(Carregando Índice)... (Carregando Índice)... |
Autores:
Fabio Pires de Souza Santos
Especialista em Hematologia pelo Hospital das Clínicas da Faculdade de Medicina da USP
Gustavo dos Santos Fernandes
Especialista em Hematologia pelo Hospital das Clínicas da Faculdade de Medicina da USP
Última revisão: 27/09/2008
Comentários de assinantes: 2
Os linfomas são neoplasias hematológicas que formam um grupo amplo de entidades malignas que podem apresentar padrão histológico e comportamento clínico diferenciado, constituindo-se muitas vezes em um desafio para o clínico. Tais diferenças baseiam-se em diversidade de evolução e prognóstico, indo desde doenças que levam anos de sua instalação a seu diagnósticos (indolentes) até entidades malignas extremamente agressivas, que podem levar ao óbito em alguns dias se não forem tratadas adequadamente. Torna-se imperativo, portanto, que tais condições tenham seu diagnóstico feito prontamente. Sendo proliferações neoplásicas malignas de linfócitos, residem predominantemente em órgãos linfóides. A classificação dos linfomas os divide em dois grandes grupos:
Linfoma não-Hodgkin.
Os linfomas não-Hodgkin (LNH) são todos aqueles não caracterizados como linfomas de Hodgkin (LH), sendo, portanto, um grupo de mais de 30 doenças diferentes, com apresentações clínicas e prognósticos muito variados. Correspondem a mais de 70% dos linfomas.
Dada a grande variedade de apresentações clínicas e achados histopatológicos, sua classificação adequada continua sendo um grande desafio para hematologistas e patologistas. Durante as últimas décadas, diversos esquemas de classificação foram propostos, e à medida que novas técnicas foram sendo desenvolvidas (por exemplo, imunofenotipagem, citogenética, biologia molecular), os esquemas de classificação eram revistos, com descrição de novos e reclassificação de subtipos antigos.
De todo modo, qualquer classificação de linfoma deve procurar definir não apenas o tipo histológico, mas também realizar correlação clínica e determinar o grau de agressividade dos diversos subtipos histológicos, haja visto que essa informação tem extrema importância no planejamento do tratamento e no prognóstico dos doentes. Tendo isto em mente, a classificação histológica mais utilizada hoje é a classificação da Organização Mundial da Saúde (OMS), uma atualização da antiga classificação REAL (do inglês Revised European American Classification of Lymphoid Neoplasms). No entanto, iremos nos guiar neste capítulo por uma classificação clínica dos LNH, em virtude sua maior praticidade e relevância na prática diária, de modo que possa se estabelecer com precisão o prognóstico do doente e iniciar as medidas terapêuticas apropriadas.
Os LNH representam cerca de 90% dos linfomas, e cerca de 4% de todas as neoplasias. Acometem todas as faixas etárias, e os linfomas mais agressivos são comuns em crianças enquanto os indolentes, mais comuns em idosos. A incidência é em torno de 8,5 casos/100.000 habitantes (< 65 anos) e 69 casos/100.000 pessoas (> 65 anos). O subtipo mais freqüente é o linfoma difuso de grandes células B, seguido pelo linfoma folicular.
A classificação dos LNH de acordo com seu comportamento clínico é composta de três grandes grupos:
linfomas indolentes: sobrevida, se não for tratado, de anos
linfomas agressivos: sobrevida, se não for tratado, de meses
linfomas muito agressivos: sobrevida, se não for tratado, de semanas
Os linfomas indolentes em geral não respondem bem à quimioterapia e não são curáveis; no entanto, mesmo os pacientes que não tiveram uma resposta adequada ao tratamento podem ter uma sobrevida longa. Já os linfomas agressivos e, especialmente os muito agressivos, respondem muito bem ao tratamento e são potencialmente curáveis, entretanto, apenas os pacientes com resposta completa à quimioterapia têm chance de obter uma cura. Alguns subtipos de linfomas não se encaixam perfeitamente nessa classificação (por exemplo, linfoma de células do manto), que, de todo modo, tem sido utilizada e norteia os estudos clínicos.
Os principais subtipos de LNH de acordo com sua classificação clínica estão listados na tabela 1. Na tabela 2 é descrita a classificação da OMS.
