo 1 (também denominada doença de von Recklinghausen) apresenta 6 ou mais manchas cor de café com leite, com diâmetros maiores que 1,5 cm. Os neuromas plexiformes são tumores maiores e mais profundos, que estão associados à hipertrofia óssea e de tecidos moles. Os neurofibrossarcomas surgem em uma pequena proporção de tumores. Ao exame da biópsia de pele, constata-se que as manchas cor de café com leite contêm macromelanossomos – grânulos gigantes de pigmento contidos em melanócitos e queratinócitos. Sardas axilares e inguinais também aparecem sob a forma de manchas pigmentadas, semelhantes a pequenas manchas cor de café com leite nas membranas interdigitais. Os nódulos de Lisch – hamartomas de íris pigmentados – também são encontrados na maioria dos pacientes com neurofibromatose.
Figura 19. Sardas inguinais, manchas cor de café com leite e neurofibromas evidentes num paciente com neurofibromatose de tipo 1.
Existem diversas variantes de neurofibromatose, incluindo a neurofibromatose segmentar, em que os pacientes apresentam uma distribuição segmentar das manchas cor de café com leite e neurofibromas cutâneos, e a neurofibromatose de tipo 2, que consiste em neuromas acústicos, schwannomas e meningiomas (sem nódulos de Lisch e com menos manchas cor de café com leite do que no tipo 1). Pacientes com neurofibromatose de tipo 2 também podem apresentar alguns neurofibromas cutâneos. As neurofibromatoses de tipos 1 e 2 são causadas por diferentes defeitos genéticos. A neurofibromatose de tipo 1 é produzida por mutações no gene NF1, codificador da neurofibromina, localizado no cromossomo 17.55 A neurofibromatose de tipo 2 foi atribuída à ocorrência de mutações ativadoras no gene supressor tumoral NF2, cujo produto (merlina) exerce vários papéis na tumorigênese.56
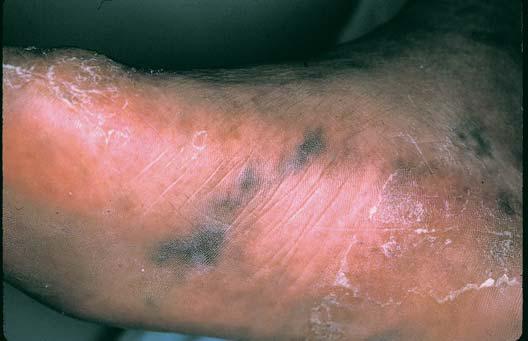
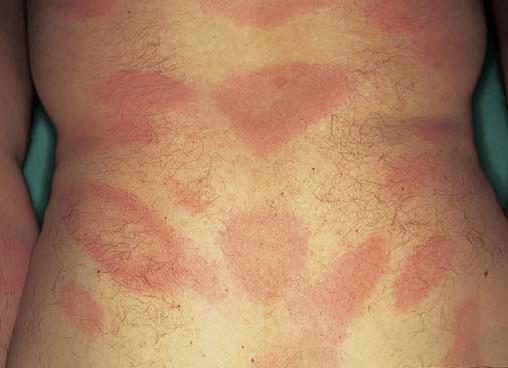
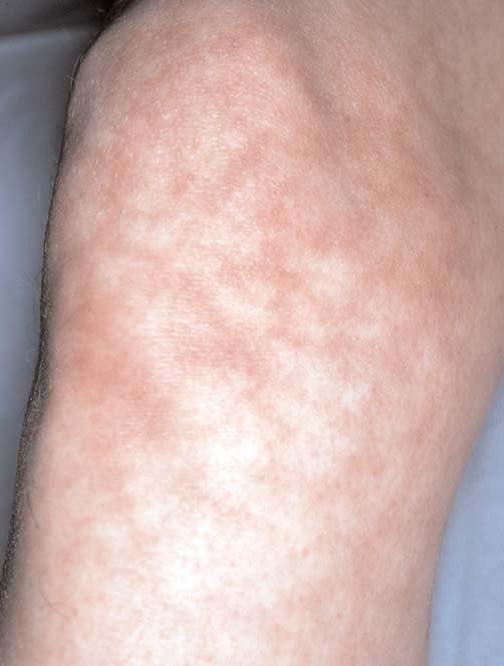
Síndrome de Sneddon é uma doença da pele e do sistema nervoso causada pela oclusão de artérias de médio calibre em indivíduos com menos de 45 anos de idade. As lesões cutâneas assemelham-se ao livedo reticular [Figura 20], tendo sido denominadas livedo racemosa. Ataques isquêmicos transitórios ou derrames são eventos comuns. O diagnóstico definitivo é estabelecido pela demonstração de alterações vasculares características em biópsias de pele de pacientes que apresentam achados neurológicos associados. O diagnóstico diferencial inclui outras causas de livedo reticular – uma condição que pode ser fisiológica ou decorrente de condições como o lúpus eritematoso sistêmico ou a poliarterite nodosa.
Figura 20. As alterações cutâneas vasculares da síndrome de Sneddon assemelham-se ao livedo reticular.
