(Carregando Índice)... (Carregando Índice)... |
Autor:
Daniel Soares Freire
Endocrinologista pelo Hospital das Clínicas da Faculdade de Medicina da USP;
Médico Colaborador do Serviço de Endocrinologia do Hospital das Clínicas da Faculdade de Medicina da USP
Última revisão: 03/09/2008
Comentários de assinantes: 1
As adrenais podem albergar tumores de diversas naturezas. Os tumores adrenais primários podem se originar no córtex ou na medula, podendo ser benignos ou malignos em ambos os casos. Além disso, a adrenal pode ser sede de implantação de tumores metastáticos. Os principais tumores adrenais estão listados na tabela 1.
Tabela 1: Principais tumores adrenais
Os tumores primários do córtex adrenal são classificados como benignos (adenomas) ou malignos (carcinomas), funcionantes ou não-funcionantes e esporádicos ou hereditários.
A diferenciação entre um adenoma e um carcinoma pode ser clínica ou histológica. Clinicamente, a ocorrência de metástases, invasão de órgãos adjacentes ou recorrência após cirurgia são indicativos de malignidade.
Contudo, é interessante que o clínico possa reconhecer, dentre os tumores inicialmente localizados, quais têm potencial de recorrência ou disseminação.
Weiss et al. propuseram um método de diferenciação entre adenomas e carcinomas adrenais baseado em critérios histológicos. Usando nove parâmetros morfológicos, denominados critérios de Weiss (tabela 2), é possível distinguir os adenomas, tumores sem qualquer potencial de malignidade, dos carcinomas, tumores com potencial de malignidade. Os adenomas apresentam menos de dois critérios de Weiss presentes, enquanto os carcinomas apresentam três ou mais.
Se por um lado o sistema de Weiss permite identificar os tumores que não apresentam risco algum de disseminação (adenomas, Weiss = 2), por outro não é capaz de identificar, dentre os carcinomas, quais terão o pior prognóstico.
Tabela 2: critérios de Weiss para classificação histológica dos Tumores corticais adrenais
|
critérios de Weiss. Pacientes com 2 ou menos critérios não apresentam risco de disseminação e são classificados como adenomas |
|
1. Grau nuclear III/IV, como descrito por Furhmann et al. para o carcinoma renal 2. Índice mitótico elevado (> 5 mitoses em 50 campos de grande aumento) 3. Presença de mitoses atípicas 4. Células claras compreendendo menos de 25% do tumor 5. Arquitetura difusa compreendendo mais de 33% do tumor 6. Necrose microscópica confluente 7. Invasão venosa 8. Invasão sinusoidal 9. Invasão capsular |
O adenoma adrenal é o tumor adrenal mais freqüente no adulto, principalmente no idoso. A grande maioria dos pequenos nódulos adrenais não-funcionantes é formada por adenomas adrenais.
Nos últimos anos, a prevalência de adenomas adrenais vem aumentando em decorrência do uso de exames de imagem abdominal com sensibilidade cada vez maior.
A maior parte dos adenomas adrenais é assintomática e não secreta qualquer hormônio. Contudo, os adenomas podem ser funcionantes, e, quando o são, geralmente secretam um único esteróide. A secreção de aldosterona determina quadro de hipertensão com hipocalemia e alcalose metabólica (hiperaldosteronismo primário, também conhecido como síndrome de Conn). Nesses pacientes demonstram-se elevação da aldosterona e bloqueio da renina plasmática, indicando secreção do mineralocorticóide independente do controle do eixo renina-angiotensina-aldosterona.
Há ainda os adenomas secretores de cortisol, que causam hipercortisolismo puro, sem secreção concomitante de andrógenos. O paciente apresenta-se com síndrome de Cushing franca, sem sinais clínicos de hiperandrogenismo e sem hiperpigmentação. De fato, a presença de síndrome de Cushing com ACTH bloqueado e andrógenos adrenais (como o sulfato de DHEA) baixos é muito sugestiva de adenoma adrenal secretor de cortisol. Uma parcela significativa dos adenomas adrenais secreta pequenas quantidades de cortisol de forma autônoma, insuficientes para causar o surgimento dos estigmas clássicos da síndrome de Cushing, mas suficientes para determinar alterações sutis no eixo hipotálamo-hipófise-adrenal. Esse quadro é conhecido como Cushing subclínico ou hipersecreção subclínica e autônoma de cortisol, e parece conferir maior risco de síndrome metabólica (obesidade, hipertensão, intolerância à glicose, dislipidemia) e redução da massa óssea.
Não existe uniformidade entre os autores na forma de avaliação do Cushing subclínico. Um consenso de 2002 do National Institute of Health sugere que o cortisol coletado pela manhã pós-dexametasona 1 mg à noite seja usado como método de rastreamento para avaliar a autonomia do eixo hipotálamo-hipófise-adrenal nos pacientes com tumores adrenocorticais. Valores superiores a 5 µg/dl sugerem secreção autônoma de cortisol (a escolha de limiares inferiores a este, como 1,8 µg/dl, aumenta a sensibilidade do teste, mas diminui sua especificidade). Quando o cortisol pós-dexametasona está elevado, recomenda-se avaliar todo o eixo hipotálamo-hipófise-adrenal, com dosagens de cortisol urinário de 24 horas e ACTH plasmático.
Os exames radiológicos representam importante ferramenta para o clínico diferenciar adenomas de carcinomas adrenais. Alguns dados são especialmente importantes para a avaliação, como o tamanho das lesões, características da tomografia computadorizada (TC) sem e com contraste e da ressonância nuclear magnética (RNM).