Tabela 1: Tipos histológicos de LNH de acordo com comportamento clínico
|
linfomas indolentes |
|
linfoma folicular Linfoma linfocítico de células pequenas (variante da leucemia linfocítica crônica – LLC) Linfoma de células B da zona marginal (por exemplo, linfoma MALT) linfoma de células do manto Mycosis fungoides/Síndrome de Sézary |
|
linfomas agressivos |
|
linfoma difuso de grandes células B |
|
linfomas muito agressivos |
|
Linfoma linfoblástico de células B/T (variante da leucemia linfocítica aguda – LLA) Linfoma/leucemia de células T do adulto |
Tabela 2: Linfomas – Classificação da OMS
|
Nome |
Exemplos |
|
Leucemia/Linfoma linfoblástico de células B/T |
Leucemia/Linfoma linfoblástico de células B precursoras Leucemia/Linfoma linfoblástico de células T precursoras |
|
LLC Macroglobulinemia de Waldenström Leucemia de células pilosas Neoplasias de plasmócitos linfoma folicular Linfoma difuso de grandes células linfoma de células do manto Linfoma de Burkitt | |
|
Neoplasias de células T maduras |
Leucemia de linfócitos grandes e granulares Leucemia/Linfoma de células T do adulto Mycosis fungoides Linfoma de grandes células anaplásico |
|
Linfoma de Hodgkin |
LH clássico LH com predomínio linfocítico nodular |
Na abordagem do doente com LNH, deve-se procurar diagnosticar, além do subtipo histológico preciso, a extensão da doença e atentar para comorbidades do doente e seu performance status, pois estes terão importância para determinar o prognóstico e o tratamento a ser iniciado.
É extremamente variável, em razão das diferentes apresentações clínicas. Alguns padrões são mais comuns, no entanto, de acordo com o comportamento clínico do LNH.
linfomas indolentes:
o evolução prolongada, de meses a anos;
o linfadenopatia em geral periférica;
o sem sintomas obstrutivos ou compressivos;
o raramente tem sintomas sistêmicos, exceto que estejam evoluindo para formas mais agressivas;
o os linfonodos podem aumentar e regridir espontaneamente;
o podem-se observar linfonodos em localizações atípicas, como epitrocleares;
o costumam ter envolvimento da medula óssea ao diagnóstico;
o exemplo: linfoma folicular.
linfomas agressivos:
o história com evolução nos últimos meses, muitas vezes com dor e sintomas obstrutivos;
o sintomas sistêmicos são comuns;
o a presença de sítios extranodais é mais prevalente neste subgrupo;
o há elevação aos exames laboratoriais, como a desidrogenase lática (DHL) e a b2-microglobulina;
o podem ser resultado da progressão de linfoma indolente para formas mais agressivas;
o exemplos: linfoma difuso de grandes células B.
linfomas muito agressivos:
o evolução rápida em algumas semanas;
o formação de grandes massas tumorais com grande deformidade; compressão de estruturas vitais, em alguns casos
o pode ter síndrome de lise tumoral espontânea; DHL e b2-microglobulina estão muito elevadas;
o em geral estão disseminados ao diagnóstico, com acometimento da medula óssea.
o exemplo: Linfoma de Burkitt – forma massas abdominais ou tumores em região mandibular, que apresentam rápido crescimento.
Outros dados da história que são de relevância clínica:
Linfadenopatia:
Mais de 60%-70% dos doentes com LNH apresentam linfadenopatia periférica. Lembrar que infecções bacterianas, virais e por micobactéria podem decorrer em linfadenopatia, especialmente em região cervical, e devem entrar na lista de diagnósticos diferenciais.
Sintomas sistêmicos:
Até 40% dos doentes com LNH podem ter sintomas sistêmicos, que são os chamados sintomas “B”. Estes incluem:
o perda de peso > 10% do peso basal nos últimos 6 meses;
o sudorese noturna;
o febre > 38ºC.
Quando presentes, esses sintomas indicam um pior prognóstico e são mais comuns em LNH agressivos/muito agressivos, ocorrendo em cerca de 47% destes e em cerca de um quarto dos pacientes com LNH indolente. Outros sintomas sistêmicos que podem ocorrer incluem fadiga, prurido e fraqueza.