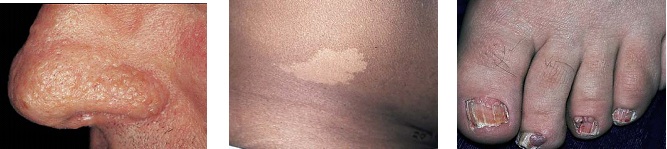
A esclerose tuberosa é uma doença autossômica dominante, que afeta a pele e o sistema nervoso. Mutações que inativam os genes supressores tumorais TSC1 ou TSC2 afetam também os respectivos produtos desses genes – hamartina e tuberina – causando esclerose tuberosa.57 Pacientes afetados podem desenvolver convulsões e retardo mental, além de lesões cerebrais (denominadas tuberosidades) detectáveis por tomografia computadorizada. O adenoma sebáceo, a manifestações cutânea mais característica da esclerose tuberosa, consiste em pápulas cor de pele que aparecem na face [Figura 21a]. Outras lesões cutâneas são:
manchas hipopigmentadas, referidas como manchas em folha de freixo [Figura 21b];
lesões hipopigmentadas menores, denominadas manchas em confete;
fibromas peri e subungueal (nódulos cor de pele que surgem em torno dos dedos da mão e das unhas dos pés) [Figura 21c];
mancha chagrém (uma placa cor de pele constituída por um espesso tecido conectivo dérmico).
a b c
Figura 21. Vários achados cutâneos característicos da esclerose tuberosa: adenoma sebáceo (a); mancha em folha de freixo (b); e fibromas periungueais (c).
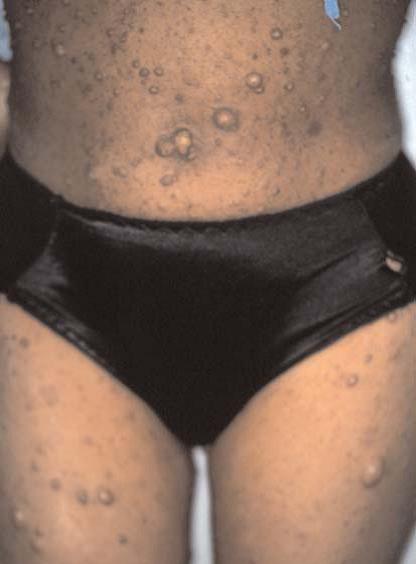
A doença de Fabry é causada por uma anormalidade envolvendo a alfa-galactosidase A, que resulta na deposição de glicoesfingolipídeos nos tecidos corpóreos. O distúrbio é herdado como caráter recessivo ligado ao cromossomo X. Diversas mutações afetando o gene da alfa-galactosidase A foram detectadas em indivíduos de famílias distintas com doença de Fabry.58 Os homens afetados com frequência se queixam de dor severa nos membros, apresentando ardência nas palmas das mãos e plantas dos pés. Os episódios de dor são passageiros, contudo os pacientes reclamam de parestesias persistentes nas mãos e nos pés.
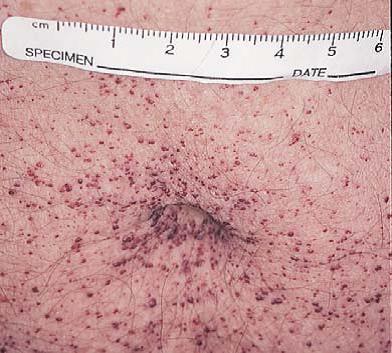
As lesões cutâneas consistem em angioceratomas, que são pontos vermelhos ou pápulas arroxeadas semelhantes a hemangiomas em cereja [Figura 22]. Os angioceratomas são mais comumente encontrados na área periumbilical, mas também podem ocorrer nas palmas das mãos, plantas dos pés, tronco, membros e membranas mucosas. Em adultos, os glicoesfingolipídeos se depositam em vasos sanguíneos e órgãos, afetando coração, válvulas cardíacas, artérias coronárias e rins. A terapia de reposição utilizando alfa-galactosidase A humana recombinante pode melhorar os sintomas cutâneos, gastrintestinais, neurológicos e psiquiátricos. Essa terapia tem se mostrado segura e pode eliminar o estoque de substrato de glicoesfingolipídeos, contudo, ainda restam dúvidas a ser esclarecidas quando à dose ideal.59
Figura 22. Angioceratomas são particularmente comuns na região periumbilical de pacientes com doença de Fabry.
A poliarterite nodosa é uma condição inflamatória que afeta as artérias musculares [ver Síndromes de vasculite sistêmica]. Há formação de aneurismas em diversas artérias, incluindo aquelas que suprem os rins e o tecido subcutâneo. O diagnóstico da forma sistêmica da poliarterite pode ser estabelecido pela demonstração dos aneurismas das artérias renais nos arteriogramas renais.
Uma forma cutânea localizada de poliarterite nodosa manifesta-se mais frequentemente como nódulos dolorosos nos membros inferiores.60 Nos casos brandos, os pacientes apresentam apenas o livedo reticular. Todavia, nos casos severos, as lesões cutâneas podem ulcerar. O distúrbio pode estar associado a uma polineuropatia. Pacientes com poliarterite clássica e microaneurismas apresentam maior incidência de antigenemia para hepatite B. Em contraste com os pacientes que apresentam outras vasculites, aqueles normalmente não possuem anticorpos anticitoplasma de neutrófilos.61
Os distúrbios perfurantes abrangem várias condições caracterizadas pela extrusão de material dérmico através da epiderme. Essas lesões com frequência se desenvolvem associadas à insuficiência renal e ao diabetes melito.62 As lesões cutâneas são caracterizadas pelo aparecimento de pápulas hiperceratóticas com crateras centrais esbranquiçadas que, histologicamente, podem apresentar conteúdo de material dérmico. A colagenose perfurante reativa, a foliculite perfurante e a doença de Kyrle exemplificam os distúrbios perfurantes associados à insuficiência renal. O diagnóstico diferencial, nesses casos, inclui a detecção de prurido nodular (conhecido como nódulos do colhedor). Diferentemente do prurido nodular, que sempre resulta do ato repetitivo de coçar, as lesões dos distúrbios perfurantes ocorrem em sítios que não podem ser facilmente coçados, podendo exibir o fenômeno de Koebner. Este, por sua vez, pode surgir em locais minimamente traumatizados, como naqueles com uma única marca de coceira.