O primeiro dado a ser avaliado é o tamanho tumoral. Ao contrário dos carcinomas, que tendem a ser maiores, os adenomas costumam ter menos de
Além do tamanho, o grau de atenuação na TC pode auxiliar. Adenomas são tumores ricos em gordura intracitoplasmática, o que lhes confere baixa atenuação na fase pré-contraste (inferior a 10 unidades Hounsfield, ou HU). A TC com contraste também fornece subsídios para a distinção da natureza dos tumores adrenais. Adenomas tendem a reter menos contraste que os carcinomas, apresentando coeficiente de atenuação inferior a 35 HU no tempo de 10-15 minutos após a injeção do contraste iodado intravenoso. Além disso, os adenomas têm contornos regulares e são homogêneos nos diversos exames de imagem.
A RNM com contraste associada a técnica do deslocamento químico (chemical shift) tem a mesma performance da TC na diferenciação de tumores adrenais benignos e malignos. Novamente, o conteúdo lipídico intracitoplasmático permite essa diferenciação. Tipicamente, os adenomas apresentam hipossinal ao fígado nas seqüências ponderadas em T1 e mostram importante queda do sinal nas seqüências “fora de fase”.
tumores adrenais com características radiológicas sugestivas de adenoma, tamanho menor que
A cirurgia está indicada quando o tumor é funcionante ou quando há dúvidas de sua natureza, quer seja por características suspeitas aos exames de imagem, quer seja por seu tamanho. Para os adenomas adrenais, a cirurgia é curativa.
Os pacientes com hipercortisolismo (mesmo que subclínico) podem apresentar insuficiência adrenal secundária por supressão da secreção de CRH e ACTH pelo hipotálamo e hipófise, respectivamente. Essa situação, grave e potencialmente fatal, se não reconhecida, deve ser evitada com administração de doses de reposição de glicocorticóide, de preferência com meia-vida curta (como acetato de cortisona ou hidrocortisona). Não há necessidade de administração de mineralocorticóide, pois o eixo renina-angiotensina-aldosterona está intacto na adrenal remanescente. Os pacientes devem receber a reposição com orientação para aumentar a dose em caso de doenças intercorrentes, trauma ou desidratação até que o eixo hipotálamo-hipófise-adrenal possa ser avaliado.
Lesões adrenais que não suscitam dúvidas quanto à sua natureza benigna podem ser abordadas por via laparoscópica. Essa técnica está associada a menor morbidade peri-operatória (especialmente no que diz respeito à dor local) e cursa com menor tempo de internação e menor necessidade de analgésicos.
Ao contrário do que se observa com os adenomas adrenais, os carcinomas adrenais são tumores raros, com incidência de 1-2 casos novos por milhão de pessoas ao ano, representando 0,05% a 0,2% das neoplasias malignas. Em crianças, a incidência de carcinoma adrenal no mundo é baixa, da ordem de 0,3 casos novos por milhão de pessoas ao ano. Contudo, nos Estados do Sul e Sudeste do Brasil (especialmente Paraná e São Paulo), essa incidência está aumentada da ordem de
O tumor adrenal mais comum na criança é o carcinoma adrenal. Todavia, alguns autores relatam um comportamento mais agressivo dos carcinomas adrenais diagnosticados nos adultos em comparação com aqueles da faixa pediátrica, embora essa observação não seja de todo uniforme.
A maior parte dos tumores adrenais é esporádica. Todavia, algumas síndromes genéticas aumentam o risco de tumorigênese adrenal, como as síndromes de Li-Fraumeni, Beckwith-Weideman e neoplasia endócrina múltipla do tipo 1 (NEM-1) (tabela 3).
Tabela 3: Síndromes genéticas associadas a tumorigênese adrenal
|
Síndrome |
Gene |
tumores adrenais |
Outros tumores |
|
Li-Fraumeni |
p53 |
Carcinomas |
Mama, sarcomas, tumores do sistema nervoso central, leucemias. |
|
Beckwith-Weideman |
IGF-2 |
Carcinomas |
Macrossomia, tumor de Wilms, neuroblastoma, hepatoblastoma |
|
NEM-1 |
Menin |
Adenomas e carcinomas |
Tumores de paratiróide, pâncreas endócrino e hipófise |
Os sintomas dos carcinomas adrenais decorrem de seu efeito de massa ou da hipersecreção de hormônios. Alguns pacientes podem apresentar dor abdominal, perda de peso e sintomas consumptivos.
Enquanto os adenomas costumam secretar um único hormônio, a secreção mista de esteróides e precursores é o principal achado laboratorial dos carcinomas adrenais. Além do aumento do cortisol e dos andrógenos, determinando concomitância de síndrome de Cushing com síndrome virilizante, observa-se também elevação de precursores da esteroidogênese adrenal como 11-deoxicortisol e 17a-hidroxiprogesterona. Raramente observa-se secreção de estradiol (determinando síndromes feminilizantes em homens) e aldosterona (determinando hipertensão mineralocorticóide). Assim como nos adenomas, a secreção de cortisol pode ser franca ou subclínica.
Os carcinomas adrenais costumam ter tamanhos grandes, às vezes deslocando o rim ipsilateral até a fossa ilíaca. Aproximadamente 25% dos tumores com mais de
Ao contrário dos adenomas, os carcinomas apresentam atenuação elevada (acima de 10 HU) na TC simples e retêm o meio de contraste por mais tempo (o que determina atenuação superior a 35 HU na fase pós-contraste). Outros dados da TC sem contraste que podem apontar para o diagnóstico de malignidade são a presença de calcificações ou áreas grosseiramente heterogêneas, que representam zonas de hemorragia ou necrose tumoral.
À RNM com contraste, os carcinomas apresentam isossinal ao fígado em T1, hipersinal em T2 e não apresentam importante queda do sinal nas seqüências “fora de fase”.