Sintomas localizatórios:
Quando presentes, dão indícios do local onde o LNH está presente. Como exemplo, podemos citar:
o tosse, desconforto respiratório: adenopatia mediastinal (vista em até 20% dos LNH);
o dor abdominal, vômitos, aumento de volume abdominal: linfadenopatia retroperitoneal, acometimento do TGI, infiltração hepática ou esplênica;
o dor óssea, fratura patológica: LNH primário do osso, acometimento secundário;
o dispnéia: infiltração pulmonar, derrame pleural;
o úlceras de pele, máculas, eritemas: linfoma primário cutâneo ou infiltração secundária;
o rinorréia, cefaléia, epistaxe: infiltração em seio nasal, nasofaringe.
Fatores de risco para LNH:
o infecção por HIV, HTLV-1, HBV, HCV, EBV, HHV-8, Helicobacter pylori, SV40. Todos os agentes anteriormente citados são fatores de risco para determinados tipos de linfomas; desta forma, o aparecimento da doença não é totalmente vinculado ao fator de risco, podendo aparecer na ausência deste. A exceção a essa regra é a doença de alto grau conhecida como leucemia/linfoma de células T do adulto (ATLL), causada pela infecção pelo vírus HTLV-1, não ocorrendo em indivíduos não infectados e aparecendo em cerca 3% daqueles infectados pelo vírus;
o colagenoses (lúpus eritematoso sistêmico, doença reumatóide, síndrome de Sjögren);
o crioglobulinemia mista;
o imunodeficiências congênitas (imunodeficiência combinada severa, Wiskott-Aldritch);
o transplante de órgãos sólidos;
o doença de Crohn, doença celíaca (para linfoma do trato gastrointestinal).
Alterações de exames laboratoriais:
Alguns pacientes com LNH podem vir à consulta médica em virtude das alterações de exames complementares que sugerem o diagnóstico de neoplasia linfóide. Dentre as mais comuns, podemos citar:
o anemia, leucopenia e/ou trombocitopenia sem etiologia definida (em geral por infiltração da medula óssea);
o hiperuricemia: pode ter sintomas de gota associada; ocorre geralmente em LNH de alto índice de proliferação (por exemplo, Linfoma de Burkitt);
o hipercalcemia: mais comum em linfoma/leucemia de células T do adulto.
Atentar para todos os possíveis sítios de linfadenopatia – cervical, axilar, inguinal, anel de Waldeyer (tonsilas, base da língua, orofaringe), supraclavicular, sítios pouco comuns (pré-auricular, epitroclear etc.). Avaliar também a presença de hepatoesplenomegalia, que pode indicar acometimento desses órgãos.
Deve-se atentar também para a detecção de sítios extranodais de acometimento, dos quais os mais comuns são o trato gastrointestinal, a pele, os testículos (a palpação é mandatória), os ossos e os rins.
Essencial para que se faça o diagnóstico do subtipo específico de LNH. Deve-se obter uma amostra intacta do linfonodo, ou uma biópsia adequada da massa tumoral quando presente. A punção por agulha fina (PAF) pode ser realizada em alguns sítios de muito difícil acesso, porém ela não substitui a biópsia linfonodal.
Essencial para estadiamento da doença, a presença de acometimento de medula óssea confirma estádio IV para o LNH (vide abaixo). Devido à heterogeneidade da infiltração, deve ser feita bilateralmente.
Importantes para determinar subtipo e dar informações prognósticas. A diferenciação entre linfomas de células B ou células T só pode ser dada por esse método, sendo a histologia B responsável por aproximadamente 85% dos casos de linfoma.
Embora não sejam específicos para LNH, dois exames laboratoriais, a b2-microglobulina e a DHL, têm muita importância, pois costumam estar elevados em pacientes com LNH e têm correlação com a massa tumoral.
Têm-se graus variados de citopenias em até 57% dos casos de LNH. A série mais comumente acometida é a vermelha (42%). A presença de neutropenia e trombocitopenia em um paciente com LNH é mais rara, porém há uma correlação muito forte entre estas e o acometimento de medula óssea (em quase 100% dos casos). Células malignas no sangue periférico são vistas mais comumente em linfomas indolentes, como o linfoma folicular.