A calcifilaxia, também conhecida como arteriopatia urêmica de calcificação, é uma condição observada em pacientes com insuficiência renal, nos quais áreas localizadas da pele tornam-se necrosadas em decorrência de um processo de calcificação vascular. A calcifilaxia tem início com o aparecimento de manchas dolorosas e de cor púrpura, que podem ser reticuladas, semelhantes ao livedo reticular. Essas manchas progridem para placas endurecidas que podem ulcerar e se tornar necróticas [Figura 23]. A calcifilaxia muitas vezes termina em amputação ou morte do paciente. A paratireoidectomia pode resultar na cicatrização da pele afetada sem necessidade de amputação.63 Recentemente, foi demonstrado que o tiossulfato de sódio, um agente quelante de cálcio e antioxidante, é altamente efetivo no tratamento da calcifilaxia para pacientes com menos de 21 anos de idade.64 Em raras ocasiões, os achados cutâneos da calcifilaxia podem ser observados em pacientes sem insuficiência renal.65
Figura 23. A calcificação das artérias em pacientes com insuficiência renal resulta em calcifilaxia. A pele afetada apresenta uma crosta negra necrosada.
As manifestações cutâneas mais bem conhecidas da dermatomiosite – um distúrbio inflamatório envolvendo os músculos e a pele – são as pápulas de Gottron e o eritema heliotrópio. As pápulas de Gottron são pápulas e manchas eritematosas de raspagem, que ocorrem no dorso das articulações [Figura 24]. O eritema heliotrópio consiste em edema e eritema periorbital. Foram descritas lesões no couro cabeludo, que podem estar associadas à alopecia.66 Com frequência, a lesões são erroneamente diagnosticadas como dermatite seborreica ou psoríase.
Figura 24. A presença de pápulas eritematosas por raspagem no aspecto dorsal das articulações dos dedos (pápulas de Gottron) constitui um sinal de dermatomiosite.
Foi estabelecida uma associação entre dermatomiosite e malignidade.67,68 Um estudo epidemiológico indicou que pacientes com dermatomiosite apresentam risco particularmente aumentado de desenvolver cânceres de ovário e de pulmão.68
As classificações da dermatomiosite incluem uma variante juvenil, caracterizada por calcificação da pele ou de músculos. Uma forma vasculítica que ocorre em crianças é agravada por infartos cutâneos e ulceração, bem como por uma vasculite gastrintestinal acompanhada de dor, sangramento ou perfuração abdominal. A forma vasculítica está associada a um prognóstico ruim, sendo que muitos pacientes morrem por causa dessa doença.
Os esclerodermas abrangem algumas síndromes distintas que compartilham um aspecto comum: a enduração da pele [ver Escleroderma e doenças relacionadas].
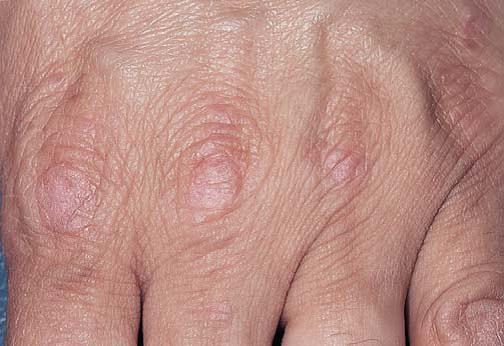
A esclerose sistêmica progressiva, também conhecida como escleroderma sistêmico, é uma doença frequentemente fatal, em que os pacientes apresentam fenômeno de Raynaud e esclerodactilia (endurecimento da pele dos dedos da mão) [Figura 25]. A enduração cutânea pode se tornar disseminada. O envolvimento da face pode produzir um aspecto característico, com lábios franzidos e pele do nariz puxada para baixo de modo semelhante a um bico. Pacientes com anticorpos anti-Scl-70 apresentam prognóstico ruim, por vezes sucumbindo à doença renal e à hipertensão maligna. Pode haver fibrose pulmonar. Pacientes com anticorpos anticentrômero apresentam a variante de escleroderma de progressão mais lenta, conhecida como síndrome CREST. Esta caracteriza-se por calcinose cutânea, fenômeno de Raynaud, dismotilidade esofágica, esclerodactilia e telangiectasia. Com o passar do tempo, o paciente desenvolve hipertensão pulmonar e insuficiência cardíaca do lado direito. Recentemente, a presença de anticorpos dirigidos contra o receptor do fator de crescimento derivado de plaquetas (PDGF) foi identificada como marcador sorológico específico do escleroderma.69
Figura 25. A esclerodactilia com úlcera digital que não cicatriza ocorre na esclerose sistêmica progressiva.
O diagnóstico diferencial da esclerose sistêmica abrange numerosas condições caracterizadas por enduração cutânea, tais como doença do enxerto versus hospedeiro crônica, escleromixedema, dermopatia fibrosante nefrogênica, fasciite eosinofílica, síndrome da eosinofilia-mialgia, escleroderma, morfeia e numerosas causas distintas fármaco-induzidas, ocupacionais, ambientais ou idiopáticas de enrijecimento da pele. A ausência do fenômeno de Raynaud em condições de diagnóstico diferencial com frequência ajuda a distinguir essas condições de um caso de esclerose sistêmica progressiva e da síndrome CREST.