Outra aplicação da RNM na avaliação de um paciente com carcinoma adrenal é o estudo da relação do tumor com os vasos (especialmente a veia cava inferior), pois este pode invadir a veia cava e produzir trombos que se estendem até o átrio direito. Essa manifestação deve ser do conhecimento do cirurgião antes da cirurgia, pois a secção inadvertida do trombo pode determinar embolia pulmonar maciça e fatal.
A medicina nuclear também pode ser empregada na diferenciação entre adenomas e carcinomas adrenais. No passado, a cintilografia com 131I-6-ß-iodometil-norcolesterol (iodo-colesterol) foi bastante usada para essa finalidade. Contudo, a baixa disponibilidade do radiotraçador e a relativa alta dose de radiação reduziram a utilização desse exame. Entretanto, estudos recentes têm demonstrado alta sensibilidade e especificidade da tomografia com emissão de pósitrons com 18F-2-flúor-D-deoxi-glicose (PET-FDG). Em virtude de sua maior taxa metabólica, os carcinomas adrenais têm maior captação do FDG que os adenomas.
Finalmente, os exames de imagem possibilitam ainda o estadiamento pré-operatório dos carcinomas adrenais. TCs de abdome e tórax permitem avaliação dos pulmões, fígado e linfonodos retroperitoniais. Na suspeita clínica de acometimento ósseo, imagens direcionadas ou cintilografia óssea permitem o diagnóstico.
A tabela 4 resume os principais achados radiológicos e hormonais dos adenomas e carcinomas adrenais.
Tabela 4: Achados radiológicos e laboratoriais dos Tumores corticais adrenais
|
|
Adenomas |
Carcinomas | |
|
Achados radiológicos | |||
|
Tamanho da lesão |
< |
> | |
|
TC sem contraste |
< 10 HU, homogênea |
> 10 HU, heterogênea, com calcificações | |
|
TC com contraste |
< 35 HU |
> 35 HU | |
|
RNM (T1 “fora de fase”) |
Perda importante do sinal |
Sem perda do sinal | |
|
RNM (T2) |
Hipossinal |
Hipersinal | |
|
PET-FDG |
Sem hipercaptação |
Com hipercaptação | |
|
Achados laboratoriais | |||
|
Perfil hormonal |
Não-funcionante hipercortisolismo puro (inclui secreção subclínica de cortisol) Hiperaldosteronismo
|
Não-funcionante Secreção mista de cortisol, andrógenos e precursores hiperandrogenismo puro hipercortisolismo puro (menos freqüente) Hiperestrogenismo como parte da síndrome mista (raro) | |
A Organização Mundial da Saúde (OMS) unificou o sistema de estadiamento do carcinoma adrenal em 2004. Segundo esse novo sistema, os carcinomas são agrupados em quatro estágios (tabela 5).
Tabela 5: Estadiamento do carcinoma adrenal
|
Estadio |
Características |
|
I |
Doença restrita à adrenal, tumor < |
|
II |
Doença restrita à adrenal, tumor > |
|
III |
Invasão local ou disseminação linfática |
|
IV |
Metástases à distância |
O tratamento do carcinoma adrenal inclui ressecção do tumor primário, ressecção das metástases e quimioterapia sistêmica. Na presença de doença disseminada, a resposta a qualquer forma de tratamento é muito ruim.
A ressecção cirúrgica do tumor primário é o tratamento de primeira linha do carcinoma adrenal localizado. A abordagem deve ser feita por via aberta, obedecendo aos preceitos da cirurgia oncológica, e deve incluir, além do tumor e dos órgãos vizinhos com suspeita de invasão, os linfonodos abdominais com aspecto alterado. Adrenalectomia laparoscópica cursa com incidência inaceitável de implantes tumorais no peritônio, e não está indicada no tratamento de um tumor com possibilidade de ser um carcinoma adrenal.
A remoção cirúrgica do tumor primário ou das metástases no paciente que se apresenta com doença disseminada é tema controverso. Uma abordagem razoável é iniciar quimioterapia sistêmica e avaliar a resposta em três a seis meses. Havendo estabilização ou regressão da doença metastática, é valido submeter o paciente a uma cirurgia, desde que esta seja tecnicamente curativa.
Também é controversa a utilidade de quimioterapia adjuvante ou radioterapia profilática do leito cirúrgico. Dois estudos retrospectivos recentes avaliaram essas abordagens, e demonstraram eficácia do mitotano profilático em prolongar a sobrevida livre de doença e da radioterapia do leito cirúrgico em reduzir a incidência de recorrência local (contudo, sem aumentar a sobrevida livre de doença).
O tratamento medicamentoso inclui o mitotano (o,p’DDD) e a quimioterapia citotóxica sistêmica (cisplatina, etoposídeo, 5-FU, estreptozotocina e doxorrubicina).
O mitotano é um agente adrenolítico específico, com efeito citotóxico sobre o córtex adrenal, principalmente as zonas fasciculada e reticular. Está disponível em comprimidos de 500 mg para administração por via oral (Lysodren®), e deve ser tomado em dose suficiente para atingir níveis séricos entre 14 e 20 µg/ml. A maior parte dos pacientes usa doses diárias de
A resposta dos pacientes em estágios III e IV à quimioterapia sistêmica é baixa, em torno de 10%. O estudo FIRM-ACT (First International Randomized trial in locally advanced and Metastatic Adrenocortical Carcinoma Treatment) é o primeiro estudo clínico controlado e randomizado para carcinoma adrenal. Sediado na Europa, esse estudo pretende comparar de forma cega dois esquemas terapêuticos em 300 pacientes: etoposídeo + doxorrubicina + cisplatina + mitotano versus estreptozotocina + mitotano.