Alguns LNH podem vir acompanhados de pico monoclonal de imunoglobulina ou hipogamaglobulinemia.
Para pacientes com sintomas neurológicos ou LNH com alto risco de infiltrar o sistema nervoso central (por exemplo, Linfoma de Burkitt, Linfoma linfoblástico).
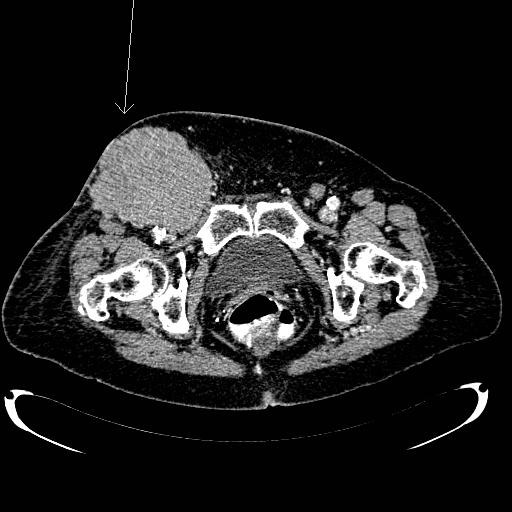
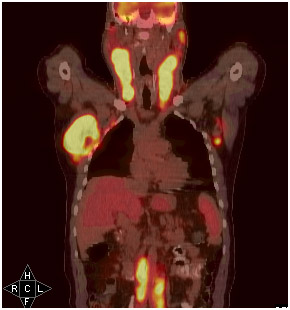
Na avaliação de um doente com suspeita de LNH, alguns exames são de grande valia, sendo a tomografia computadorizada (TC) de pescoço, tórax, abdômen e pelve (figura 1) considerada o exame-padrão. O uso do PET-TC scan (figura 2) incrementa a avaliação, fornecendo uma avaliação funcional da doença e por vezes modificando o estadiamento. Exames como cintilografia com gálio, SPECT, TC de crânio, endoscopia digestiva alta, cintilografia óssea etc. podem ser utilizados de acordo com a necessidade e disponibilidade.
Figura 1: TC de pelve mostrando massa inguinal em paciente com linfoma de alto grau.

Figura 2: PET-TC scan de um paciente com LNH agressivo captando em massas linfonodais cervicais, axilares e inguinais
Após a realização de todos os exames indicados, deve-se determinar o estadiamento do tumor, por meio da escala de Ann Arbor (tabela 3), inicialmente criada para LH, mas posteriormente adaptada para LNH.
Tabela 3: Escala de Ann Arbor para estadiamento de linfomas
|
Estádio |
Definição |
|
I |
Linfoma acometendo uma única cadeia (IE) ou único sítio extranodal (IE) |
|
II |
Linfoma acometendo mais de uma cadeia linfonodal do mesmo lado do diafragma (II) ou com acometimento de um sítio extranodal por contigüidade (IIE) |
|
III |
Linfoma acometendo mais de uma cadeia linfonodal, de ambos os lados do diafragma (III), podendo ter acometimento de órgão extr-nodal por contigüidade (IIIE), ou baço (IIIS), ou ambos (IIIES) |
|
IV |
Linfoma disseminado, acometendo medula óssea e/ou acometimento de sítios extranodais sem contigüidade |
|
Designações: A – Sem sintomas sistêmicos; B – Com sintomas Sistêmicos (febre, perda de peso, sudorese noturna); E – Doença extranodal. | |
O diagnóstico diferencial de LNH (tabela 4) se dá principalmente com LH e outras doenças que cursam com linfadenopatia. Geralmente, se confirma o diagnóstico de LNH por estudo da biópsia linfonodal, e, em alguns casos, por imunofenotipagem e estudo genético. Manifestações mais raras que podem levar a outros diagnósticos diferenciais incluem úlceras gástricas, massas pulmonares, massas abdominais etc.