A morfeia, também denominada escleroderma localizado, caracteriza-se pelo aparecimento de pequenas áreas agudamente delimitadas de pele endurecida que se tornam generalizadas. Distingue-se da esclerose sistêmica progressiva pela ausência do fenômeno de Raynaud, esclerodactilia ou das complicações do escleroderma. Foram introduzidas inovações no tratamento da esclerose sistêmica progressiva e da morfeia. Foi demonstrado que a exposição ao psoraleno e à radiação ultravioleta A (PUVA) melhora dramaticamente ambas as condições.20 A exposição à radiação UVA1 (o espectro mais longo de UVA, de 340 a 400 nm) resultou em benefícios para paciente com escleroderma localizado.71 Evidências anedóticas sugerem que o uso tópico de calcipotrieno constitui um tratamento efetivo da morfeia.72 Novos estudos se fazem necessários para confirmar a eficácia desses tratamentos. Relatos anedóticos indicaram que a minociclina pode beneficiar pacientes com esclerose sistêmica progressiva, embora ainda seja preciso realizar triagens controladas.73
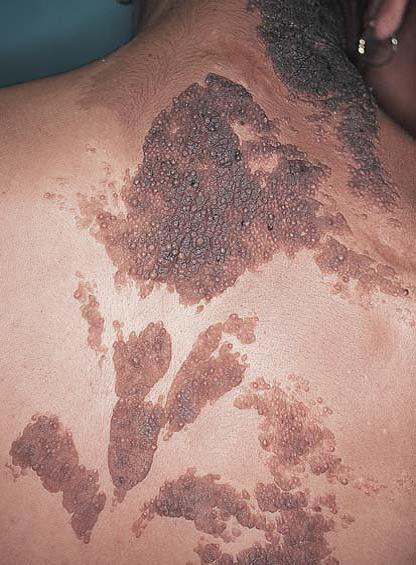
Conforme o transplante de órgãos se torna mais comum, outra doença similar ao escleroderma – a doença do enxerto versus hospedeiro – vai se tornando mais frequente, em particular após transplantes de medula [ver Medicina Transfusional; e Transplante de células hematopoiéticas]. A doença do enxerto versus hospedeiro apresenta dois estágios. O primeiro estágio – doença do enxerto versus hospedeiro aguda – desenvolve-se em 10 a 40 dias após o transplante e consiste no aparecimento de erupções cutâneas eritematosas maculares e papulares, com frequência associadas a febre, hepatomegalia, linfadenopatia ou sintomas gastrintestinais. A doença do enxerto versus hospedeiro crônica costuma se desenvolver em 3 meses após o transplante, mas pode ocorrer mais tarde. Essa fase da doença consiste no aparecimento de pápulas arroxeadas, semelhantes ao líquen plano [Figura 26]. As alterações cutâneas esclerodermatosas, com telangiectasia, hiperpigmentação reticulada e alopecia, são mais características. O mesmo marcador sorológico que foi implicado no desenvolvimento da esclerose sistêmica progressiva (isto é, anticorpos antirreceptor de PDGF) também está presente nos pacientes com doença do enxerto versus hospedeiro crônica.74 Tanto a ciclosporina como o PUVA mostraram-se úteis na prevenção e no tratamento da doença do enxerto versus hospedeiro.75,76 O infliximabe foi utilizado com bastante sucesso no tratamento da doença do enxerto versus hospedeiro aguda.77
Figura 26. Pápulas planas são observadas nesta reação de enxerto versus hospedeiro crônica liquenoide.
O enrijecimento da pele similar ao escleroderma também ocorre na fasciite eosinofílica. É típico haver enrugamento da pele nos membros associado à dor. O sinal do sulco (sulcos suaves ao longo das veias) foi descrito em pacientes com fasciite eosinofílica. Em contraste com a esclerose sistêmica progressiva, não há fenômeno de Raynaud. O diagnóstico definitivo requer exame de biópsia da pele e da fáscia que recobre o músculo afetado. Em alguns casos de fasciite eosinofílica, há desenvolvimento de anomalias hematológicas, tais como anemia aplástica, trombocitopenia, doença de Hodgkin e leucemias.78
O lúpus eritematoso sistêmico (LES) abrange numerosas manifestações, incluindo manifestações inespecíficas de fenômeno de Raynaud, fotossensibilidade, alopecia e úlceras mucosas. As manifestações cutâneas mais específicas do LES são o lúpus discoide (caracterizado pelo aparecimento de lesões cutâneas arredondadas com cicatriz, hipopigmentação central e uma borda hiperpigmentada) e o eritema malar [ver Lúpus eritematoso sistêmico]. Conforme aumenta nosso conhecimento acerca do lúpus, o espectro de doenças cutâneas associadas a esse distúrbio continua em expansão. O lúpus cutâneo subagudo, uma variante sorologicamente caracterizada pela presença de anticorpos anti-Ro e anti-La, está associado à formação de lesões cutâneas anulares ou psoriasiformes [Figura 27].
Figura 27. Manchas anulares eritematosas de raspagem são características do lúpus eritematoso cutâneo subagudo.