A medula adrenal, constituída por células cromafins derivadas da crista neural e responsável pela síntese de catecolaminas, é local de origem de tumores, sendo os feocromocitomas os tumores mais comuns.
Os feocromocitomas são tumores raros originários das células cromafins que produzem, estocam, metabolizam e secretam catecolaminas. A classificação dos tumores endócrinos da OMS reserva o termo “feocromocitoma” para os tumores produtores de catecolaminas originários da medula adrenal, e “paraganglioma” para os tumores com as mesmas características que se desenvolvem no tecido cromafin extra-adrenal, como os gânglios simpáticos e parassimpáticos.
A prevalência do feocromocitoma foi estimada em 0,05-0,1% dos hipertensos. Contudo, é provável que esse número seja muito maior (talvez o dobro), pois somente 50% dos pacientes com feocromocitomas têm hipertensão mantida. O tumor afeta igualmente o sexo masculino e feminino e pode ocorrer em qualquer idade, concentrando-se na terceira a quinta décadas de vida.
A maior parte dos casos é esporádica. Contudo, o feocromocitoma pode se apresentar como parte de uma doença genética. A tabela 6 lista as doenças genéticas associadas com o feocromocitoma.
Tabela 6: Síndromes genéticas associadas ao feocromocitoma
|
Síndrome |
Mutação |
Clínica |
Risco de Feo |
|
Ret |
Carcinoma medular de tiróide feocromocitoma Hiperparatiroidismo |
50% | |
|
Ret |
Carcinoma medular de tiróide feocromocitoma Ganglioneuromatose de mucosa e intestino Hábito marfanóide |
50% | |
|
Von Hipplel-Lindau |
VHL |
Hemangioblastoma de sistema nervoso central Angiomas de retina Carcinoma e cistos renais Cistos e tumores pancreáticos feocromocitoma |
10-30% |
|
Síndrome paraganglioma familiar |
SDHB SDHD |
Paragangliomas cervicais feocromocitoma |
20% |
|
NF-1 |
Neurofibromas de nervos periféricos Manchas “café-com-leite” Feocromocitomas |
1-5% |
As manifestações clínicas do feocromocitoma constituem o principal motivo de investigação da doença. Contudo, um número crescente de feocromocitomas silentes ou oligossintomáticos vem sendo diagnosticado nos pacientes com massas adrenais detectadas por exames de imagem solicitados para avaliação de doença extra-adrenal ou por exames de check-up (“incidentalomas adrenais”).
A maior parte dos sinais e sintomas é conseqüência direta dos efeitos cardiovasculares, metabólicos e viscerais das catecolaminas, embora nem sempre o seu nível plasmático se correlacione com a magnitude da manifestação clínica. A tabela 7 lista os principais sinais e sintomas dos feocromocitomas e sua freqüência em um estudo retrospectivo realizado no Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo.
Tabela 7: Sinais e sintomas dos feocromocitomas
|
Sinal / sintoma |
Freqüência |
|
Hipertensão nas crises |
95% |
|
Palpitações |
77% |
|
Cefaléia |
76% |
|
Sudorese |
70% |
|
Palidez |
23% |
|
Náuseas |
22% |
|
Dor abdominal |
20% |
|
Dispnéia |
19% |
|
Tontura |
19% |
|
Vômitos |
17% |
|
Tremores |
14% |
|
Angina |
14% |
|
Embaçamento visual |
10% |
|
Poliúria pós-crise |
3% |
|
Convulsão |
3% |
|
Rubor facial |
3% |
|
Flushing |
1,5% |
|
Acidente vascular cerebral hemorrágico |
1,5% |
Adaptado de Albergaria Maa et al., 2004
Hipotensão postural é um achado relativamente freqüente nos pacientes com feocromocitoma, e resulta da hipovolemia decorrente de vasoconstrição mantida e sudorese persistente, diminuição da sensibilidade e do número de receptores adrenérgicos, e eventual secreção de substâncias vasodilatadoras pelo tumor.
Os feocromocitomas também podem causar síndromes paraneoplásicas, como síndrome de Cushing ACTH-dependente (por secreção ectópica de ACTH), hipercalcemia (por secreção de peptídeo relacionado ao paratormônio) e policitemia (por secreção de fatores estimuladores da eritropoiese).
Após uma suspeita clínica, o diagnóstico bioquímico deve ser realizado antes de uma investigação topográfica.
O objetivo da investigação laboratorial numa suspeita de feocromocitoma é demonstrar a presença de produção excessiva de catecolaminas. Isto é realizado com a dosagem de adrenalina e noradrenalina e de seus metabólitos (metanefrinas e ácido vanilmandélico) em urina ou plasma.
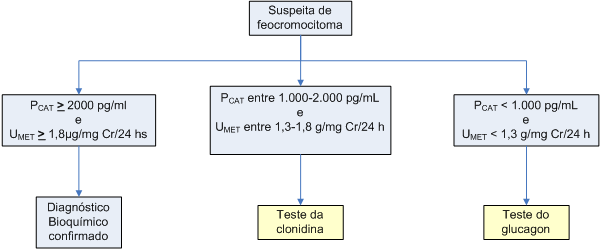
O fluxograma para diagnóstico bioquímico de feocromocitoma, proposto por Bravo et al., está esquematizado no algoritmo 1.