Tabela 4: Diagnóstico diferencial de LNH
|
Doença |
Comentário |
|
Linfoma de Hodgkin |
Disseminação linfática por contigüidade; raramente extranodal; mais comum ter apresentação mediastinal |
|
Doença linfoproliferativa benigna |
Doença de Castleman |
|
Infecções |
HIV, mononucleose infecciosa, tuberculose, histoplasmose, paracoccidioidomicose |
|
Imunológicas |
Reações a drogas (por exemplo, pseudolinfoma por hidantal), lúpus eritematoso sistêmico, síndrome de Sjögren, doença de Still, sarcoidose |
|
Metástases de neoplasias sólidas |
Linfonodos endurecidos, pétreos, fixos à planos profundos |
De uma maneira prática e simplificada, no entanto acurada, a avaliação prognóstica dos linfomas mais freqüentemente encontrados na prática clínica pode ser feita utilizando-se dois índices prognósticos extensamente validados internacionalmente: o IPI e o FLIPI.
O índice prognóstico internacional (IPI) foi desenvolvido para fornecer uma previsão prognóstica em pacientes com LNH agressivos tratados com esquema quimioterápico-padrão (CHOP), que será descrito adiante. Cada fator apresentado pelo paciente recebe um ponto quando presente e zero quando ausente, podendo a pontuação total variar de 0 a 5 pontos, sendo a gravidade da doença diretamente proporcional ao número de pontos recebidos. A partir da pontuação, os pacientes são divididos em grupos prognósticos: aqueles pacientes que apresentam 0 ou 1 ponto são ditos de baixo risco; aqueles com 2 pontos são estratificados como risco intermediário baixo; os que atingem escore 3 são denominados como risco intermediário alto; e aqueles que recebem pontuação
Os fatores contabilizados (1 ponto cada) no IPI são:
Tabela 5: Escala de performance – Eastern Cooperative Oncology Group (ECOG)
|
performance status |
Definição |
|
0 |
Plenamente ativo, sem restrição |
|
1 |
Atividade física extenuante limitada, mas deambula sem dificuldade e pode realizar trabalhos leves |
|
2 |
Capaz de cuidados pessoais, mas incapaz de vida produtiva. Acamado < 50% do dia |
|
3 |
Capaz somente de cuidados pessoais limitados. Confinado a cama ou poltrona > 50% do dia |
|
4 |
Completamente incapaz: não pode realizar cuidados pessoais e totalmente confinado a leito ou poltrona |
Baseado no mesmo princípio de utilização do IPI, foi desenhado e validado um índice prognóstico para os linfoma indolentes, especialmente aquele mais prevalente deste grupo, o linfoma folicular. O índice prognóstico internacional para linfoma folicular (FLIPI) considera os mais importantes fatores que podem determinar desfecho clínico destes pacientes permitindo, da mesma forma que no índice anterior, a estratificação de risco dos pacientes. Nesse grupo de pacientes, a estratificação prognóstica é feita em três grupos: aqueles pacientes de baixo risco (0 ou 1 fator de risco) apresentam sobrevida em 10 anos de aproximadamente 85%; aqueles com risco intermediário (2 fatores de risco) apresentam sobrevida com o mesmo seguimento de aproximadamente 60%; já aqueles de alto risco (3 ou mais fatores de risco) apresentam sobrevida em 10 anos de 40%.
Os pontos a serem contados no FLIPI são:
Além dos índices principais que foram discutidos anteriormente e que são disseminadamente utilizados por hematologistas e oncologistas em todo o mundo, existem outras mecanismos de avaliação prognóstica para doenças específicas, como linfoma T periférico, linfoma de células do manto etc., bem como outras variações do IPI, como IPI ajustado à idade ou ajustado ao uso de rituximabe (anticorpo anti-CD20). Tais índices são menos utilizados e, portanto, não serão discutidos.
O tratamento dos LNH depende de vários fatores, incluindo tipo histológico, estádio clínico, idade do paciente, estado geral, comorbidades, velocidade de progressão das doenças, além do índice prognóstico (IPI/FLIPI). A abordagem terapêutica dos linfomas pode ser extremamente diferente; por exemplo, um paciente jovem com linfoma Burkitt extremamente agressivo deve ter seu tratamento iniciado o mais rápido possível, enquanto um paciente idoso com um linfoma de curso indolente como o folicular pode ser apenas seguido durante muitos anos sem que surjam indicações de tratamento. Essa abordagem diferenciada visa tratar rapidamente aqueles pacientes com bom estado geral e que são potencialmente curáveis, e postergar o tratamento daqueles pacientes idosos e incuráveis que podem padecer mais do tratamento do que da doença naquele momento. Descreveremos o tratamento de uma maneira didática, visando englobar as patologias mais freqüentes e os principais tratamentos de primeira linha.