A síndrome do anticorpo anticardiolipina, que pode ocorrer em pacientes com LES, foi descrita em pacientes que haviam sofrido repetidos episódios de flebite, trombose arterial e abortos repetidos. Infartos cutâneos são manifestações comuns e também pode haver livedo reticular. Os pacientes podem apresentar resultados falso-positivos em testes sorológicos para sífilis, enquanto o anticoagulante lúpico circulante pode estar presente. A ocorrência de anticorpos antifosfolipídeos circulantes constitui a principal característica sorológica dessa síndrome. Entretanto, além de muitos indivíduos assintomáticos possuírem anticorpos antifosfolipídeos,79 o teste de detecção desses anticorpos pode resultar falso-negativo. Para alguns pacientes, pode ser necessário realizar uma bateria de testes para estabelecer o diagnóstico. O tempo de reação ao veneno da víbora de Russell diluído, um ensaio para detecção do anticoagulante lúpico circulante, é considerado um dos testes mais sensíveis.80
Outro distúrbio associado ao lúpus é a vasculite em livedo, caracterizada pelo desenvolvimento de dolorosas úlceras recorrentes sobre a parte inferior de pernas e tornozelos. Essas úlceras cicatrizam, deixando no local cicatrizes escleróticas esbranquiçadas. Os pacientes afetados com frequência apresentam livedo reticular. Essa condição, também conhecida como atrofia branca, foi atribuída à ocorrência de processos trombóticos, em vez da deposição de imunocomplexos ou da vasculite leucocitoclástica.81
O lúpus neonatal constitui uma síndrome distinta, em que pápulas e manchas eritematosas anulares aparecem na face de bebês recém-nascidos. Esse distúrbio foi atribuído à passagem transplacentária de anticorpos anti-Ro e, ocasionalmente, de anticorpos anti-La. As mães costumam ser assintomáticas, mas algumas podem ter lúpus ou síndrome de Sjögren. O bloqueio cardíaco congênito é a complicação mais grave associada a esse distúrbio.82
A dermopatia fibrosante nefrogênica é uma síndrome recém-descrita, caracterizada por um endurecimento cutâneo progressivo que afeta especialmente os membros de pacientes com doença renal. O enrijecimento da pele pode ser tão severo que o paciente pode apresentar contraturas por flexão, com consequente limitação da mobilidade.83 A fibrose pode afetar órgãos internos, entre os quais miocárdio, pulmões e vasculatura. A condição foi atribuída ao gadolínio contido nos contrastes empregados nos exames de imagem por ressonância magnética. Por isso, recomenda-se evitar o uso de gadolínio para pacientes com doença renal.84
O autor contou com apoio financeiro para fins educacionais da Abbott Laboratories, Inc., Amgen, Inc. e Centocor, Inc.
O FDA ainda não aprovou os seguintes fármacos para os usos específicos descritos nesse capítulo: infliximabe e bloqueadores de TNF-alfa, para tratamento da sarcoidose e da granulomatose de Wegener; e calcipotrieno, para tratamento da morfeia.
1. Baughman RP, Drent M, Kavuru M, Judson MA, Costabel U, du Bois R, et al. Infliximabe therapy in patients with chronic sarcoidosis and pulmonary involvement. Am J Respir Crit Care Med. 2006 Oct 1;174(7):795-802. Epub 2006 Jul 13.
2. Heffernan MP, Smith DI. Adalimumab for treatment of cutaneous sarcoidosis. Arch Dermatol. 2006 Jan;142(1):17-9.
3. Haley H, Cantrell W, Smith K. Infliximabe therapy for sarcoidosis (lupus pernio). Br J Dermatol. 2004 Jan;150(1):146-9.
4. Menon Y, Cucurull E, Reisin E, Espinoza LR. Interferon-alpha-associated sarcoidosis responsive to infliximabe therapy. Am J Med Sci. 2004 Sep;328(3):173-5.
5. English JC 3rd, Patel PJ, Greer KE. Sarcoidosis. J Am Acad Dermatol. 2001 May;44(5):725-43.
6. Kleinert J, Lorenz M, Köstler W, Hörl W, Sunder-Plassmann G, Soleiman A. Refractory Wegener's granulomatosis responds to
7. Stone JH, Holbrook JT, Marriott MA, Tibbs AK, Sejismundo LP, Min YI, et al. Solid malignancies among patients in the Wegener's Granulomatosis Etanercepte
8. Aries PM, Lamprecht P, Gross WL. Biological therapies: new treatment options for ANCA-associated vasculitis? Expert Opin Biol Ther. 2007 Apr;7(4):521-33.
9. Davis MD, Daoud MS, McEvoy MT, Su WP. Cutaneous manifestations of Churg-Strauss syndrome: a clinicopathologic correlation. J Am Acad Dermatol. 1997 Aug;37(
10. Seo P, Stone JH. The antineutrophil cytoplasmic antibody-associated vasculitides. Am J Med. 2004 Jul 1;117(1):39-50.
11. Gong F, Shiraishi H, Momoi MY. Follow-up of coronary artery lesions caused by Kawasaki disease and the value of coronary angiography. Chin Med J (Engl). 2002 May;115(5):681-4.
12. Curtis N. Kawasaki disease and toxic shock syndrome--at last the etiology is clear? Adv Exp Med Biol. 2004;549:191-200.
13. Newburger JW, Takahashi M, Gerber MA, Gewitz MH, Tani LY, Burns JC, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Circulation. 2004 Oct 26;110(17):2747-71.
14. Oates-Whitehead RM, Baumer JH, Haines L, Love S, Maconochie IK, Gupta A, et al. Intravenous immunoglobulin for the treatment of Kawasaki disease in children. Cochrane Database Syst Rev. 2003;(4):CD004000.
15. Ringpfeil F, Lebwohl MG, Christiano AM, Uitto J. Pseudoxanthoma elasticum: mutations in the MRP6 gene encoding a transmembrane ATP-binding cassette (ABC) transporter. Proc Natl Acad Sci U S A. 2000 May 23;97(11):6001-6.
16. Lebwohl MG, Distefano D, Prioleau PG, Uram M, Yannuzzi LA, Fleischmajer R. Pseudoxanthoma elasticum and mitral-valve prolapse. N Engl J Med. 1982 Jul 22;307(4):228-31.
17. Lebwohl M, Halperin J, Phelps RG. Brief report: occult pseudoxanthoma elasticum in patients with premature cardiovascular disease. N Engl J Med. 1993 Oct 21;329(17):1237-9.