Algoritmo 1: Diagnóstico laboratorial do feocromocitoma

PCAT: catecolaminas (noradrenalina + adrenalina) plasmáticas, UMET: metanefrinas urinárias de 24 horas. Adaptado de Bravo et al., 2003
O teste da clonidina está indicado para aqueles pacientes com níveis intermediários de catecolaminas e metanefrinas, para distinguir os pacientes com feocromocitoma daqueles com uma hiperatividade do sistema simpático. O racional do teste se baseia no fato de que a clonidina, um agonista a2-adrenérgico, provoca redução do tônus simpático, determinando queda da concentração de catecolaminas em pessoas normais ou com hiperatividade simpática, mas não em portadores de feocromocitoma. Assim, o teste consiste na dosagem de catecolaminas plasmáticas antes e depois (tempos 1 e 2 horas) da administração de 300 mg de clonidina por via oral. No indivíduo sem feocromocitoma, as catecolaminas plasmáticas caem para níveis abaixo de 500 pg/ml, e se acompanham de queda da pressão arterial. Já nos pacientes com feocromocitoma, não se observa essa redução das catecolaminas, embora a pressão arterial até possa cair.
O teste do glucagon está indicado para pacientes com níveis mais baixos de catecolaminas plasmáticas. O glucagon tem um efeito estimulatório para a liberação de catecolaminas pelos feocromocitomas, mas não pela medula adrenal normal ou pelo sistema nervoso simpático. Administra-se 1 mg de glucagon por via IV e dosam-se as catecolaminas plasmáticas antes e após a injeção (1, 2 e 3 minutos). Nos pacientes com feocromocitoma, as catecolaminas plasmáticas ultrapassam 2.000 pg/ml. O teste deve ser feito em ambiente hospitalar e com monitorização da pressão arterial e equipe preparada para a eventual ocorrência de crise hipertensiva.
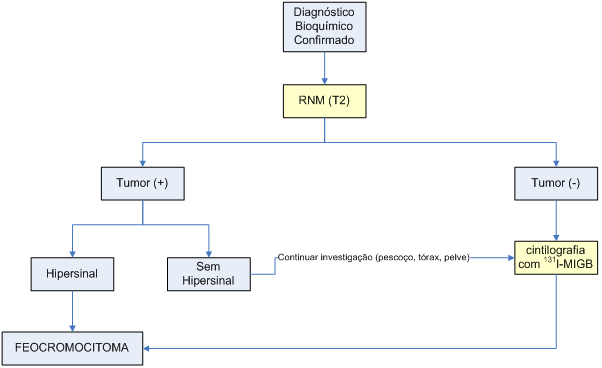
Feito o diagnóstico laboratorial, inicia-se o diagnóstico topográfico do feocromocitoma.
A localização do tumor é fundamental para a decisão cirúrgica, pois os feocromocitomas podem ser múltiplos, extra-adrenais ou metastáticos. Deve-se considerar que o feocromocitoma tem localização adrenal em 90% dos casos, e que os tumores de localização extra-adrenal podem ocorrer desde a base do crânio até a pelve.
Os métodos mais freqüentemente empregados são TC, RNM e mapeamento do corpo inteiro com 131I-meta-iodo-benzil-guanidina (131I-MIGB).
A sensibilidade da TC em identificar massas adrenais é comparável à da RNM. Contudo, o hipersinal nas seqüências ponderadas em T2 é um achado típico de feocromocitomas, o que garante maior especificidade da RNM. Além disso, a RNM tem maior sensibilidade que a TC para diagnóstico de tumores extra-adrenais.
A meta-iodo-benzil-guanidina apresenta similaridade estrutural com a noradrenalina, e assim é captada e armazenada em vesículas adrenérgicas. A marcação do composto com 131I permite a localização dos tecidos com maior quantidade dessas vesículas. A cintilografia com 131I-MIGB apresenta especificidade muito alta, e mostra-se útil principalmente na investigação de doença extra-adrenal ou metastática.
Outros radiotraçadores podem ser empregados no diagnóstico topográfico do feocromocitoma, como a 18F-deoxi-glicose (FDG) e a 18F-fluordopamina associadas à tomografia com emissão de pósitrons (PET). A PET-FDG é sensível, porém inespecífica para o diagnóstico de feocromocitoma. Já a 18F-fluordopamina é um método com alta sensibilidade e especificidade, mas não está disponível na maioria dos centros.
O algoritmo 2 ilustra como deve ser feita a investigação topográfica do feocromocitoma.
Algoritmo 2: Diagnóstico topográfico do feocromocitoma
RNM(T2): imagem de ressonância nuclear magnética das adrenais, com seqüências ponderadas em T2.
Adaptado de Bravo et al., 2003
O tratamento do feocromocitoma é cirúrgico. Contudo, é importante que a cirurgia seja precedida por um tratamento clínico para minimizar o risco de intercorrências cirúrgicas.
Preparo pré-operatório
O objetivo do tratamento clínico pré-operatório é tratar a hipertensão arterial e corrigir a hipovolemia relativa que os pacientes podem apresentar. Caso isto não seja feito, o paciente pode apresentar choque hipovolêmico após a retirada do tumor e interrupção abrupta da vasoconstrição.
A dieta dos pacientes deve ser rica em sal e o paciente deve ter livre acesso à água. Reposição de volume por via parenteral deve se restringir aos pacientes com evidências de hipovolemia a despeito do tratamento clínico com bloqueadores a-adrenérgicos, manifestada pela persistência de hipotensão postural e hematócrito elevado.
A classe de drogas utilizada no preparo são os bloqueadores a-adrenérgicos. A fenoxibenzamina é um bloqueador não-específico (a1 + a2), não-competitivo e de ação prolongada. É eficaz para o preparo dos pacientes, mas causa mais taquicardia reflexa que os agentes mais novos como prazosin e doxazosin (bloqueadores a1-específicos, competitivos e com meia-vida curta). O preparo deve ser feito por pelo menos 15 dias e deve ser interrompido 48 horas antes da cirurgia no caso da fenoxibenzamina, e 8 horas antes no caso do prazosin.