Em geral, o tratamento não é curativo, embora mesmo aqueles pacientes que não apresentem resposta adequada possam ter sobrevida de vários anos.
Em casos avançados (estádio III-IV), o tratamento se faz em geral com esquemas baseados em ciclofosfamida, vincristina, fludarabina e prednisona em combinações diversas. Pacientes com mais de 60 anos, assintomáticos e sem nenhuma citopenia significativa podem ser observados de perto sem tratamento, visto que se trata de patologia incurável neste estádio e que indivíduos idosos podem sofrer, com maior freqüência, conseqüências indesejáveis do tratamento quimioterápico.
Os pacientes com doença localizada (estádio I-II) podem ser tratados com radioterapia localizada apenas. Aparentemente esta abordagem propicia a possibilidade de cura nesse subgrupo de doentes, embora recidivas tardias (após mais de 10 anos) possam ser observadas. É cada vez menos recomendada a abordagem de somente observar estes pacientes.
O anticorpo monoclonal rituximab (anti-CD 20) é uma arma terapêutica recente, e bons resultados têm sido obtidos, tanto em casos refratários quanto em tratamento de primeira linha. Os dados comprovam que o tratamento claramente incrementa a sobrevida livre de doença, no entanto os dados de sobrevida global ainda não foram atingidos devido ao curso lento da doença. Em nossa prática, recomendamos o uso de rituximab em primeira linha de tratamento sempre que disponível, devendo ser combinado a outros agentes como fludarabina, ciclofofamida ou ambos.
O subtipo linfoma MALT gástrico com H. pylori+, em estádio precoce, responde bem ao tratamento da bactéria com claritromicina, metronidazol e omeprazol.
O tratamento-padrão atual para os estádios avançados (III-IV) é CHOP (ciclofosfamida, 750 mg/m² EV; doxorrubicina, 50 mg/m² EV; vincristina, 1,4 mg/m² EV, máximo de 2 mg; e prednisona, 100 mg VO, do D1 ao D5 repetidos a cada 21 dias) associado a rituximab (anticorpo anti-CD 20) por 8 ciclos. O anticorpo está indicado em todas as faixas etárias, no entanto seus benefícios são especialmente evidentes nos idosos (> 60 anos). A radioterapia está indicada na consolidação daquelas áreas que inicialmente apresentavam doença volumosa (bulky).
Para estádios mais iniciais (I-II), o esquema de tratamento básico é CHOP com rituximab por 4 ciclos, seguido de radioterapia para o campo envolvido.
É importante lembrar que os pacientes que receberão doxorubicina no esquema CHOP devem ter sua função cardíaca avaliada antes do início do tratamento, devido à toxicidade cardíaca deste fármaco.
O tratamento deste grupo de patologias deve ser feito de maneira rápida, visto ser uma doença extremamente agressiva e com potencial de óbito em dias caso não tratada com tempo e intensidade adequados.
O tratamento se faz com esquemas quimioterápicos em altas doses, para se obter citorredução inicialmente, e depois eliminação do clone maligno.
Os esquemas são os mais variados, utilizando alguma combinação destas diversas drogas: ciclofosfamida, vincristina, doxorubicina, prednisona, metotrexate, asparaginase, citarabina. Alguns dos esquemas mais utilizados para Linfoma de Burkitt e linfoblástico são o COPADM, CODOX-M/IVAC e o hiper-CVAD (ciclofosfamida, vincristina, doxorubicina, dexametasona alternando com metotrexate e citarabina).
Os LNH são neoplasias malignas de linfócitos que podem ter uma ampla gama de apresentações clinicas, e cuja classificação histológica é de alta complexidade e sempre foi tema de debate.
A classificação dos LNH de acordo com seu comportamento clínico divide os linfomas em três grandes grupos: os linfomas indolentes, os linfomas agressivos e os linfomas muito agressivos.
Os LNH mais agressivos são comuns em crianças e adolescentes e, embora tenham rápida evolução, respondem bem a quimioterapia e podem ser curados. Já os linfomas mais indolentes são comuns em idosos e, embora tenham evolução insidiosa, não respondem bem à quimioterapia e dificilmente são curáveis.