18. Lebwohl M, Phelps RG, Yannuzzi L, Chang S, Schwartz I, Fuchs W. Diagnosis of pseudoxanthoma elasticum by scar biopsy in patients without characteristic skin lesions. N Engl J Med. 1987 Aug 6;317(6):347-50.
19. D'Alessandro A, Muzi G, Monaco A, Filiberto S, Barboni A, Abbritti G. Yellow nail syndrome: does protein leakage play a role? Eur Respir J. 2001 Jan;17(1):149-52.
20. Litonjua P, Piñero-Piloña A, Aviles-Santa L, Raskin P. Prevalence of acanthosis nigricante in newly-diagnosed type 2 diabetes. Endocr Pract. 2004 Mar-Apr;10(2):101-6.
21. Verrotti A, Chiarelli F, Amerio P, Morgese G. Necrobiosis lipoidica diabeticorum in children and adolescents: a clue for underlying renal and retinal disease. Pediatr Dermatol. 1995 Sep;12(3):220-3.
22. Rho YW, Suhr KB, Lee JH, Park JK. A clinical observation of scleredema adultorum and its relationship to diabetes. J Dermatol. 1998 Feb;25(2):103-7.
23. Dogra S, Handa S, Kanwar AJ. Dexamethasone pulse therapy for scleredema. Pediatr Dermatol. 2004 May-Jun;21(3):280-1.
24. Bowen AR, Smith L, Zone JJ. Scleredema adultorum of Buschke treated with radiation. Arch Dermatol. 2003 Jun;139(6):780-4.
25. Janiga JJ, Ward DH, Lim HW. UVA-1 as a treatment for scleredema. Photodermatol Photoimmunol Photomed. 2004 Aug;20(4):210-1.
26. Hecker MS, Lebwohl MG. Recalcitrant pyoderma gangrenosum: treatment with thalidomide. J Am Acad Dermatol. 1998 Mar;38(3):490-1.
27. Tan MH, Gordon M, Lebwohl O, George J, Lebwohl MG. Improvement of Pyoderma gangrenosum and psoriasis associated with Crohn disease with anti-tumor necrosis factor alpha monoclonal antibody. Arch Dermatol. 2001 Jul;137(7):930-3.
28. Reichrath J, Bens G, Bonowitz A, Tilgen W. Treatment recommendations for pyoderma gangrenosum: an evidence-based review of the literature based on more than 350 patients. J Am Acad Dermatol. 2005 Aug;53(2):273-83.
29. Kanaseki T, Torigoe T, Hirohashi Y, Hirai I, Himi T, Sato N. Identification of germline mutation of PTEN gene and analysis of apoptosis resistance of the lymphocytes in a patient with Cowden disease. Pathobiology. 2002;70(1):34-9.
30. Barax CN, Lebwohl M, Phelps RG. Multiple hamartoma syndrome. J Am Acad Dermatol. 1987 Aug;17(2 Pt 2):342-6.
31. García JM, Silva J, Peña C, Garcia V, Rodríguez R, Cruz MA, et al. Promoter methylation of the PTEN gene is a common molecular change in breast cancer. Genes Chromosomes Cancer. 2004 Oct;41(2):117-24.
32. Collin P, Pukkala E, Reunala T. Malignancy and survival in dermatitis herpetiformis: a comparison with coeliac disease. Gut. 1996 Apr;38(4):528-30.
33. Scott RJ, Crooks R, Meldrum CJ, Thomas L, Smith CJ, Mowat D, et al. Mutation analysis of the STK11/LKB1 gene and clinical characteristics of an Australian series of Peutz-Jeghers syndrome patients. Clin Genet. 2002 Oct;62(4):282-7.
34. Mallipeddi R. Epidermolysis bullosa and cancer. Clin Exp Dermatol. 2002 Nov;27(8):616-23.
35. Christiano AM, LaForgia S, Paller AS, McGuire J, Shimizu H, Uitto J. Prenatal diagnosis for recessive dystrophic epidermolysis bullosa in 10 families by mutation and haplotype analysis in the type VII collagen gene (COL7A1). Mol Med. 1996 Jan;2(1):59-76.
36. Gardella R, Castiglia D, Posteraro P, Bernardini S, Zoppi N, Paradisi M, et al. Genotype-phenotype correlation in italian patients with dystrophic epidermolysis bullosa. J Invest Dermatol. 2002 Dec;119(6):1456-62.
37. Dubrey SW, Burke MM, Hawkins PN, Banner NR. Cardiac transplantation for amyloid heart disease: the United Kingdom experience. J Heart Lung Transplant. 2004 Oct;23(10):1142-53.
38. Palladini G, Perfetti V, Perlini S, Obici L, Lavatelli F, Caccialanza R, et al. The combination of thalidomide and intermediate-dose dexamethasone is an effective but toxic treatment for patients with primary amyloidosis (AL). Blood. 2005 Apr 1;105(7):2949-51. Epub 2004 Nov 30.
39. Topar G, Staudacher C, Geisen F, Gabl C, Fend F, Herold M, et al. Urticaria pigmentosa: a clinical, hematopathologic, and serologic study of 30 adults. Am J Clin Pathol. 1998 Mar;109(3):279-85.
40. Horny HP, Ruck P, Kröber S, Kaiserling E. Systemic mast cell disease (mastocytosis). General aspects and histopathological diagnosis. Histol Histopathol. 1997 Oct;12(4):1081-9.
41. Kiszewski AE, Durán-Mckinster C, Orozco-Covarrubias L, Gutiérrez-Castrellón P, Ruiz-Maldonado R. Cutaneous mastocytosis in children: a clinical analysis of 71 cases. J Eur Acad Dermatol Venereol. 2004 May;18(3):285-90.