Outros anti-hipertensivos, como bloqueadores de canais de Ca+2 e inibidores da enzima conversora de angiotensina (IECA) podem ser usados para o tratamento da hipertensão arterial dos pacientes com feocromocitoma. Somente os b-bloqueadores devem ser usados com cuidado e somente nos pacientes que já estejam tratados com a-bloqueadores, pois o bloqueio isolado dos receptores b magnifica a resposta a-adrenérgica, exacerbando os sintomas decorrentes do excesso de catecolaminas.
A tabela 8 resume os pontos importantes do preparo para a cirurgia de feocromocitoma.
Tabela 8: Preparo dos pacientes para a cirurgia
|
Dieta |
Rica em sódio Acesso livre à água |
|
Administração de volume IV |
Reservada aos pacientes com sinais de hipovolemia (hipotensão postural, hematócrito elevado) a despeito de tratamento clínico e dieta rica em sal |
|
Bloqueio a-adrenérgico |
Fenoxibenzamina: iniciar 10 mg VO 12/12 h; titular até 20 mg VO 8/8 h prazosin: iniciar 1 mg VO 8/8 h; titular até 4 mg VO 8/8 h |
|
Duração do preparo |
Aproximadamente 15 dias (observar sinais de hipovolemia) Interromper droga antes da cirurgia: fenoxibenzamina (48 horas) e prazosin (8 horas) |
|
Outras drogas |
Bloqueadores de canal de Ca+2, IECAs: eficazes no controle da pressão b-bloqueadores: usar somente no paciente com bloqueio a. Podem ser úteis para controle da taquicardia |
Tratamento cirúrgico
A cirurgia é o tratamento curativo dos feocromocitomas. A cirurgia por via videolaparoscópica é uma alternativa segura à via aberta, e que cursa com menor morbidade peri-operatória.
A equipe médica deve estar familiarizada com as possíveis intercorrências que possam vir a acontecer. É recomendada monitorização invasiva de pressão arterial e pressão capilar pulmonar (ou PVC, na ausência desta), ritmo e freqüência cardíaca. As crises hipertensivas podem ocorrer durante a manipulação do tumor, e devem ser tratadas com o bloqueador a-adrenérgico fentolamina ou com nitroprussiato de sódio em infusão contínua. As taquiarritmias devem ser tratadas com um b-bloqueador IV.
No pós-operatório, os pacientes podem apresentar hipotensão, devido à falta de correção da hipovolemia no preparo cirúrgico. Essa situação costuma responder a volume, não sendo necessário uso de drogas vasoativas na maioria dos casos. Hipoglicemia também pode ser observada nas primeiras 48 horas após a cirurgia, e ocorre pela retirada do efeito bloqueador das catecolaminas sobre a secreção de insulina. Assim, é recomendada monitorização periódica da glicemia capilar no período pós-operatório.
Tratamento do feocromocitoma maligno
Cerca de 10% a 15% dos feocromocitomas são malignos, e os principais sítios de metástases são os linfonodos regionais, ossos, fígado e pulmões. Em muitos pacientes, a doença tem evolução lenta e indolente, só sendo diagnosticada anos após a cirurgia. Assim, é recomendável a realização de cintilografia de corpo inteiro com 131I-MIBG após a cirurgia para rastreamento de doença metastática (pelo menos nos pacientes com tumores grandes, com mais de
O tratamento do feocromocitoma maligno deve se voltar ao controle da hipertensão e da doença oncológica. Agentes anti-hipertensivos em combinação devem ser utilizados, de forma semelhante ao descrito para o preparo pré-operatório.
Quimioterapia com ciclofosfamida, vincristina e dacarbazina pode ser realizada, mas a taxa de resposta é baixa. A medicina nuclear também oferece algumas alternativas terapêuticas, como a dose ablativa com 131I-MIBG e o uso terapêutico de análogos de somatostatina marcados com 111In ou 90Y.
As glândulas adrenais são sítios freqüentes de instalação de metástases. Os principais tumores associados com acometimento secundário das adrenais são melanomas (50%), carcinomas de mama (30%-40%), carcinomas de pulmão (30%-40%) e carcinomas de rim (10%-20%). Aproximadamente 75% dos nódulos adrenais em pacientes com câncer representam acometimento secundário da glândula.
O acometimento pode ser unilateral ou bilateral. Geralmente, as lesões são assintomáticas, mas há risco de insuficiência adrenal devido a metástases bilaterais.
As características da imagem podem auxiliar a diferenciar uma metástase para a adrenal de um adenoma silente, como mostrado na tabela 9.
Tabela 9: Diagnóstico diferencial entre metástases para a adrenal e adenomas corticais adrenais
|
|
Metástase |
Adenoma |
|
Tamanho da lesão |
Variável |
< |
|
Lateralidade |
Unilateral ou bilateral |
Unilateral |
|
TC sem contraste |
> 10 HU |
< 10 HU |
|
TC com contraste |
> 35 HU |
< 35 HU |
|
RNM (T2) |
Hipersinal |
Hipossinal |
|
PET-FDG |
Com hipercaptação |
Sem hipercaptação |
A biópsia adrenal guiada por TC pode dar o diagnóstico do sítio primário. Contudo, é um procedimento com risco de complicações (dor, hematúria, pneumotórax).
A indicação de tratamento para as metástases adrenais depende da extensão da doença de base. Obviamente, os pacientes portadores de tumores multimetastáticos não irão se beneficiar de adrenalectomia. Contudo, há dados que mostram que a adrenalectomia poderia aumentar a sobrevida livre de doença em pacientes com metástase adrenal isolada, especialmente se o tumor primário for um adenocarcinoma e se a ressecção for completa. Os estudos desenhados para avaliar essa abordagem mostram sobrevida em cinco anos em aproximadamente 30%.