A apresentação clínica dos linfomas é extremamente variável: as formas indolentes costumam se apresentar com linfadenomegalia periférica, poucos sintomas sistêmicos e raramente levam a sintomas compressivos ou obstrutivos. Já as formas mais agressivas podem causar acometimento extranodal, além de freqüentemente causar sintomas sistêmicos, compressivos ou obstrutivos.
Os principais exames na avaliação de um paciente com suspeita de linfoma são: 1) biópsia linfonodal ou do tecido suspeito, essencial para o diagnóstico; 2) biópsia de medula óssea, necessária para o estadiamento; 3) imunofenotipagem/citogenética/biologia molecular, importantes para determinar subtipo e dar informações prognosticas; 4) TC de pescoço, tórax, abdômen e pelve, essencial para avaliação e estadiamento; e 5) exames laboratoriais como b2-microglobulina, DHL, hemograma e eletroforese de proteínas/dosagem de imunoglobulinas.
O diagnóstico diferencial de LNH se dá principalmente com LH e outras doenças que cursam com linfadenopatia, como infecções, alterações imunológicas e metástases linfonodais de neoplasias sólidas.
O tratamento do LNH é individualizado e baseia-se em comportamento clínico do tumor, estadiamento e fatores prognósticos.
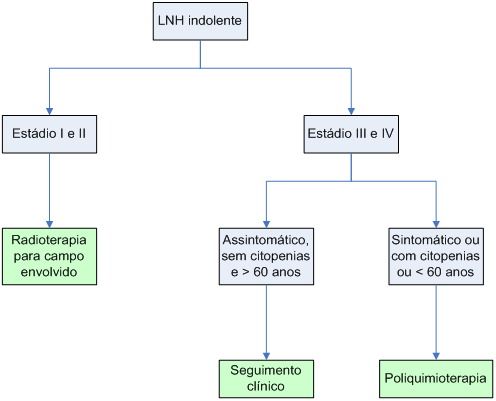
Algoritmo1: Tratamento das formas mais comuns de LNH indolente
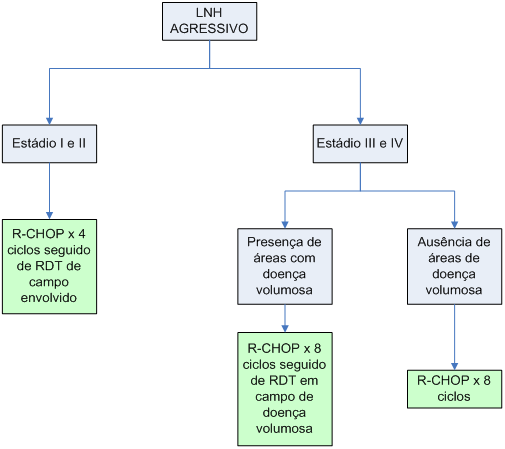
Algoritmo 2: Tratamento de primeira linha de linfoma agressivo de células B
1. Connors JM. Radioimmunotherapy: hot new treatment for lymphoma. N Engl J Med 2005; 352:496.
2. Evans LS, Hancock BW. Non-Hodgkin’s lymphoma. Lancet 2003;362:139-46.
3. Ezdinli EZ, Anderson JR, Melvin F et al. Moderate versus aggressive chemotherapy of nodular lymphocytic poorly differentiated lymphoma. J Clin Oncol 1985;3:769.
4. Ezdinli EZ, Harrington DP, Kucuk O et al. The effect of intensive intermittent maintenance therapy in advanced low-grade non-Hodgkin's lymphoma. Cancer 1987;60:156.
5. Gallagher CJ, Gregory WM, Jones AE et al. Follicular lymphoma: prognostic factors for response and survival. J Clin Oncol 1986;4:1470.
6. Glick JH, Barnes JM, Ezdinli EZ et al. Nodular mixed lymphoma: results of a randomized trial failing to confirm prolonged disease-free suvival with COPP chemotherapy. Blood 1981;58:920.
7. Hoffman R, Benz Jr. EJ, Shattil SJ, Furie B, Cohen HJ, Silberstein LE, McGlave P. Hematology: basic principles and practice. 3 ed. New York: Churchill Livingstone; 2000.