42. Pottage JC Jr, Kessler HA. Herpes simplex virus resistance to acyclovir: clinical relevance. Infect Agents Dis. 1995 Sep;4(3):115-24.
43. Chibo D, Mijch A, Doherty R, Birch C. Novel mutations in the thymidine kinase and DNA polymerase genes of acyclovir and foscarnet resistant herpes simplex viruses infecting an immunocompromised patient. J Clin Virol. 2002 Aug;25(2):165-70.
44. Lalezari J, Schacker T, Feinberg J, Gathe J, Lee S, Cheung T, et al. A randomized, double-blind, placebo-controlled trial of cidofovir gel for the treatment of acyclovir-unresponsive mucocutaneous herpes simplex virus infection in patients with Aids. J Infect Dis. 1997 Oct;176(4):892-8.
45. Chang CC, Chomel BB, Kasten RW, Tappero JW, Sanchez MA, Koehler JE. Molecular epidemiology of Bartonella henselae infection in human immunodeficiency virus-infected patients and their cat contacts, using pulsed-field gel electrophoresis and genotyping. J Infect Dis. 2002 Dec 15;186(12):1733-9. Epub 2002 Nov 22.
46. Agan BK, Dolan MJ. Laboratory diagnosis of Bartonella infections. Clin Lab Med. 2002 Dec;22(4):937-62.
47. Resneck JS Jr, Van Beek M, Furmanski L, Oyugi J, LeBoit PE, Katabira E, et al. Etiology of pruritic papular eruption with HIV infection in Uganda. JAMA. 2004 Dec 1;292(21):2614-21.
48. Albrecht D, Meyer T, Lorenzen T, Stoehr A, Arndt R, Plettenberg A. Epidemiology of HHV-8 infection in HIV-positive patients with and without Kaposi sarcoma: diagnostic relevance of serology and PCR. J Clin Virol. 2004 Jun;30(2):145-9.
49. Parsonnet J. Nonmenstrual toxic shock syndrome: new insights into diagnosis, pathogenesis, and treatment. Curr Clin Top Infect Dis. 1996;16:1-20.
50. Childers BJ, Potyondy LD, Nachreiner R, Rogers FR, Childers ER, Oberg KC, et al. Necrotizing fasciitis: a fourteen-year retrospective study of 163 consecutive patients. Am Surg. 2002 Feb;68(2):109-16.
51. Nakafusa J, Misago N, Miura Y, Kayaba M, Tanaka T, Narisawa Y. The importance of serum creatine phosphokinase level in the early diagnosis, and as a prognostic factor, of Vibrio vulnificus infection. Br J Dermatol. 2001 Aug;145(2):280-4.
52. Philipp MT, Masters E, Wormser GP, Hogrefe W, Martin D. Serologic evaluation of patients from Missouri with erythema migrans-like skin lesions with the C6 Lyme test. Clin Vaccine Immunol. 2006 Oct;13(10):1170-1.
53. Lam CW, Leung CY, Lee KC, Xie J, Lo FM, Au TS, et al. Novel mutations in the PATCHED gene in basal cell nevus syndrome. Mol Genet Metab. 2002 May;76(1):57-61.
54. Smahi A, Courtois G, Rabia SH, Döffinger R, Bodemer C, Munnich A, et al. The NF-kappaB signalling pathway in human diseases: from incontinentia pigmenti to ectodermal dysplasias and immune-deficiency syndromes. Hum Mol Genet. 2002 Oct 1;11(20):2371-5.
55. Dasgupta B, Dugan LL, Gutmann DH. The neurofibromatosis 1 gene product neurofibromin regulates pituitary adenylate cyclase-activating polypeptide-mediated signaling in astrocytes. J Neurosci. 2003 Oct 1;23(26):8949-54.
56. Xiao GH, Chernoff J, Testa JR. NF2: the wizardry of merlin. Genes Chromosomes Cancer. 2003 Dec;38(4):389-99.
57. Nellist M, Sancak O, Goedbloed MA, Rohe C, van Netten D, Mayer K, et al. Distinct effects of single amino-acid changes to tuberin on the function of the tuberin-hamartin complex. Eur J Hum Genet. 2005 Jan;13(1):59-68.
58. Germain DP, Shabbeer J, Cotigny S, Desnick RJ. Fabry disease: twenty novel alpha-galactosidase A mutations and genotype-phenotype correlations in classical and variant phenotypes. Mol Med. 2002 Jun;8(6):306-12.
59. Desnick RJ, Brady R, Barranger J, Collins AJ, Germain DP, Goldman M, et al. Fabry disease, an under-recognized multisystemic disorder: expert recommendations for diagnosis, management, and enzyme replacement therapy. Ann Intern Med. 2003 Feb 18;138(4):338-46.
60. Daoud MS, Hutton KP, Gibson LE. Cutaneous periarteritis nodosa: a clinicopathological study of 79 cases. Br J Dermatol. 1997 May;136(5):706-13.
61. Guillevin L, Lhote F, Amouroux J, Gherardi R, Callard P, Casassus P. Antineutrophil cytoplasmic antibodies, abnormal angiograms and pathological findings in polyarteritis nodosa and Churg-Strauss syndrome: indications for the classification of vasculitides of the polyarteritis Nodosa Group. Br J Rheumatol. 1996 Oct;35(10):958-64.
62. Poliak SC, Lebwohl MG, Parris A, Prioleau PG. Reactive perforating collagenosis associated with diabetes mellitus. N Engl J Med. 1982 Jan 14;306(2):81-4.