A via laparoscópica é segura para o tratamento das metástases para as adrenais, e cursa com morbidade inferior à da cirurgia aberta.
Até meados da década de 1970, as neoplasias adrenais eram relativamente incomuns. Contudo, após a melhoria técnica e disseminação em ampla escala de métodos de imagem, o diagnóstico de nódulos adrenais aumentou consideravelmente. Estes “incidentalomas adrenais”, definidos como “lesões com mais de
A maioria dos incidentalomas adrenais são lesões benignas e não-funcionantes (tabela 10). A avaliação clínica, bioquímica e radiológica deverá, portanto, identificar as lesões com maior risco de serem tumores malignos e nódulos funcionantes, os quais deverão ser tratados cirurgicamente. Os tumores com características benignas e que não produzam hormônios devem ser observados clinicamente.
Tabela 10: Distribuição dos diagnósticos etiológicos em 380 incidentalomas adrenais (confirmados histologicamente)
|
Diagnóstico |
Prevalência |
|
Adenoma |
52% |
|
Adenocarcinoma |
12% |
|
feocromocitoma |
11% |
|
Mielolipoma |
8% |
|
Cisto adrenal |
5% |
|
Ganglioneuroma |
4% |
|
Metástases |
2% |
|
Outros |
6% |
Adaptado de Mantero et al., 2000
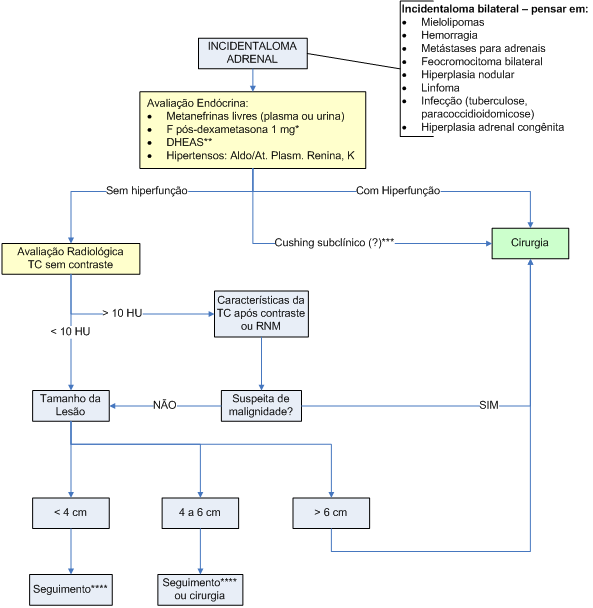
Para avaliar o potencial de malignidade, são usados dados de história clínica, como o conhecimento de uma neoplasia prévia, e dados do exame de imagem. O tamanho tumoral e as características da imagem são cruciais para decisão terapêutica. A maior parte dos serviços recomenda tratamento cirúrgico para todo nódulo com mais de
A avaliação funcional é feita com dosagem de cortisol pós-dexametasona 1 mg às 24 horas para todos os pacientes, e avaliação do eixo mineralocorticóide somente para os pacientes hipertensos. O rastreamento de hiperaldosteronismo primário é feito com dosagem concomitante de aldosterona e atividade plasmática de renina. Uma relação superior a 30 tem boa sensibilidade para identificar os casos suspeitos, que devem ser encaminhados para confirmação diagnóstica.
O diagnóstico diferencial com feocromocitoma deve ser feito, idealmente, por meio da dosagem de metanefrinas plasmáticas, e, na ausência desse teste, dosagens de metanefrinas em urina de 24 horas e as catecolaminas plasmáticas. Mais detalhes sobre a investigação laboratorial do feocromocitoma estão descritos anteriormente.
Os tumores com mais de
Os pacientes com tumores menores que
O exame de imagem deve ser repetido em
Os pacientes com nódulos que não apresentam funcionalidade ou aumento de tamanho durante 4 anos podem ser dispensados de seguimento adicional.
O algoritmo 3 ilustra a avaliação e conduta diante de incidentalomas adrenais.
Algoritmo 3: Avaliação dos incidentalomas adrenais
*Alguns autores utilizam doses maiores de dexametasona para aumentar a especificidade do teste em identificar pacientes com Cushing subclínico (CSC).
**Níveis muito elevados de DHEAS sugerem carcinoma adrenal virilizante; níveis baixos estão presentes na maioria dos adenomas e em alguns carcinomas que secretam exclusivamente cortisol.
***Não há recomendação formal de indicar tratamento cirúrgico baseado exclusivamente na presença do Cushing subclínico (CSC). Contudo, em situações especiais, esse achado pode contribuir para a decisão terapêutica (ver texto).
****Quando se opta por uma conduta conservadora, o paciente deverá ser avaliado quanto ao crescimento tumoral ou mudança no padrão funcional (ver texto).
A maior parte dos adenomas adrenais é assintomática e não secreta qualquer hormônio. Contudo, os adenomas podem ser funcionantes, secretando aldosterona ou cortisol.
tumores adrenais com características radiológicas sugestivas de adenoma, tamanho menor que
A cirurgia está indicada em tumores funcionantes, com características suspeitas aos exames ou maiores que 4 cm no maior eixo.
Os sintomas dos carcinomas adrenais decorrem de seu efeito de massa ou da hipersecreção de hormônios. Pode haver ainda dor abdominal, perda de peso e sintomas consumptivos.
Os achados dos exames de imagem e o perfil hormonal costumam diferenciar carcinomas dos adenomas de adrenal.