8. Johnson PW, Rohatiner AZ, Whelan JS et al. Patterns of survival in patients with recurrent follicular lymphoma: a 20-year study from a single center. J Clin Oncol 1995;13:140.
9. Jones SE, Grozea PN, Metz EN et al. Superiority of adriamycin-containing combination chemotherapy in the treatment of diffuse lymphoma: a Southwest Oncology Group study. Cancer 1979;43:417.
10. Jones SE, Grozea PN, Miller TP et al. Chemotherapy with cyclophosphamide, doxorubicin, vincristine, and prednisone alone or with levamisole or with levamisole plus BCG for malignant lymphoma: a Southwest Oncology Group Study. J Clin Oncol 1985;3:1318.
11. Kaminski MS, Tuck M, Estes J et al. 131I-tositumomab therapy as initial treatment for follicular lymphoma. N Engl J Med 2005;352:441.
12. Leonard JP, Coleman M, Kostakoglu L et al. Abbreviated chemotherapy with fludarabine followed by tositumomab and iodine I 131 tositumomab for untreated follicular lymphoma. J Clin Oncol 2005; 23:5696.
13. Maloney DG, Press OW, Braziel RM et al. A phase II trial of CHOP followed by rituximab chimeric monoclonal anti-CD20 antibody for treatment of newly diagnosed follicular non-Hodgkin's lymphoma: SWOG 9800. Blood 2001;98:843. [Abstract]
14. Morrison VA, Peterson BA. Combination chemotherapy in the treatment of follicular low-grade lymphoma. Leuk Lymphoma 1993;10(Suppl):29.
15. Nickenig C, Dreyling M, Hoster E et al. Combined cyclophosphamide, vincristine, doxorubicin, and prednisone (CHOP) improves response rates but not survival and has lower hematologic toxicity compared with combined mitoxantrone, chlorambucil, and prednisone (MCP) in follicular and mantle cell lymphomas: results of a prospective randomized trial of the German Low-Grade Lymphoma Study Group. Cancer 2006;107:1014.
16. Peterson BA, Petroni GR, Frizzera G et al. Prolonged single-agent versus combination chemotherapy in indolent follicular lymphomas: a study of the cancer and leukemia group B. J Clin Oncol 2003;21:5.
17. Press OW, Unger JM, Braziel RM et al. A phase 2 trial of CHOP chemotherapy followed by tositumomab/iodine I 131 tositumomab for previously untreated follicular non-Hodgkin lymphoma: Southwest Oncology Group Protocol S9911. Blood 2003;102:1606.
18. Press OW, Unger JM, Braziel RM et al. Phase II trial of CHOP chemotherapy followed by tositumomab/iodine I-131 tositumomab for previously untreated follicular non-Hodgkin's lymphoma: five-year follow-up of Southwest Oncology Group Protocol S9911. J Clin Oncol 2006; 24:4143.
19. Seyfarth B, Josting A, Dreyling M, Schmitz N. Relapse in common lymphoma subtypes: salvage treatment options for follicular lymphoma, diffuse large cell lymphoma and Hodgkin disease. Br J Haematol 2006;133:3.
20. Wiernik PH, Goldman JM, Dutcher JP, Kyle RA. Neoplastic diseases of the blood. 4 ed. Cambridge: Cambridge University Press; 2003.
21. Witzig TE, White CA, Gordon LI et al. Safety of Yttrium-90 ibritumomab tiuxetan radioimmunotherapy for relapsed low-grade, follicular, or transformed non-Hodgkin's lymphoma. J Clin Oncol 2003;21:1263.
22. Young RC, Johnson RE, Canellos GP et al. Advanced lymphocytic lymphoma: randomized comparisons of chemotherapy and radiotherapy, alone or in combination. Cancer Treat Rep 1977; 61:1153.
"Prezado Dr. Fransergio Martins de Oliveira, agradecemos seu contato e informamos que os gráficos e as imagens da revisão intitulada “Linfomas não- Hodgkin” já estão disponíveis no portal. Atenciosamente, Os Editores."
"Algumas imagens e graficos não estão aparecendo. teste em todos navegadores, e realmente não estão carregando na pagina nem pra impressão."
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.