63. Arch-Ferrer JE, Beenken SW, Rue LW, Bland KI, Diethelm AG. Therapy for calciphylaxis: an outcome analysis. Surgery. 2003 Dec;134(6):941-4.
64. Araya CE, Fennell RS, Neiberger RE, Dharnidharka VR. Sodium thiosulfate treatment for calcific uremic arteriolopathy in children and young adults. Clin J Am Soc Nephrol. 2006 Nov;1(6):1161-6. Epub 2006 Aug 30.
65. Guldbakke KK, Khachemoune A. Calciphylaxis. Int J Dermatol. 2007 Mar;46(3):231-8.
66. Kasteler JS, Callen JP. Scalp involvement in dermatomyositis. Often overlooked or misdiagnosed. JAMA. 1994 Dec 28;272(24):1939-41.
67. Buchbinder R, Forbes A, Hall S, Dennett X, Giles G. Incidence of malignant disease in biopsy-proven inflammatory myopathy. A population-based cohort study. Ann Intern Med. 2001 Jun 19;134(12):1087-95.
68. Hill CL, Zhang Y, Sigurgeirsson B, Pukkala E, Mellemkjaer L, Airio A, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001 Jan 13;357(9250):96-100.
69. Baroni SS, Santillo M, Bevilacqua F, Luchetti M, Spadoni T, Mancini M, et al. Stimulatory autoantibodies to the PDGF receptor in systemic sclerosis. N Engl J Med. 2006 Jun 22;354(25):2667-76.
70. Kanekura T, Fukumaru S, Matsushita S, Terasaki K, Mizoguchi S, Kanzaki T. Successful treatment of scleroderma with PUVA therapy. J Dermatol. 1996 Jul;23(7):455-9.
71. Kerscher M, Volkenandt M, Gruss C, Reuther T, von Kobyletzki G, Freitag M, et al. Low-dose UVA phototherapy for treatment of localized scleroderma. J Am Acad Dermatol. 1998 Jan;38(1):21-6.
72. Tay YK. Topical calcipotriol ointment in the treatment of morphea. J Dermatolog Treat. 2003 Dec;14(4):219-21.
73. Le CH, Morales A, Trentham DE. Minocycline in early diffuse scleroderma. Lancet. 1998 Nov 28;352(9142):1755-6.
74. Svegliati S, Olivieri A, Campelli N, Luchetti M, Poloni A, Trappolini S, et al. Stimulatory autoantibodies to PDGF receptor in patients with extensive chronic graft-versus-host disease. Blood. 2007 Jul 1;110(1):237-41. Epub 2007 Mar 15.
75. Zikos P, Van Lint MT, Frassoni F, Lamparelli T, Gualandi F, Occhini D, et al. Low transplant mortality in allogeneic bone marrow transplantation for acute myeloid leukemia: a randomized study of low-dose cyclosporin versus low-dose cyclosporin and low-dose methotrexate. Blood. 1998 May 1;91(9):3503-8.
76. Vogelsang GB, Wolff D, Altomonte V, Farmer E, Morison WL, Corio R, et al. Treatment of chronic graft-versus-host disease with ultraviolet irradiation and psoralen (PUVA). Bone Marrow Transplant. 1996 Jun;17(6):1061-7.
77. Yamane T, Yamamura R, Aoyama Y, Nakamae H, Hasegawa T, Sakamoto C, et al. Infliximabe for the treatment of severe steroid refractory acute graft-versus-host disease in three patients after allogeneic hematopoietic transplantation. Leuk Lymphoma. 2003 Dec;44(12):2095-7.
78. Kim SW, Rice L, Champlin R, Udden MM. Aplastic anemia in eosinophilic fasciitis: responses to immunosuppression and marrow transplantation. Haematologia (Budap). 1997;28(3):131-7.
79. Tektonidou MG, Sotsiou F, Nakopoulou L, Vlachoyiannopoulos PG, Moutsopoulos HM. Antiphospholipid syndrome nephropathy in patients with systemic lupus erythematosus and antiphospholipid antibodies: prevalence, clinical associations, and long-term outcome. Arthritis Rheum. 2004 Aug;50(8):2569-79.
80. Proven A, Bartlett RP, Moder KG, Chang-Miller A, Cardel LK, Heit JA, et al. Clinical importance of positive test results for lupus anticoagulant and anticardiolipin antibodies. Mayo Clin Proc. 2004 Apr;79(4):467-75.
81. McCalmont CS, McCalmont TH, Jorizzo JL, White WL, Leshin B, Rothberger H. Livedo vasculitis: vasculitis or thrombotic vasculopathy? Clin Exp Dermatol. 1992 Jan;17(1):4-8.
82. Brucato A, Franceschini F, Buyon JP. Neonatal lupus: long-term outcomes of mothers and children and recurrence rate. Clin Exp Rheumatol. 1997 Sep-Oct;15(5):467-73.
83. Introcaso CE, Hivnor C, Cowper S, Werth VP. Nephrogenic fibrosing dermopathy/nephrogenic systemic fibrosis: a case series of nine patients and review of the literature. Int J Dermatol. 2007 May;46(5):447-52.
84. Centers for Disease Control and Prevention (CDC). Nephrogenic fibrosing dermopathy associated with exposure to gadolinium-containing contrast agents – St. Louis, Missouri, 2002-2006. MMWR Morb Mortal Wkly Rep. 2007 Feb 23;56(7):137-41.
"Prezada Brenda, Sua observação está correta. De fato, o tópico é sobre "hipotireoidismo". O texto já foi corrigido. Agradecemos seu comentário. Atenciosamente, Atendimento MedicinaNET"
"Onde está o tópico "hipertireoidismo", acredito que o certo seria "hipotireoidismo", por toda a descrição dos sinais."
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.