Os feocromocitomas são os tumores mais comuns da medula adrenal e produzem, estocam, metabolizam e secretam catecolaminas.
Os sinais e sintomas dos feocromocitomas são conseqüência dos efeitos das catecolaminas, sendo os mais comuns hipertensão, palpitações, cefaléia e sudorese.
Para o diagnóstico deve haver confirmação bioquímica por meio da dosagem de adrenalina e noradrenalina e seus metabólicos (metanefrinas e ácido vanilmandélico) na urina ou plasma, seguida de exames de imagem.
O tratamento do feocromocitoma é cirúrgico e deve ser precedido de tratamento clínico para minimizar o risco de intercorrências.
As glândulas adrenais são sítios freqüentes de metástases, sendo os principais tumores associados os melanomas e tumores de mama, pulmão e rim.
Aproximadamente 75% dos nódulos adrenais em pacientes com câncer representam acometimento secundário da glândula.
Os tumores adrenais constituem um desafio para o endocrinologista, pois podem representar desde lesões sem qualquer significado para a saúde até tumores agressivos e refratários ao tratamento. O aumento da prevalência dos nódulos diagnosticados em exames de imagem trouxe o problema ao médico generalista, que freqüentemente é o responsável pela investigação e decisão terapêutica.
Seguindo um algoritmo de fácil execução, é possível decidir com segurança a conduta na grande maioria dos tumores.
1. Allolio B, Fassnacht M. Adrenocortical carcinoma: clinical update. J Clin Endocrinol Metab 2006;91:2027-37.
2. Bravo EL, Tagle R. Pheochromocytoma: state-of-the-art and future prospects. Endocr Rev 2003;24:539-553.
3. Fassnacht M, Hahner S, Polat B, Koschker AC, Kenn W, Flentje M et al. Efficacy of adjuvant radiotherapy of the tumor bed on local recurrence of adrenocortical carcinoma. J Clin Endocrinol Metab 2006;91:4501-4.
4. Grumbach MM, Biller BMK, Braunstein GD, Campbell KK, Carney JA, Godley PA et al. Management of the clinically inapparent adrenal mass ("incidentaloma"). Ann Intern Med 2003; 138:424-9.
5. Lee MJ, Hahn PF, Papanicolaou N, Egglin TK, Saini S, Mueller PR et al. Benign and malignant adrenal masses: CT distinction with attenuation coefficients, size, and observer analysis. Radiology 1991;179:415-8.
6. Liou LS, Kay R. Adrenocortical carcinoma in children. Review and recent innovations. Urol Clin North Am 2000;27:403-21.
7. Mantero F, Terzolo M, Arnaldi G, Osella G, Masini AM, Ali A et al. A survey on adrenal incidentaloma in Italy. J Clin Endocrinol Metab 2000;85:637-44.
8. Mansmann G, Lau J, Balk E, Rothberg M, Miyachi Y, Bornstein SR. The clinically inapparent adrenal mass: update in diagnosis and management. Endocr Rev 2004;25:309-40.
9. Namimoto T, Yamashita Y, Mitsuzaki K, Nakayama Y, Makita O, Kadota M et al. Adrenal masses: quantification of fat content with double-echo chemical shift in-phase and opposed-phase flash MR images for differentiation of adrenal adenomas. Radiology 2001;218:642-6.
10. Outwater EK, Siegelman ES, Huang AB, Birnbaum BA. Adrenal masses: correlation between CT attenuation value and chemical shift ratio at MR imaging with in-phase and opposed-phase sequences. Radiology 1996;200:749-52. [Erratum in Radiology 1996;201(3):880]
11. Pereira RM, Michalkiewicz E, Sandrini F, Figueiredo BC, Pianovski M, Franca SN et al. Childhood adrenocortical tumors. Arq Bras Endocrinol Metabol 2004;48:651-8.
12. Sandrini R, Ribeiro RC, DeLacerda L. Childhood adrenocortical tumors. J Clin Endocrinol Metab 1997;82:2027-31.
13. Sarela AI, Murphy I, Coit DG, Conlon KCP. Metastasis to the adrenal gland: the emerging role of laparoscopic surgery. Ann Surg Oncol 2003;10:1191-6.
14. Terzolo M, Angeli A, Fassnacht M, Daffara F, Tauchmanova L, Conton PA et al. Adjuvant mitotane treatment for adrenocortical carcinoma. N Engl J Med 2007;356:2372-80.
15. Wajchenberg BL, Albergaria Pereira MA, Medonça BB, Latronico AC, Campos Carneiro P, Alves VA et al. Adrenocortical carcinoma: clinical and laboratory observations. Cancer 2000;88:711-36.
16. Weiss LM. Comparative histologic study of 43 metastasizing and nonmetastasizing adrenocortical tumors. Am J Surg Pathol 1984;8:163-9.
17. Yun M, Kim W, Alnafisi N, Lacorte L, Jang S, Alavi A. 18F-FDG PET in characterizing adrenal lesions detected on CT or MRI. J Nucl Med 2001;42:1795-9.
"Gostei muito da abordagem. Gostaria de passar meu caso clínico: Tenho 48 a, fui submetida à adrenalectomia esquerda em 13/12/2014, com histopatológico de hiperplasia medular da adrenal. Apesar da cirurgia, persisto com picos pressóricos, dor abdominal intensa, cefaléia, vômitos, perda importante de peso.CT-Tórax:Nódulo sólido, não calcificado, 3mm, LID.CT-Abdome:Área focal hipervascular hepática,indeterminada e imagem nodular hiperdensa, 9mm,no ceco, indeterminada. Solicito locais p tratamento. Grata, Nídia."
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.